PENGEMBANGAN DAN VALIDASI PROSEDUR ANALISIS
KESERAGAMAN KANDUNGAN TABLET GLIPIZID
LEPAS DIPERPANJANG
TESIS
Karya tulis sebagai salah satu syarat untuk memperoleh gelar Magister dari
Institut Teknologi Bandung
Oleh
INDHAH FATMAWATI
NIM : 20712318
(Program Studi Magister Farmasi)
PENGEMBANGAN DAN VALIDASI PROSEDUR ANALISIS
KESERAGAMAN KANDUNGAN TABLET GLIPIZID
LEPAS DIPERPANJANG
Oleh
INDHAH FATMAWATI
NIM : 20712318
(Program Studi Magister Farmasi)
Institut Teknologi Bandung
Menyetujui Tim Pembimbing Tanggal 25 Februari 2015
Pembimbing utama
Dr. Ilma Nugrahani
Pembimbing serta
i ABSTRAK
PENGEMBANGAN DAN VALIDASI PROSEDUR ANALISIS KESERAGAMAN KANDUNGAN TABLET GLIPIZID
LEPAS DIPERPANJANG
Oleh
Indhah Fatmawati NIM : 20712318
Glipizid merupakan obat antidiabetik oral generasi kedua golongan sulfonilurea dengan mekanisme aksi mengeblok kanal kalium yang sensitif terhadap ATP dalam sel β Langerhans pankreas sehingga dapat menstimulasi pelepasan insulin. Sediaan tablet glipizid lepas diperpanjang atau Extended Release (ER) yang ada di pasaran, memiliki matriks yang sangat kompleks dan mengganggu dalam proses analisis dibandingkan dengan sediaan glipizid lepas segera atau Immediate Release (IR). Berdasarkan hal tersebut, diperlukan suatu prosedur analisis yang spesifik, mudah dan cepat dalam memisahkan tablet glipizid ER dengan matriks pembawanya. Sampel dipreparasi dengan ekstraksi fase padat menggunakan sorben HLB (Hydrophilic-Lipophilic Balance), kemudian dilarutkan dalam fase gerak dan dianalisis menggunakan Kromatografi Cair Kinerja Tinggi (KCKT) fase terbalik. Fase gerak yang digunakan adalah campuran dapar fosfat monobasa 0,1M pH 6,00 ± 0,05 dan metanol perbandingan 55:45, laju alir 1,0 mL/menit, suhu kolom 30ºC dan detektor UV pada panjang gelombang 225 nm dengan kolom YMC Triart C18 (150 x 4,6 mm, ID S-5 µm 12 nm). Prosedur analisis yang dikembangkan memberikan linearitas yang baik pada rentang konsentrasi 0,01 – 0,07 mg/mL dengan persamaan regresi y = 58985,35x + 13,88 dan koefisien korelasi r = 0,9995. Metode ini mempunyai batas deteksi dan batas kuantitasi secara statistik sebesar 0,0025 mg/mL dan 0,0075 mg/mL. Presisi inter day glipizid ER nilai % SBR berturut-turut sebesar 0,90%, 1,40% dan 0,86%, sedangkan presisi intra day sebesar 1,23%. Rata-rata persen perolehan kembali plasebo yang di-spike dengan baku glipizid adalah 100,68%. Untuk menguji kelaikan metode, dilakukan pengujian terhadap dua dosis sediaan tablet glipizid ER (5 dan 10 mg/tablet). Berdasarkan hasil yang diperoleh bisa disimpulkan bahwa prosedur analisis yang dikembangkan memenuhi persyaratan parameter validasi dan dapat diterapkan dalam penetapan keseragaman kandungan tablet glipizid ER.
ii ABSTRACT
DEVELOPMENT AND VALIDATION OF CONTENT UNIFORMITY ANALYTICAL PROCEDURE OF GLIPIZIDE
EXTENDED RELEASE TABLET
By
Indhah Fatmawati NIM : 20712318
Glipizide is an oral antidiabetic drug belonging to the class of second-generation sulfonylureas. It acts by blocking adenosine triphosphate-sensitive potassium channels in β-cells of islet of Langerhans on pancreatic cells, which stimulates release of insulin. The extended release (ER) dosage form of glipizide with its matrix often brings problem in analytical process that shows different yield with the immediate release (IR) one. Based on this case, a specific, easy, and fast analytical procedure suitable for extraction of glipizide in its ER matrices is needed. Samples were prepared by Solid Phase Extraction (SPE) using HLB (Hydrophilic-Lipophilic Balance) sorbent and dissolved in the mobile phase than analysed by A Reversed Phase High Performance Liquid Chromatographic (RP-HPLC). The chromatographic separation was achieved on a HPLC YMC Triart C18 (150 x 4.6 mm, ID S-5 µm 12nm) column. The mobile phase used was 0,1M buffer sodium dihydrogen phosphate monobase pH 6.00 ± 0.05 - methanol in the ratio 55:45 with flow rate of 1.0 mL/min and column temperatue was maintained at 30ºC. The eluted compound was monitored at a wavelength of 225 nm using a UV detector. The method described herein separated glipizid from all other formulation components within a run time of 23 min. Analytical procedure development was obtain good linearity at range concentration 0.01 – 0.07 mg/mL with the calibration curve of y = 58985.35x + 13.88 and the correlation coefficient of r = 0.9995. The limit of detection (LOD) was 0.0025 mg/mL, while the limit of quantitation (LOQ) was 0.0075 mg/mL. The % RSD the inter-day precision was obtained 0.90%, 1.40% and 0.86%, while the % RSD the intra-day precision was obtained 1.23%. The mean recovery of glipizide placebo spike was 100,68%. Furthermore the method was tried to use in the analysis 2 dose of glipizide ER tablets (5 and 10 mg/tablet). Based on the result of validation method evaluation, it was concluded that the proposed procedure is valid and can be applied for determination content uniformity of glipizide in ER tablet dosage form.
iii
PEDOMAN PENGGUNAAN TESIS
Tesis S2 yang tidak dipublikasikan terdaftar dan tersedia di Perpustakaan Institut Teknologi Bandung, dan terbuka untuk umum dengan ketentuan bahwa hak cipta ada pada pengarang dengan mengikuti aturan HaKI yang berlaku di Institut Teknologi Bandung. Referensi kepustakaan diperkenankan dicatat, tetapi pengutipan atau peringkasan hanya dapat dilakukan seizin pengarang dan harus disertai dengan kebiasaan ilmiah untuk menyebutkan sumbernya.
iv
v
KATA PENGANTAR
Segala puji dan syukur penulis panjatkan kepada Allah SWT, dzat pemilik segala ilmu yang tersirat maupun yang tersurat, atas limpahan kasih sayang-Nya, sehingga penulis bisa menyelesaikan penelitian tesis yang berjudul “Pengembangan dan Validasi Prosedur Analisis Keseragaman Kandungan Tablet Glipizid Lepas Diperpanjang”. Tesis ini disusun sebagai salah satu syarat untuk meraih gelar Master Sains dari Program Studi Magister Farmasi, Sekolah Farmasi Institut Teknologi Bandung.
Pada kesempatan ini penulis ingin menyampaikan ungkapan rasa terimakasih yang sebesar-besarnya kepada yang terhormat :
1. Ibu Dr. Ilma Nugrahani selaku pembimbing utama yang telah memberikan bimbingan dan pengarahan untuk menyelesaikan tesis ini.
2. Bapak Prof. Dr. Slamet Ibrahim Surantaatmadja selaku pembimbing serta yang telah memberikan bimbingan dan pengarahan untuk menyelesaikan tesis ini 3. Pimpinan dan seluruh staf Balai Besar POM di Bandung, khususnya staf
Laboratorium Pengujian Teranokoko BBPOM di Bandung.
4. Suami tercinta Yuda Prawira, S.Hut atas doa dan dukungannya, serta penyemangat terbesarku ananda Farras Hasanain Asshidiq.
5. Yang tercinta ibunda Hj.Siti Fatonah, S.Ag dan ayahanda H.Mashudi Arief, ibu Hj.Emma Sutrisno dan bapak Drs.H.Sutrisno yang telah memberikan bantuan moril, doa, dan juga semangat selama melakukan penelitian ini.
6. Teman-teman seperjuangan atas segala bantuan, dukungan dan kerjasamanya 7. Semua pihak yang telah membantu dalam menyelesaikan penelitian ini.
Dengan segala kerendahan hati dan keterbatasan penulis, penulis mengharapkan tesis ini bisa bermanfaat bagi kita semua.
Februari, 2015
vi DAFTAR ISI
Halaman
ABSTRAK ... i
ABSTRACT ... ii
PEDOMAN PENGGUNAAN TESIS ... iii
KATA PENGANTAR ... iv
LEMBAR PERSEMBAHAN ... v
DAFTAR ISI ... vi
DAFTAR GAMBAR ... vii
DAFTAR TABEL ... ix
DAFTAR SINGKATAN DAN LAMBANG ... x
Bab I Pendahuluan ... 1
Bab II Tinjauan Pustaka ... 4
II.1 Glipizid ... 4
II.1.1 Sifat fisikokimia ... 4
II.1.2 Farmakologi ... 4
II.2 Sediaan extended release ... 5
II.2.1 Tujuan sediaan extended release ... 5
II.2.2 Matriks extended release ... 6
II.2.3 Glipizid extended release ... 7
II.3 Solid Phase Extraction (SPE) ... 7
II.4 Keseragaman kandungan ... 10
II.5 Kromatografi Cair Kinerja Tinggi (KCKT) ... 11
II.5.1 Batasan kromatografi ... 11
II.5.2 Sistem dan instrument KCKT ... 12
II.5.3 Parameter dalam KCKT ... 13
II.6 Pengembangan Metode Analisis ... 16
II.7 Uji Kesesuaian Sistem ... 17
II.8 Validasi Metode Analisis ... 17
vii
II.8.2 Linearitas ... 18
II.8.3 Kecermatan (Akurasi) ... 18
II.8.4 Keseksamaan (Presisi) ... 19
II.8.5 Batas deteksi dan batas kuantisasi ... 21
Bab III Metodologi Penelitian ... 22
Bab IV Percobaan ... 23
IV.1 Bahan ... 23
IV.2 Alat ... 23
IV.3 Tahapan Penelitian ... 23
IV.3.1 Penyiapan larutan ... 23
IV.3.2 Penyiapan fase gerak ... 24
IV.3.3 Optimasi ekstraksi ... 24
IV.4 Uji Kesesuaian Sistem ... 25
IV.5 Validasi Metode ... 25
IV.5.1 Kekhasan (Spesifisitas) ... 25
IV.5.2 Linearitas ... 25
IV.5.3 Batas deteksi dan batas kuantisasi ... 26
IV.5.4 Kecermatan (Akurasi) ... 26
IV.5.5 Keseksamaan (Presisi) ... 26
IV.6 Uji Keseragaman Kandungan Glipizid dari Sampel di Perdagangan ... 26
Bab V Hasil dan Pembahasan ... 28
Bab VI Kesimpulan ... 41
viii
DAFTAR GAMBAR
Halaman
Gambar II.1 Struktur molekul glipizid ... 4
Gambar II.2 Skema prosedur umum SPE ... 9
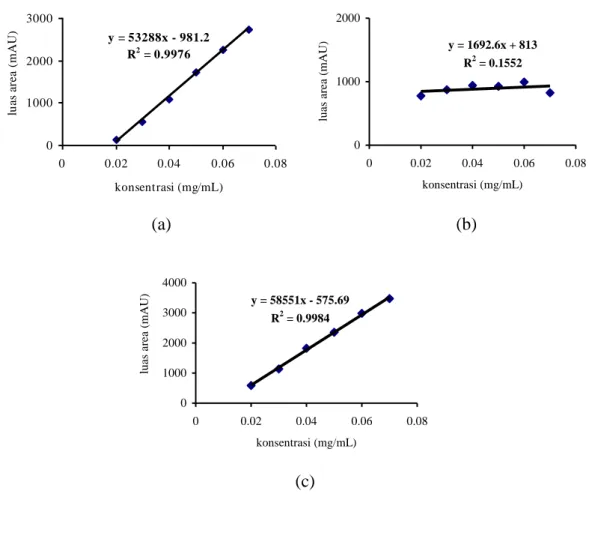
Gambar V.1 Kurva kalibrasi (a) tahap loading : baku induk glipizid dalam metanol, plasebo dalam metanol ... 30
(b) tahap elusi : baku induk glipizid dalam metanol, plasebo dalam metanol ... 30
(c) tahap elusi : baku induk glipizid dalam metanol, plasebo dalam dapar fosfat ... 30
Gambar V.2 Kromatogram baku glipizid ... 32
Gambar V.3 Kromatogram (a) pelarut ... 33
(b) larutan plasebo ... 33
(c) baku induk glipizid ... 33
(d) plasebo yang di-spike ... 33
ix
DAFTAR TABEL
Halaman
Tabel V.1 Perbandingan prosedur analisis glipizid IR dan ER ... 28
Tabel V.2 Hasil optimasi pemilihan sorben SPE ... 31
Tabel V.3 Data hasil uji kesesuaian sistem larutan baku glipizid 0,05 mg/mL ... 32
Tabel V.4 Parameter uji ... 34
Tabel V.5 Konsentrasi dan luas area sampel simulasi glipizid ... 35
Tabel V.6 Parameter regresi linier dan kuadratik glipizid ... 36
Tabel V.7 Presisi intra hari glipizid dalam sampel simulasi ... 37
Tabel V.8 Presisi antar hari glipizid dalam sampel simulasi ... 38
Tabel V.9 Akurasi glipizid dalam sampel simulasi ... 38
Tabel V.10 Uji keseragaman kandungan tablet Glipizid ER 5 mg ... 39
x
DAFTAR SINGKATAN DAN LAMBANG
SINGKATAN NAMA Pemakaian pertama
kali pada halaman
IR Immediate Release 1
ER Extended Release 1
HPMC Hydroxypropyl methylcellulose 1
BPOM Badan Pengawas Obat dan Makanan 1
KCKT Kromatografi Cair Kinerja Tinggi 2
SPE Solid Phase Extraction 2 USP United State Pharmacopoea 7 HLB Hydrophilic-Lipophilic Balance 8 MAX Mixed-mode Anion Exchange 8 MCX Mixed-mode Cation Exchange 8 WAX Weak Anion Exchange 8 WCX Weak Cation Exchange 8
SBR Simpangan Baku Relatif 17
SB Simpangan Baku 19
KV Koefisien variasi 19
HorRat Horwitz Ratio 20
UKS Uji Kesesuaian Sistem 22
UV Ultra Violet 22
xi LAMBANG
k‟ Faktor kapasitas 14
α Selektivitas 14
N Jumlah lempeng teoritik 15
tR Waktu retensi 15
Rs Resolusi 15
r Koefisien korelasi 18
Vx0 Koefisien variansi fungsi regresi 18
1
BAB I PENDAHULUAN
Diabetes mellitus adalah penyakit degeneratif dan merupakan salah satu penyakit dengan prevalensi cukup tinggi di Indonesia. Salah satu pengobatannya dengan obat-obat dari golongan sulfonilurea. Glipizid adalah generasi kedua golongan sulfonilurea dengan mekanisme aksi mengeblok kanal kalium dalam sel β -Langerhans. Glipizid ditemukan di pasaran baik dalam bentuk tablet immediate release (IR) atau lepas segera maupun extended release (ER) atau lepas diperpanjang. Keduanya memiliki matriks yang berbeda, terutama adanya matriks hidroksipropil metilselulosa (HMPC) yang merupakan polimer hidrofilik. Pada umunya HPMC digunakan sebagai polimer yang mengontrol kecepatan pelepasan obat. Adanya perbedaan matriks antara dua jenis sediaan tablet membawa konsekuensi pada beberapa hal seperti aspek farmakoekonomi, farmakologi, farmakokinetik sampai pada masalah analisis (Brunton dan Parker, 2008).
Peran BPOM dalam pengawasan obat dan makanan merupakan bagian integral dari upaya pembangunan kesehatan di Indonesia. Visi dan misi BPOM dalam melindungi masyarakat dari produk obat dan makanan yang membahayakan kesehatan dituangkan dalam sistem pengawasan full spectrum mulai dari pre-market hingga post-market yang disertai dengan upaya penegakan hukum dan pemberdayaan masyarakat (community empowerment). Pelaksanaan pengawasan ini salah satunya dilakukan oleh Unit-Unit Pelaksana Teknis Laboratorium yang tersebar di Indonesia yang mempunyai tugas melaksanakan pemeriksaan secara laboratorium, pengujian dan penilaian mutu produk terapetik, narkotika, psikotropika dan zat adiktif lain, alat kesehatan, obat tradisional, kosmetika, produk komplimen, pangan dan bahan berbahaya sesuai dengan peraturan perundang-undangan yang berlaku.
2
juga untuk tujuan analisis, preparasi tablet ER juga tidak dapat diperlakukan sama dengan tablet IR karena matriks yang digunakan lebih kompleks.
Masuknya air ke dalam sistem matriks hidrofilik akan membentuk suatu lapisan gel yang kental yang dapat memperlambat penetrasi air sehingga dapat memperlambat pelepasan obat. Polimer HPMC bila kontak dengan medium sering menyulitkan proses analisis karena terjadi swelling dengan membentuk lapisan hidrogel yang viskositasnya tinggi yang kemudian menurun kekentalannya saat mulai terjadi erosi terhadap polimer (Maderuelo, dkk, 2011). Penyaringan sampel obat dengan matriks HPMC seringkali menyulitkan dan harus dibantu dengan penyaring vakum, lebih jauh lagi dapat merusak kolom dalam sistem KCKT. Oleh karena itu preparasi yang tepat menjadi salah satu faktor penentu keberhasilan analisis (Nickerson, 2011).
Salah satu parameter mutu yang ditetapkan dalam pengujian obat yakni uji keseragaman kandungan yang didefinisikan sebagai derajat keseragaman jumlah zat aktif dalam tiap satuan sediaan. Untuk memastikan konsistensi kadar bahan aktif per unit sediaan, masing-masing unit harus mengandung zat aktif sesuai dengan jumlah yang dipersyaratkan dalam monografi (The USP, 2011). Farmakope mensyaratkan untuk tablet bersalut dan tablet yang mengandung zat aktif kurang dari 25 mg atau bobot zat aktif lebih kecil dari 25% bobot sediaan harus memenuhi syarat uji keseragaman kandungan (Suplemen I FI IV, 2009).
3
diekstraksi, analit yang sudah terpisah dari matriks pembawanya diinjeksikan ke dalam sistem KCKT untuk dianalisis dan divalidasi. Selanjutnya untuk menguji kelaikan metode, dilakukan pengujian keseragaman kandungan terhadap sampel yang ada di pasaran.
Penelitian ini bertujuan untuk mendapatkan prosedur analisis keseragaman kandungan tablet glipizid ER yang cepat, mudah dan handal dengan ekstraksi secara SPE dan analisis secara KCKT.
4
BAB II TINJAUAN PUSTAKA
II.1 Glipizid
II.1.1 Sifat fisikokimia
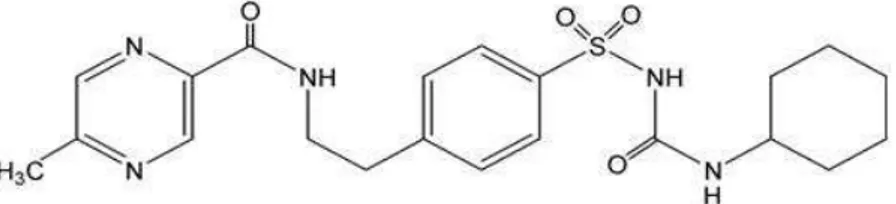
Glipizid memiliki pemerian serbuk hablur putih atau hampir putih. Rumus molekul dari glipizid adalah C21H27N5O4S, berat molekul 445,54 gram/mol, nama
kimia 1-Sikloheksil-3-[[p-[2-(5-metilpirazin karboksamido)etil]fenil]sulfonil]urea. Glipizid mengandung tidak kurang dari 98,0% dan tidak lebih dari 102,0% C21H27N5O4S, dihitung terhadap zat yang telah dikeringkan. Glipizid praktis tidak
larut dalam air dan dalam etanol 96%; sangat sukar larut dalam metilen klorida dan dalam aseton; larut dalam larutan alkali hidroksida encer. Titik leleh glipizide 200 – 203 ⁰C, pKa 5,9 (The USP, 2012). Glipizid harus disimpan dalam wadah yang tertutup rapat dan terlindung dari cahaya (Suplemen I FI IV, 2009).
Gambar II.1 Struktur molekul glipizid (The USP, 2012).
II.1.2 Farmakologi
5
Glipizid dosis 5 mg termasuk dalam golongan daftar obat esensial yang direkomendasikan sebagai obat antidiabetes oral selain glibenklamid dan metformin, sesuai Kepmenkes Nomor 312/Menkes/SK/IX/2013 tentang Daftar Obat Esensial Nasional 2013. Pemakaian yang luas dari obat-obat antidiabetik oral dapat meningkatkan kemungkinan terjadinya overdosis (Tran D, 2010). Dengan demikian maka pengawasan terhadap glipizid menjadi hal yang penting, terutama pengawasan terhadap kadar zat aktif yang terkandung dalam sediaan.
II.2 Sediaan Extended Release
II.2.1 Tujuan Sediaan Extended Release
Bentuk sediaan pelepasan dimodifikasi adalah sistem penghantaran obat yang berdasarkan formulasi dan desain produk, memberikan pelepasan obat dalam bentuk yang dimodifikasi. Pelepasan obat dapat ditunda (delay release) atau diperpanjang (extended release). Sediaan extended release dibagi menjadi beberapa jenis seperti controlled release, sustained release dan prolong release
(Qiu & Zhang, 2000).
Sediaan extended release dirancang untuk melepaskan obatnya dengan cara yang terkendali baik kecepatan, waktu, maupun lokasi pelepasannya agar kadar obat dalam darah dapat dipertahankan dan pengobatannya optimum. Pada sedian ER terjadi pengurangan frekeuensi pemberian obat hampir dua kali lipat dari sediaan konvensional. Beberapa karakteristik obat yang bisa dibuat sediaan ER seperti : tidak menunjukkan tingkat absorpsi dan eksresi yang terlalu lambat maupun terlalu cepat; diabsorbsi dari saluran pencernaan; diberikan dalam dosis kecil; mempunyai indeks terapetik yang aman dan lebih digunakan untuk pengobatan kronis daripada akut (Allen, dkk, 2011).
6 II.2.2 Matriks extended release
Dalam sistem penghantaran obat, sistem matriks hidrofilik paling sering digunakan dalam mengontrol sistem pelepasan obat karena formulasinya yang sederhana, murah, proses produksi yang mudah, mempunyai korelasi in-vivo dan
in-vitro yang bagus dan memungkinkan formulasi dengan obat yang berbobot molekul besar. Matriks hidrofilik merupakan dispersi homogen obat dalam satu kerangka bersama satu atau beberapa eksipien berupa polimer hidrofilik yang bergabung, seperti derivat selulosa, Na-alginat, xantan gum, polietilen oksid yang akan mengembang bila kontak dengan air (Maderuelo C, dkk, 2011).
Eksipien atau matriks yang digunakan pada produk Glipizid ER terdiri dari polietilen oksida, hidroksi propil metil selulosa (HPMC), magnesium stearat, natrium klorida, etil selulosa, polietilen glikol, opadry-white (Roerig, 2013). Polietilen oksida digunakan sebagai bahan pengikat tablet pada konsentrasi 5– 85%. Semakin besar tingkat bobot molekul maka akan berfungsi sebagai matriks hidrofil yang dapat menunda pelepasan obat (Rowe, dkk, 2009). HMPC sebagai polimer yang paling sering digunakan sebagai matriks karena beberapa kelebihan seperti dapat diterima dalam persyaratan regulasi secara global, stabil dan non-ionik (tidak tergantung pH), mudah diproduksi baik secara pengempaan langsung maupun granulasi, tidak berwarna dan berbau, mudah disediakan, cocok untuk berbagai macam profil pelepasan obat yakni mempunyai berbagai varian sifat fisikokimia dan derajat viskositas (Tiwari dan Rajabi, 2008). Selulosa asetat digunakan sebagai membran semi permeabel pada tablet, terutama pada tipe pompa osmotik yang memungkinkan tablet dilepaskan secara terkendali (Rowe, dkk, 2009).
7
sekaligus larut (erosion). Proses disolusi inilah yang mengontrol pelepasan obat (Maderuelo C, dkk, 2011).
II.2.3 Glipizid Extended Release
Adanya bentuk sediaan ER diharapkan kadar glipizid dalam darah dapat dipertahankan secara terkontrol untuk mencegah hipoglikemia, mengurangi efek samping dan meningkatkan kepatuhan pasien (Brunton, dkk, 2008).
Selain tablet biasa, di pasaran sudah ada sediaan tablet glipizid ER dosis 5 dan 10 mg. Saat ini, Glucotrol XL® merupakan satu-satunya paten di Indonesia untuk tablet glipizid sediaan ER yang dirancang untuk memberikan tingkat pelepasan glipizid yang terkendali ke dalam saluran pencernaan yang tidak tergantung dari pH atau motilitas pencernaan.
Selama ini metode analisa keseragaman kandungan terhadap glipizid yang ada di laboratorium pengujian Badan POM mengacu pada metode standar USP Edisi 35 untuk tablet glipizid IR dengan cara memasukkan 1 tablet dalam labu ukur yang berisi campuran pelarut dapar fosfat dan metanol, dikocok mekanik dan diencerkan sampai konsentrasi tertentu, selanjutnya disaring dan diukur secara KCKT pada panjang gelombang 225 nm (The USP, 2012), namun metode ini tidak menyebutkan penggunaanya untuk penetapan sediaan ER dengan matriks khusus, sehingga diperlukan suatu pengembangan terhadap metode analisis yang sudah ada.
II.3 Solid Phase Extraction (SPE)
Keberhasilan analisis suatu sediaan obat tidak lepas dari teknik preparasi sampel yang digunakan. Informasi tentang bahan aktif obat seperti pKa, kelarutan, polaritas, dan potensi interaksi antara analit dengan eksipien penting dipertimbangkan selama preparasi (Nickerson, 2011).
8
Proses ekstraksi dilakukan dengan memasukkan analit dalam pelarut dengan kekuatan elusi yang rendah, ke dalam suatu sorben (penyerap). Analit yang diinginkan akan terkonsentrasi pada sorben. Analit kemudian dicuci dengan sejumlah pelarut dengan daya elusi rendah dan kemudian dielusi dari sorben dengan sejumlah kecil pelarut dengan daya elusi yang kuat (Watson, 2005).
Mekanisme retensi dan elusi dalam SPE ini merupakan proses distribusi antara fase gerak dan fase diam seperti yang terjadi pada kromatografi cair, hanya dalam sebuah kolom pendek, dengan sejumlah kecil lempeng teoritis dan menyertakan perbedaan koefisien distribusi senyawa. Laju alir sampel dalam kolom harus diperhatikan agar perolehan kembali analit optimal (Camel, 2003).
Berdasarkan tipe fase diam atau penyerap yang dikemas dalam cartridge, SPE dibagi menjadi empat, yakni fase normal (normal phase), fase terbalik (reversed phase), adsorbsi (adsorption) dan pertukaran ion (ion exchange). Massa sorben dipilih dengan mempertimbangkan volum sampel dan konsentrasi analit. Pemilihan massa sorben yang sesuai menjadi tahap kritis karena sorben yang tidak tepat menyebabkan kolom overload dan recovery analit yang rendah, serupa dengan „stacked injection‟ pada KCKT (Pavlovic, dkk, 2009). Beberapa tipe
sorben yang umum digunakan adalah tC18, HLB (Hydrophilic-Lipophilic Balance), MAX (Mixed-mode Anion Exchange), MCX (Mixed-mode Cation Exchange), WAX (Weak Anion Exchange) dan WCX (Weak Cation Exchange). Besarnya nilai perolehan kembali analit dengan SPE dipengaruhi oleh pH, volume dan konsentrasi sampel, jenis sorben, volume dan kekuatan larutan pengelusi (Hennion, 1999). Mekanisme retensi yang umum terjadi di ekstraksi jenis fase padat berdasarkan gaya van der Waals (interaksi non polar), ikatan hidrogen, gaya dipol-dipol (interaksi polar) dan interaksi ionik.
9
hanya membawa sampel melewati tahap SPE tetapi harus cukup lemah agar analit bisa terikat ke sorben.
Kelebihan SPE yakni lebih praktis dan cepat, hanya dibutuhkan sejumlah kecil pelarut, pengoperasiannya mudah, adanya tipe sorben (cartridge) yang bervariasi sehingga selektif untuk sejumlah analit dengan gugus fungsional tertentu, dan saat ini sudah terdapat peralatan SPE yang bisa dikopling secara online dengan instrumen lain seperti KCKT (Hennion, 1999). Sementara keterbatasan penggunaan SPE terletak pada harga sorben yang relatif mahal dan hanya untuk satu kali pemakaian, jenis sorben sangat beragam sesuai dengan jenis pabrik, ukuran dan isi sorben, juga dapat terjadi adsorpsi yang irreversibel terhadap analit pada sorben yang tidak dapat dielusi oleh pelarut/eluen (Watson, 2005). Permasalahan yang bisa terjadi selama SPE seperti elusi analit yang tidak sempurna, analit tidak tertahan di sorben (breaktrough), pemilihan pelarut yang tidak selektif sehingga efektivitas ekstraksi rendah.
Prosedur dalam penggunaan SPE terlihat pada skema gambar II.3 di bawah ini :
Gambar II.2 Skema prosedur umum SPE (Harris, D.C., 2007).
Keterangan :
a. Pengkondisian kolom : dengan cara melewatkan pelarut tertentu ke dalam
cartridge yang bertujuan untuk meningkatkan daya serap sorben.
b. Loading sampel : sampel yang akan dipisahkan dimasukkan ke dalam
10
c. Pencucian : membilas komponen lain / pengotor yang tertahan di sorben dengan sejumlah pelarut tertentu.
d. Elusi analit dengan pelarut tertentu yang lebih kuat.
SPE secara luas digunakan sebagai metode ekstraksi yang potensial dalam berbagai bidang penelitian seperti biologi, pangan, farmasi, klinis dan lingkungan sebagai alternatif dari ekstraksi cair-cair yang dinilai lebih praktis dan cepat.
II.4 Keseragaman Kandungan
Keseragaman dosis per unit digambarkan dengan dua metode yaitu keseragaman kandungan (content uniformity) dan keragaman bobot (weight variation) (The USP, 2011). Tablet harus memenuhi uji keragaman bobot jika zat aktif merupakan bagian terbesar dari tablet dan jika uji keragaman bobot cukup mewakili keseragaman kandungan. Keragaman bobot bukan merupakan indikasi yang cukup dari keseragaman kandungan jika zat aktif merupakan bagian kecil dari tablet atau jika tablet bersalut gula. Penerapan uji keseragaman kandungan berlaku untuk tablet salut baik salut film maupun yang lain, dengan dosis bahan aktif kurang dari 25 mg atau perbandingan kadar bahan aktif dengan bobot tablet kurang dari 25%.
Prosedur uji keseragaman kandungan tablet glipizid dengan menetapkan kadar 10 satuan satu per satu seperti yang tertera dalam monografi glipizid. Tablet glipizid dalam bentuk sediaan ER mempunyai dosis 5 dan 10 mg, memenuhi syarat untuk dilakukan pengujian keseragaman kandungan untuk mengetahui kadar dan homogenitas bahan aktif dalam setiap tabletnya.
11
II.5 Kromatografi Cair Kinerja Tinggi (KCKT)
Pembahasan KCKT meliputi: batasan, sistem dan instrumentasi KCKT serta parameter KCKT.
II.5.1 Batasan kromatografi
Dalam industri farmasi yang modern, KCKT merupakan instrumen analisis utama dan menyeluruh yang digunakan pada semua tahap mulai dari penemuan, pengembangan sampai produksi. Prinsipnya adalah teknik pemisahan analit atau campuran analit yang melibatkan dua fase, yakni fase diam dan fase gerak. Fase gerak membawa campuran analit melewati suatu media dengan permukaan berpori (fase diam). Analit akan terdispersi dalam fase gerak pada level molekular dan memungkinkan terjadinya transport serta interaksi antara fase diam dan fase gerak (Kazakevich dan Lobrutto, 2007).
Untuk mencapai tujuan pemisahan analit dengan analisis menggunakan KCKT, beberapa hal yang harus diperhatikan diantaranya adalah:
a. Sampel harus terlarut, karena jika tidak akan mengakibatkan rendahnya hasil perolehan kembali. Ketidaklarutan analit ini bisa diantisipasi dengan preparasi sampel yang tepat sebelum masuk ke sistem KCKT.
b. Analit harus dapat diretensi di dalam kolom dan memiliki laju migrasi yang berbeda dengan analit lainnya.
c. Pemilihan komposisi fase gerak yang tepat agar analit dapat terpisah dari komponen lainnya dalam sampel.
d. Pelarut akhir dari analit sebaiknya sama dengan fase gerak yang digunakan atau pelarut yang lebih lemah dibandingkan dengan fase gerak (Ahuja dan Dong, 2005).
Teknik KCKT menawarkan perbaikan besar terhadap kromatografi kolom klasik dan memiliki beberapa keunggulan yang signifkan jika dibandingkan teknik yang lebih baru, seperti kromatografi cair superkritis, elektroforesis kapiler dan kapiler
12
ketepatan, kecepatan dan kemampuan untuk memisahkan komponen yang sulit dipisahkan (Hanai, 1999).
II.5.2 Sistem dan Instrument KCKT
Sistem KCKT terdiri dari dua bagian, yaitu sistem pemisahan dan sistem pendeteksi. Sistem pemisahan dengan bagian utamanya pompa yang mengalirkan pelarut dan sampel (yang diinjeksikan melalui injektor) ke dalam kolom, sedangkan sistem pendeteksi adalah detektor yang dihubungkan pada ujung akhir kolom.
a. Pompa
Fungsi pompa di dalam sistem KCKT adalah untuk mendorong fase gerak masuk ke dalam kolom. Biasanya menggunakan katup inlet (terhubung ke pelarut) dan katup outlet (terhubung ke kolom). Pada dasarnya pompa KCKT mempunyai syarat sebagaimana syarat wadah pelarut yaitu harus inert terhadap pelarut organik (fase gerak), dapat memompakan fase gerak secara konstan, mempunyai tekanan maksimum yang cukup tinggi (400 psi) dan mempunyai noise yang rendah (Skoog dkk, 2007).
b. Sistem injektor
Adanya injektor pada KCKT memungkinkan volum sampel yang tepat masuk ke dalam kolom. Injektor manual terdiri dari katup enam port dengan rotor, loop sampel dan jarum port. Pada saat posisi load, aliran datang dari pompa melalui salah satu port dan keluar melalui port yang lain menuju ke kolom (Ahuja dan Dong, 2005). Loop yang digunakan bisa disesuaikan berdasarkan ukuran sampel dengan kisaran loop antara 5 sampai 500 μL (Skoog dkk, 2004). Sistem injeksi dapat dilakukan secara manual atau diotomatisasi melalui autosampler (Skoog dkk, 2007).
c. Kolom
13
jumlah lempeng teoritis sebanyak 100.000 lempeng/meter. Instrumen KCKT modern sudah dilengkapi dengan pengatur suhu kolom (heater) agar suhu kolom tetap konstan (Skoog dkk, 2004).
d. Detektor
Detektor KCKT seringkali berupa modifikasi spektofotometer yang memantau konsentrasi (atau massa) dari analit yang terelusi (Ahuja dan Dong, 2005). Detektor yang paling banyak digunakan untuk KCKT yakni berdasarkan absorbsi sinar ultra violet – sinar tampak, fluoresens, indeks bias dan detektor elektrokimia. Detektor spektrometri massa saat ini sudah cukup populer, akan tetapi pada campuran analit yang kompleks, perpaduan KCKT dengan detektor spektrometri massa memberikan resolusi yang kurang bagus (Skoog dkk, 2007).
Secara umum terdapat empat jenis teknik kromatografi cair yang sering digunakan yaitu kromatografi fase normal, kromatografi fase terbalik, kromatografi pertukaran ion, kromatografi ekslusi ukuran. Kromatografi fase terbalik merupakan jenis kromatografi yang paling banyak digunakan, hampir 90% analisis sampel dengan bobot molekul rendah menggunakam kromatografi fase terbalik. Pemisahannya berdasarkan gaya hidrofobik atau interaksi van der Waals. Permukaan dari fase diam pada tipe ini bersifat hidrofobik dan bersifat non polar (Kazakevich dan Lobrutto, 2007).
II.5.3 Parameter dalam KCKT
Tiap-tiap analit yang spesifik dalam sebuah kromatogram ditampilkan dalam bentuk puncak. Adanya interaksi yang kuat antara analit dan fase diam pada konsentrasi analit yang relatif rendah akan menghasilkan puncak yang simetris dan mengikuti distribusi normal (tipe kurva Gaussian). Teori kolom dapat digunakan sebagai petunjuk dalam mendesain pengoperasian KCKT (Kazakevich dan Lobrutto, 2007).
a. Waktu retensi (tR)
14
dalam sampel serta sifat dari analit tersebut. Waktu retensi merupakan parameter yang mudah diukur. Waktu retensi tergantung dari laju alir fase gerak dan dimensi kolom, dimana semakin cepat laju alir yang digunakan makan semakin kecil (singkat) waktu retensinya. Selain itu, waktu retensi tergantung dari kestabilan laju alir.
b. Faktor kapasitas
Faktor kapasitas merupakan parameter penting yang dapat digunakan untuk menjelaskan laju migrasi analit dalam kolom. Untuk spesi A, faktor kapasitas k‟A didefinisikan sebagai berikut :
Faktor kapasitas, k‟ =
0 0 t
t tR
………. (Persamaan II.1) Jika k‟ bernilai 0 (nol) berarti bahwa komponen atau analit tidak diretensi dan dielusi bersama dengan pelarut. Jika k‟ bernilai satu artinya komponen diretensi secara lemah oleh fase diam dalam kolom, sementara jika k‟ bernilai duapuluh artinya komponen diretensi secara kuat dan berinteraksi dengan fase diam cukup lama. Pada sebagian besar analisis, nilai k‟ untuk analit yang terelusi berkisar antara 1 sampai 20, sehingga mempunyai peluang yang cukup untuk berinteraksi dengan fase diam menghasilkan pemisahan yang baik (Ahuja dan Dong, 2005). tR adalah waktu retensi, dan t0 adalah waktu yang
tidak teretensi. Beberapa pustaka merekomendasikan faktor kapasitas yang baik adalah 1< k‟<10.
c. Faktor selektivitas
15
Besarnya α harus > 1 untuk pemisahan yang baik. Selektifitas tergantung dari komposisi fase diam dan fase gerak yang bisa ditingkatkan besarnya dengan melakukan modifikasi antara keduanya (Ahuja dan Dong, 2005).
d. Efisiensi kolom
Jumlah lempeng teoritik (N) merupakan ukuran kuantitatif dari efisiensi kolom dan besarnya merupakan perbandingan antara waktu retensi (tR) dengan
standar deviasi lebar puncak (σ), sedangkan besarnya Wb setara dengan 4σ
yang dapat dihitung secara empiris dari kromatogram dengan rumus berikut ini : Kolom yang efisien dapat mencegah pelebaran pita sehingga menghasikan pita yang sangat sempit. Semakin kecil dan seragam ukuran partikel dalam kolom maka semakin besar efisiensinya (Ahuja dan Dong, 2005).
e. Resolusi
Tujuan akhir dari setiap analisis menggunakan KCKT yakni pemisahan satu atau lebih analit dari komponen lain (matriks) dalam sampel agar didapatkan informasi yang kuantitatif dari masing-masing analit. Resolusi dari suatu kolom adalah kemampuan kolom untuk memisahkan dua analit. Resolusi dinyatakan dengan rumus sebagai berikut :
16
Dimana tR1 dan tR2 adalah waktu retensi spesi 1 dan 2, sedangkan Wb1 dan Wb2
adalah lebar alas puncak spesi 1 dan 2. Nilai Rs > 1,5 menunjukkan puncak 1
dan 2 terpisah dengan sempurna (Ahuja dan Dong, 2005).
II.6 Pengembangan Metode Analisis
Hal-hal yang perlu di pertimbangkan sebagai tahapan dalam pengembangan metode analisis secara KCKT meliputi (Kazakevich dan Lobrutto, 2007) :
1. Karakterisasi dan pengumpulan informasi analitik
Mencakup pengumpulan informasi analitik analit maupun sampel (termasuk matriksnya), sifat fisikokimia sampel, metode analisis yang telah ada, metode baku, perlakuan awal atau pemisahan sebelum dilakukan analisis, informasi mengenai formula dan komposisi sampel serta matriks yang digunakan dan persyaratan spesifik yang telah ditentukan.
2. Penilaian kebutuhan metode
Penilaian kebutuhan metode tergantung pada tujuan analisis yang dilakukan dengan kriteria yang harus ditentukan sebelum percobaan dimulai diantaranya kecermatan, keseksamaan, kepekaan, selektivitas, linearitas dan batas deteksi. Faktor yang perlu dipertimbangkan meliputi waktu, jumlah sampel, biaya dan tenaga, kemudahan dan kepraktisan metode serta ketersediaan instrumen. 3. Penelusuran pustaka
Penelusuran pustaka yang berhubungan dengan analisis dan metode analisis yang sesuai.
4. Pemilihan metode analisis
Terlebih dahulu mengkategorikan golongan masalah analitik. Jika memungkinkan, metode analisis sebelumnya bisa diadopsi sehingga lebih efisien, atau dengan memodifikasi metode analisis yang sudah ada. Adaptasi dapat mencakup kondisi preparasi maupun kondisi instrumentasi. Akan tetapi jika masalah analitiknya sama sekali baru, maka perlu dibuat metode/ prosedur baru dengan pendekatan senyawa analog yang mempunyai kemiripan sifat fisikokimia.
17
Menyiapkan instrumen yang sudah terkalibrasi sesuai dengan prosedur operasional baku alat. Uji pendahuluan biasanya memakan waktu yang cukup lama sebelum kondisi optimum didapatkan.
6. Optimasi Prosedur Analisis
Bertujuan untuk mencari aras-aras yang optimum jika hasil studi pendahuluan jauh dari yang diharapkan, dilakukan dengan cara mengubah parameter-parameter yang mempengaruhi analisis sampai diperoleh kondisi yang paling baik.
7. Evaluasi data hasil optimasi 8. Validasi metode
Validasi metode mengacu pada parameter-parameter validasi pada pustaka. Validasi diawali dengan melakukan uji kesesuaian sistem untuk memastikan bahwa sistem analisis berjalan secara baik dan benar.
II.7 Uji Kesesuaian Sistem
Uji kesesuaian sistem merupakan serangkaian uji untuk memastikan efektivitas sistem pemisahan yang digunakan. Parameter-parameter yang digunakan meliputi bilangan lempeng teoritis (N) > 2000, faktor ikutan ≤ 2,0, kapasitas (k‟ atau α) >
2,0, resolusi (Rs > 1,5) dan nilai simpangan baku relatif (SBR) < 2,0% dari waktu retensi dan luas area dari 5 kali injeksi (Elmer dan Miller, 2004).
II.8 Validasi Metode Analisis
Validasi metode analisis adalah suatu tindakan penilaian terhadap parameter tertentu, berdasarkan percobaan laboratorium, untuk membuktikan bahwa parameter tersebut memenuhi persyaratan untuk penggunaannya. Parameter analisis yang ditentukan pada validasi adalah spesifikasi, linieritas dan rentang kadar, akurasi, presisi, limit deteksi dan limit kuantisasi (ICH, 1994).
II.8.1 Kekhasan (Spesifisitas)
18
metode tidak ada atau kurang baik, metode dapat dilengkapi dengan prosedur analisis pendukung yang memadai seperti pemisahan (Ermer dan Miller, 2005).
Selektivitas metode ditentukan dengan membandingkan hasil analisis sampel yang mengandung cemaran, hasil urai, senyawa sejenis, senyawa asing lainnya atau pembawa plasebo dengan hasil analisis sampel tanpa penambahan bahan-bahan tadi. Penyimpangan hasil jika ada merupakan selisih dari hasil uji keduanya. Pada metode analisis yang melibatkan kromatografi, selektivitas ditentukan melalui perhitungan daya resolusinya (Rs) (ICH, 1994).
II.8.2 Linearitas
Linearitas adalah kemampuan metode analisis yang memberikan respon yang secara langsung atau dengan bantuan transformasi matematik yang baik, proporsional terhadap konsentrasi analit dalam sampel. Pengujian kelinieran dilakukan untuk membuktikan bahwa larutan sampel memberikan respon analit yang berbanding lurus dengan konsentrasi (Ibrahim, 2005).
Parameter linearitas ini diuji dengan membuat kurva baku, dimana kelinieran kurva baku yang baik dapat dilihat dari nilai koefisien korelasi (r) yang ≥ 0,999 serta nilai koefisien variansi fungsi regresi (Vx0) ≤ 2,0% untuk kurva baku
penetapan kadar obat dalam sediaan atau bahan baku, dan ≤ 5,0% untuk analisis obat dalam kajian metabolit dan bahan biologis. Nilai koefisien korelasi (r) > 0,999 sudah cukup dan dapat digunakan untuk membuktikan kelinieran kurva baku (Ibrahim, 2005).
II.8.3 Kecermatan (Akurasi)
19
Akurasi ditentukan dengan dua cara yaitu metode simulasi (spiked-placebo recovery) atau metode penambahan baku (standard addition method). Dalam metode simulasi, sejumlah analit bahan murni ditambahkan ke dalam campuran bahan pembawa sediaan farmasi (plasebo) lalu campuran tersebut dianalisis dan hasilnya dibandingkan dengan kadar analit yang ditambahkan (kadar yang sebenarnya). Dalam metode penambahan baku, sampel dianalisis lalu sejumlah tertentu analit yang diperiksa ditambahkan ke dalam sampel dicampur dan dianalisis lagi. Selisih kedua hasil dibandingkan dengan kadar yang sebenarnya (hasil yang diharapkan). Dalam kedua metode tersebut, persen peroleh kembali dinyatakan sebagai rasio antara hasil yang diperoleh dengan hasil yang sebenarnya.
Perhitungan persen perolehan kembali dinyatakan dengan rumus :
A CF = konsentrasi total sampel yang diperoleh dari pengukuran
CA = konsentrasi sampel sebenarnya
C*A = konsentrasi analit yang ditambahkan (Harmita, 2004).
II.8.4 Keseksamaan (Presisi)
Presisi atau keseksamaan adalah ukuran yang menunjukkan derajat kesesuaian antara hasil uji individual, diukur melalui penyebaran hasil individual dari rata-rata jika prosedur diterapkan secara berulang pada sampel-sampel yang diambil dari campuran yang homogen. Presisi diukur sebagai simpangan baku atau simpangan baku relatif (koefisien variasi). Presisi dapat dinyatakan sebagai keterulangan (repeatability) atau ketertiruan (reproducibility).
20
seksama diberikan jika metode memberikan simpangan baku relatif atau koefisien variasi (KV) 2% atau kurang (ICH, 1994).
Presisi diukur sebagai simpangan baku (SB) atau simpangan baku relatif (SBR) Dari nilai % SBR atau % KV yang diperoleh dibandingkan dengan KV Horwitz, yaitu suatu kurva berbentuk terompet yang menghubungkan ketertiruan (reproducibility) (presisi yang dinyatakan sebagai % KV) dengan konsentrasi analit. Presisi metode analisis dinyatakan sebagai fungsi dari konsentrasi melalui persamaan :
KVHorwitz = 2 1-0,5 log C
………. (Persamaan II.7) Dengan C adalah konsentrasi yang dinyatakan dengan sebagai fraksi desimal. Dengan menggunakan pembanding KV Horwitz nilai yang dapat diterima untuk keterulangan (repeatability) adalah :
SBR ≤ KVHorwitz
21 II.8.5 Batas deteksi dan batas kuantisasi
Batas deteksi dari suatu metode analisis adalah konsentrasi analit terendah dalam sampel yang dapat dideteksi yang masih memberikan respon signifikan dibandingkan dengan blanko, tetapi tidak dikuantisasi pada kondisi percobaan yang dilakukan. Sedangkan batas kuantisasi adalah konsentrasi analit terendah dalam sampel yang dapat ditentukan dengan presisi dan akurasi yang dapat diterima pada kondisi percobaan yang telah ditentukan (Harmita, 2004).
Penentuan batas deteksi dan batas kuantisasi diperoleh dari perhitungan statistik data hasil pengujian linearitas (Ibrahim, 2004) dengan persamaan sebagai berikut :
Batas deteksi =
22
BAB III METODOLOGI PENELITIAN
Sistem KCKT yang digunakan pada pengembangan prosedur analisis keseragaman kandungan tablet glipizid ER pada penelitian ini mengadopsi dari metode standar penetapan kadar glipizid tablet biasa yang tercantum dalam Farmakope Indonesia. Adanya matriks tablet yang lebih kompleks membawa konsekuensi perlunya proses ekstraksi yang berbeda untuk sediaan lepas diperpanjang.
Penelitian diawali dengan uji pendahuluan yakni mencari metode ekstraksi yang paling sesuai untuk memisahkan glipizid dari matriksnya sebelum dianalisis secara KCKT. Selama orientasi pemilihan metode ekstraksi mempertimbangkan hal-hal seperti sifat fisikokimia eksipien dan analit, ketersediaan alat dan bahan untuk ektraksi hingga didapatkan kondisi ekstraksi yang optimum.
23
BAB IV PERCOBAAN
IV.1 Bahan
Bahan-bahan yang digunakan pada penelitian ini meliputi baku pembanding glipizid (PPOMN), Metanol pro KCKT (Merck), Natrium dihidrogen fosfat monobasa (Merck), air pro KCKT, Natrium Hidroksida (KCKT), Glucotrol ER 5 mg dan 10 mg, Etilselulosa (AqualonTM EC-N50 Pharm), Opadry® Complete Film Coating System YS-2-7063 White (Colorcon), Polietilen oksida (Sentry Polyox WSR N750 – Colorcon), PEG 4000, NaCl (Merck), HMPC E5 Premium LV (PT.Dexa Medica), Magnesium stearat (Merck), sorben ekstraksi terdiri dari : Oasis® HLB 3cc (60mg), Oasis® MAX 3cc (60mg), Oasis® MCX 3cc (60mg) dan Sep-Pak® Vac. 3cc (500mg) tC18.
IV.2 Alat
Alat-alat yang digunakan pada penelitian ini meliputi Kromatografi Cair Kinerja Tinggi (Agilent 1260 Infinity–G1316A 1260 TCC), Kolom YMC Triart C18 (150 x 4.6 mm, ID S-5 µm 12nm), pH meter (Mettler Toledo), gelas ukur, pipet ukur 1mL, 2mL, 2,5mL, 4mL, 5mL, labu tentukur 10mL, 20mL, 50mL dan 100mL, buret 10mL, timbangan semimikro, timbangan analitik, penyaring membran nilon 0,45 µm, diameter 13mm (WhatmannTM), penyaring vakum, sonikator dan degassing unit (FALC), pipet Eppendorf, vakum manifold SPE (Agilent), magnetic stirer.
IV.3 Tahapan Peneltian IV.3.1 Penyiapan larutan
a. Pembuatan larutan dapar fosfat 0,1M pH 6,0
Dapar fosfat 0,1M pH 6,0 dibuat dengan menimbang 13,8 gram NaH2PO4.H2O,
24 b. Penyiapan larutan baku induk
Larutan baku induk glipizid dibuat dengan melarutkan senyawa baku glipizid dalam metanol sehingga diperoleh larutan baku induk glipizid dengan konsentrasi 1mg/mL. Baku induk disimpan pada suhu 2-8⁰C.
c. Penyiapan larutan plasebo
Matriks tablet glipizid ER yang digunakan mengacu pada informasi produk Glucotrol XL® (PT. Pfizer) yang terdiri dari polietilen oksida, hidroksi propil metil selulosa (HPMC), magnesium stearat, natrium klorida, etil selulosa, polietilen glikol, opadry-white (Roerig, 2013). Campuran matriks dilarutkan dalam dapar fosfat kemudian disaring. Selanjutnya disimpan pada suhu 2-8⁰C sebagai larutan plasebo.
d. Penyiapan larutan baku kerja
Larutan baku induk glipizid yang telah dibuat dengan konsentrasi 1 mg/mL, dipipet sejumlah tertentu sesuai konsentrasi baku kerja (sampel simulasi) yang diinginkan ke dalam labu tentukur 10 mL yang berisi 4 mL larutan plasebo, diencerkan dengan dapar fosfat sampai tanda, dipipet 1 mL kemudian diekstraksi fase padat dan hasil elusi diencerkan lagi dengan dapar fosfat hingga 5 mL.
IV.3.2 Penyiapan fase gerak
Fase gerak merupakan campuran dapar fosfat pH 6,0 : metanol dengan perbandingan 55:45.
IV.3.3 Optimasi Ekstraksi
a. Penentuan pelarut sampel
25 b. Penentuan sorben ekstraksi fase padat
Pemilihan sorben atau penyerap yang digunakan berdasarkan kemampuannya berikatan dengan analit, dimana ikatan antara analit dengan sorben harus lebih kuat dibandingkan antara ikatan analit dengan matriks sampel. Kondisi ini bertujuan agar analit dapat tertahan dalam sorben dan dapat dilepaskan ikatannya dengan sorben mengguakan pelarut yang selektif pada tahap elusi.
IV.4 Uji kesesuaian sistem
Uji kesesuaian sistem dilakukan dengan menyuntikkan larutan baku induk yang telah diencerkan dengan dapar fosfat sehingga didapatkan konsentrasi 0,05 mg/mL sebanyak 6 kali, kemudian dilihat resolusi (Rs > 1,5), faktor ikutan(Tf < 2), jumlah lempeng teoritis (N > 2000), dan RSD luas puncak (RSD < 2%).
IV.5 Validasi Metode
Validasi prosedur analisis tablet glipizid ER meliputi spesifisitas/selektivitas, linearitas, batas deteksi (LOD) dan batas kuantisasi (LOQ), kecermatan/akurasi dan keseksamaan/presisi.
IV.5.1 Kekhasan (Spesifisitas)
Parameter spesifisitas/selektivitas ditentukan dengan melakukan analisis terhadap pelarut, larutan plasebo, larutan baku, larutan sampel (placebo-spiked). Pengujian ini untuk memastikan bahwa prosedur analisis ini spesifik untuk glipizid tanpa ada gangguan dari matriks ataupun pelarut. Kriteria keberterimaan ditentukan dengan melihat tidak adanya gangguan pada waktu retensi senyawa uji dan resolusi senyawa uji (Rs >1,5).
IV.5.2 Linearitas
26
yang linear digunakan koefisien korelasi (r ≥ 0,999), koefisien variansi fungsi regresi (Vxo ≤ 2,0%) pada analisis regresi linier y = bx + a dan homogenitas variansi (uji F).
Disamping parameter linearitas di atas, dilakukan juga perhitungan dan uji kaji statistik meliputi homogenitas variansi, batas deteksi, dan batas kuantisasi menggunakan metode yang digunakan oleh Gottwald (Gottwald, 2000).
IV.5.3 Batas deteksi dan batas kuantisasi
Pada penelitian ini, penentuan batas deteksi dan batas kuantisasi diperoleh dari perhitungan statistik data hasil pengujian linearitas (Ibrahim, 2004).
IV.5.4 Kecermatan (Akurasi)
Akurasi ditentukan dengan menghitung persen perolehan kembali melalui metode simulasi (spiked-placebo recovery). Penetapan dilakukan dengan membuat tiga tingkat kadar glipizid 70%, 100% dan 130% sesuai dengan persyaratan rentang
dosis minimum untuk penetapan keseragaman kandungan obat antara 70-130%
(Ermer and Miller, 2005). Masing-masing konsentrasi dianalisis 3 kali pengulangan dan dihitung persen perolehan kembali.
IV.5.5 Keseksamaan (Presisi)
Presisi diukur dengan mengulang pengukuran suatu konsentrasi sampel simulasi glipizid sebanyak 6 kali (konsentrasi 100%, 0,05 mg/mL). Pengukuran keseksamaan ini dilakukan untuk satu hari analisis yang sama dan untuk beberapa hari analisis yang berbeda. Hasil pengukuran keseksamaan dinyatakan sebagai simpangan baku relatif (SBR), dimana kriteria keberterimaannya SBR < 2% (Harmita, 2004).
27
28
BAB V HASIL DAN PEMBAHASAN
Keseragaman kandungan didefinisikan sebagai derajat keseragaman jumlah zat aktif dalam tiap satuan sediaan. Untuk memastikan konsistensi dosis glipizid per unit, masing-masing tablet harus mengandung zat aktif sesuai dengan rentang yang dipersyaratkan dalam label (The USP, 2012). Pengembangan prosedur analisis menggunakan KCKT dilakukan untuk mendapatkan prosedur analisis yang mampu menetapkan keseragaman kandungan tablet glipizid sediaan ER. Sistem KCKT pada penelitian ini diadopsi dari penetapan kadar glipizid tablet biasa yang tercantum dalam Suplemen I Farmakope Indonesia Edisi IV, tahun 2009 seperti yang tercantum pada tabel V.1.
Tabel V.1. Perbandingan prosedur analisis glipizid IR dan ER
Parameter Kondisi KCKT untuk Sediaan Tablet Glipizid IR Fase gerak Dapar natrium fosfat
monobasa 13,8 gram/liter : Preparasi sampel Dilarutkan langsung dalam
fase gerak
Ektraksi fase padat
29
pelarut yang sesuai dan diukur secara KCKT (Venkata R, dkk, 2011). Ekstraksi glipizid dalam plasma darah manusia juga pernah dilakukan dengan terlebih dahulu mengendapkan protein plasma menggunakan HCl, dilanjutkan ekstraksi pelarut menggunakan toluen, kemudian fase organik diuapkan dan direkonstitusi dengan fase gerak kemudian diukur secara KCKT (Atif, dkk, 2013). Kedua penelitian tersebut diperoleh hasil yang memenuhi persyaratan validasi. Hasil dari ekstraksi pelarut menggunakan diklorometan untuk glipizid dengan matriks tablet ER didapatkan efisiensi ekstraksi di bawah 80% dan hasil perolehan kembali yang tidak konsisten dengan SBR > 2%.
Selanjutnya dilakukan ekstraksi secara SPE berdasarkan penelitian sebelumnya yang memisahkan campuran delapan obat antidiabetes termasuk salah satunya glipizid, dalam matriks plasma darah manusia secara SPE dan dilanjutkan dengan KCKT juga diperoleh hasil yang memenuhi persyaratan parameter validasi (Lakshmi dan Rajesh, 2011). Sehingga untuk glipizid dalam matriks tablet ER ini dilakukan ekstraksi secara SPE dan didapatkan hasil efisiensi ekstraksi di atas 80%, hasil perolehan kembali yang konsisten dengan SBR < 2%.
30 baku induk glipizid dalam metanol, plasebo dalam dapar fosfat
Setelah didapatkan pelarut yang sesuai, berikutnya menentukan jenis sorben dengan kondisi ekstraksi yang sama (pelarut dan pH). Parameter yang diamati meliputi jumlah lempeng teoritis, faktor ikutan, resolusi dan persen perolehan kembali. Pemilihan sorben ekstraksi fase padat yang sesuai tergantung pada mekanisme interaksi antara sorben dengan analit.
31
adalah tC18, HLB, MAX dan MCX. Hal mendasar yang membedakan jenis sorben tersebut diantaranya sifat fisika kimia sorben, ukuran massa sorben, ukuran partikel dan ukuran pori. Hasil optimasi pemilihan sorben SPE bisa dilihat pada tabel V.2.
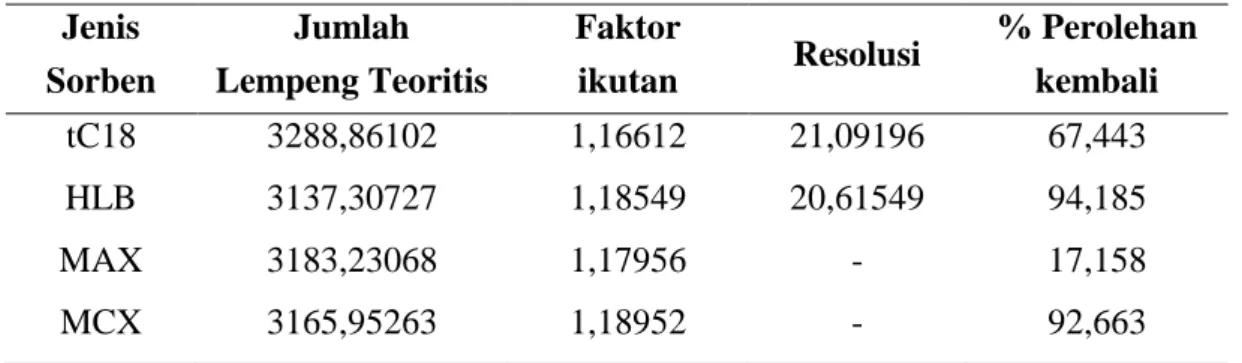
Tabel V.2 Hasil optimasi pemilihan sorben SPE
Jenis
tC18 3288,86102 1,16612 21,09196 67,443
HLB 3137,30727 1,18549 20,61549 94,185
MAX 3183,23068 1,17956 - 17,158
MCX 3165,95263 1,18952 - 92,663
Penggunaan sorben HLB menunjukkan hasil yang paling optimum dari sisi persen perolehan kembali dan lebih selektif karena bisa mendeteksi 2 puncak dengan resolusi > 1,5 bila dibandingkan dengan sorben tC18, MAX dan MCX. Keempat jenis sorben yang dioptimasi merupakan sorben dengan tipe fase terbalik, dimana fase diam bersifat non polar. Fase diam pada sorben HLB berupa copolymer, yakni N-vinylpirolidone yang bersifat hidrofilik dan divinylbenzene yang bersifat lipofilik dalam jumlah yang seimbang (Anonim, 2014). Ditinjau secara struktur kimia, gugus utama glipizid berupa sulfonilurea merupakan moietas hidrofilik dan substitusi R1 dan R2 sebagai moietas hidrofobik, dengan demikian sorben HLB paling selektif untuk analit glipizid. Analisis yang lain, karena matriks hidrofilik akan mengembang jika bertemu dengan air, maka pada saat tahap loading, dimungkinkan matriks teretensi secara irreversibel dalam sorben dan tidak terelusi saat washing sehingga hanya glipizid yang terelusi dengan metanol pada tahap
akhir SPE.
32
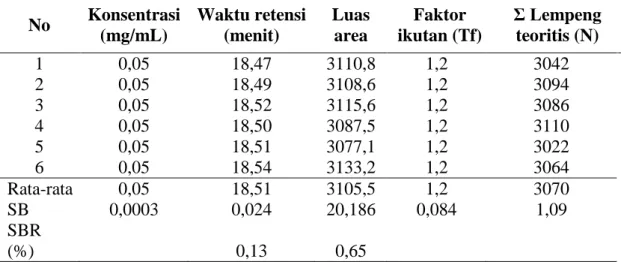
keberulangan penyuntikan, waktu retensi, luas area, faktor ikutan dan jumlah lempeng teoritis. Hasil uji kesesuaian sistem dapat dilihat pada tabel V.3.
Tabel V.3 Data hasil uji kesesuaian sistem larutan baku glipizid 0,05 mg/ml
No Konsentrasi (mg/mL)
Waktu retensi (menit)
Luas area
Faktor ikutan (Tf)
Σ Lempeng teoritis (N)
1 0,05 18,47 3110,8 1,2 3042
2 0,05 18,49 3108,6 1,2 3094
3 0,05 18,52 3115,6 1,2 3086
4 0,05 18,50 3087,5 1,2 3110
5 0,05 18,51 3077,1 1,2 3022
6 0,05 18,54 3133,2 1,2 3064
Rata-rata 0,05 18,51 3105,5 1,2 3070
SB 0,0003 0,024 20,186 0,084 1,09
SBR
(%) 0,13 0,65
Tabel V.3 menunjukkan hasil keberulangan penyuntikan larutan baku glipizid 0,05 mg/mL sebanyak 6 kali. Nilai SBR dari area dan waktu retensi secara berturut-turut adalah 0,65% dan 0,13%. Hal ini memenuhi persyaratan keberulangan penyuntikan yaitu SBR lebih kecil dari 2%. Faktor ikutan sebesar 1,2, hal ini menunjukan bahwa bentuk puncak cukup simetris karena nilai faktor ikutan mendekati 1 (Tf < 2). Jumlah lempeng teoritis (N) lebih besar dari 2000, hal ini menunjukan efisiensi kolom baik (CDER,1994). Kromatogram baku glipizid ditunjukkan pada gambar V.2 dengan waktu retensi 18,65 menit.
33
Setelah memenuhi persyaratan uji kesesuaian sistem kemudian dilakukan validasi berdasarkan pedoman International Conference of Harmonization (ICH) dengan parameter spesifisitas/selektivitas, linearitas, batas deteksi, batas kuantisasi, presisi intra dan antar hari dan akurasi.
Pengujian spesifisitas dilakukan untuk memastikan bahwa metode analisis yang digunakan spesifik untuk analit tertentu dan tidak terganggu dengan adanya pelarut, matriks maupun keberadaan zat selain analit. Spesifisitas dalam penelitian ini ditentukan dengan membandingkan antara pelarut, plasebo, larutan baku, dan larutan sampel.
(a) (b)
(c) (d)
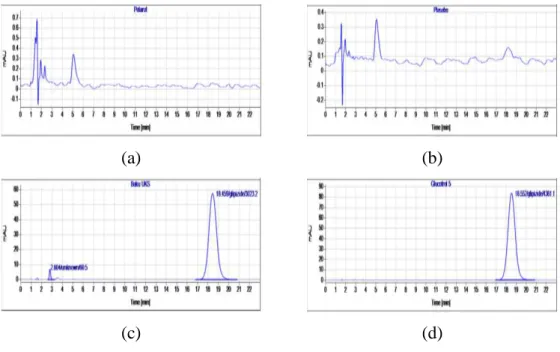
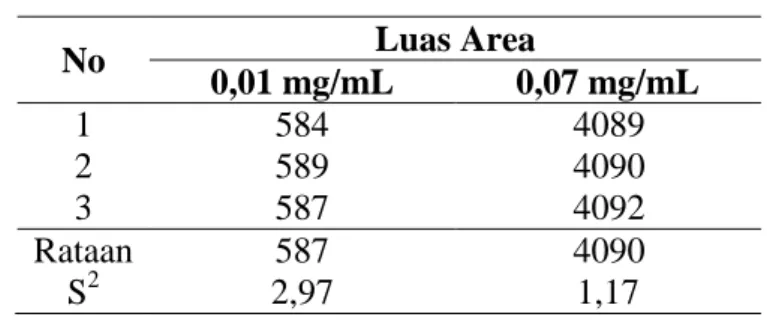
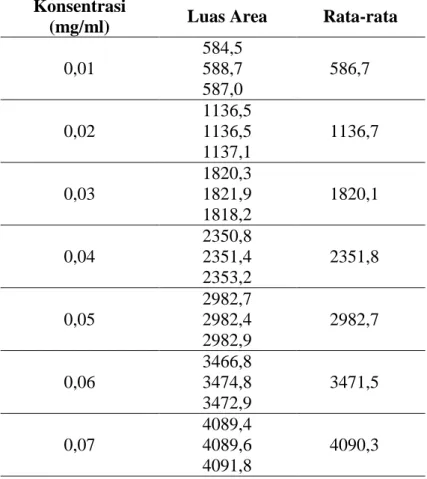
Gambar V.3 Kromatogram (a) pelarut (b) larutan plasebo (c) baku induk glipizid (d) plasebo yang di-spike.
Pada gambar V.3 terlihat bahwa pada kromatogram pelarut dan plasebo tidak terdapat puncak dengan waktu retensi yang sama dengan baku glipizid. Hal ini mengindikasikan bahwa tidak ada gangguan dari pelarut maupun matriks dan prosedur analisis yang dikembangkan spesifik untuk glipizid.
34
dengan respon instrumen yang dinyatakan dengan luas area, yang terdiri dari 7 level konsentrasi dan masing-masing level konsentrasi disuntikkan sebanyak 6 kali. Sebagai parameter adanya hubungan linier digunakan koefisien korelasi (r) dan koefisien variansi fungsi regresi pada analisis regresi linier y = bx + a. Berdasarkan kurva kalibrasi yang diperoleh, dilakukan perhitungan dan uji kaji statistik meliputi homogenitas variansi, linieritas, batas deteksi, dan batas kuantisasi menggunakan metode yang digunakan oleh Gottwald (Gottwald, 2000).
Respon luas area yang digunakan untuk pembuatan kurva kalibrasi tidak boleh menunjukkan perbedaan homogenitas variansi pada berbagai konsentrasi larutan baku kerja yang digunakan. Untuk menguji homogenitas variansi, dilakukan pengujian F terhadap larutan baku kerja konsentrasi terendah dan tertinggi dengan cara menghitung variansi (S2 ) dari pengulangan pengukuran larutan baku dengan konsentrasi terendah dan tertinggi, kemudian ditentukan Parameter Uji (PU):
PU = 2
PG dibandingkan dengan nilai F tabel.
35
perhitungan semua parameter regresi kuadratik maupun linear kemudian parameter tersebut dibandingkan.
Tabel V.5 Konsentrasi dan luas area sampel simulasi glipizid
Konsentrasi
36
Persamaan regresi linier yang diperoleh adalah y = 58985,35 x + 13,88 dengan koefisien korelasi r = 0,9995.
Tabel V.6 Parameter regresi linier dan kuadratik glipizid
Keterangan Linier Kuadratik
Persamaan regresi y = 58985,35 x + 13,88
y = -52361,29x2 + 63130,32x – 47,64 Kemiringan garis regresi/slope (b) 58985,32 -
Perpotongan garis dengan sumbu y (a)
13,88 -
X rata-rata 0,0396 0,0396
Sy/x (simpangan baku residu) 44,191 43,463
(Sy/x)/b 0,001 0,001
Vx0 /koefisien variansi regresi (%) 1,893 1,862
E (sensitivitas) - 58985,32
r (koefisien korelasi) 0,9995 -
LOD/ Batas Deteksi (mg/mL) 0,0025 -
LOQ/ Batas Kuantisasi (mg/mL) 0,0075 -
Dari gambar kurva linearitas glipizid V.4 dan tabel diatas dapat dilihat bahwa parameter regresi linier pada rentang konsentrasi 0,01 - 0,07 mg/mL seluruhnya menunjukkan hasil yang baik dengan persamaan garis regresi y = 58985,35 x + 13,88, nilai Vx0 1,893 dan koefisien korelasi 0,9995. Sedangkan hasil perhitungan
parameter regresi kuadratik menghasilkan persamaan kuadratik y = -361,29x2 + 63130,31x – 47,64 dengan nilai Vx0 1,862. Pada tabel dapat dilihat bahwa Vxo
regresi linear > Vxo regresi kuadratik yang menunjukkan bahwa distribusi titik kalibrasi lebih sesuai mengikuti regresi kuadratik, maka diperlukan uji linearitas lanjutan dan kurva regresi linier tidak dapat digunakan untuk kurva kalibrasi. Batas deteksi (BD) dan batas kuantisasi (BK) dihitung secara statistik dari kurva kalibrasi menggunakan metode Deutsches Institut für Normung (DIN) 38402 yaitu BD 0,0025 mg/mL dan BK 0,0075 mg/mL.
37
Uji presisi dilakukan intra dan antar hari selama tiga hari berturut-turut dan dihitung simpangan baku relatifnya harus memenuhi syarat keberterimaan < 2%.
Tabel V.7 Presisi intra hari glipizid dalam sampel simulasi
No
Waktu retensi (menit) Luas Area Kadar (%)
Hari ke Hari ke Hari ke
1 2 3 1 2 3 1 2 3
1 18,57 18,27 19,33 2837 3074 2957 95,00 100,66 97,18 2 18,61 18,18 19,32 2896 2991 2970 97,01 97,93 97,61 3 18,54 18,27 19,33 2849 3022 2976 95,41 98,94 97,81 4 18,55 18,24 20,27 2875 2950 3018 96,28 96,60 99,17 5 18,75 18,22 20,26 2846 2987 2948 95,32 97,82 96,90 6 18,53 18,56 19,84 2893 2993 2954 96,90 98,00 97,10
Rata-rata kadar (%) 95,99 98,32 97,63
SB 0,86 1,37 0,83
SBR (%) 0,90 1,40 0,86
Dari seluruh hasil presisi diatas diperoleh SBR berturut-turut pada hari ke-1, ke-2 dan ke-3 adalah sebesar 0,90%, 1,40% dan 0,86%, dimana seluruhnya lebih kecil dari 2,0% sehingga dikatakan bahwa metode yang digunakan memenuhi syarat presisi.
Tabel V.8 Presisi antar hari glipizid dalam sampel simulasi
Hari ke Kadar (%)
1 95,99
2 98,32
3 97,63
Rata-rata 97,31
SB 1,20
SBR (%) 1,23
38
Selanjutnya dilakukan uji akurasi menggunakan larutan plasebo yang ditambahkan baku dengan tiga rentang konsentrasi yaitu 70, 100 dan 130%. Hasil akurasi bisa di lihat dalam tabel V.9.
Tabel V.9 Akurasi glipizid dalam sampel simulasi
No Persentase Baku (%)
Luas Area
Analit sebenarnya (mg)
Analit diperoleh (mg)
% Perolehan kembali
1 70 2049,63 1,77 1,70 95,88
2 70 2076,60 1,77 1,72 97,14
3 70 2117,33 1,77 1,75 100,97
4 100 2951,93 2,53 2,44 103,46
5 100 2971,80 2,53 2,46 102,77
6 100 2992,97 2,53 2,47 102,04
7 130 3939,53 3,28 3,26 100,78
8 130 3907,40 3,29 3,23 101,61
9 130 3912,60 3,29 3,23 101,47
Rata-rata 100,68
Dari tabel V.9 dapat dilihat bahwa nilai persen perolehan kembali berkisar antara 95,888 – 103,46% dengan rata-rata 100,68%. Syarat persen perolehan kembali untuk glipizid 0,05 mg/mL adalah 90 – 107%. Hasil ini menunjukkan bahwa prosedur analisis yang digunakan memenuhi syarat akurasi.
39
Tabel V.10 Uji keseragaman kandungan tablet Glipizid ER 5 mg
Uraian Bobot (mg) fp Kons
Tabel V.11 Uji keseragaman kandungan tablet Glipizid ER 10 mg
40
41
BAB VI KESIMPULAN
Prosedur analisis keseragaman kandungan glipizid ER dapat dilakukan secara KCKT dengan sistem : fase gerak campuran dapar fosfat monobasa 0,1M pH 6,00 ± 0,05 dan metanol (55:45), laju alir 1,0 mL/menit, suhu kolom 30ºC dan detektor UV pada 225 nm dengan kolom YMC Triart C18 (150 x 4,6 mm, ID S-5 µm 12 nm); didahului dengan preparasi ekstraksi fase padat SPE – menggunakan sorben jenis HLB, pengkondisian dengan 1 mL metanol dan 1 ml aquadest, loading
sampel 1 mL, pembilasan dengan 1 mL aquadest dan pengelusian dengan 1 mL metanol. Hasil pengembangan dan validasi metode analisis tersebut memenuhi syarat keberterimaan dengan data linearitas y = 58985,35x + 13,88; r = 0,9995; batas deteksi 0,0025 mg/mL, dan batas kuantisasi 0,0075 mg/mL; SBR presisi 1,232% dan rata-rata % recovery 100,68%. Prosedur analisis ini dapat digunakan pada pengujian keseragaman kandungan produk obat glipizid ER yang beredar di pasaran.
42
DAFTAR PUSTAKA
Ahuja, S., M. W. Dong (2005) : Handbook of Pharmaceutical Analysis by HPLC, Elsevier Academic Press, New York.
Allen, L.V., Popovich, N.G., Ansel, H.C. (2011) : Ansel’s Pharmaceutical
Dosage Forms and Drug Delivery Systems: Solid oral modified-release dosage forms and drug delivery systems, Wolter Kluwer, Lippincott Williams & Wilkins., Philadelpia, 9th Ed, p 257-270.
Anonim. (2014) : Care and Use Manual, Oasis HLB Catridges and 96 Well Plate, Waters Corporation, Milford, USA.
AOAC. (2012) : Guidelines for Standard Method Performance Requirements,
AOAC Official Methods of Analysis, Appendix F.
Atif, M., Khalid, S.H., Kit, G.L.Onn., Sulaiman, S.A.S., Asif, M. dan Chandersekaran., A. (2013) : Development and validation of RP-HPLC-UV method for the determination of glipizide in human plasma, Journal of Young Pharmacists, Reed Elsevier India Pvt. Ltd, 5 (2013) 26-29.
Brunton, L., Parker, K., Blumenthal, D., dan Buxton, I. (2008) : Goodman &
Gilman’s, Manual of pharmacology and therapeutics, McGraw-Hill
Companies, USA, p. 1039-1060.
Camel, Valeriae. (2003) : Solid Phase Extraction of Trace Elements in review, Elsevier, Spectrochimica Acta Part B 58 (2003) 1177 – 1233, France. Centre for Drug Evaluation and Research. (1994) : Reviewer Guidance.
Validation of Chromatographic Methods, p. 21-28.
Chan, C. (2004) : Analytical Methode Validation and Instrument Performance Verification. John Wiley and Sons, Inc. Publication, Canada.
Depkes. (2009) : Farmakope Indonesia Ed IV Suplemen I, hal 1425.
Ermer, J., dan J.H. McB Miller (2005) : Method Validation in Pharmaceutical Analylis. Wiley-VCH Verlag GmbH & Co. KgaA, Weinheim.
Gottwald, W. (2000) : Statistik fuer Anwender, 1. Auflage, Wiley-VCH, Weinheim, Bundesrepublik Deutschland, 2000, s. 89–146.
43
Harris, D.C. (2007) : Quantitative chemical analysis: Sample preparation, W.H. Freeman and Company, England, 7th Ed, 28 : 656-659.
Harmita. (2004) : Petunjuk Pelaksanaan Validasi Metode dan Cara Perhitungannya. Majalah Ilmu Kefarmasian, 1: 117-135.
Hennion, M.C. (1999) : Solid-phase extraction: method development, sorbents, and coupling with liquid chromatography, Journal of Chromatography A, Elsevier Science B.V., France, 856 (1999) 3-54.
Ibrahim, S. (2004) : Berbagai Pendekatan pada Penaksiran Batas Deteksi dan Batas Kuantisasi Suatu Metode Analisis Instrumental, Acta Pharm. Ind, 29 (4): 153-159.
Ibrahim, S. (2005) : Berbagai Pendekatan Pengujian Kelinieran Kurva Baku pada Metode Analisis Instrumental. Acta Pharm. Ind, 30 (1): 30-34.
ICH. (1994) : Validation of analytical procedures: text and methodology, International Conference on Harmonization, IFPMA, Geneva.
Kazakevich, Y. dan Lobrutto, R. (2007) : HPLC for Pharmaceutical Scientist, John Wiley & Sons, Inc., Canada, p.361-384.
Kobylinska M, Bukowska-Kiliszek M, Barlinska M, Sobik B dan Kobylinska K,. (2000) : Bioequivalence Study Of Two Brands Of Glipizide Tablets, Polish Pharmaceutical Society, Acta Poloniae Pharmaceutica – Drug Research, Poland, 57 (2) p.101-104.
Lakshmi, K.S., dan Rajesh, T. (2011) : Separation and quantification of eight antidiabetic drugs on a high-performance liquid chromatography: Its application to human plasma assay, ISRN Pharmaceutics, India, Vol 2011, ID 521353, p 7.
Maderuelo, C., Zarzuelo, A. dan Lanao, J.M. (2011) : Critical Factors In The Release Of Drug From Sustained Release Hydrophilic Matrices, Elsevier B.V., Spain, Journal of Controlled Release 154 (2011) p.2-19.
Nickerson, B. (2011) : Sample Preparation of Pharmaceutical Dosage Forms, Sample Preparation for Solid Oral Dosage Form, Springer, New York, 7, p.145–174.
Qiu, Y., Zhang, G. (2000) : Research and development aspects of oral controlled-release dosage forms. In : Handbook of Pharmaceutical Controlled Release Technology, Wise DL. Eds. Marcel Dekker Inc.