Practical Immunology, Fourth Edition
Bebas
409
0
0
Teks penuh
Gambar
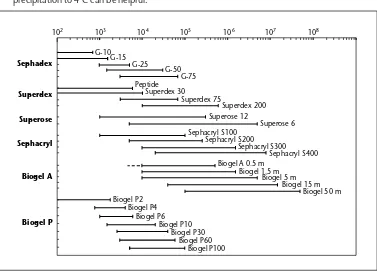
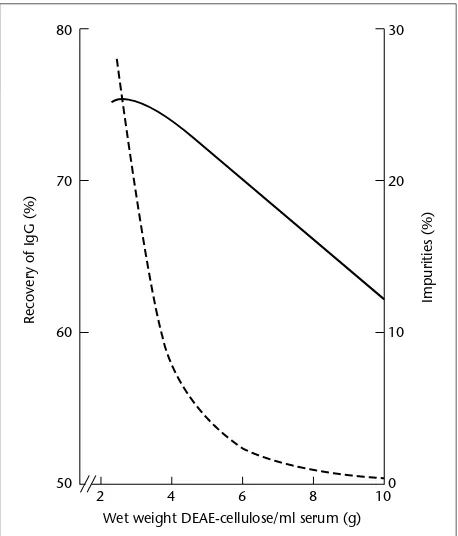
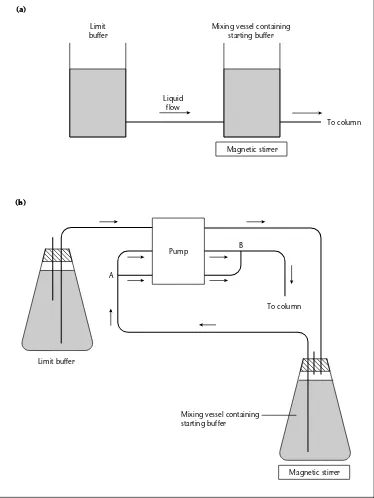

+7
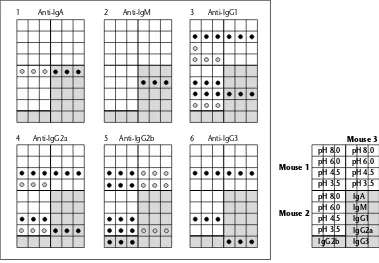
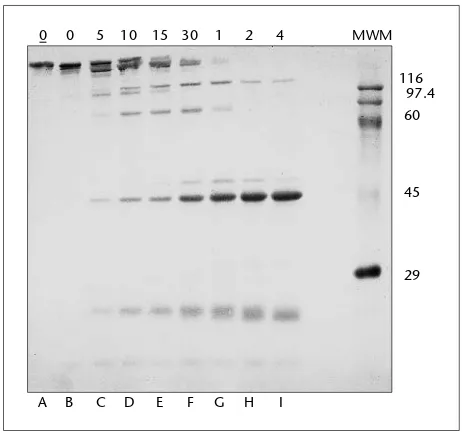
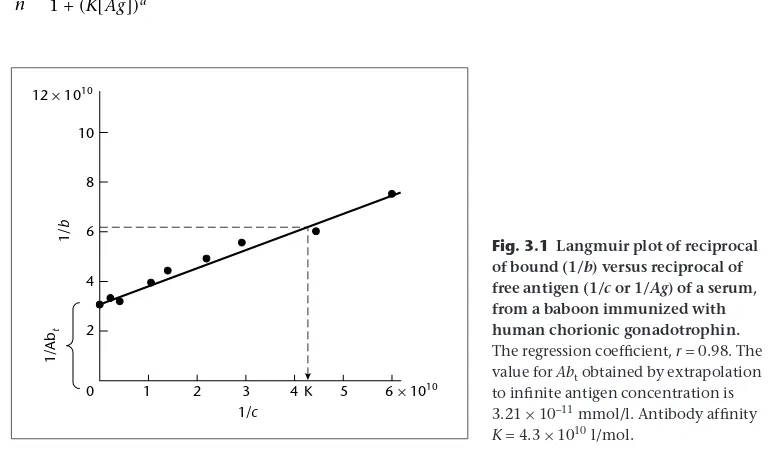
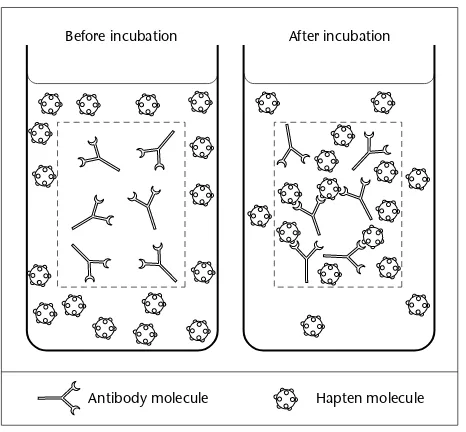


Garis besar
Dokumen terkait
