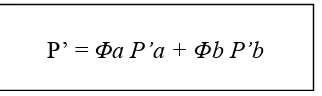i
VALIDASI METODE PENETAPAN KADAR KURKUMIN DALAM SEDIAAN CAIR OBAT HERBAL TERSTANDAR MERK KIRANTI® SECARA KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Farmasi
Oleh: Marsella Widjaja NIM: 078114030
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
i SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Farmasi
Oleh: Marsella Widjaja NIM: 078114030
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
ii
Persetujuan Pembimbing
VALIDASI METODE PENETAPAN KADAR KURKUMIN DALAM SEDIAAN CAIR OBAT HERBAL TERSTANDAR MERK KIRANTI® SECARA KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK
Skripsi yang diajukan oleh: Marsella Widjaja NIM : 078114030
telah disetujui oleh:
Pembimbing
iv
Karya untuk yang terkasih . . .
TUHAN-ku, ALLAH yang Maha Mulia;
Keluargaku tersayang;
v
PERNYATAAN KEASLIAN KARYA
Saya menyatakan dengan sesungguhnya bahwa skripsi yang telah saya tulis ini tidak memuat karya atau bagian orang lain, kecuali yang telah disebutkan dalam kutipan dan daftar pustaka, sebagaimana layaknya karya ilmiah.
Apabila di kemudian hari ditemukan indikasi plagiarisme dalam naskah ini, maka saya bersedia menanggung segala sanksi sesuai peraturan perundang-undangan yang berlaku.
Yogyakarta, 29 Desember 2010 Penulis
vi
LEMBAR PERNYATAAN PERSETUJUAN PUBLIKASI KARYA ILMIAH UNTUK KEPENTINGAN AKADEMIS
Yang bertanda tangan di bawah ini, saya mahasiswa Universitas Sanata Dharma: Nama : Marsella Widjaja
Nomor Mahasiswa : 07 8114 030
Demi perkembangan ilmu pengetahuan, saya memberikan kepada Perpustakaan Universitas Sanata Dharma karya ilmiah saya yang berjudul:
VALIDASI METODE PENETAPAN KADAR KURKUMIN DALAM SEDIAAN CAIR OBAT HERBAL TERSTANDAR MERK KIRANTI® SECARA KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK beserta perangkat yang diperlukan (bila ada). Dengan demikian saya memberikan kepada Perpustakaan Universitas Sanata Dharma hak untuk menyimpan, mengalihkan dalam bentuk media lain, mengelolanya dalam bentuk pangkalan data, mendistribusikan secara terbatas, dan mempublikasikannya di internet atau media lain untuk kepentingan akademis tanpa perlu meminta ijin dari saya ataupun memberi royalti kepada saya selama tetap mencantumkan nama saya sebagai penulis.
Demikian pernyataan ini yang saya buat dengan sebenarnya. Dibuat di Yogyakarta
Pada tanggal: 4 Februari 2011 Yang menyatakan
vii PRAKATA
Segala hormat dan puji syukur penulis panjatkan kepada Tuhan yang Maha Pengasih atas hikmat, berkat, kasih karunia, dan penyertaan-Nya sehingga skripsi yang berjudul “Validasi Metode Penetapan Kadar Kurkumin dalam Sediaan Cair Obat Herbal Terstandar Merk Kiranti® secara Kromatografi Cair Kinerja Tinggi Fase Terbalik” dapat diselesaikan dengan baik. Skripsi ini disusun untuk memenuhi salah satu syarat memperoleh gelar Sarjana Farmasi (S. Farm.) di Fakultas Farmasi Universitas Sanata Dharma, Yogyakarta.
Proses penyusunan skripsi ini tidak lepas dari bantuan dan dukungan banyak pihak, oleh karena itu pada kesempatan ini penulis ingin mengucapkan terima kasih sebesar-besarnya kepada:
1. Ipang Djunarko, M.Sc., Apt. selaku Dekan Fakultas Farmasi Universitas Sanata Dharma Yogyakarta sekaligus dosen pembimbing akademik penulis. 2. Christine Patramurti, M.Si., Apt. selaku dosen pembimbing yang telah
banyak meluangkan waktu, mendampingi dan memberi masukan, kritik, solusi, serta semangat kepada penulis selama penelitian dan penyusunan skripsi. Terima kasih untuk ilmu dan pengalaman yang dibagikan kepada penulis.
3. Jefrry Julianus, M.Si. selaku dosen penguji atas saran dan kritik yang diberikan. Terima kasih untuk “semangat yang tidak pernah padam”.
viii
5. Rini Dwi Astuti, M.Sc., Apt. selaku Kepala Laboratorium Farmasi Universitas Sanata Dharma yang telah memberikan ijin kepada penulis untuk melakukan penelitian di laboratorium.
6. Prof. Dr. Sudibyo Martono, M.S., Apt. yang telah memberikan senyawa baku untuk penelitian yang dilakukan oleh penulis.
7. C.M. Ratna Rini Nastiti, M. Pharm., Apt. dan Phebe Hendra, M.Si., Ph.D., Apt. untuk bantuan, semangat, dan perhatian yang diberikan.
8. Seluruh staf laboratorium kimia: Mas Bimo, Pak Parlan, dan Mas Kunto yang telah membantu penulis selama penelitian di laboratorium.
9. Staf sekretariat Farmasi: Mas Dwi, Pak Mukimin, dan Mas Narto atas bantuanya.
10. Teman seperjuangan, Nana “Upil” dan “Pakde” Toro atas kebersamaan, kerja sama, dan bantuan yang diberikan. Terima kasih untuk semua masukan dan kritik.
11. Katrin, Benny, Pace, Tere, Seno, Lilis, Eliz, Yunita, dan Veny atas dukungan dan kebersamaan selama penelitian di laboratorium.
12. Fransisca Ayu Ningtyas Wiranti yang selalu setia mendengar setiap keluh kesah penulis. Terima kasih sudah menjadi sahabat yang baik.
13. Kelompok belajar malam: Dika, Ius, Daniel, Yudi, dan Wawan untuk kebersamaan yang indah di tiap malam selama ujian. Harus tetap semangat, teman.
ix
menempuh studi S1 di Farmasi USD. Terima kasih untuk suka duka yang pernah terjadi.
15. Teman-teman FST 2007 atas hari-hari penuh kebersamaan yang tidak akan pernah terlupakan.
16. Teman-teman satu atap di Kost Putri Wulandari yang telah menjadi keluraga baru bagi penulis selama menempuh studi di Yogyakarta.
17. Eurike Chrtistiani Hutauruk dan Andrias Pratiwi yang telah menjadi saudara yang baik bagi penulis. Terima kasih untuk kasih sayang dan motivasi yang tidak pernah putus.
18. Debora Inggraini dan Ade William Widjaja yang selalu menjadi motivasi bagi penulis.
19. Semua orang yang mungkin tidak dapat disebutkan satu per satu oleh penulis, terima kasih atas semua bantuan yang telah diberikan.
x DAFTAR ISI
HALAMAN JUDUL ...……….. i
HALAMAN PERSETUJUAN PEMBIMBING ……… ii
HALAMAN PENGESAHAN ………... iii
HALAMAN PERSEMBAHAN ……… iv
PERNYATAAN KEASLIAN KARYA ……… v
PERNYATAAN PERSETUJUAN PUBLIKASI ILMIAH ………. vi
xi
B. Obat Herbal Terstandar ………. 9
C. Sediaan Cair ………... 10
D. Spektrofotometri Visibel ……… 11
E. Kromatografi Cair Kinerja Tinggi ………. 14
1. Definisi ………. 14
2. Instrumentasi ……… 16
3. Pemisahan puncak dalam kromatografi ………. 20
4. Pengembangan metode KCKT ………. 22
F. Validasi Metode Analisis ……….. 23
1. Selektivitas ………... 24
BAB III. METODE PENELITIAN ……….. 31
A. Jenis dan Rancangan Penelitian ……… 31
B. Variabel Penelitian ……… 31
C. Definisi Operasional ………. 32
D. Bahan-bahan Penelitian ……… 32
E. Alat-alat Penelitian ………. 32
F. Tata Cara Penelitian ……….. 33
xii
2. Pembuatan pelarut metanol pH 4 ……… 33
3. Pembuatan larutan baku kurkumin ………. 34
4. Penentuan panjang gelombang maksimum kurkumin ….. 34
5. Preparasi sampel ………... 35
6. Validasi metode analisis ………... 35
G. Analisis Hasil ………. 37
B. Pembuatan Larutan Baku Kurkumin ……… 40
C. Penentuan Panjang Gelombang Maksimum Kurkumin ……. 41
D. Pengamatan Waktu Retensi (tR) Kurkumin ……… 43
E. Pembuatan Kurva Baku Kurkumin ……… 46
F. Validasi Metode Analisis ………... 48
xiii
B. Saran ………... 55
DAFTAR PUSTAKA ……… 56
LAMPIRAN ………... 61
xiv
DAFTAR TABEL
Tabel I. Nilai indeks polaritas beberapa pelarut pada KCKT fase
terbalik ……….. 19
Tabel II. Kategori metode pengujian ………... 23
Tabel III. Parameter analisis yang dibutuhkan dalam validasi metode analisis ………... 24
Tabel IV. Kriteria rentang recovery yang dapat diterima ……... 27
Tabel V. Kriteria akurasi dan presisi yang masih dapat diterima …… 29
Tabel VI. Data perolehan AUC seri baku kurkumin ……….... 47
Tabel VII. Hasil perhitungan resolusi (Rs) ……….... 49
Tabel VIII. Hasil penetapan recovery baku kurkumin ……….... 50
Tabel IX. Hasil penetapan recovery baku yang diadisi ……….... 53
xv
DAFTAR GAMBAR
Gambar 1. Struktur kurkumin ……… 6
Gambar 2. Reaksi degradasi kurkumin dalam suasana alkali ………… 7
Gambar 3. Logo OHT ……… 10
Gambar 4. Diagram tingkat energi elektronik ……… 12
Gambar 5. Diagram alir instrumentasi spektrofotometer visibel ……... 13
Gambar 6. Reaksi silanisasi ……… 16
Gambar 7. Reaksi pembuatan kolom oktadesilsilan ……….. 16
Gambar 8. Skema instrumen KCKT ……….. 17
Gambar 9. Pemisahan dua puncak ………. 21
Gambar 10. Langkah pengembangan metode KCKT ……….. 22
Gambar 11. Reaksi degradasi kolom C18 dalam suasana asam ………… 39
Gambar 12. Gugus metilen aktif pada kurkumin ………. 40
Gambar 13. Gugus kromofor dan auksokrom pada kurkumin …………. 41
Gambar 14. Spektra panjang gelombang maksimum kurkumin ……….. 42
Gambar 15. Gugus polar dan nonpolar kurkumin ……… 44
Gambar 16. Interaksi kurkumin dengan fase gerak metanol : asam asetat glasial 2% ……….. 44
Gambar 17. Interaksi kurkumin dengan fase diam oktadesilsilan (C18) ... 45
Gambar 18. Kromatogram baku kurkumin dan sampel ………... 46
Gambar 19. Kurva baku kurkumin ………... 48
xvi
DAFTAR LAMPIRAN
Lampiran 1. Pernyataan jaminan keaslian bahan kurkumin standar hasil sintesis ………... 62 Lampiran 2. Data penimbangan bahan ………... 63 Lampiran 3. Spektra panjang gelombang maksimum kurkumin ……… 64 Lampiran 4. Kromatogram baku kurkumin ……… 65 Lampiran 5. Contoh perhitungan konsentrasi baku kurkumin ………... 76 Lampiran 6. Perolehan AUC seri baku kurkumin ……….. 77 Lampiran 7. Persamaan dan gambar kurva baku kurkumin …………... 78 Lampiran 8. Kromatogram baku kurkumin untuk validasi metode …… 79 Lampiran 9. Perolehan nilai AUC dan contoh perhitungan konsentrasi
terukur baku kurkumin ………... 87 Lampiran 10. Contoh perhitungan persen perolehan kembali (recovery)
dan koefisien variasi (KV) baku kurkumin ……… 88 Lampiran 11. Kromatogram sampel dan sampel adisi ………. 89 Lampiran 12. Perolehan nilai AUC sampel dan sampel adisi, contoh
perhitungan konsentrasi terukur, perhitungan recovery
dan KV baku kurkumin adisi ………. 94 Lampiran 13. Contoh perhitungan resolusi pemisahan kurkumin dalam
sampel ……… 96
xvii
VALIDASI METODE PENETAPAN KADAR KURKUMIN DALAM SEDIAAN CAIR OBAT HERBAL TERSTANDAR MERK KIRANTI® SECARA KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK
INTISARI
Kurkumin merupakan senyawa alam yang banyak terkandung dalam obat tradisional. Kurkumin dapat ditetapkan kadarnya menggunakan metode Kromatografi Cair Kinerja Tinggi (KCKT) fase terbalik. Untuk menjamin bahwa karakteristik kinerja metode yang digunakan memenuhi persyaratan aplikasi analitik, maka perlu dilakukan tahap validasi terlebih dahulu.
Penelitian yang dilakukan bersifat non eksperimental deskriptif. Kurkumin dianalisis secara kuantitatif menggunakan sistem KCKT fase terbalik dengan detektor visibel pada panjang gelombang 432 nm menggunakan fase diam oktadesilsilan (C18) dan fase gerak metanol : asam asetat glasial 2 % (90:10 v/v) dengan kecepatan alir 0,5 ml/menit.
xviii
VALIDATION OF CURCUMIN QUANTIFICATION METHOD IN LIQUID DOSAGE FORM OF STANDARDIZED HERBAL MEDICINE
KIRANTI® USING HIGH PERFORMANCE LIQUID CHROMATOGRAPHY REVERSE PHASE
ABSTRACT
Curcumin is a natural substance contained in many traditional medicines. Curcumin can quantify using High Performance Liquid Chromatography (HPLC) reversed phase. To ensure that the performance characteristics of the methods used meet the requirements for analytic applications, it is necessary to advance the validation stage.
Research conducted in non experimental descriptive. Curcumin was analyzed quantitatively using reverse phase HPLC system with visible detector at a wavelength of 432 nm using octadecylsylane stationary phase (C18) and a mobile phase of methanol: 2% glacial acetic acid (90:10 v / v) with flow rate 0.5 ml / minutes.
The validity of the method used is indicated by the parameters selectivity, linearity, accuracy, and precision. The results showed that the method has good selectivity with the resolution (Rs) 1.4383 and a good linearity with correlation coefficient (r) of 0.9992 at a concentration of 1.515 to 4.545 ppm. Recovery value and CV for raw curcumin at the concentration 1.515 ppm, 3.030 ppm and 4.545 ppm respectively 101.9208-107.5049% and 2.3435%, 99.1947-101.9703% and 1.1346%, 92.3524-108.4202% and 5.8678%, while for the raw curcumin which added in the sample is from 102.9600-106.8267% and 1.4504%. Based on these results, the method of determination of curcumin in liquid dosage form of standardized herbal medicine Kiranti® using reverse phase HPLC has good validity.
1 BAB I PENGANTAR
A. Latar Belakang
Semakin banyak manusia yang memilih gaya hidup back to nature
berpengaruh terhadap meningkatnya minat konsumsi obat tradisional. Hal ini mendorong dilakukannya pengembangan terhadap obat tradisional sehingga penggunaannya dapat diterima dalam pengobatan formal di kalangan masyarakat. Salah satu hasil pengembangan obat tradisional tersebut adalah obat herbal terstandar (OHT) yang khasiat dan keamanannya telah terbukti secara ilmiah melalui uji praklinik dan bahan bakunya telah distandarisasi.
Sebagian besar sediaan OHT yang beredar di pasaran banyak mengandung kurkumin sebagai kandungan utamanya. Salah satunya adalah sediaan cair OHT merk Kiranti® yang banyak dikonsumsi masyarakat dan sangat dipercaya berkhasiat sebagai anti nyeri. Akan tetapi, dibalik khasiat yang dimiliki, kurkumin bermasalah dalam stabilitasnya. Kurkumin akan terdegradasi pada pH di atas 6,5 atau karena terpapar cahaya berlebih (Tonnesen dan Karlsen, 1985b). Karena sifatnya yang tidak stabil tersebut, maka besar kemungkinan terjadinya penurunan kandungan kurkumin selama proses distribusi dan penyimpanan sehingga perlu dilakukan upaya penjaminan keseragaman kadar sediaan. Untuk itu, diperlukan suatu metode analisis yang tepat.
memiliki sensitivitas dan selektivitas yang lebih baik dibandingkan dengan metode analisis yang lain. Hal ini ditandai oleh kemampuan daya pisah yang baik dan kemampuan untuk mendeteksi analit dalam jumlah kecil.
Beberapa penelitian analisis kurkumin secara KCKT yang pernah dilakukan antara lain menggunakan fase diam C18 serta fase gerak campuran asetonitril dan asam trifluoro asetat dengan detektor visibel, fase diam C18 dengan detektor UV, fase diam Nucleosil NH2 dengan detektor UV, serta fase diam
amino-bonded dengan detektor visibel. Hal yang membedakan penelitan ini dengan beberapa penelitian sebelumnya terletak pada fase gerak yang digunakan. Pada penelitian ini fase gerak yang digunakan merupakan campuran metanol dan asam asetat glasial 2%. Metanol dan asam asetat glasial 2% dipilih karena kurkumin memiliki kelarutan yang baik dalam keduanya sehingga dapat mengelusi kurkumin dari kolom lebih cepat.
kinerja metode yang perlu dipertimbangkan dalam validasi meliputi selektivitas, linearitas, akurasi, dan presisi(United States Pharmacopeial Convention, 2007).
1. Permasalahan
Berdasarkan uraian latar belakang tersebut, maka permasalahan yang muncul adalah apakah metode penetapan kadar kurkumin dalam sediaan cair OHT merk Kiranti® secara KCKT fase terbalik menggunakan fase diam C
18 dan fase gerak metanol : asam asetat glasial 2% dengan perbandingan 90:10 (v/v) memenuhi parameter validitas yang baik, meliputi selektivitas, linearitas, akurasi, dan presisi?
2. Keaslian penelitian
Sakariah, 2002), KCKT dengan kolom ODS menggunakan detektor UV (Smith dan Witowska, 1984), KCKT menggunakan kolom HiQ-Sil C18 (Rungphanichkul, 2004), KCKT menggunakan kolom C18 detektor UV (Heath, Pruitt, Brenner, Begun, Frautschy, dan Roch, 2005), KCKT fase terbalik dengan detektor visibel (Jadhav, Mahadik, dan Paradkar, 2007), KCKT dengan kolom amino-bonded
detektor visibel (Sumule, 2007), kromatografi high-speed countercurrent (Inoue, Nomura, Ito, Nagatsu, Hino, dan Oka, 2008), Kromatografi Lapis Tipis Kinerja Tinggi (KLTKT) dengan detektor visibel (Paramasivam, Aktar, Poi, Banerjee, dan Bandyopadhyay, 2008). Akan tetapi sejauh pengamatan penulis, validasi metode penetapan kadar kurkumin dalam sediaan cair OHT merk Kiranti® secara KCKT fase terbalik dengan fase diam C18 dan fase gerak berupa campuran metanol dan asam asetat glasial 2% belum pernah dilakukan sebelumnya.
3. Manfaat penelitian
a. Manfaat teoritis. Penelitian ini diharapkan dapat memberi sumbangan informasi ilmiah mengenai validasi metode kadar kurkumin dalam sediaan obat cair OHT merk Kiranti® secara KCKT fase terbalik.
B. Tujuan Penelitian
6 BAB II
PENELAAHAN PUSTAKA
A. Kurkumin
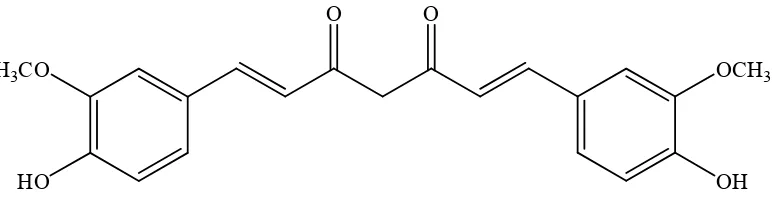
Kurkumin merupakan senyawa α-β-diketon tak jenuh dengan beberapa pusat elektrofilik (Martono, 1996). Kurkumin terdiri atas dua molekul asam ferulat yang dihubungkan dengan jembatan metilen pada atom karbon dari gugus karboksilnya (Gambar 1).
HO
H3CO OCH3
OH
O O
Gambar 1. Struktur kurkumin (Aggarwal dkk., 2006)
Kristal kurkumin berbentuk batang atau prisma dan berwarna kuning jingga. Jumlah atom karbon pada kurkumin kurang dari 40 namun dapat dikelompokkan dalam karotenoid (pigmen yang berstruktur tetraterpenoid dan bersifat larut lemak) dengan memberikan warna kuning sampai merah. Karena kekhasan dalam strukturnya, senyawa ini disebut berasal dari penguraian karotenoid dan bukan terbentuk dari satuan yang lebih kecil (Robinson, 1995).
gelombang 430 nm dalam pelarut metanol dan pada 415 sampai 420 nm dalam pelarut aseton. Kurkumin akan berfluoresensi pada panjang gelombang 524 nm dalam pelarut asetonitril dan pada 549 nm dalam pelarut etanol (Aggarwal dkk., 2006).
Stabilitas kurkumin dipengaruhi oleh pH dan cahaya. Kurkumin dalam larutan air akan mengalami reaksi hidrolisis yang sangat tergantung pada pH lingkungan. Pada larutan asam (pH rendah), kurkumin berwarna kuning, sedangkan dalam suasana alkali kurkumin menghasilkan warna coklat kemerahan yang pekat sampai kuning muda (Tonnesen dan Karlsen, 1995a). Kurkumin stabil pada pH di bawah 6,5 dan akan terdegradasi pada pH di atas 6,5. Hal ini disebabkan adanya gugus metilen aktif. Produk degradasi kurkumin dalam suasana alkali (pH 7-10) akan menghasilkan asam ferulat dan feruloil metan
Kurkumin disintesis pertama kali oleh Lampe pada tahun 1913 (Majeed, Badmaev, Shivakumar, dan Rajendran, 1995). Kurkumin dan turunannya dapat disintesis dari vanillin atau turunan benzaldehid dan asetil aseton (Hakim, 2002). Isolasi kurkumin pertama kali dilakukan pada tahun 1815. Pada tahun 1910, Daube berhasil memperoleh bentuk kristalnya. Walaupun demikian, potensi kurkumin dalam bidang kesehatan baru diteliti pada era tahun 1970 dan 1980 (Majeed dkk., 1995).
Aktivitas kurkumin antara lain sebagai antiinflamsi, analgesik, antipiretik, antimikroba, antimutagenik, antioksidan, dan antikanker (Jankun dkk., 2003; Jayaprakasha dkk., 2002). Kurkumin juga memiliki efek hepatoprotektif, neuroprotektif, hipoglikemik, dan antireumatik (Anand, Kunnumakkara, Newman, dan Anggarwal, 2007). Kurkumin juga poten menghambat aktivitas siklooksigenase dan lipoksigenase serta memiliki efek inhibitor kuat pada DNA dan RNA terhadap karsinogenesis dan pertumbuhan tumor dengan menghambat sintesis DNA pada beberapa jenis sel kanker (Huang dkk., 1997).
Kurkumin dapat diperoleh dari ekstrak tanaman dari famili Zingiberaceae khususnya Curcuma seperti Curcuma longa, Curcuma aromatica, Curcuma amada, Curcuma zedoaria, Curcuma caesia, Curcuma aerugiosa, Curcuma
angustifolia, Curcuma leucorrhiza, Curcuma pierreana, Curcuma domestica,
kondisi tempat tumbuh tanaman tersebut. Sebagai contoh, kandungan kurkumin di dalam tanaman Curcuma zedoaria berkisar 0,5-0,73% sedangkan kandungan kurkumin di dalam tanaman Curcuma xanthorrhiza ROXB berkisar 1,6-2,2% (Zahro, Cahyono, dan Hastuti, 2009).
Analisis kurkumin dapat dilakukan dengan KCKT fase normal maupun terbalik (Tonnesen dan Karlsen, 1983), namun karena sifat kurkumin yang sangat labil maka KCKT fase terbalik dengan kolom C18 lebih disukai dalam analisis (Khurana dan Ho, 1988). Pelarut yang sering digunakan dalam KCKT fase terbalik dalam analisis kurkumin adalah metanol, asetonitril, dan tetrahidrofuran (Smith dan Witowska, 1984).
B. Obat Herbal Terstandar (OHT)
Bahan baku sediaan OHT dapat berupa simplisia maupun ekstrak dari simplisia nabati maupun hewani. Mutu simplisia dan ekstrak yang digunakan sangat berpengaruh terhadap kualitas produk OHT yang dihasilkan. Untuk itu maka perlu dilakukan standarisasi. Standarisasi adalah serangkaian parameter, prosedur, dan cara pengukuran yang hasilnya berupa paradigma mutu sesuai standar dan jaminan stabilitas produk. Simplisia dan ekstrak yang telah distandarisasi memiliki kadar senyawa aktif yang ajeg serta memenuhi parameter standarisasi yang diperbolehkan (Badan Pengawas Obat dan Makanan RI, 2005b).
Gambar 3. Logo OHT (Badan Pengawas Obat dan Makanan RI, 2004)
C. Sediaan Cair
D. Spektrofotometri Visibel
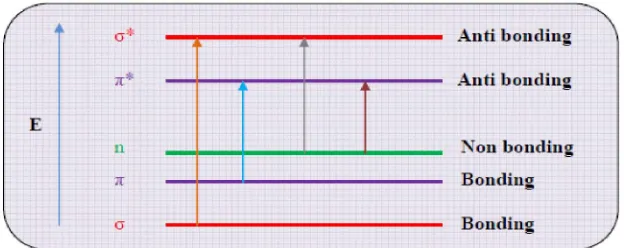
Spektrofotometri visibel merupakan teknik analisis spektroskopik menggunakan sumber radiasi elektromagnetik sinar tampak dalam rentang panjang gelombang 380-780 nm. Prinsip kerja spektrofotometri berdasarkan atas interaksi antara radiasi elektromagnetik dengan atom atau molekul. Interaksi tersebut menyebabkan terjadinya absorpsi, yaitu perpindahan energi dari sinar radiasi ke molekul. Akibat absorpsi radiasi elektromagnetik oleh molekul tersebut maka terjadi eksitasi ke tingkat energi yang lebih tinggi yang dikenal sebagai orbital elektron antibonding (Mulja dan Suharman, 1995). Hasil interaksi antara radiasi elektromagnetik dengan atom atau molekul dapat digambarkan oleh suatu grafik yang menghubungkan banyaknya radiasi elektromagnetik yang diserap dengan panjang gelombangnya, yang disebut dengan spektrum absorpsi (Rohman, 2007).
Gambar 4. Diagram tingkat energi elektronik (Mulja dan Suharman, 1995)
Transisi elektronik yang berguna dalam penelitian adalah transisi n→π* dan π→π* karena memberikan spektra pada 200-700 nm. Kedua transisi ini membutuhkan adanya kromofor dan auksokrom dalam struktur molekulnya. Kromofor adalah suatu gugus fungsional tidak jenuh yang menyediakan orbital π yang dapat menyerap pada daerah ultraviolet. Molekul yang mengandung kromofor disebut kromogen. Sedangkan auksokrom merupakan gugus jenuh yang bila terikat pada kromofor mengubah panjang gelombang dan intensitas serapan maksimum, cirinya adalah heteroatom yang langsung terikat pada kromofor. Gugus auksokrom paling sedikit memiliki sepasang elektron bebas yang dapat berinteraksi dengan elektron π, misalnya -OH, -NH2 (Christian, 2004; Sastrohamidjojo, 2002).
Instrumen yang digunakan disebut spektrofotometer. Spektrofotometer adalah suatu instrumen yang akan memisahkan radiasi polikromatis menjadi beberapa panjang gelombang yang berbeda. Instrumentasi dari spektrofotometer meliputi sumber radiasi kontinyu pada λ tertentu, monokromator untuk mendapatkan berkas sempit dari sumber spektrum, sel sampel, detektor, dan
Gambar 5. Diagram alir instrumentasi spektrofotometer visibel
Spektrofotometer visibel dapat digunakan untuk analisis kualitatif maupun kuantitatif. Dalam analisis kuantitatif, suatu berkas radiasi dikenakan pada cuplikan (larutan sampel) dan intensitas sinar radiasi yang diteruskan diukur besarnya. Radiasi yang diserap oleh cuplikan ditentukan dengan membandingkan intensitas sinar yang diteruskan bila spesies penyerap tidak ada dengan intensitas sinar yang diteruskan bila spesies penyerap ada. Intensitas sinar yang diteruskan bila tidak ada spesies penyerap merupakan intensitas sinar yang masuk dikurangi dengan yang hilang karena penghamburan, pemantulan, dan serapan konstituen lain (Sastrohamidjojo, 2002).
Analisis kuantitatif selalu melibatkan pembacaan serapan radiasi elektromagnetik oleh molekul yang dikenal dengan absorban (A) tanpa satuan atau radiasi elektromagnetik yang diteruskan yang dikenal dengan transmitan dengan satuan persen (% T). Bouger, Lambert, dan Beer membuat formula secara matematik hubungan antara transmitan atau absorban terhadap intensitas radiasi atau konsentrasi zat yang dianalisis dan tebal larutan, sebagai berikut:
Dimana:
T = persen transmitan
I0 = intensitas radiasi yang datang It = intensitas radiasi yang diteruskan ε = daya serap molar (L mol-1 cm-1)
C = konsentrasi (mol L-1) b = tebal larutan (cm) A = serapan/absorbans
(Mulja dan Suharman, 1995).
E. Kromatografi Cair Kinerja Tinggi 1. Definisi
Kromatografi adalah prosedur pemisahan senyawa berdasarkan perbedaan kecepatan migrasi karena adanya perbedaan koefisien distribusi masing-masing senyawa di antara dua fase yang saling bersinggungan dan tidak saling campur, yang disebut sebagai fase gerak (mobile phase) yang berupa zat cair atau gas dan fase diam (stationary phase) yang berupa zat cair atau zat padat (Noegrohati, 1994). Pada Kromatografi Cair Kinerja Tinggi (KCKT) fase geraknya dialirkan menuju kolom secara cepat dengan bantuan tekanan dari pompa, kemudian hasilnya dideteksi dengan detektor (Hendayana, 2006). KCKT merupakan teknik analisis yang paling sering digunakan dalam analisis farmasi untuk pemisahan, identifikasi, dan determinasi dalam campuran yang kompleks (Skoog, Holler, dan Nieman, 1998).
atmosfer dan tekanan pada bagian atas kolom kurang dari 70 atmosfer (Direktorat Jenderal Pengawasan Obat dan Makanan RI, 1995). Pada akhir tahun 1970, perkembangan instrumen ini dapat menghasilkjan poemisahan yang baik sehingga sistem ini lebih dikenal dengan Kromatografi Cair Kinerja Tinggi (Kromidas, 2000).
KCKT merupakan kromatografi partisi. Prinsip kromatografi partisi didasarkan pada partisi solut di antara dua fase yang tidak saling campur karena adanya perbedaan koefisien distribusi dari masing-masing senyawa. Jika solut ditambahkan ke dalam sistem yang terdiri dari dua pelarut tidak saling campur dan keseluruhan sistem dibiarkan setimbang, maka solut akan tersebar di antara dua fase menurut persamaan:
(3) Dimana K adalah koefisien distribusi, Cs adalah konsentrasi solut dalam
fase diam dan Cm adalah konsentrasi solut dalam fase gerak (Johnson dan Stevenson, 1978).
Kolom yang biasa digunakan dalam kromatografi partisi fase terbalik adalah kolom yang fase diamnya terikat secara kimia pada penyangga sehingga tidak mudah terbawa oleh fase gerak. Penyangga yang digunakan biasanya terbuat dari silika yang sudah diseragamkan, berpori, dan umunya terdiri dari partikel berdiameter 3,5 atau 10 µm (Skoog dkk., 1998).
KCKT partisi fase terbalik biasanya mengandung bagian organik yang terikat secara kimia dengan gugus silanol pada permukaan silika. Bagian organik
K=
C
C
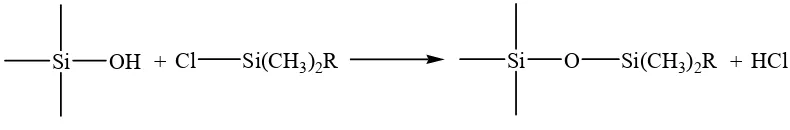
stersebut umumnya berupa hidrokarbon rantai panjang. Gugus silanol permukaan dapat direkasikan dengan berbagai cara untuk menempelkan berbagai jenis gugus organik. Kemasan fase terikat dengan tipe ikatan siloksan (Si-O-Si-O) dibuat dengan mereaksikan organosiloksan dengan gugus silanol pada permukaan silika gel yang terhidrolisis (Gambar 6).
Si OH + Cl Si(CH3)2R Si O Si(CH3)2R + HCl
Gambar 6. Reaksi silanisasi (Harris, 1999)
Reaksi tersebut (Gambar 7) digunakan untuk membuat isian kolom oktadesilsilan dari gugus silanol dan oktadesilklorosilan sepertipada Gambar 7.
Si OH + Cl Si (CH2)17CH3 Si O Si (CH2)17CH3 + HCl
Gambar 7. Reaksi pembuatan kolom oktadesilsilan
Pada kromatografi parisi fase terbalik dengan kemasan fase terikat, R pada siloksan biasanya berupa gugus C18 atau C8. Panjang atau pendeknya rantai karbon mempengaruhi tertambatnya suatu senyawa pada fase diam (Skoog dkk., 1998).
2. Instrumentasi
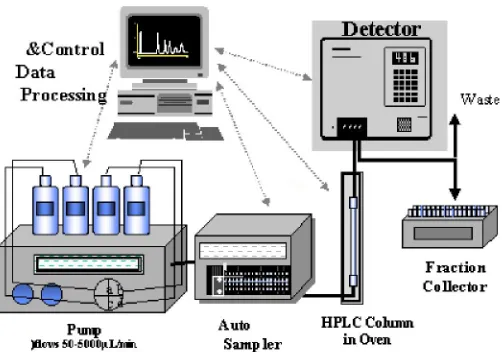
Gambar 8. Skema instrumen KCKT
Menurut Gritter, Bobbit, dan Schwarting (1991) sistem KCKT terdiri dari tiga variabel utama yang harus diperhatikan, yaitu fase diam, fase gerak, dan detektor.
a. Fase diam. Fase diam dalam KCKT berupa kolom yang merupakan bagian sangat penting dalam pemisahan komponen-komponen sampel. Keberhasilan pemisahan komponen sampel bergantung pada keadaan kolom (Mulja dan Suharman, 1995). Kolom dapat berupa gelas atau baja anti karat yang dapat diisi dengan silika gel, alumina, dan elit. Panjang kolom bervariasi antara 15-150 cm (Khopkar, 1990).
end-capping, yaitu suatu proses penutupan residual silanol dengan gugus trimetilsilil (Rohman dan Gandjar, 2007).
b. Fase gerak. Kemampuan KCKT untuk memisahkan senyawa tergantung pada fase gerak yang digunakan, terutama dalam hal tambatan dan pemisahan senyawa (Munson, 1984). Fase gerak dapat berupa pelarut tunggal atau pelarut campuran. Pengembangan KCKT menggunakan pelarut campuran yang susunannya terus menerus berubah, biasanya terdiri dari dua atau tiga pelarut disebut elusi gradien (Gritter dkk., 1991). Fase gerak untuk analisis secara KCKT harus murni, tanpa cemaran, tidak bereaksi dengan kemasan, dapat melarutkan cuplikan (solute), viskositas rendah, memungkinkan memperoleh kembali cuplikan dengan mudah, dan harganya wajar (Johnson dan Stevenson, 1978). Menurut Gritter dkk. (1985), fase gerak untuk KCKT harus bebas dari gas terlarut karena adanya gas dapat mempengaruhi respon detektor sehingga menghasilkan sinyal palsu dan mempengaruhi kolom.
dapat bercampur dengan air. Pemodifikasi organik yang banyak digunakan adalah metanol, asetonitril, dan tetrahidrofuran (Gritter dkk., 1985; Munson, 1984).
Kepolaran dinyatakan dalam indeks polaritas (P’) yang dapat dihitung dengan persamaan berikut ini:
(4) Dengan Φa dan Φb adalah fraksi pelarut a dan b dalam campuran, sedangkan P’a dan P’b adalah angka P’ pelarut murni (Grtitter dkk., 1991). Berikut ini adalah nilai indeks polaritas (P’) dari beberapa pelarut :
Tabel I. Nilai indeks polaritas beberapa pelarut pada KCKT fase terbalik (Snyder, Kirkland, dan Glajch, 1997)
Tabel I tersebut menunjukkan bahwa semakin besar eluotropic values
dari pelarut menunjukkan semakin mudah untuk mengelusi sampel. Semakin besar indeks polaritas yang dimiliki pelarut, maka semakin polar pelarut yang digunakan (Snyder dkk., 1997).
c. Detektor. Menurut John dan Stevenson (1978), detektor diperlukan untuk mendeteksi adanya komponen cuplikan yang terdapat dalam kolom serta untuk mengukur jumlah komponen yang ada dalam cuplikan. Detektor yang baik adalah detektor yang memenuhi persyaratan sensitivitas yang tinggi dengan rentang sensitivitas 10-8 -10-15 gram solut per detik, kestabilan dan reprodusibilitas yang sangat baik, respon yang linear terhadap konsentrasi solut, dapat bekerja dari temperatur kamar sampai 400oC, tidak dipengaruhi perubahan temperatur dan kecepatan pelarut pengembang, mudah didapat, mudah dipakai operator, selektif terhadap macam-macam linarut dalam pelarut pengembang dan tidak merusak sampel (Mulja dan Suharman, 1995).
Secara umum detektor dibagi menjadi 2 kategori, yaitu: 1. Bulk property detector
Jenis detektor ini mengukur sifat solut dan fase gerak. Contohnya adalah detektor indeks bias. Kelemahan detektor ini adalah kurang sensitif dan tidak cocok untuk kondisi elusi landaian (Munson, 1991).
2. Solute property detector
Detektor ini merupakan detektor yang selektif mengukur sifat solut dan lebih sensitif dibandingkan bulk property detector. Contohnya adalah detektor UV-Vis dan detektor fluoresensi (Settle, 1997).
3. Pemisahan puncak dalam kromatografi
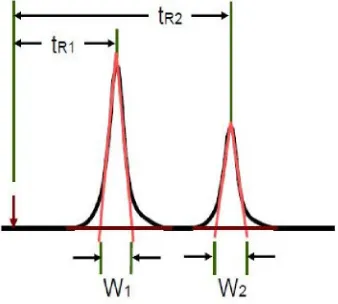
kolom dalam memisahkan senyawa dan menghasilkan puncak yang sempit (Johnson dan Stevenson, 1978). Ukuran pemisahan dari dua puncak adalah nilai resolusi yang dapt diukur dengan persamaan:
(5)
Keterangan:
tR1 dan tR2 = waktu retensi senyawa diukur pada titik maksimum puncak Δt = selisih waktu antara tR2 dan tR1
W1 dan W2 = lebar alas puncak
Pemisahan dua puncak berari pemisahan dua senyawa, dapat digambarkan sebagai berikut:
Gambar 9. Pemisahan dua puncak (Jasco International, 2004).
Harga Rs > 1,5 disebut baseline resolution, yaitu pemisahan sempurna dari dua puncak dengan ukuran yang sama. Dalam prakteknya, pemisahan dengan harga Rs = 1,0 dianggap memadai (Pescok, Shields, dan Cains, 1976).
4. Pengembangan metode KCKT
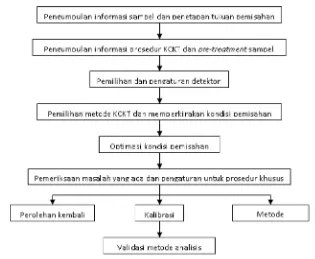
Pengembangan metode KCKT diharapkan menghasilkan suatu metode analisis yang memiliki waktu analisis singkat untuk analisis secara rutin, menghasilkan puncak yang sempit untuk rasio signal to noise yang besar, dan meminimalkan penggunaan fase gerak. Oleh karena itu validasi metode analisis setelah pengembangan metode merupakan tahap terpenting.
Namun sebelum masuk pada tahap validasi, tahap yang juga sangat penting adalah menentukan kesesuaian sistem KCKT, antara lain verifikasi pompa, sistem injeksi, dan detektor. Ini sangat penting dilakukan untuk meyakinkan bahwa sistem KCKT telah layak digunakan dalam validasi dan penetapan kadar suatu senyawa (Miller dan Miller, 1988). Berikut ini adalah langkah-langkah umum dalam pengembangan metode analisis menggunakan KCKT:
F. Validasi Metode Analisis
Validasi metode analisis adalah proses yang dibangun melalui studi laboratorium untuk meyakinkan bahwa performa karakteristik suatu metode memenuhi persyaratan untuk aplikasi analisis (United States Pharmacopeial Convention, 2007). Dengan kata lain, validasi metode analisis merupakan penilaian terhadap parameter analisis tertentu. Parameter analisis tersebut adalah ketepatan (akurasi), ketelitian (presisi), selektivitas, limit deteksi, limit kuantitasi, linearitas, dan rentang (United States Pharmacopeial Convention, 2007).
Tujuan utama validasi metode adalah untuk menjamin bahwa metode analisis yang dikembangkan dapat memberikan hasil pengukuran yang cermat dan sesuai dengan tujuan yang diharapkan. Validasi metode perlu dilakukan karena adanya kesalahan pada analisis seperti kesalahan sistematik dan kesalahan acak.
Metode pengujian yang berbeda membutuhkan validasi yang berbeda pula. Terdapat 4 kategori metode pengujian dengan parameter validasi metode analisis berbeda (Tabel II dan III).
Tabel II. Kategori metode pengujian (United States Pharmacopeial Convention, 2007)
Kategori Keterangan
I Metode untuk penetapan kadar komponen utama bahan baku atau bahan aktif (termasuk pengawet) dalam produk jadi sediaan farmasi
II Metode analisis untuk penetapan ketidakmurnian bahan baku atau hasil degradasi dalam produk jadi sediaan farmasi
III Metode analisis untuk penetapan karakteristik performa (seperti disolusi, pelepasan obat).
Tabel III. Parameter analisis yang dibutuhkan dalam validasi metode analisis (United States Pharmacopeial Convention, 2007)
Karakteristik
Selektivitas suatu metode analisis adalah kemampuan untuk mengukur analit secara cermat dan seksama dengan adanya komponen yang mungkin ada dalam sampel. Selektivitas sering dinyatakan sebagai derajat bias dari hasil yang diperoleh dengan membandingkannya terhadap impurities, produk degradasi, atau senyawa kimia yang mirip. Bias dapat dinyatakan sebagai perbedaan antara hasil uji antara 2 kelompok sampel. Selektivitas juga merupakan ukuran derajat interferensi dalam analisis campuran sampel yang kompleks. Selektivitas ditentukan dengan menginjeksikan sampel pada sistem kromatografi. Puncak yang muncul tidak boleh terpengaruh oleh puncak lain yang dibuktikan dengan perhitungan resolusi (Yuwono dan Indrayanto, 2005).
(kedua puncak berhimpit lebih kurang 2%) dianggap memadai (Pescok dkk.,
1976).
2. Linearitas dan rentang
Linearitas adalah kemampuan metode analisis untuk menunjukkan hasil uji yang secara langsung atau dengan persamaan matematis proporsional terhadap konsentrasi analit dalam sampel pada rentang tertentu. Linearitas dapat ditentukan dengan pengukuran pada beberapa konsentrasi analit. Hasil slope (b), intersep (a) dan koefisien korelasi (r) menggambarkan informasi linearitas. Sebagai parameter adanya hubungan linear digunakan koefisien korelasi (r) pada analisis regresi linear y = bx + a. Hubungan linear yang ideal dicapai apabila nilai a = 0 dan r = +1 atau –1 tergantung pada arah garis. Nilai a menunjukkan kepekaan analisis terutama instrumen yang digunakan (Harmita, 2004). Nilai koefisien korelasi 0,999 diterima untuk sebagian besar metode khususnya komponen dalam jumlah besar pada metode pengujian. Jika koefisien korelasi memiliki nilai kurang dari 0,999 maka perlu dilakukan perhitungan terhadap parameter lain yaitu Vxo ≤ 5 % (Yuwono dan Indrayanto, 2005).
3. Ketepatan (accuracy)
Ketepatan adalah ukuran kedekatan antara hasil analisis dan kadar analit yang sebenarnya. Ketepatan dinyatakan sebagai persen perolehan kembali (recovery). Ketepatan hasil analisis sangat tergantung pada sebaran galat sistematik di dalam keseluruhan tahapan analisis. Oleh karena itu, untuk mencapai ketepatan yang tinggi dapat dilakukan dengan mengurangi galat sistematik tersebut seperti menggunakan peralatan yang terkalibrasi, menggunakan pereaksi dan pelarut yang dapat melarutkan senyawa dengan sempurna, pengontrolan suhu, pelaksanaan yang cermat dan taat asas serta sesuai prosedur (Harmita, 2004).
Kesulitan utama yang dihadapi pada evaluasi ketepatan suatu metode analisis adalah fakta bahwa nilai sebenarnya kadar analit biasanya tidak diketahui. Secara internasional, dikenal tiga macam cara yang umum digunakan untuk mengevaluasi ketepatan metode analisis kimia, yaitu dengan menggunakan bahan rujukan baku (Standard Reference Material / SRM), menggunakan baku sebagai pembanding (standard method), dan recovery dengan menempatkan analit plasebo (spiked placebo recovery) (Snyder dkk., 1997).
memiliki ketepatan yang tinggi sehingga menghasilkan data yang dapat dianggap sebagai hasil yang sebenarnya. Metode spiked placebo recovery dilakukan dengan menganalisis sampel suatu obyek yang diperkaya dengan sejumlah analit baku yang telah ditetapkan. Berat total analit yang diperoleh dari analisis sampel yang diperkaya dikurangi dengan berat analit dalam sampel yang tidak diperkaya, dibandingkan terhadap jumlah analit baku yang ditambahkan (Snyder dkk., 1997).
Akurasi dinyatakan sebagai persen perolehan kembali (recovery) analit yang ditambahkan (Harmita, 2004). Persen perolehan kembali yang dapat diterima bergantung pada matriks analit, prosedur pengolahan analit dan konsentrasi analit (Anonim, 2004). Berikut ini adalah rentang recovery yang masih dapat diterima:
Tabel IV. Kriteria rentang recovery yang dapat diterima (United States Pharmacopeial Convention, 2007)
4. Ketelitian (precision)
homogen (Snyder dkk., 1997). Ketelitian diukur sebagai simpangan baku atau simpangan baku relatif/koefisien variasi (Harmita, 2004).
Pengertian presisi suatu metode dapat dikelompokkan menjadi tiga, yaitu keterulangan (repeatability), intermediet presisi, dan ketertiruan (reproducibility). Keterulangan adalah ketelitian metode analisis dalam kondisi operasi yang sama pada laboratorium yang sama pada interval waktu yang singkat dengan analis dan peralatan yang berbeda. Keterulangan terbagi lagi dalam dua aspek yaitu presisi instrumental dan intra-assay precision. Keterulangan dinilai melalui penetapan terpisah lengkap terhadap sampel-sampel identik dari batch yang sama. Keterulangan memberikan ukuran ketelitian pada kondisi normal (Harmita, 2004). Intermediet presisi adalah keseuaian pengukuran ketika metode analisis yang sama diaplikasikan beberapa kali pada hari, instrumen atau analis yang berbeda pada laboratorium yang sama (Snyder dkk., 1997). Ketertiruan adalah ketelitian metode jika dikerjakan pada kondisi yang berbeda (Harmita, 2004).
Tabel V. Kriteria akurasi dan presisi yang masih dapat diterima (United States
Kurkumin merupakan senyawa alam berwarna kuning atau kuning jingga yang banyak ditemukan dalam obat tradisional. Kurkumin terdiri atas dua molekul asam ferulat yang dihubungkan dengan jembatan metilen pada atom karbon dari gugus karboksilnya. Kurkumin stabil pada pH di bawah 6,5 dan akan terdegradasi pada pH di atas 6,5.
Keberhasilan analisis menggunakan metode KCKT sangat tergantung pada pemilihan kolom dan kondisi kerja yang optimum. Kondisi optimum tersebut diperoleh dari hasil optimasi. Sistem KCKT yang telah dioptimasi selanjutnya divalidasi. Validasi dilakukan untuk meyakinkan bahwa metode yang digunakan telah memenuhi persyaratan aplikasi analisis. Validasi suatu metode analisis ditentukan oleh parameter validasi yang meliputi akurasi, presisi, linearitas, sensitivitas, dan selektivitas. Selektivitas ditentukan dari nilai resolusi, linearitas dinyatakan dengan nilai koefisien korelasi, akurasi dinyatakan sebagai persen perolehan kembali, dan presisi dinyatakan dengan koefisien variasi.
H. Hipotesis
31 BAB III
METODE PENELITIAN
A. Jenis dan Rancangan Penelitian
Penelitian yang dilakukan mengikuti jenis penelitian non eksperimental dengan rancangan penelitian deskriptif, sebab pada penelitian ini tidak dilakukan manipulasi pada subjek uji dan hanya mendeskripsikan keadaan yang ada.
B. Variabel Penelitian 1. Variabel bebas
Variabel bebas pada penelitian ini adalah sistem KCKT yang telah dioptimasi.
2. Variabel tergantung
Variabel tergantung pada penelitian ini adalah parameter-parameter validasi yang meliputi selektivitas, linearitas, akurasi, dan presisi.
3. Variabel pengacau terkendali
a. Pelarut yang digunakan, berupa pelarut pro analysis yang memiliki tingkat kemurnian tinggi.
C. Definisi Operasional
1. Sistem KCKT fase terbalik yang digunakan terdiri atas fase diam berupa kolom oktadesilsilan (C18) serta fase gerak berupa campuran metanol p.a dan asam asetat glasial p.a 2 % (90:10 v/v) dengan kecepatan alir 0,5 ml/menit. 2. Kadar kurkumin dinyatakan dalam satuan part per million (ppm).
3. Parameter validasi yang digunakan pada penelitian ini meliputi selektivitas, linearitas, akurasi, dan presisi.
D. Bahan-bahan Penelitian
Bahan yang digunakan adalah baku kurkumin hasil sintesis Prof. Dr. Sudibyo Martono, M.S., Apt. yang telah dikonfirmasi strukturnya dengan metode spektroskopi 1H-NMR dan Mass Spectra dengan titik lebur 181,2-182,4o-C, metanol p.a EMSURE® ACS, ISO, Reag. Ph Eur (E. Merck), asam asetat glasial
p.a EMPARTA® ACS (E. Merck), aquabidestilata dan OHT merk Kiranti®.
E. Alat-alat Penelitian
Alat yang digunakan adalah organic solvent membrane filter (Whatman) ukuran pori 0,45μm; diameter 47mm, indikator pH, penyaring Millipore,
mikropipet Socorex, neraca kasar, neraca analitik (Ohaus PAJ1003), ultrasonikator (Retsch tipe T460 no V935922013 Ey), vaccum (Gaast model DOA-P104-BN), magnetic stirrer, seperangkat alat spektrofotometri UV-VIS merk Milton Ray Spectronic 3000 Array yang dihubungkan dengan printer merk
228-46703-38, SERIAL No. C21254706757 LP, Shimadzu Corporaion), kolom oktadesilsilan (C18) berukuran 250 x 4,6 mm merk KNAUER No. 25EE181KSJ (B115Y620), seperangkat komputer (merk Dell B6RDZ1S Connexant System
RD01-D850 A03-0382 JP France S.A.S, printer HP Deskjet D2566 HP-024-000 625 730), dan alat-alat gelas yang umum digunakan dalam analisis (Pyrex).
F. Tata Cara Penelitian 1. Pembuatan fase gerak KCKT
Fase gerak yang digunakan terdiri dari asam asetat glasial p.a 2% dan metanol p.a Masing-masing komponen fase gerak disaring melalui organic solvent membrane filter (Whatman) berukuran pori 0,45 μm; diameter 47 mm dengan bantuan pompa vakum, selanjutnya diawaudarakan dengan ultrasonikator selama 15 menit. Pencampuran kedua komponen fase gerak dilakukan di dalam instrument KCKT dengan perbandingan metanol : asam asetat glasial 2% sebesar 90:10 v/v.
2. Pembuatan pelarut metanol pH 4
3. Pembuatan larutan baku kurkumin
a. Pembuatan larutan stok kurkumin. Ditimbang lebih kurang seksama 10,0 mg baku kurkumin dilarutkan dengan metanol pH 4 dalam labu takar 10,0 ml hingga tanda sehingga diperoleh konsentrasi 1000 ppm.
b. Pembuatan larutan intermediet kurkumin. Larutan stok kurkumin diambil sebanyak 1,0 ml dan diencerkan dengan metanol pH 4 dalam labu takar 10,0 ml hingga tanda sehingga diperoleh konsentrasi sebesar 100 ppm.
c. Pembuatan seri larutan baku kurkumin. Dibuat seri larutan baku kurkumin dengan konsentrasi 1,5; 2,0; 2,5; 3,0; 3,5; 4,0 dan 4,5 ppm dengan mengambil 150; 200; 250; 300; 350; 400 dan 450 µl larutan intermediet kurkumin, masukkan dalam labu takar 10,0 ml dan tambahkan metanol pH 4 hingga tanda. Larutan kemudian disaring dengan Millipore dan diawaudarakan dengan ultrasonikator selama 15 menit.
4. Penentuan panjang gelombang (λ) maksimum kurkumin
5. Preparasi sampel
Sebanyak 1,0 ml sampel sediaan cair OHT merk Kiranti® dimasukkan ke dalam labu takar 10,0 ml dan diencerkan dengan metanol pH 4 hingga tanda. Kemudian diekstraksi menggunakan ultrasonikator selama 15 menit. Ekstrak disaring dan filtrat diencerkan dengan metanol pH 4 sampai 10,0 ml.
6. Validasi metode analisis
a. Penentuan resolusi sampel. Sebanyak 20 µl larutan ekstrak sampel yang telah diawudarakan selama 15 menit diinjeksikan pada sistem KCKT fase terbalik menggunakan fase diam C18 dan fase gerak metanol : asam asetat glasial 2% (90:10 v/v) dengan kecepatan alir 0,5 ml/menit. Dilakukan repetisi sebanyak 5 kali. Resolusi dihitung dengan memasukkan selisih waktu retensi dan lebar peak
ke dalam rumus perhitungan resolusi.
c. Penentuan persen perolehan kembali dan koefisien variasi baku kurkumin. Sebanyak 20 µl larutan baku kurkumin 1,5; 3,0 dan 4,5 ppm yang telah disaring dengan Millipore dan diawaudarakan selama 15 menit diinjeksikan pada sistem KCKT fase terbalik menggunakan fase diam C18 dan fase gerak metanol : asam asetat glasial 2% (90:10 v/v) dengan kecepatan alir 0,5 ml/menit. Dilakukan replikasi sebanyak 5 kali. Konsentrasi baku kurkumin dihitung dengan memasukkan nilai AUC yang diperoleh ke dalam persamaan kurva baku.
G. Analisis Hasil
Kesahihan metode penetapan kadar kurkumin dalam sediaan cair OHT secara KCKT fase terbalik dapat ditentukan berdasarkan parameter berikut:
1. Selektivitas
Menurut Harmita (2004), selektivitas ditentukan dengan parameter resolusi (Rs). Rumus perhitungan resolusi:
Resolusi (Rs) = 1(tR2− tR1)
2(W1+ W2) (6)
Keterangan: Rs = resolusi
tR1 = waktu retensi puncak analit pertama tR2 = waktu retensi puncak analit kedua W1 = lebar dasar puncak pertama W2 = lebar dasar puncak kedua
2. Linearitas
Linearitas dinyatakan dengan koefisien korelasi (r). Konsentrasi larutan baku kurkumin yang diperoleh diplotkan terhadap luas area pada kromatogram sehingga diperoleh nilai koefisien korelasi (r) dari persamaan y = Bx + A.
3. Akurasi
Akurasi dinyatakan sebagai persen perolehan kembali (recovery) dan dapat dihitung dengan rumus:
4. Presisi
Presisi dihitung sebagai simpangan deviasi relatif (RSD) atau koefisien variasi (KV). Rumus perhitungan koefisien variasi
KV = simpangan deviasi (SD)rata-rata (x) x 100% (8)
Keterangan: KV = koefisien variasi SD = simpangan deviasi
39 BAB IV PEMBAHASAN
A. Pembuatan Fase Gerak KCKT
Sistem kromatografi yang digunakan pada penelitian ini merupakan sistem kromatografi partisi fase terbalik karena menggunakan fase gerak yang bersifat lebih polar dibandingkan fase diamnya. Fase gerak yang digunakan pada penelitian ini adalah campuran metanol dan asam asetat glasial 2% dengan perbandingan 90:10 (v/v). Baik metanol maupun asam asetat glasial dipilih sebagai fase gerak karena keduanya dapat melarutkan kurkumin dengan baik sehingga diharapkan dapat mengelusi kurkumin lebih cepat.
Campuran fase gerak metanol dan asam asetat glasial 2% (90:10 v/v) memiliki nilai pH 4 sehingga tidak akan merusak kolom kromatografi. Jika pH fase gerak ≤ 2, maka kolom oktadesilsilan (C18) akan melepaskan gugus oktadesilnya kembali ke bentuk silanol (Gambar 11).
Si O Si (CH2)17CH3 Si OH + HO Si (CH2)17CH3 + H+
H2O/H+
Gambar 11. Reaksi degradasi kolom C18 dalam suasana asam
Pencampuran masing-masing komponen fase gerak dilakukan di dalam instrument KCKT (sistem gradien).
B. Pembuatan Larutan Baku Kurkumin
Larutan baku kurkumin dibuat dengan melarutkan baku kurkumin dalam pelarut metanol p.a. yang telah diatur pada pH 4 untuk menjaga stabilitas kurkumin. Pengaturan pH metanol pada pH 4 dilakukan dengan menambahkan asam asetat glasial 2% ke dalam metanol dengan perbandingan (9:1). Pengaturan pH ini dilakukan karena kurkumin bersifat pH-sensitive. Kurkumin stabil pada pH di bawah 6,5 dan akan terdegradasi pada pH di atas 6,5 menghasilkan asam ferulat dan feruloil metan. Hal ini disebabkan adanya gugus metilen aktif (Tonnesen dan Karlsen, 1995b).
Gambar 12. Gugus metilen aktif pada kurkumin
Larutan baku yang dibuat pada penelitian ini terdiri dari tiga macam, yaitu larutan stok, larutan intermediet, dan larutan seri baku. Larutan stok dibuat dengan konsentrasi 1000 ppm, sedangkan larutan intermediet dibuat dengan konsentrasi 100 ppm. Larutan seri baku dibuat dalam tujuh konsentrasi berbeda yaitu 1,5; 2,0; 2,5; 3,0; 3,5; 4,0 dan 4,5 ppm.
C. Penentuan Panjang Gelombang Maksimum Kurkumin
Panjang gelombang maksimum suatu senyawa adalah panjang gelombang dimana senyawa memiliki absorbansi atau serapan maksimum. Penetapan panjang gelombang maksimum pada penelitian ini dilakukan dengan mengukur serapan analit kurkumin menggunakan spektrofotometer visibel. Kurkumin memiliki gugus kromofor yang panjang dan gugus auksokrom pada strukturnya sehingga dapat memberikan serapan pada daerah sinar tampak (Gambar 13). Kromofor adalan ikatan rangkap terkonjugasi yang mengandung elektron π yang mudah terkesitasi ke tingkat energi yang lebih tinggi yaitu orbital π* jika terkena radiasi elektromagnetik, sedangkan auksokrom adalah gugus yang terikat pada kromofor dan dapat mengubah atau meningkatkan intensitas maksimum dari senyawa.
Kurkumin diukur serapannya dalam bentuk larutan menggunakan pelarut metanol dengan tiga konsentrasi berbeda, yaitu 0,4; 1,0 dan 1,6 ppm. Pelarut yang digunakan dalam penetapan panjang gelombang maksimum sama dengan pelarut yang digunakan untuk analisis pada sistem KCKT. Pembacaan serapan dilakukan pada rentang panjang gelombang 300-500 nm yang berada pada daerah visibel. Rentang pembacaan panjang gelombang ditetapkan berdasarkan panjang gelombang maksimum teoritis kurkumin dalam pelarut metanol yaitu 430 nm (Aggarwal dkk., 2006).
Spektra yang dihasilkan pada penetapan panjang gelombang kurkumin menggunakan tiga konsentrasi berbeda ditunjukkan pada gambar di bawah ini:
Gambar 14. Spektra panjang gelombang maksimum kurkumin
nm sehingga ditetapkan panjang gelombang maksimum kurkumin dalam penelitian ini sebesar 432 nm. Panjang gelombang hasil pengukuran dapat digunakan apabila besarnya tidak lebih dari ± 2 nm terhadap panjang gelombang teoritis (Direktorat Jenderal Pengawasan Obat dan Makanan RI, 1979). Panjang gelombang maksimum yang diperoleh dari pengukuran ini menunjukkan pergeseran sebesar 2 nm dari panjang gelombang maskimum teoritis sehingga dapat digunakan selanjutnya untuk mengukur serapan larutan baku maupun sampel yang akan dianalisis.
D. Pengamatan Waktu Retensi (tR) Kurkumin
Waktu retensi (tR) untuk senyawa tertentu pada kondisi tertentu bersifat spesifik sehingga dapat digunakan untuk analisis kualitatif. Waktu retensi suatu senyawa dipengaruhi oleh interaksi senyawa tersebut terhadap fase diam dan fase gerak yang digunakan. Pada penelitian ini digunakan metode KCKT fase terbalik dimana fase diamnya bersifat lebih nonpolar daripada fase geraknya. Fase diam yang digunakan adalah oktadesilsilan (C18) sedangkan fase geraknya berupa campuran metanol dan asam asetat glasial 2%. Oleh karena itu, senyawa yang bersifat lebih polar akan terelusi lebih dahulu, sedangkan senyawa yang bersifat lebih nonpolar akan tertambat lebih lama pada fase diam.
Gambar 15. Gugus polar dan nonpolar kurkumin
Gugus polar berinteraksi dengan fase gerak melalui ikatan hidrogen, sedangkan gugus nonpolar berinteraksi dengan fase diam melalui interaksi Van Der Waals.
O
HO
Interaksi Van Der Waals (Watson, 2003)
Gambar 17. Interaksi kurkumin dengan fase diam oktadesilsilan (C18)
Dari gambar 16 dan 17 diketahui bahwa interaksi kurkumin dengan fase gerak lebih kuat dibandingkan dengan fase diam. Hal ini disebabkan oleh adanya ikatan hidrogen antara kurkumin dengan fase gerak dengan energi disosiasi sebesar 5 Kkal/mol yang bersifat lebih kuat daripada interaksi Van Der Waals
antara kurkumin dengan fase diam dengan energi disosiasi sebesar 1 Kkal/mol (Fessenden dan Fessenden, 1986).
(a)
(b)
Gambar 18. Kromatogram baku kurkumin (a) dan sampel (b)
E. Pembuatan Kurva Baku Kurkumin
Pembuatan kurva baku bertujuan untuk mendapatkan persamaan regresi linear yang selanjutnya digunakan untuk analisis kuantitatif. Persamaan yang diperoleh menyatakan korelasi yang linear antara konsentrasi analit dengan respon
Penelitian ini menggunakan tujuh seri konsentrasi larutan baku kurkumin yaitu 1,515; 2,020; 2,525; 3,030; 3,535; 4,040dan 4,545 ppm yang masing-masing dibuat replikasi 3 kali. Dari ketiganya dibuat kurva hubungan antara konsentrasi dan respon AUC sehingga dapat dipilih salah satu kurva baku yang akan digunakan selanjutnya. Pemilihan kurva baku didasarkan pada nilai koefisien korelasi (r). Nilai r yang masih dapat diterima untuk sebagian besar pengujian adalah lebih besar dari 0,999 (Yuwono dan Indrayanto, 2005).
Gambar 19. Kurva baku kurkumin
Hubungan yang linear antara seri konsentrasi kurkumin dengan respon AUC ditunjukkan dalam gambar 19. Dari kurva tersebut dapat dilihat bahwa respon AUC meningkat seiring dengan meningkatnya konsentrasi.
F. Validasi Metode Analisis
Validasi metode analisis merupakan penilaian terhadap parameter analisis yang terdiri dari ketepatan (akurasi), ketelitian (presisi), selektivitas, limit deteksi, limit kuantitasi, linearitas, dan rentang (United States Pharmacopeial Convention, 2007). Validasi dilakukan untuk membuktikan dan menjamin bahwa suatu metode analisis memiliki validitas yang baik sehingga hasilnya dapat dipercaya dan dipertanggungjawabkan. Parameter analisis yang dibutuhkan dalam validasi metode ditentukan oleh kategori metode analisis yang digunakan. Pada penelitian ini metode analisis yang digunakan mengikuti kategori I karena
0
0 1,000 2,000 3,000 4,000 5,000
AUC
Konsentrasi (ppm)
merupakan metode untuk analisis kuantitatif komponen utama bahan baku dalam produk jadi sediaan farmasi. Dengan demikian, parameter analisis yang dibutuhkan dalam penelitian ini adalah selektivitas, linearitas, akurasi, dan presisi.
1. Selektivitas
Selektivitas suatu metode berkaitan dengan kemampuannya untuk mengukur analit secara cermat dan seksama dengan adanya komponen lain yang mungkin terdapat dalam matriks sampel. Penentuan selektivitas pada metode KCKT dapat diamati dari pemisahan peak kurkumin dalam sampel dan dinyatakan sebagai nilai resolusi (Rs). Metode KCKT dikatakan memiliki selektivitas yang baik apabila nilai resolusi yang dihasilkan (Rs) > 1,5. Namun dalam prakteknya, pemisahan dengan harga Rs = 1,0 (kedua puncak berhimpit lebih kurang 2%) dianggap memadai (Pescok dkk., 1976).
Berikut ini adalah hasil perhitungan resolusi (Rs) sampel yang diperoleh:
Tabel VII. Hasil perhitungan resolusi (Rs) sampel Repetisi Resolusi (Rs) Rata-rata
1 1,3371
2. Linearitas
Linearitas merupakan kemampuan suatu metode analisis untuk menunjukkan hubungan yang proporsional antara konsentrasi analit dengan respon yang dihasilkan. Linearitas ditunjukkan oleh besarnya nilai koefisien korelasi (r) kurva baku. Suatu metode dikatakan memiliki linearitas yang baik jika memiliki nilai r lebih besar dari 0,999 (Yuwono dan Indrayanto, 2005). Berdasarkan hasil pembuatan kurva baku diperoleh nilai r sebesar 0,9992 sehingga dapat disimpulkan bahwa metode yang digunakan memiliki linearitas yang baik.
3. Akurasi
Akurasi suatu metode dinyatakan sebagai persen perolehan kembali (recovery) konsentrasi analit yang terukur terhadap konsentrasi sebenarnya. Pada penelitian ini dilakukan penetapan recovery baku menggunakan 3 konsentrasi larutan baku kurkumin yang direplikasi sebanyak 5 kali. Karena analit yang diukur merupakan baku dan konsentrasinya dalam matriks sebesar 100% maka rentang recovery yang dapat diterima adalah 98-102% (United States Pharmacopeial Convention, 2007). Hasil penetapan recovery baku kurkumin disajikan pada tabel berikut:
Tabel VIII. Hasil penetapan recovery baku kurkumin
Konsentrasi
(ppm) Replikasi 1 Replikasi 2 Replikasi 3 Replikasi 4 Replikasi 5 Recovery (%) 1,515 102,0462 101,9208 107,5049 105,1287 105,8746
3,030 101,9703 101,6898 99,1947 101,4917 101,7888
Dari tabel VIII dapat diketahui bahwa rentang recovery baku kurkumin pada konsentrasi rendah (1,515 ppm) adalah sebesar 101,9208-107,5049%, pada konsentrasi tengah (3,030 ppm) sebesar 99,1947-101,9703%, dan pada konsentrasi tinggi (4,545 ppm) sebesar 92,5324-108,4202%. Dari ketiga level konsentrasi baku kurkumin tersebut, hanya konsentrasi tengah (3,030 ppm) saja yang masuk pada rentang 98-102%. Dengan demikian dapat dikatakan bahwa metode yang digunakan memiliki akurasi yang baik pada konsentrasi tengah, yaitu 3,030 ppm.
(a)
(b)
(c)
Berikut ini adalah hasil penetapan recovery menggunakan metode penambahan baku:
Tabel IX. Hasil penetapan recovery baku yang diadisi
Repetisi
sampel Sampel kurkumin Baku adisi
Rentang yang dapat diterima pada penetapan recovery menggunakan metode penambahan baku adalah 80-110% karena konsentrasi baku kurkumin yang ditambahkan dalam matriks sampel kurang dari 1 ppm. Berdasarkan hasil yang diperoleh, diketahui rentang recovery baku yang diadisi sebesar 102,9600-106,8267% masuk dalam rentang recovery yang ditetapkan. Dengan demikian dapat dipastikan bahwa konsentrasi tengah (3,030 ppm) pada metode yang digunakan memiliki akurasi yang baik.
4. Presisi
Tabel X. Data koefisien variasi baku kurkumin
Konsentrasi
(ppm) rata-rata Konsentrasi (x) deviasi (SD) Simpangan Koefisien variasi (KV)
1,515 1,5831 0,0371 2,3435 %
3,030 3,0672 0,0348 1,1346 %
4,545 4,5571 0,2674 5,8678 %
55 BAB V
KESIMPULAN DAN SARAN
A. Kesimpulan
Metode penetapan kadar kurkumin dalam sediaan cair OHT merk Kiranti® secara KCKT fase terbalik menggunakan fase diam C
18 dan fase gerak metanol : asam asetat glasial 2% (90:10 v/v) memenuhi parameter validitas yang baik, meliputi selektivitas (Rs = 1,4383), linearitas (r = 0,9992), akurasi dan presisi (pada konsentrasi 3,030 ppm).
B. Saran
56
DAFTAR PUSTAKA
Aggarwal, B. B., Bhatt, I. D., Ichikawa, H., Ahn, K.S., Sethi, G., Sandur, S. K., dkk., 2006, Curcumin-Biological and Medical Properties, http://www.indsaff.com/10%20Curcumin%20biological.pdf, diakses tanggal 23 September 2010.
Anand, P., Kunnumakkara, A. B., Newman, R. A. and Anggarwal, B. B., 2007, Bioavailability of Curcumin: Problems and Promises, Mol. Pharm., 4 (6), 807-818.
Badan Pengawas Obat Dan Makanan, 2004, Keputusan Kepala Badan Pengawas Obat Dan Makanan Republik Indonesia Nomor: HK.00.05.4.2411 tentang Ketentuan Pokok Pengelompokan Dan Penandaan Obat Bahan
Alam Indonesia, BPOM RI, Jakarta,
http://www.pom.go.id/public/hukum_perundangan/pdf/Penandaan_OAI. pdf, diakses tanggal 13 Desember 2010.
Badan Pengawas Obat dan Makanan, 2005a, Peraturan Kepala Badan Pengawas Obat dan Makanan Nomor HK.00.05.41.1384 tentang Kriteria dan Tata Laksana Pendaftaran Obat Tradisional, Obat Herbal Terstandar dan
Fitofarmaka, BPOM RI, Jakarta,
http://www.pom.go.id/public/hukum_perundangan/pdf/KRITCARA%20 PENDAFT.OT.pdf, diakses tanggal 13 Desember 2010.
Badan Pengawas Obat dan Makanan, 2005b, Standarisasi Ekstrak Tumbuhan Obat Indonesia, Salah Satu Tahapan Penting dalam Pengembangan Obat Asli
Indonesia, InfoPOM, 6 (4), 1-5,
http://perpustakaan.pom.go.id/KoleksiLainnya/Buletin.pdf, diakses tanggal 13 Desember 2010.
Christian, G. D., 2004, Analytical Chemistry, 6th ed., Jhon Willey & Sons, Inc., USA, 65, 66, 483, 484.
Direktorat Jenderal Pengawasan Obat dan Makanan RI, 1979, Farmakope Indonesia, Edisi III, Departemen Kesehatan RI, Jakarta, 773.
Direktorat Jenderal Pengawasan Obat dan Makanan RI, 1995, Farmakope Indonesia, Edisi IV, Departemen Kesehatan RI, Jakarta, 6, 17, 1009. Dwivedi, A. K., Raman, M., Seth, R. K. and Sarin, J. P. S., 1992, Combined Thin
Fessenden, R. J. and Fessenden, J. S., 1986, Organic Chemistry, jilid 1, edisi 3, diterjemahkan oleh Pudjaatmaka, A. H., Penerbit Erlangga, Jakarta, 23-26.
Gritter, R. J., Bobbit, J. M., and Schwarting, A. E., 1991, Introduction to Chromatography, diterjemahkan oleh Kosasih Padmawinata, edisi II, ITB, Bandung, 205-219.
Hakim, A. R., 2002, Sintesis Kurkumin, Demetoksikurkumin, Bis -demetoksikurkumin an Pentagamavunon-O serta Pengaruhnya terhadap Farmakokinetika Teofilin pada Tikus, Tesis, Sekolah Pascasarjana, UGM, Yogyakarta, 38-41.
Harris, D. C., 1999, Quantitative Chemical Analysis, 2nd ed., W. H. Freeman Company, New York, 643, 648, 716-717.
Harmita, 2004, Petunjuk Pelaksanaan Validasi Metode dan Cara Perhitungannya,
Majalah Ilmu Kefarmasian, 1 (3), 117-134, Departemen Farmasi FMIP, Universitas Indonesia, Jakarta.
Heath, D. D., Pruitt, M. A., Brenner, D. E., Begun, A. N., Frautschy, S. A. and Roch, C. L., 2005, Tetrahydrocurcumin in Plasma and Urine: Quantitation by High Performance Liquid Chromatography, J. Chrom. B. Hendayana, S., 2006, Kimia Pemisahan Metode Kromatografi dan Elektroforesis
Modern, PT Remaja Rosdakarya, Bandung, 21-25.
Huang, M., Ma, W., Yen, P., Guo-Xie, J., Han, J., Frenkel, K., Grunberger, B. and Conney, A. H., 1997, Inhibitory Effect of Topical Application of Low Dose of Curcumin on 12-O-tetradecanoylphorbol-13-acetate Induced Tumor Promotion and Oxidized DNA Bases in Mouse Epidermis,
Carcinogenesis, 18 (1), 83-88.
Inoue, K., Nomura, C., Ito, S., Nagatsu, A., Hino, T. and Oka, H., 2008, Purification of Curcumin, Demethoxycurcumin, and Bisdemethoxycurcumin by High-Speed Countercurrent Chromatography,
J. Agric. Food. Chem., 56 (20), 9328-9336.
Jadhav, B.K., Mahadik, K.R. and Paradkar, A.R., 2007, Development and Validation of Improved Reversed-Phase HPLC Method for Simultaneous Determination of Curcumin, Demethoxycurcumin, and Bis-Demethoxycurcumin, Chrom., 65 (7/8), 483-488.