VALIDASI PENETAPAN KADAR CAMPURAN PARASETAMOL,
PROPIFENAZON, DAN KAFEIN DENGAN METODE KROMATOGRAFI
CAIR KINERJA TINGGI FASE TERBALIK
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Ilmu Farmasi
Oleh :
Adrian Rendy Irmanto
NIM : 058114010
FAKULTAS FARMASI UNIVERSITAS SANATA DHARMA
ii
VALIDASI PENETAPAN KADAR CAMPURAN PARASETAMOL,
PROPIFENAZON, DAN KAFEIN DENGAN METODE KROMATOGRAFI
CAIR KINERJA TINGGI FASE TERBALIK
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Ilmu Farmasi
Oleh :
Adrian Rendy Irmanto
NIM : 058114010
FAKULTAS FARMASI UNIVERSITAS SANATA DHARMA
v
Halaman Persembahan
You still are blind if you see a winding road
Because there is always a straight way to the point you
see
Don’t try to looked so wise
Don’t cry ‘cause you’re so right
Don’t try with fakes or fears
Because you will hate yourself in the end
(Akeboshi)
Kupersembahkan karyaku ini untuk :
Tuhan Yang Maha Kuasa
Papa dan Mamaku tercinta
Adik
–
adikku
Sahabat
–
sahabatku
vi
PRAKATA
Puji dan syukur kepada Tuhan Yang Maha Esa yang telah menyertai dan
melimpahkan kasih karunia-Nya kepada penulis, sehingga penulis dapat
menyelesaikan skripsi yang berjudul VALIDASI PENETAPAN KADAR
CAMPURAN PARASETAMOL, PROPIFENAZON, DAN KAFEIN
DENGAN METODE KROMATOGRAFI CAIR KINERJA TINGGI FASE
TERBALIK, sebagai salah satu syarat untuk memperoleh gelar Sarjana Farmasi
(S.Farm) pada Fakultas Farmasi Universitas Sanata Dharma Yogyakarta.
Keberhasilan penulis dalam menyusun skripsi ini tidak bisa lepas dari bantuan dan
dukungan dari banyak pihak, baik berupa material, moral, maupun spiritual.
Penulis mengucapkan terima kasih yang sebesar-besarnya kepada :
1. Ibu Rita Suhadi,M.Si.,Apt. selaku Dekan Fakultas Farmasi Universitas
Sanata Dharma.
2. Ibu Christine Patramurti, M.Si., Apt, selaku dosen pembimbing dan dosen
penguji. Terima kasih atas segala bimbingan, masukan, waktu, kesabaran
dan perhatiannya yang besar selama penelitian dan penyusunan skripsi ini.
3. Ibu Lucia Wiwid Wijayanti, M.Si. dan Bapak Jeffry Julianus, M.Si. selaku
dosen penguji atas segala masukan berupa kritik dan saran demi
kesempurnaan skripsi ini.
4. Papa, Mama, Vania, Nana, dan Juan atas doa dan dukungannya.
5. Mas Thomas Arian Adrianto, S.Farm. atas bantuan, masukan, dan
vii
6. Mas Bimo, Mas Kunto, Mas Parlan, Mas Wagiran, serta Mas Ottok atas
bantuan dan dukungan selama pelaksanaan penelitian ini
7. My best partner Happy yang sekuat tenaga membantu penelitian ini dari awal sampai selesai.
8. Sahabat –sahabatku yang terbaik, Dewi ”Sutok”, Mia, Aster, Tyas `Ndut,
dan Widdy terima kasih atas doa dan dukungannya.
9. Rio atas pinjaman scannernya yang sangat membantu penyusunan skripsi ini.
10.Teman-teman angkatan 2005, terutama kelas FST; Yoyok, Fian, Feli, Lina
Chang, Ong, Totok, Made, Berto, dan Reni, terima kasih atas kebersamaan
selama ini serta doa dan dukungannya.
11.Semua pihak yang tidak dapat penulis sebutkan satu persatu yang telah
banyak membantu.
Semoga Tuhan melimpahkan berkat dan rahmatNya atas segala kebaikan dan
ketulusan yang telah diberikan.
Penulis menyadari masih banyak kekurangan dalam penyusunan skripsi ini. Oleh
karena itu, penulis mengharapkan kritik dan saran demi penyempurnaan skripsi
ini. Akhirnya besar harapan penulis semoga skripsi ini dapat bermanfaat bagi
perkembangan ilmu pengetahuan dan bermanfaat bagi orang banyak.
Yogyakarta, Desember2008
viii
PERNYATAAN KEASLIAN KARYA
Saya menyatakan dengan sesungguhnya bahwa skripsi yang saya tulis ini
tidak memuat karya atau bagian karya orang lain, kecuali yang telah disebutkan
dalam kutipan dan daftar pustaka, sebagaimana layaknya karya ilmiah
Yogyakarta, Desember 2008
LEMBAR PERNYATAAN PERSETUJUAN
PUBLIKASI KARYA ILMIAH UNTUK KEPENTINGAN AKADEMIS
Yang bertanda tangan di bawah ini, saya mahasiswa Universitas Sanata Dharma :
Nama : Adrian Rendy Irmanto
Nomor Mahasiswa : 058114010
Demi pengembangan ilmu pengetahuan, saya memberikan kepada Perpustakaan
Universitas Sanata Dharma karya ilmiah saya yang berjudul :
VALIDASI PENETAPAN KADAR CAMPURAN PARASETAMOL,
PROPIFENAZON, DAN KAFEIN DENGAN METODE KROMATOGRAFI
CAIR KINERJA TINGGI FASE TERBALIK
Dengan demikian saya memberikan kepada Perpustakaan Universitas Sanata
Dharma hak untuk menyimpan, mengalihkan dalam bentuk media lain,
mengelolanya dalam bentuk pangkalan data, mendistribusikan secara terbatas, dan
mempublikasikannya di internet atau media lain untuk kepentingan akademis
tanpa perlu meminta ijin dari saya maupun memberikan royalti kepada saya
selama tetap mencantumkan nama saya sebagai penulis.
Demikian pernyataan ini yang saya buat dengan sebenarnya.
Dibuat di Yogyakarta
Pada tanggal 16 Februari 2009
Yang menyatakan
ix
INTISARI
Salah satu kombinasi zat aktif yang umum digunakan dalam obat analgesik – antipiretik adalah parasetamol, propifenazon, dan kafein yang memiliki kelarutan dalam etanol yang hampir sama dan serapan maksimum pada daerah UV yang berdekatan. Metode yang dapat digunakan untuk memisahkan sekaligus menetapkan kadar ketiga komponen tersebut yaitu metode KCKT fase terbalik dengan detektor UV.
Kondisi KCKT fase terbalik yang optimal untuk menetapkan kadar ketiga komponen tersebut yaitu kolom DuPont Instruments Zorbax ODS 4,6mm x 25cm; fase gerak metanol : aquabidest (40 : 60) pada flow rate 2 ml/menit serta detektor UV pada panjang gelombang 272 nm. Parameter validitas yang diuji meliputi akurasi, presisi, spesifisitas, linearitas, dan range.
Akurasi ditunjukkan oleh nilai rentang recovery sebesar95,741 – 98,759% untuk parasetamol; 97,760 – 101,220% untuk propifenazon; dan 105,556 – 109,397% untuk kafein. Presisi ditunjukkan oleh nilai CV sebesar 0,978% untuk parasetamol; 1,132% untuk propifenazon; dan 1,128% untuk kafein. Spesifisitas ditunjukkan oleh profil pemisahan ketiga analit dalam campuran. Linearitas ditunjukkan oleh nilai koefisien korelasi (r) sebesar 0,9999 untuk parasetamol; 0,9991 untuk propifenazon; dan 0,9991 untuk kafein. Range untuk parasetamol antara 239,3998 – 246,6509 ppm; untuk propifenazon antara 148,3515 – 151,6788 ppm; dan untuk kafein antara 50,0863 – 83,9953 ppm.
x
ABSTRACT
One of active ingredients combination commonly used in analgesic– antipiretic drug is paracetamol, propyphenazone, and caffeine which are have almost similar solubility in ethanol and maximum absorbance at nearly UV range. The method which can be used to separate and quantify those three active ingredients is Reversed Phase High Performance Liquid Chromatography with UV detector.
The optimum condition of RP-HPLC for quantifying those three active ingredients were DuPont Instruments Zorbax ODS 4,6mm x 25cm column; methanol : aquabidest (40 : 60) mobile phase at 2 ml/minute flow rate and UV detector at the wavelength of 272 nm. Validity parameters which were tested including accuracy, precision, specificity, linearity, and range.
Accuracy was proved by recovery range of 95.741 – 98.759% for paracetamol, 97.760 – 101.220% for propyphenazone, and 105.556 – 109.397% for caffeine. Precision was proved by CV of 0.978% for paracetamol; 1.132% for propyphenazone; and 1.128% for caffeine. Specificity was showed by separation profile of those three analytes in mixture. Linearity was proved by correlation coefficient (r) of 0.9999 for paracetamol; 0.9991 for propyphenazone; and 0.9991 for caffeine. Range for paracetamol were between 239.3998 – 246.6509 ppm; for propyphenazone were between 148.3515 – 151.6788 ppm; and for caffeine were between 50.0863 – 83.9953 ppm.
xi
DAFTAR ISI
HALAMAN SAMPUL ... . i
HALAMAN JUDUL ... ii
HALAMAN PERSETUJUAN PEMBIMBING ... iii
HALAMAN PENGESAHAN ... iv
HALAMAN PERSEMBAHAN ... v
PRAKATA ... vi
PERNYATAAN KEASLIAN KARYA ... viii
INTISARI ... ix
ABSTRACT ... x
DAFTAR ISI ... xi
DAFTAR TABEL ... xv
DAFTAR GAMBAR ... xvii
DAFTAR LAMPIRAN ... xx
BAB I. PENGANTAR ... 1
A. Latar Belakang ………... 1
1. Permasalahan... 2
2. Keaslian penelitian ... 3
3. Manfaat penelitian ... 4
B. Tujuan Penelitian ... 4
BAB II. PENELAAHAN PUSTAKA ... 5
A. Parasetamol... 5
xii
C. Kafein ... 6
D. Kromatografi Cair Kinerja Tinggi... 7
1. Peralatan KCKT... 7
2. Pembagian Jenis Kromatografi... 12
3. Kromatografi Partisi Fase Balik... 14
4. Analisis Kualitatif dan Kuantitatif... 16
E. Spektrofotemetri Ultraviolet... 24
F. Kesahihan Metode Analisis Instrumental... 25
1. Akurasi... 26
2. Presisi... 26
3. Limit of Detection... 27
4. Limit of Quantitation... 27
5. Linieritas... 28
6. Spesifisitas... 28
7. Range…………. 28
G. Kesalahan Metode Analisis Instrumental……….…………... 30
1. Kesalahan Sistematik... 30
2. Kesalahan Tidak Sistematik... 31
H. Keterangan Empiris …... 31
BAB III. METODOLOGI PENELITIAN ... 33
A. Jenis Rancangan Penelitian ... 33
B. Variabel Penelitian ... 33
xiii
2. Variabel Pengacau Terkendali... 34
C. Definisi Operasional... 34
D. Bahan Penelitian... 34
E. Alat Penelitian ... 35
F. Tata Cara Penelitian ... 36
1. Pembuatan Larutan Baku Parasetamol, Propifenazon, dan Kafein... 36
2. Pembuatan Fase Gerak... 37
3. Pengamatan Panjang Gelombang Pengamatan antara Parasetamol, Propifenazon, dan Kafein dengan Spektrofotometer UV... 38
4. Pengamatan Waktu Retensi Parasetamol, Propifenazon, dan Kafein... 38
5. Optimasi Pemisahan Parasetamol, Propifenazon, dan Kafein dengan KCKT... 39
6. Validasi Metode Analisis... 39
F. Analisis Hasil... 40
BAB IV. HASIL DAN PEMBAHASAN ... 42
A. Penyiapan Fase Gerak... 42
B. Pembuatan Larutan Baku... 43
C. Optimasi Metode KCKT... 44
1. Penentuan Panjang Gelombang Pengamatan Dengan Spektofotometri UV... 44
xiv
D. Penetapan Kadar Parasetamol, Propifenazon, dan Kafein dalam Sampel
Simulasi... 60
1. Pembuatan Kurva Baku... 60
2. Penetapan Kadar Parasetamol, Propifenazon, dan Kafein dalam Campuran Sampel Simulasi dan Validasi Metode... 63
BAB V. KESIMPULAN DAN SARAN ... 71
A. Kesimpulan ... 71
B. Saran ... 72
DAFTAR PUSTAKA ... 73
LAMPIRAN ... 76
xv
DAFTAR TABEL
Tabel I. Nilai indeks polaritas pelarut... 11
Tabel II. Parameter analitik...……… 29
Tabel III. Pengamatan waktu retensi parasetamol, propifenazon, dan kafein pada berbagai perbandingan fase gerak dan flow rate tertentu... 47
Tabel IV. Data kurva baku parasetamol………... 60
Tabel V. Data kurva baku propifenazon………... 61
Tabel VI. Data kurva baku kafein…………... 62
Tabel VII. Hasil penetapan kadar parasetamol dalam sampel simulasi kadar rendah………... 64
Tabel VIII. Hasil penetapan kadar propifenazon dalam sampel simulasi kadar rendah ………... 64
Tabel IX. Hasil penetapan kadar kafein dalam sampel simulasi kadar rendah...………... 64
Tabel X. Hasil penetapan kadar parasetamol dalam sampel simulasi kadar sedang...………... 66
Tabel XI. Hasil penetapan kadar propifenazon dalam sampel simulasi kadar sedang...………... 66
Tabel XII. Hasil penetapan kadar kafein dalam sampel simulasi kadar sedang...………... 67
xvi
Tabel XIV. Hasil penetapan kadar propifenazon dalam sampel simulasi
kadar tinggi...………... 68
Tabel XV. Hasil penetapan kadar kafein dalam sampel simulasi kadar
xvii
DAFTAR GAMBAR
Gambar 1. Rumus struktur parasetamol... 5
Gambar 2. Rumus struktur propifenazon... 6
Gambar 3. Rumus struktur kafein... 6
Gambar 4. Skema peralatan KCKT... 7
Gambar 5. Skema pemilihan kolom... 10
Gambar 6. Pemilihan jenis KCKT... 13
Gambar 7. Reaksi antara gugus silanol dan gugus klorosilan... 15
Gambar 8. Reaksi pembuatan kolom oktadesilsilan... 15
Gambar 9. Resolusi antara dua peak yang berdekatan... 17
Gambar 10. Difusi Eddy... 20
Gambar 11. Transfer massa fase diam dan fase gerak... 21
Gambar 12. Distribusi analit dalam fase gerak dan fase diam... 22
Gambar 13. Pengukuran peak asymmetry factor dan peak tailing factor... 23
Gambar14. Spektra panjang gelombang maksimum parasetamol, propifenazon, dan kafein... 44
xviii
Gambar 17. Kromatogram pemisahan parasetamol, propifenazon, dan
kafein dengan fase gerak metanol : aquabidest (60 : 40) flow
rate 0,5 ml/menit... 50
Gambar 18. Kromatogram pemisahan parasetamol, propifenazon, dan
kafein dengan fase gerak metanol : aquabidest (50 : 50) flow
rate 0,5 ml/menit ………... 51
Gambar 19. Kromatogram pemisahan parasetamol, propifenazon, dan
kafein dengan fase gerak metanol : aquabidest (50 : 50) flow
rate 1,0 ml/menit... 52
Gambar 20. Penggaraman kafein oleh asam asetat glasial ………... 53
Gambar 21. Kromatogram pemisahan parasetamol, propifenazon, dan
kafein dengan fase gerak metanol : aquabidest : asam asetat
glasial (70 : 28,5 : 1,5) flow rate 0,5 ml/menit ………. 53
Gambar 22. Kromatogram pemisahan parasetamol, propifenazon, dan
kafein dengan fase gerak metanol : aquabidest : asam asetat
glasial (60 : 37 : 3) flow rate 0,5 ml/menit ………... 54
Gambar 23. Penggaraman propifenazon oleh asam asetat glasial ……… 55
Gambar 24. Kromatogram pemisahan parasetamol, propifenazon, dan
kafein dengan fase gerak metanol : aquabidest : asam asetat
glasial (50 : 49 : 1) flow rate 0,5 ml/menit ………... 56
Gambar 25. Kromatogram pemisahan parasetamol, propifenazon, dan
kafein dengan fase gerak metanol : aquabidest (40 : 60) flow
xix
Gambar 26. Kromatogram pemisahan parasetamol, propifenazon, dan
kafein dengan fase gerak metanol : aquabidest (40 : 60) flow
rate 1,5 ml/menit ………... 58
Gambar 27. Kromatogram pemisahan parasetamol, propifenazon, dan kafein dengan fase gerak metanol : aquabidest (40 : 60) flow rate 2,0 ml/menit ………... 59
Gambar 28. Kurva baku parasetamol………...……… 61
Gambar 29. Kurva baku propifenazon………...………... 61
xx
DAFTAR LAMPIRAN
Lampiran 1. Sertifikat analisis parasetamol... 76
Lampiran 2. Sertifikat analisis propifenazon... 77
Lampiran 3. Sertifikat analisis kafein... 79
Lampiran 4. Skema pembuatan larutan baku parasetamol dan contoh perhitungan kadar larutan baku yang digunakan ... 80
Lampiran 5. Skema pembuatan larutan baku propifenazon dan contoh perhitungan kadar larutan baku yang digunakan ... 81
Lampiran 6. Skema pembuatan larutan baku kafein dan contoh perhitungan kadar larutan baku yang digunakan ... 82
Lampiran 7. Kromatogram larutan kurva baku parasetamol……... 83
Lampiran 8. Kromatogram larutan kurva baku propifenazon... 87
Lampiran 9. Kromatogram larutan kurva baku kafein... 91
lampiran 10. Skema pembuatan sampel simulasi dan contoh perhitungan kadar parasetamol, propifenazon, dan kafein dalam sampel simulasi... 95
1
BAB I
PENGANTAR
A. Latar Belakang
Dewasa ini, obat seolah – olah sudah menjadi suatu kebutuhan penting
dalam hidup sehari – hari. Hal ini didukung oleh kecenderungan masyarakat untuk
melakukan self medication terutama untuk penyakit pada tingkat keparahan yang tidak serius (Azizahwati, 2000). Salah satu penyakit yang sering menjadi objek
self medication oleh masyarakat adalah influenza, yang umumnya diobati dengan obat analgesik – antipiretik.
Salah satu kombinasi zat aktif dalam obat analgesik – antipiretik yang
umum digunakan adalah kombinasi parasetamol, propifenazon, dan kafein.
Kombinasi ini berfungsi untuk mengoptimalkan efek terapi obat serta
meminimalisasi efek merugikan (adverse effect) yang mungkin terjadi bila zat aktif tersebut dipejankan dalam bentuk tunggal dengan dosis besar untuk
mencapai intensitas efek terapi yang diinginkan (Raffa, 2006).
Kini, banyak obat analgesik – antipiretik yang berupa kombinasi
parasetamol, propifenazon, dan kafein diproduksi dalam berbagai merek dagang,
di antaranya Bodrex migra, Paramex, dan Saridon yang berbentuk tablet. Banyaknya produksi obat – obat ini perlu diimbangi dengan peningkatan
pengawasan mutu, agar obat yang beredar tersebut dapat dijamin keamanan dan
khasiatnya. Belum adanya suatu metode yang sederhana, cepat, dan tepat yang
simultan, mendasari perlunya dilakukan suatu penelitian untuk mendapatkan
kondisi yang optimal untuk menetapkan kadar ketiga komponen tersebut secara
simultan.
Metode kromatografi cair kinerja tinggi fase terbalik dipilih karena dengan
metode ini dapat dilakukan pemisahan zat aktif parasetamol, propifenazon, dan
kafein sekaligus penetapan kadar tiap zat aktif tersebut dalam campuran. Ketiga
komponen zat aktif tersebut memiliki sifat fisika – kimia yang mirip, antara lain
kelarutan dalam etanol yang hampir sama dan serapan maksimum pada daerah
UV yang berdekatan (Clarke, 1986).
Hasil yang diperoleh dari optimasi yang dilakukan merupakan suatu
metode analisa yang baru sehingga perlu dilakukan validasi agar metode ini
memiliki hasil yang dapat dipertanggungjawabkan. Dalam USP 28, parameter
yang diuji pada validasi metode meliputi akurasi, presisi, spesifisitas, linieritas,
dan range (Anonim, 2005). Melalui penelitian ini, diharapkan dapat diperoleh informasi mengenai metode analisis multikomponen dari campuran parasetamol,
propifenazon, dan kafein menggunakan kondisi KCKT yang teruji validitasnya.
1. Permasalahan
Berdasarkan latar belakang tersebut, maka dapat disusun permasalahan
sebagai berikut :
a. Bagaimanakah kondisi yang optimal untuk memisahkan dan menetapkan kadar
parasetamol, propifenazon, dan kafein secara simultan dengan metode KCKT
b. Apakah metode KCKT fase terbalik yang digunakan untuk penetapan kadar
parasetamol, propifenazon, dan kafein secara simultan memiliki akurasi,
presisi, spesifisitas, linieritas, dan range yang baik?
2. Keaslian Penelitian
Analisis multikomponen dari campuran parasetamol, propifenazon, dan
kafein dalam tablet sediaan obat pernah dilakukan oleh Dimitrovska,
Trajkovic-Jolevska, Nancovska, dan Ilievska (1995) dengan metode KLT dan spektrofometri
UV. Penelitian lain yang pernah dilakukan adalah Program Komputer Analisis
Multikomponen Untuk Obat Flu dan Analgesik – Antiinflamasi (Yanuar, Hayun,
Suryadi, Henry, dan Wulandari, 2003) yang menggabungkan teknik
spektrofotometri UV dan matriks matematika, serta Optimasi Penetapan Kadar
Obat Analgesik Multikomponen dengan Metode KCKT Fase Balik (Ivanovic,
Medenica, Malenovic, Jancic, dan Misljenovic, 2003) yang menggunakan fase
gerak metanol : aquabidest pada berbagai rasio berkisar dari (30 : 70 v/v) hingga
(65 : 35 v/v) pada detektor UV 265 nm dan kolom Beckman Ultrasphere ODS
4,6 mm x 150 mm dengan ukuran partikel 5 µm. Namun tidak diketahui kondisi
yang optimal untuk penelitian tersebut karena tidak dipublikasikan secara bebas.
Metode KCKT banyak digunakan untuk menganalisis sediaan obat
multikomponen. Namun validasi penetapan kadar campuran parasetamol,
propifenazon, dan kafein dengan metode KCKT dengan fase gerak, flow rate, dan kolom DuPont Instruments Zorbax ODS 4,6 mm x 25 cm P.N 880952-702 yang
3. Manfaat Penelitian
a. Manfaat metodologis
Manfaat metodologis dari penelitian ini adalah dapat memberikan sumbangan
ilmiah mengenai metode penetapan kadar parasetamol, propifenazon, dan
kafein secara simultan yang teruji validitasnya.
b. Manfaat praktis
Manfaat praktis dari penelitian ini adalah dapat digunakan sebagai metode
untuk analisis multikomponen dari campuran parasetamol, propifenazon, dan
kafein.
B. Tujuan Penelitian
Berdasarkan latar belakang dan permasalahan yang ada, maka dapat
disusun tujuan penelitian ini, yaitu :
1. Mengetahui kondisi yang optimal untuk memisahkan dan menetapkan kadar
parasetamol, propifenazon, dan kafein secara simultan dengan metode KCKT
fase terbalik menggunakan kolom C18.
2. Mengetahui akurasi, presisi, spesifisitas, linieritas, dan range metode KCKT fase terbalik yang digunakan untuk penetapan kadar parasetamol,
5
BAB II
PENELAAHAN PUSTAKA
A. Parasetamol
Parasetamol mempunyai sinonim asetaminofen dan p-asetamidifenol,
dengan rumus molekul C8H9NO2 dan berat molekul 151,6 (Anonim,1995).
O
N H
OH
Gambar 1. Rumus struktur parasetamol (Anonim,1995)
Menurut Clarke (1986) 1 gram parasetamol larut dalam 70 ml air, 20 ml
air mendidih, 7 ml etanol, dan 50 ml kloroform. Parasetamol larut dalam
dimetilforfamid, metanol, etilendiklorida, aseton, etil asetat, dan natrium
hidroksida 1 N. Parasetamol tidak larut dalam eter, petroleum eter, pentana, dan
benzen (Anonim, 1995). Parasetamol memberikan serapan maksimum dalam
etanol pada panjang gelombang 250 nm dengan nilai A % 1
1cmsebesar 913.
Parasetamol memberikan serapan maksimum dalam metanol pada panjang
gelombang 250 nm dengan nilai A % 1
1cm sebesar 900 (Autherhoff,1987).
B. Propifenazon
Propifenazon mempunyai sinonim 4-isopropilantipirin, isopropilfenazon,
Baukal, dan Causyth, dengan rumus molekul C14H18N2O dan berat molekul
O
N
N
Gambar 2. Rumus struktur propifenazon (Anonim,1989)
Propifenazon berbentuk kristal dengan rasa agak pahit. Titik didih
propifenazon pada 1030C. Propifenazon mudah larut dalam alkohol dan eter.
Kelarutan propifenazon dalam air sebesar 0,24 g / 100 ml pada 16,50C (Anonim,
1989). Propifenazon memberikan serapan maksimum dalam etanol pada panjang
gelombang 248 nm dengan nilai A % 1
1cmsebesar 483; dan pada panjang gelombang
277 nm dengan nilai A % 1
1cm sebesar 493 (Clarke, 1986).
C. Kafein
Kafein mempunyai sinonim 1,3,7-trimetilxantin atau
1,3,7-trimetil-2,6-dioksopurin, dengan rumus molekul C8H10N4O2. Pemeriannya berupa serbuk
putih atau bentuk jarum mengkilat, biasanya menggumpal, tidak berbau, dan
berasa pahit (Anonim,1995).
Gambar 3. Rumus struktur kafein (Anonim,1995)
Menurut Clarke (1986) 1 gram kafein larut dalam 60 ml air, 75 ml etanol,
50 ml aseton, 900 ml eter, dan 8 ml kloroform. Kafein agak sukar larut dalam air
(Anonim, 1995). Kafein memberikan serapan maksimum dalam HCl 0,1 N pada
panjang gelombang 272 nm dengan nilai A % 1
1cmsebesar 470. Kafein memberikan
serapan maksimum dalam etanol pada panjang gelombang 273 nm dengan nilai
A % 1
1cmsebesar 519 (Clarke, 1986).
D. Kromatografi Cair Kinerja Tinggi
1. Peralatan KCKT
KCKT merupakan kondisi kromatografi yang fase geraknya dialirkan
menuju kolom secara cepat dengan bantuan tekanan dari pompa dan hasilnya
dapat dideteksi dengan detektor (Hendayana, 2006). Tujuan dari KCKT adalah
memperoleh hasil pemisahan yang baik dalam waktu relatif singkat (Mulja dan
Suharman, 1995). Peralatan KCKT biasanya terdiri dari beberapa komponen
seperti yang dapat dilihat pada gambar di bawah ini :
Menurut Gritter, Bobbit, dan Schwarting (1991), ada tiga variabel utama
pada kondisi KCKT yang harus diperhatikan, yaitu :
a. Detektor
Detektor diperlukan untuk mendeteksi adanya komponen cuplikan yang
terdapat dalam kolom dan untuk mengukur jumlah komponen yang ada dalam
cuplikan (Johnson dan Stevenson, 1978). Beberapa persyaratan detektor
menurut Mulja dan Suharman ialah sensitivitas yang sangat tinggi dengan
rentang sensitivitas 10-8 hingga 10-15 gram solut per detik, kestabilan dan
reprodusibilitas yang sangat baik, memberikan respon yang linier terhadap
konsentrasi solut, dapat bekerja dari temperatur kamar hingga 400 0C, tidak
dipengaruhi perubahan temperatur dan kecepatan pelarut pengembang, mudah
didapat dan mudah dipakai oleh operator, selektif terhadap macam – macam
linarut dalam pelarut pengembang dan tidak merusak sampel. Detektor untuk
KCKT dibagi dalam dua kategori, yaitu :
1)Bulk Property Detector
Detektor ini merupakan jenis detektor yang mengukur sifat solut dan fase
gerak. Contohnya adalah detektor indeks bias. Detektor indeks bias adalah
suatu jenis detektor universal yang menangkap sinyal pada setiap solut yang
memiliki indeks bias berbeda dengan indeks bias fase gerak. Kelemahannya
adalah indeks bias sangat dipengaruhi oleh suhu. Selain itu, detektor indeks
bias juga kurang sensitif dan tidak cocok untuk kondisi elusi landaian
2)Solute Property Detector
Detektor ini merupakan detektor yang selektif mengukur sifat solut.
Detektor ini lebih sensitif dibanding bulk property detector. Contohnya : detektor UV-Vis dan detektor fluoresensi. Detektor pada KCKT yang sering
digunakan dalam analisis farmasi ialah detektor UV-Vis. Hal ini disebabkan
kebanyakan senyawa obat memiliki struktur yang dapat menyerap sinar
UV-Vis. Detektor UV digunakan untuk mendeteksi senyawa – senyawa
yang memiliki gugus kromofor dengan atau tanpa adanya gugus auksokrom,
sedangkan detektor visibel digunakan untul mendeteksi senyawa berwarna
yaitu senyawa yang memiliki gugus kromofor yang sangat panjang maupun
merupakan senyawa kompleks (Settle, 1997).
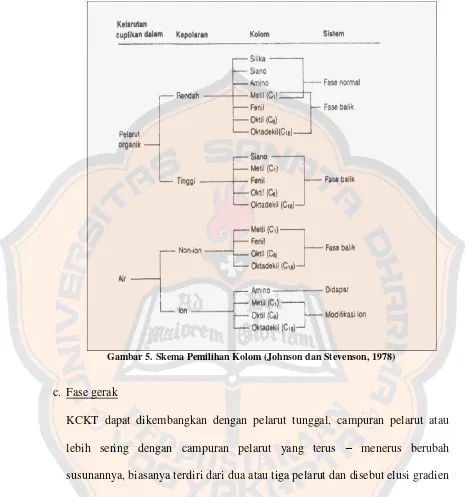
b. Kolom
Kolom pada kondisi KCKT merupakan bagian yang sangat penting karena
pemisahan komponen – komponen sampel akan terjadi di dalam kolom.
Keberhasilan pemisahan komponen – komponen sampel akan sangat
bergantung pada keadaan kolom (Mulja dan Suharman, 1995). Pemilihan
kolom yang tepat sangat menentukan keberhasilan pemisahan. Berikut ini
Gambar 5. Skema Pemilihan Kolom (Johnson dan Stevenson, 1978)
c. Fase gerak
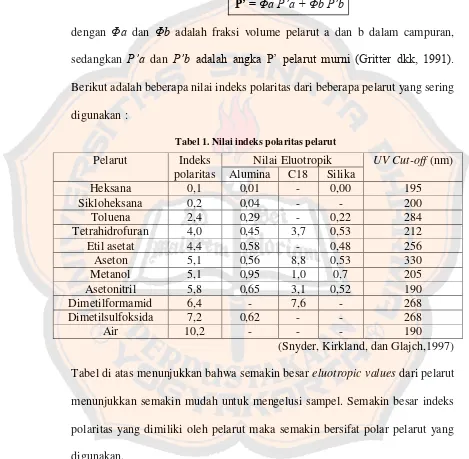
KCKT dapat dikembangkan dengan pelarut tunggal, campuran pelarut atau
lebih sering dengan campuran pelarut yang terus – menerus berubah
susunannya, biasanya terdiri dari dua atau tiga pelarut dan disebut elusi gradien
(Gritter dkk, 1991). Pada KCKT, fase gerak harus mempunyai sifat : murni dan
tanpa cemaran, tidak bereaksi dengan kemasan, sesuai dengan detektor, dapat
melarutkan sampel, viskositas rendah, memungkinkan memperoleh kembali
sampel dengan mudah (jika diperlukan), dan harganya wajar (Johnson dan
campuran pelarut yang digunakan yang bersifat linier dengan kepolaran pelarut
murninya. Nilai kepolaran antara dua campuran sembarang pelarut dapat
dihitung dengan persamaan di bawah ini :
P’ = Φa P’a + Φb P’b
dengan Φa dan Φb adalah fraksi volume pelarut a dan b dalam campuran,
sedangkan P’a dan P’b adalah angka P’ pelarut murni (Gritter dkk, 1991).
Berikut adalah beberapa nilai indeks polaritas dari beberapa pelarut yang sering
digunakan :
Tabel 1. Nilai indeks polaritas pelarut
Pelarut Indeks
polaritas
Nilai Eluotropik UV Cut-off (nm)
Alumina C18 Silika
(Snyder, Kirkland, dan Glajch,1997)
Tabel di atas menunjukkan bahwa semakin besar eluotropic values dari pelarut menunjukkan semakin mudah untuk mengelusi sampel. Semakin besar indeks
polaritas yang dimiliki oleh pelarut maka semakin bersifat polar pelarut yang
2. Pembagian jenis kromatografi
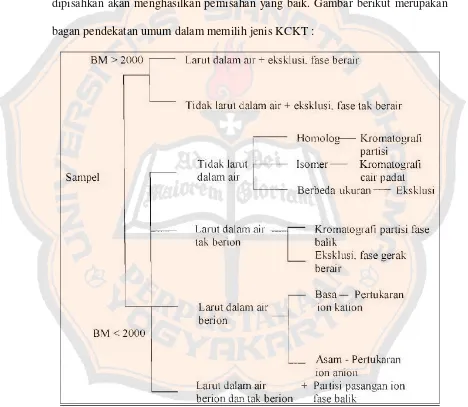
Menurut Harris (1999), KCKT dibagi menjadi 5 jenis, yaitu :
a. Kromatografi partisi
Pada kromatografi partisi, fase diam dapat polar atau non polar. Bila fase diam
polar dan fase gerak non polar disebut kromatografi partisi fase normal,
sedangkan bila fase diam non polar dan fase gerak polar dinamakan
kromatografi partisi fase terbalik. Solut berkeseimbangan di antara fase diam
dan fase gerak.
b. Kromatografi adsorpsi
Kromatografi ini menggunakan fase diam padat dan fase gerak cair atau gas.
Solut dapat diadsorpsi pada permukaan partikel padat.
c. Kromatografi pertukaran ion
Anion atau kation diikatkan secara kovalen pada fase diam padat, biasanya
disebut resin. Ion – ion solut muatan berlawanan menyerang fase diam dengan
kekuatan elektrostatik dan fase geraknya berupa zat cair.
d. Kromatografi eksklusi
Pada kromatografi ini tidak ada interaksi tarik – menarik antara fase diam dan
solut. Fase gerak cair atau gas melalui gel berpori. Ukuran pori cukup kecil
untuk mengeluarkan solut yang besar. Molekul solut yang kecil akan masuk ke
dalam pori gel, sedangkan molekul yang besar akan mengalir tanpa memasuki
e. Kromatografi afinitas
Digunakan untuk interaksi yang spesifik antara suatu jenis molekul solut dan
sebuah molekul yang lain yang secara kovalen terikat pada fase diam. Misalnya
untuk pemisahan komponen protein.
Pemilihan jenis KCKT yang tepat dan sesuai dengan sampel yang
dipisahkan akan menghasilkan pemisahan yang baik. Gambar berikut merupakan
bagan pendekatan umum dalam memilih jenis KCKT :
3. Kromatografi partisi fase balik
Istilah fase normal dan fase terbalik digunakan dalam kromatografi partisi
untuk menggambarkan polaritas efektif antara fase diam dan fase gerak (Settle,
1997). Kromatografi dengan fase diam polar dan fase gerak kurang polar atau non
polar disebut kromatografi fase normal. Sebaliknya, kromatografi yang
menggunakan fase diam relatif non polar seperti hidrokarbon dan fase gerak polar
seperti air atau metanol disebut kromatografi fase terbalik (Gritter dkk, 1991).
Menurut Gritter dkk. (1991), konsep pada pengembangan kromatografi
cair partisi yaitu perlakuan sampel dalam kondisi cair – cair tergantung pada
kelarutannya di dalam kedua cairan yang terlibat. Jika solut ditambahkan ke
dalam kondisi yang terdiri atas dua pelarut yang tidak bercampur dan keseluruhan
kondisi dibiarkan seimbang, solut akan tersebar antara kedua fase itu menurut
persamaan :
Cm Cs
K
K adalah koefisien distribusi, Cs adalah konsentrasi solut dalam fase diam dan
Cm adalah konsentrasi solut dalam fase gerak (Skoog, West, dan Holler,1994).
Hal – hal yang harus diperhatikan dalam pemilihan metode kromatografi
partisi fase balik adalah :
a. Kolom
Kolom yang digunakan pada jenis kromatografi ini ialah kemasan fase terikat.
Penyanga pada kemasan fase terikat terbuat dari silika (Gritter dkk, 1991).
Kemasan fase terikat bersifat stabil karena fase diamnya terikat secara kimia
terikat mempunyai tipe ikatan siloksan (Si-O-Si-C), dibuat dari reaksi antara
gugus klorosilan dengan gugus silanol yang terdapat pada permukaan silika
gel, seperti pada gambar berikut :
Si OH + ClSi(CH3)2R Si O Si(CH3)2R + HCl
Gambar 7. Reaksi antara gugus silanol dan gugus klorosilan
Tertambatnya sampel pada fase diam dipengaruhi oleh panjang pendeknya
rantai karbon. Kelebihan kolom dengan rantai karbon yang lebih panjang
adalah sifatnya yang lebih retensif, sehingga sampel yang mempunyai sifat
mirip dengan kolom akan tertambat lebih lama (Skoog dkk, 1994). Menurut
Willard, Merritt, Dean, dan Settle (1988), kolom oktadesilsilan digunakan pada
aplikasi yang membutuhkan retensi yang maksimal, sehingga pemisahan
komponen sampel dapat lebih optimal. Oktadesilsilan dapat dibuat dari reaksi
berikut :
Si OH + Cl Si (CH2)17CH3 Si O Si (CH2)17CH3 + HCl
Gambar 8. Reaksi pembuatan kolom oktadesilsilan (Gritter dkk, 1991)
Fase diam yang biasa digunakan pada kromatografi partisi fase balik adalah
oktadesilsilan (ODS). Selain ODS, dikenal pula silika dengan substitusi oktil
(C8) (Munson, 1991). Interaksi antara senyawa dengan sisa silanol dapat
mengganggu penggunaan kolom fase terbalik. Akibatnya waktu retensi dari
senyawa standar menjadi lebih sulit untuk ditafsirkan. Dalam beberapa kasus,
senyawa ditahan akan tergantung pada lipofilisitas dalam kasus kolom fase
terbalik seperti ODS silika gel. Fase gerak yang lebih hidrofil akan lebih cepat
mengelusi suatu senyawa dari kolom fase terbalik (Watson, 1999). Ukuran
kinerja kolom dilihat dari kemampuan kolom untuk memisahkan komponen
senyawa yang akan dianalisis. Batasan yang banyak digunakan adalah sebagai
berikut yaitu jumlah lempeng teoritik (N), dan nilai H atau HETP (Height Equivalent to a Theoritical Plate) yang merupakan penentu ukuran efisiensi kolom, faktor resolusi, serta bentuk peak. Kolom yang efisien dapat mencegah pelebaran peak yang sempit.
b. Fase gerak
Kemampuan KCKT dalam memisahkan banyak senyawa terutama bergantung
pada tambatan sampel dan pemisahan komponen dalam campuran. Pada fase
balik, kandungan utama fase geraknya adalah air. Pelarut yang dapat campur
dengan air seperti metanol, etanol, asetonitril, dan tetrahidrofuran ditambahkan
untuk mengatur kepolaran fase gerak. Menurut Munson (1991), pemodifikasi
fase gerak yang paling banyak digunakan ialah metanol, asetonitril, dan
tetrahidrofuran. Metanol sering digunakan karena merupakan pelarut yang
sangat murni, mudah didapat, dan berhasil baik pada banyak pemisahan
(Johnson dan Stevenson, 1978).
4. Analisis kualitatif dan analisis kuantitatif
Waktu retensi adalah selang waktu yang diperlukan oleh linarut (solut)
dan dinyatakan sebagai tr ( Mulja dan Suharman, 1995). Waktu retensi ini bersifat
sangat khas untuk senyawa tertentu pada kondisi tertentu (kolom, suhu, laju
aliran, dan sebagainya). Beberapa senyawa mungkin mempunyai waktu retensi
berdekatan tetapi tiap senyawa hanya mempunyai satu waktu retensi saja. Waktu
retensi tidak terpengaruh oleh adanya komponen lain (Nair dan Bonelli, 1988).
Di dalam setiap pemisahan yang dipentingkan adalah kemampuan suatu
kondisi untuk memisahkan dua komponen dalam campuran. Kemampuan tersebut
dinamakan resolusi (Rs) yang didefinisikan sebagai jarak antara dua puncak
dibagi dengan rata – rata lebar dasar puncak, seperti terlihat pada gambar berikut :
Gambar 9. Resolusi antara dua peak yang berdekatan (Johnson dan Stevenson, 1991)
Berdasarkan definisi di atas, maka persamaan resolusi adalah sebagai
berikut :
telah mencapai 99,7% (Sastrohamidjojo, 2002). Resolusi juga dipengaruhi oleh
Rs =
dioptimasi. Optimasi efisiensi kolom (a) dilakukan dengan menambah jumlah
lempeng teoritis (N), yaitu dengan memperpanjang kolom (N=L/H) dengan L
adalah panjang kolom yang digunakan, dan N adalah jumlah pelat teoritis dari
suatu kolom sehingga diperoleh puncak yang kecil dan resolusi yang baik.
Optimasi faktor selektivitas (b) dilakukan dengan mengganti pelarut atau
mengubah komposisi pelarut sehingga efisiensi pelarut bertambah dan resolusi
juga meningkat. Optimasi faktor kapasitas (c) dilakukan dengan memvariasi
kekuatan pelarut sehingga fase gerak dapat memberikan harga k’ suatu komponen
sampel menjadi lebih besar atau lebih kecil. Dengan meningkatkan harga k’ maka
akan memperbaiki resolusi (Noegrohati, 1994).
Ada dua teori yang dapat menerangkan tentang efisiensi kolom yaitu :
a. Teori pelat
Teori ini menyatakan bahwa pelat (atau lebih baik disebut HETP) merupakan
tinggi atau panjang dari kolom yang cukup dapat mencapai kesetimbangan
antara solut dalam fase gerak dan fase diam. Semakin banyak pelat yang
dimiliki kolom maka akan memberikan puncak yang lebih sempit atau dapat
dikatakan efisiensi kolom menjadi lebih baik.
HETP =
N L
HETP adalah ketinggian ekivalen terhadap jumlah pelat teoritis, L adalah
panjang kolom yang digunakan, dan N adalah jumlah pelat teoritis dari suatu
kolom (Sastrohamidjojo, 2002). Menurut Mulja dan Suharman (1995), dapat
dikatakan bahwa makin kecil harga L/N atau makin kecil harga HETP maka
makin baik efisiensi kolom yang digunakan. Konsekuensi dari penambahan
lempeng teoritis yaitu semakin lama sampel untuk terelusi yang berakibat
waktu retensi semakin lama (Skoog dkk, 1994). Daya pisah dapat diperbaiki
apabila efisiensi kolom rendah yang dapat diukur secara kuantitatif seperti pada
persamaan berikut :
sedangkan W1/2 adalah lebar peak pada setengah tinggi peak (Anonim, 1995).
b. Teori laju
Teori ini didasarkan pada parameter – parameter transfer massa antara fase
diam dan fase gerak, laju difusi solut di sepanjang kolom, laju alir fase gerak,
dan dinamika fase gerak. Ada 3 faktor yang sangat menentukan berdasarkan
teori laju ini yaitu :
1)Difusi Eddy
Difusi ini disebabkan karena banyaknya kemungkinan celah dalam partikel
terpacking yang dapat dilewati oleh molekul solut. Dengan demikian molekul solut ada yang melewati bagian kolom yang dekat dengan dinding
tersebut dapat lebih cepat keluar dari kolom. Sedangkan untuk molekul yang
melalui bagian tengah kolom yang merupakan suatu daerah packing lebih tinggi akan keluar kolom dengan kecepatan yang lebih rendah. Hal ini
menyebabkan elusi untuk tiap solut menjadi kurang efisien (Hendayana,
2006).
Gambar 10. Difusi Eddy
2)Difusi longitudinal
Difusi ini merupakan gerakan molekul solut yang cenderung untuk berdifusi
ke segala arah secara acak karena adanya perbedaan konsentrasi
(Noegrohati, 1994). Semakin lama solut berada dalam kolom maka semakin
besar pula kecenderungan untuk berdifusi yang dapat mengakibatkan
melebarnya peak kromatogram. 3)Transfer massa non ekuilibrium
Terjadi karena aliran fase gerak yang terlalu cepat sementara sebagian
molekul solut tidak dapat keluar dari fase diam secara cepat sehingga
sebagian solut akan terlambat meninggalkan kolom sehingga dapat terjadi
A B
Gambar 11. Transfer massa fase diam (A) dan fase gerak (B)
Demikian pula yang terjadi pada cekungan kolom, solut dalam fase diam
bertemu dengan fase gerak yang masih baru, karena laju transfer solut tidak
terjadi dengan segera, masih ada solut yang tertinggal dalam fase diam. Efek
netto yang terjadi dari kedua keadaan ini adalah pelebaran peak solut pada kedua ujungnya.
Hubungan antara difusi, kesetimbangan dan kecepatan dinyatakan dalam
persamaan Van Deemter yaitu :
HETP = A + B + C ...………. (5)
HETP adalah tinggi lempeng teoritis. Suku A adalah difusi Eddy. Untuk
mendapatkan harga A yang kecil maka diameter dalam packing kolom harus dibuat kecil dan kerapatannya seragam. Suku B adalah difusi longitudinal.
Peran difusi ini tidak terlalu penting dalam kromatografi cair tetapi sangat
berperan terutama dalam kromatografi gas. Pelebaran peak dapat dikurangi dengan meningkatkan flow rate fase gerak dan menjadi penting bila flow rate
fase gerak sangat lambat (Watson, 1999). Tanda berarti kecepatan rata-rata
dari fase gerak. Suku C adalah transfer massa yang merupakan hasil
penjumlahan dari nilai transfer massa fase gerak dan fase diam (Noegrohati,
fase gerak, di samping pelebaran peak yang tergantung pada keadaan aliran dalam kolom, difusi longitudinal dan laju transfer massa.
Bentuk peak yang dihasilkan dari hasil pemisahan merupakan ukuran efisiensi kolom. Kolom yang menghasilkan pemisahan dengan peak yang simetris selalu lebih disukai. Peak yang kurang simetris dapat mengakibatkan ketidakakuratan pengukuran resolusi, ketidaktelitian hasil pengukuran kuantitatif,
memperkecil resolusi, dan tidak dapat mendeteksi sinyal yang kecil, dan waktu
retensi tidak reprodusibel (Noegrohati, 1994). Bentuk peak yang tidak simetris dengan front di belakang turun dengan landai disebut tailing. Sedangkan pada keadaan sebaliknya di mana pada bagian depan naik dengan landai disebut
fronting atau leading (Kuwana, 1980). Distribusi analit dalam fase gerak dan fase diam pada saat terjadi tailing dan leading dapat dilihat sebagai berikut :
Gambar 12. Distribusi analit dalam fase gerak dan fase diam
Terjadinya peak yang asimetri dapat disebabkan oleh jumlah solut yang terlalu besar dalam kolom, dekomposisi solut, analit teradsorpsi kuat dalam sisi
Salah satu cara untuk menilai bentuk peak adalah dengan peak asymmetry factor (As). Nilai As diukur pada 10% dari tinggi peak. Kolom yang baik akan menghasilkan nilai As sebesar 0,9 – 1,1 (Snyder dkk, 1997). Harga As > 1 berarti
kromatogram tersebut mengekor. Semakin besar harga As maka makin tidak
efisien kolom yang dipakai (Mulja dan Suharman, 1995). Cara lain untuk menilai
bentuk peak adalah dengan peak tailing factor (PTF). Pengukuran dengan menggunakan peak tailing factor lebih disukai. Pengukuran dengan PTF diukur pada 5% dari tinggi peak. Peak asymmetry factor dan peak tailing factor dapat dihitung seperti disajikan pada gambar di bawah ini. Kedua hal tersebut dapat
disebabkan karena kolom yang buruk, sampel overload, pemilihan pelarut yang tidak tepat, efek kimia, dan efek tambatan sekunder dari silanol (Snyder dkk,
1997).
Gambar 13. Pengukuran peak asymmetry factor dan peak tailing factor
Analisis kualitatif bertujuan untuk membuktikan ada tidaknya senyawa
tertentu dalam sampel dengan cara membandingkan waktu retensi senyawa murni
Respon yang berupa tinggi peak maupun luas area peak dapat digunakan untuk analisis kuantitatif. Analisis berdasarkan tinggi peak dapat memberikan ketelitian yang tinggi jika keadaan kolom tidak menyebabkan pelebaran peak. Pada kromatogram yang memiliki bentuk peak relatif lebar, analisis berdasarkan luas area peak lebih disukai dibanding analisis berdasarkan tinggi peak
(Noegrohati,1994).
E. Spektrofotometri Ultraviolet
Spektrofotometri UV-Vis merupakan bagian dari teknik analisis
spektroskopik yang memakai sumber radiasi elektromagnetik ultraviolet dekat
(190 – 380 nm) dan sinar tampak (380 – 780 nm) dengan menggunakan instrumen
spektrofotometer. Spektrum UV-Vis merupakan korelasi serapan (sebagai ordinat)
dan panjang gelombang (sebagai absis) berupa pita spektrum. Terbentuknya pita
tersebut disebabkan transisi energi yang tidak sejenis dan terjadi eksitasi
elektronik lebih dari satu macam pada gugus molekul yang kompleks.
Analisis dengan spektrofotometri UV-Vis selalu melibatkan pembacaan
serapan radiasi elektromagnetik oleh molekul atau radiasi elektromagnetik yang
diteruskan. Keduanya dikenal sebagai serapan (A) tanpa satuan dan transmitan
dengan satuan persen (%T).
Bouguer Lambert dan Beer membuat rumus hubungan antara transmitan
atau serapan terhadap intensitas radiasi atau konsentrasi zat yang dianalisis dan
T =
Dengan T adalah persen transmitan, I0 dan It adalah intensitas radiasi yang datang
dan yang diteruskan. A adalah serapan, ε adalah koefisien ekstingsi molar atau
daya serap molar (L mol-1cm-1), yaitu serapan suatu larutan dibagi dengan tebal
larutan b dalam cm dan konsentrasi molar c dalam mol. L-1. a adalah daya serap
dengan satuan L g-1cm-1, yang merupakan hasil bagi serapan (A) dibagi dengan
hasil perkalian kadar c yang dinyatakan dalam gram per liter zat dan panjang sel
dalam cm (b). Nilai daya serap molar dapat dihitung dengan persamaan sebagai
F. Kesahihan Metode Analisis Instrumental
Persoalan analisis terkait dengan kecilnya kadar senyawa yang dianalisis
dan kompleksnya kandungan sampel yang dianalisis. Untuk mengatasi hal
tersebut, metode analisis instrumental yang dipilih harus dapat menyelesaikan
kecermatan dan ketelitian alat. Untuk itu, diperlukan suatu pedoman mengenai
kesahihan metode analisis. Parameter – parameter yang digunakan sebagai
pedoman kesahihan metode analisis antara lain :
1. Akurasi
Akurasi adalah ukuran kedekatan nilai hasil percobaan dengan nilai yang
sesungguhnya, dinyatakan dengan persen recovery (Anonim, 2005). Akurasi untuk bahan obat dengan kadar kecil disepakati 90 – 110%, akurasi untuk kadar
obat yang lebih besar disepakati 95 – 105%, sedangkan akurasi untuk bahan baku
disepakati 98 – 102%. Sedangkan untuk bioanalisis rentang akurasi 80 – 120%
masih bisa diterima (Mulja dan Hanwar, 2003).
2. Presisi
Presisi adalah suatu ukuran kedekatan nilai data satu dengan data lainnya
dalam suatu pengukuran pada kondisi analisis yang sama. Menurut United State Pharmacopeia (USP) 28, presisi didefinisikan sebagai tingkat kesesuaian di antara masing – masing hasil analisis yang dihasilkan dengan menggunakan metode
analitik secara berulang – ulang untuk pengambilan sampel homogen yang
berulang kali. Presisi seringkali diukur sebagai persen Relative Standard Deviation (RSD) atau Coefficient of Variation (CV) untuk sejumlah sampel yang berbeda bermakna secara statistik. Kriteria presisi diberikan jika metode
memberikan nilai CV 2% atau kurang. Akan tetapi nilai ini fleksibel tergantung
deviasi relatif antara laboratorium ialah sekitar 2,5%. Pada kadar satu per sejuta,
RSD-nya adalah 16% (Harmita ,2004)
3. Limit of Detection (LOD)
LOD adalah konsentrasi terrendah dari analit yang dapat diukur pada
kondisi percobaan tertentu tetapi tidak perlu secara kuantitatif. LOD merupakan
parameter uji batas pengukuran dan menentukan apakah analit berada di atas atau
di bawah suatu nilai tertentu. Menurut USP 28, untuk metode instrumental, signal to noise ratio ditentukan dengan membandingkan hasil uji dari sampel yang telah diketahui konsentrasinya dengan hasil uji blanko dan menetapkan konsentrasi
analit terrendah yang dapat dideteksi. Konsentrasi analit yang mampu
memberikan respon 2-3 kali respon blanko inilah yang kemudian ditetapkan
sebagai LOD.
4. Limit of Quantification (LOQ)
LOQ adalah konsentrasi terrendah dari analit dalam sampel yang dapat
ditentukan dengan presisi dan akurasi yang baik pada kondisi percobaan tertentu
dari suatu metode. LOQ ditentukan dengan membandingkan sinyal terukur dari
sampel dengan konsentrasi analit yang rendah dengan sinyal dari blankonya.
5. Linieritas
Linieritas suatu metode analitik adalah kemampuannya untuk memperoleh
hasil uji yang proporsional dengan konsentrasi analit pada sampel yang
dinyatakan dengan koefisien korelasi (r). Linieritas yang baik ialah nilai r yang
lebih besar dari nilai r tabel (Snyder dkk,1997). Persyaratan data linieritas yang
bisa diterima jika memenuhi nilai koefisien korelasi (r) > 0.999 (Harmita, 2004).
6. Spesifitas
Spesifitas merupakan kemampuan suatu metode untuk mengukur dengan
akurat respon analit di antara seluruh komponen sampel yang mungkin ada dalam
matriks sampel (Mulja dan Hanwar,2003).
7. Range
Range adalah interval antara kadar terrendah sampai kadar tertinggi dari
suatu analit yang masih dapat diukur secara kuantitatif menggunakan metode
tertentu yang masih dapat menghasilkan akurasi dan presisi yang mencukupi.
Biasanya range memiliki satuan yang sama dengan satuan yang digunakan pada
USP 28 mencantumkan beberapa kategori uji umum yang harus memenuhi
validitas data, yaitu :
a. Kategori I
Meliputi metode analitik yang digunakan untuk mengukur secara kuantitatif
sejumlah besar komponen dari serbuk obat atau senyawa aktif (termasuk
preservatif) dalam sediaan obat jadi.
b. Kategori II
Meliputi metode analitik yang digunakan untuk penentuan kemurnian dalam
serbuk obat atau penentuan senyawa degradasi dalam sediaan obat jadi.
c. Kategori III
Meliputi metode analitik yang digunakan untuk penentuan sifat – sifat khusus
seperti kecepatan disolusi dan pelepasan obat.
d. Kategori IV
Meliputi metode analitik yang digunakan untuk mengidentifikasi sediaan
farmasi.
*Mungkin diperlukan, tergantung sifat uji spesifik yang dilakukan
G. Kesalahan Metode Analisis Instrumental
Kesalahan atau galat pada metode pada umumnya dapat disebabkan oleh
beberapa faktor yaitu metode dan prosedur analisis zat yang ditentukan, instrumen
yang dipakai, dan faktor individu yang mengerjakan. Kesalahan pada analisis
kimia dibagi menjadi dua macam, yaitu :
1. Kesalahan sistematik
Kesalahan ini disebut juga kesalahan prosedur yakni kesalahan yang
menyimpang secara tetap dari harga kadar yang sebenarnya karena proses
pelaksanaan prosedur analisis. Kesalahan sistematik ini dibagi lagi menjadi dua
macam berdasarkan sumber kesalahan, yaitu :
a. Kesalahan pada metode analisis
Kesalahan ini agak sulit dideteksi karena kesalahan pada metode analisis ini
antara lain disebabkan sifat fisika dan kimia dari reagen yang dipakai tidak
memadai secara ideal. Demikian juga dapat disebabkan oleh reaksi yang tidak
sempurna.
b. Kesalahan individual
Kesalahan ini timbul karena kesalahan individu dalam mengamati dan
Untuk menghindari kemungkinan terjadinya kesalahan sistematik ini ada
beberapa hal yang harus diperhatkan, yaitu :
1)Kalibrasi instrumen secara berkala
2)Pemilihan metode dan prosedur standar dari badan resmi
3)Pemakaian bahan kimia yang memiliki derajat pro analysis
4)Peningkatan pengetahuan dan kemampuan dari individu yang bekerja di
laboratorium analisis.
2. Kesalahan tidak sistematik
Kesalahan ini disebut juga kesalahan acak yaitu penyimpangan yang tidak
tetap dari hasil penentuan kadar dengan instrumen yang disebabkan oleh fluktuasi
dari instrumen yang digunakan. Kesalahan acak yang disebabkan oleh derau
instrumen tidak dapat diketahui penyebabnya dan juga tidak dikontrol. Pemakaian
instrumen dengan kualitas baik akan dapat menekan harga galat tidak sistematik
ini. Demikian juga pemakaian ilmu statistik untuk perhitungan hasil analisis
diharapkan dapat memperkecil perbedaan dalam menentukan kadar dengan harga
yang sebenarnya (Mulja dan Suharman, 1995).
H. Keterangan Empiris
Salah satu kombinasi obat yang banyak beredar adalah campuran dari
parasetamol, propifenazon, dan kafein yang memiliki kelarutan dalam etanol yang
mirip dan serapan maksimum pada panjang gelombang yang berdekatan sehingga
memisahkan sekaligus menetapkan kadar dari tiap komponen yang sudah
dipisahkan. Campuran ketiganya tersebut akan ditetapkan dengan menggunakan
kondisi KCKT yang optimal. Parameter validitas metode yang diuji meliputi
akurasi (ditinjau dari nilai recovery), presisi (ditinjau dari nilai CV), spesifisitas (ditinjau dari profil pemisahan pada kromatogram yang menunjukkan pemisahan
hingga baseline untuk ketiga analit), linieritas (ditunjukkan oleh nilai koefisien korelasi (r) yang diperoleh dari penentuan persamaan kurva baku dengan analisis
33
BAB III
METODE PENELITIAN
A. Jenis dan Rancangan Penelitian
Penelitian ini merupakan jenis rancangan penelitian eksperimental
deskriptif dengan dua variabel bebas dalam perlakuan pada subyek uji.
B. Variabel Penelitian
1. Variabel utama
a. Variabel bebas
1) Jenis dan perbandingan fase gerak yaitu metanol : aquabidest dan
metanol : aquabidest : asam asetat glasial.
2) Flow rate fase gerak yang digunakan. b. Variabel tergantung
1) Pemisahan peak dari parasetamol, propifenazon, dan kafein yang dapat dilihat dari waktu retensi masing – masing.
2) Validitas metode yang ditinjau dari akurasi, presisi, spesifisitas, linieritas,
2. Variabel pengacau terkendali
Variabel pengacau terkendali pada percobaan ialah kemurnian pelarut dan
kemurnian senyawa baku yang digunakan. Untuk mengatasinya digunakan pelarut
pro analysi dan bahan kualitas working standard.
C. Definisi Operasional
1. Larutan induk sampel simulasi adalah campuran 50 mg parasetamol, 30 mg
propifenazon, dan 10 mg kafein yang dilarutkan dalam metanol hingga 10 ml.
2. Sampel simulasi kadar rendah adalah campuran parasetamol, propifenazon, dan
kafein yang dibuat dari larutan induk sampel simulasi yang dipipet sebanyak
125 µl dan diencerkan dengan metanol hingga 10 ml.
3. Sampel simulasi kadar sedang adalah campuran parasetamol, propifenazon,
dan kafein yang dibuat dari larutan induk sampel simulasi yang dipipet
sebanyak 500 µl dan diencerkan dengan metanol hingga 10 ml.
4. Sampel simulasi kadar tinggi adalah campuran parasetamol, propifenazon, dan
kafein yang dibuat dari larutan induk sampel simulasi yang dipipet sebanyak
750 µl dan diencerkan dengan metanol hingga 10 ml.
5. Parameter validitas metode analisis yang digunakan yaitu akurasi, presisi,
spesifisitas, linieritas, dan range.
D. Bahan – bahan Penelitian
Bahan yang digunakan dalam penelitian adalah parasetamol kualitas
working standard (Vani Chemicals & Intermediates Limited), kafein kualitas
working standard (Brataco Chemika), metanol p.a. (E. Merck), asam asetat glasial
p.a (E.Merck), dan aquabidest (Laboratorium Kimia Organik Fakultas Farmasi Universitas Sanata Dharma).
E. Alat – Alat Penelitian
Alat – alat yang digunakan dalam penelitian ini adalah :
1. Spektrofotometer UV/Vis merek Perkin Elmer Lambda 20
2. Kuvet.
3. Seperangkat sistem KCKT yang terdiri dari :
a. Pompa merek Shimadzu LC-10 AD No. C20293309457 J2.
b. Detektor UV/Vis merek Shimadzu SPD-10 AV No. C20343503697 KG. c. CBM 101 merek Shimadzu No. C50363502311.
d. Seperangkat komputer merek ACER.
e. Printer merek Hewlett Packard Deskjet 670 C. f. Injektor jenis katup suntik model 77251.
g. Kolom ODS merek DuPont Instruments Zorbax berdimensi 4,6 mm x 25 cm P.N 880952-702.
4. Syringe merek Hamilton Part. No. 2933087.
5. Alat degasing ultrasonik merek RetschT640 No. 935922012 EY. 6. Penyaring Whatmann anorganik dan organik.
9. Vakum merek GastDOA-P104-BN. 10.Penyaring Milipore.
11.Mikropipet 100 – 1000 µl merek Biohit. 12.Seperangkat alat gelas.
F. Tatacara Penelitian
1. Pembuatan larutan baku parasetamol, propifenazon, dan kafein
a. Larutan baku parasetamol
1) Pembuatan larutan baku induk parasetamol
Lebih kurang 50 mg baku parasetamol yang ditimbang seksama
dilarutkan dalam metanol hingga 10 ml.
2) Pembuatan seri larutan baku parasetamol
Larutan baku induk parasetamol dari langkah di atas dipipet 125 µl;
250 µl; 375 µl; 500 µl; 625 µl; dan 750 µl lalu dimasukkan dalam labu
ukur 10 ml dan diencerkan dengan metanol hingga tanda sehingga
didapatkan kadar 62,5 ppm; 125,0 ppm; 187,5 ppm; 250,0 ppm; 312,5
ppm; dan 375,0 ppm.
b. Larutan baku propifenazon
1) Pembuatan larutan baku induk propifenazon
Lebih kurang 30 mg baku propifenazon yang ditimbang seksama
2) Pembuatan seri larutan baku propifenazon
Larutan baku induk propifenazon dari langkah di atas dipipet 125 µl;
250 µl; 375 µl; 500 µl; 625 µl; dan 750 µl lalu dimasukkan dalam labu
ukur 10 ml dan diencerkan dengan metanol hingga tanda sehingga
didapatkan kadar 37,5 ppm; 75,0 ppm; 112,5 ppm; 150,0 ppm; 187,5
ppm; dan 225,0 ppm.
c. Larutan baku kafein
1) Pembuatan larutan baku induk kafein
Lebih kurang 10 mg baku kafein yang ditimbang seksama dilarutkan
dalam metanol hingga 10 ml.
2) Pembuatan seri larutan baku kafein
Larutan baku induk kafein dari langkah di atas dipipet 125 µl; 250 µl;
375 µl; 500 µl; 625 µl; dan 750 µl lalu dimasukkan dalam labu ukur 10
ml dan diencerkan dengan metanol hingga tanda sehingga didapatkan
kadar 12,5 ppm; 25,0 ppm; 37,5 ppm; 50,0 ppm; 62,5 ppm; dan 75,0
ppm.
2. Pembuatan fase gerak
Fase gerak yang digunakan dalam penelitian ialah campuran :
a. Metanol : aquabidest dengan perbandingan 40 : 60; 50 : 50; 60 : 40; dan
70 : 30.
b. Metanol : aquabidest : asam asetat glasial dengan perbandingan 50 : 49 : 1;
Masing – masing perbandingan fase gerak dibuat sesuai dengan volume yang
dibutuhkan kemudian digojog dan disaring dengan penyaring Whatman anorganik
dengan bantuan pompa vakum. Fase gerak kemudian dihilangkan gelembungnya
dengan degassing selama 15 menit.
3. Penentuan panjang gelombang pengamatan antara parasetamol,
propifenazon, dan kafein dengan spektrofotometer UV
Lebih kurang 10 mg baku parasetamol, propifenazon, dan kafein yang
ditimbang seksama dilarutkan dalam metanol hingga 10 ml. Larutan tersebut
diencerkan hingga kadar 10 ppm untuk tiap zat dan dibaca absorbansinya pada
panjang gelombang 200 – 300 nm dengan spektrofotometer UV. Berdasarkan
kurva panjang gelombang vs absorbansi parasetamol, propifenazon, dan kafein
yang diperoleh, diamati dan ditentukan panjang gelombang overlapping.
4. Pengamatan waktu retensi parasetamol. propifenazon, dan kafein
Larutan baku induk parasetamol, propifenazon, dan kafein masing –
masing dipipet 500 µl lalu diencerkan dengan metanol hingga 10 ml. Larutan
hasil pengenceran tiap zat tersebut disaring dengan millipore dan dihilangkan gelembungnya dengan degassing selama 15 menit. Kemudian sebanyak 40 μl
larutan tersebut disuntikkan ke dalam KCKT dengan kolom ODS
(4,6 mm x 25 cm); fase gerak yang telah dibuat pada langkah no.2 dan flow rate
tertentu pada panjang gelombang pengamatan yaitu 272 nm. Waktu retensi tiap
5. Optimasi pemisahan parasetamol, propifenazon, dan kafein
Lebih kurang 50 mg parasetamol, 30 mg propifenazon, dan 10 mg kafein
ditimbang seksama, lalu dicampur dan dilarutkan dalam metanol hingga 10 ml.
Larutan tersebut dipipet 500 μl dan diencerkan dengan metanol hingga 10 ml,
sehingga didapatkan larutan campuran parasetamol, propifenazon, dan kafein
dengan perbandingan kadar masing – masing ialah 250 ppm : 150 ppm : 50 ppm.
Kemudian larutan tersebut disaring dengan millipore dan dihilangkan gelembungnya dengan degassing selama 15 menit. Sebanyak 40 μl larutan campuran disuntikkan ke dalam KCKT dengan kolom ODS (4,6 mm x 25 cm);
fase gerak yang telah dibuat pada langkah no.2 dan flow rate tertentu pada panjang gelombang pengamatan yaitu 272 nm. Dari hasil kromatogram diamati
waktu retensi masing – masing senyawa pada berbagai perbandingan fase gerak
serta flow rate yang digunakan.
6. Validasi metode analisis
a. Pembuatan kurva baku
Seri kadar larutan baku parasetamol, propifenazon, dan kafein dari langkah
no. 1 yang telah disaring dengan penyaring milipore dan dihilangkan gelembungnya dengan degassing selama 15 menit diinjeksikan pada sistem KCKT dengan fase gerak metanol : aquabidest dengan rasio 40 : 60 dan
flow rate 2 ml/menit. AUC (Area Under Curve) untuk tiap peak yang muncul diamati dari kromatogram yang didapat. Lalu, ditentukan persamaan
b. Pembuatan larutan induk campuran parasetamol, propifenazon, dan kafein
Lebih kurang 50 mg parasetamol, 30 mg propifenazon, dan 10 mg kafein
ditimbang seksama, dicampur, dan dilarutkan dalam metanol hingga 10 ml.
c. Penetapan kadar parasetamol, propifenazon, dan kafein dalam campuran
Larutan campuran parasetamol, propifenazon, dan kafein dari langkah
no.6b. dipipet 125 μl; 500 μl; dan 750 μl dan diencerkan dengan metanol
hingga 10 ml. Larutan tersebut disaring dengan milipore dan dihilangkan gelembungnya dengan degassing selama 15 menit, lalu diinjeksikan pada sistem KCKT dengan fase gerak metanol : aquabidest dengan rasio 40 : 60
dan flow rate 2 ml/menit. AUC (Area Under Curve) tiap peak yang muncul diamati dari kromatogram yang didapat. Kemudian kadar analit dihitung
dengan memasukkan nilai AUC yang diperoleh dari tiap analit ke dalam
persamaan kurva baku yang telah diperoleh dari analisis regresi linear.
G. Analisis Hasil
Kondisi KCKT yang optimal untuk mendapatkan pemisahan yang baik
dari parasetamol, propifenazon, dan kafein dalam campuran dapat dilihat dari
profil pemisahan dari kromatogram yang diperoleh dan perhitungan resolusi
(jarak antara dua puncak dibagi dengan rata – rata lebar dasar puncak) dengan
rumus sebagai berikut :
Hasil optimasi ini lalu digunakan untuk menentukan kesahihan metode,
yang dinyatakan dengan parameter berikut :
1. Akurasi ditentukan dengan nilai recovery
Recovery = x 100%
diketahui kadar
kur kadar teru
2. Presisi diukur dengan Coefficient of variance (CV)
CV = x 100%
rata -rata
baku simpangan
recovery
3. Spesifisitas ditentukan dari profil pemisahan pada kromatogram yang
menunjukkan pemisahan hingga baseline untuk ketiga analit.
4. Linieritas ditunjukkan oleh nilai koefisien korelasi (r) yang diperoleh dari
penentuan persamaan kurva baku dengan analisis regresi linear.
42
BAB IV
HASIL DAN PEMBAHASAN
A. Penyiapan Fase Gerak
Fase gerak yang digunakan dalam penelitian ialah campuran dari metanol :
aquabidest dan metanol : aquabidest : asam asetat glasial yang bersifat polar.
Pemilihan fase gerak tersebut didasarkan pada kondisi kromatografi yang dipilih
yaitu kromatografi partisi fase terbalik, karena ketiga senyawa analit bersifat polar
sehingga untuk mengelusinya dengan cepat digunakan fase gerak yang polar
sesuai dengan kepolaran ketiga senyawa analit, serta menggunakan kolom C-18
yang bersifat non polar agar ketiga analit dapat terpisah akibat perbedaaan
interaksi tiap analit dengan fase diam. Pemilihan fase gerak ini sangat penting
karena dapat mempengaruhi waktu retensi dan pemisahan dari komponen –
komponen yang akan dianalisis. Kedua jenis fase gerak yang digunakan pada
penelitian ini mengandung metanol yang termasuk golongan alkohol karena
ketiga analit mudah larut dalam etanol yang juga termasuk golongan alkohol.
Fase gerak sebelum digunakan harus disaring untuk menghilangkan
partikel yang dapat menyebabkan kerusakan pada pompa serta menyumbat kolom.
Selanjutnya, fase gerak didegas untuk menghilangkan gelembung – gelembung gas yang terlarut dalam fase gerak, agar tidak terjadi sinyal palsu pada detektor.
Fase gerak yang digunakan ini mengacu pada studi pustaka terhadap jurnal