VALIDASI METO DENGAN AG
SPE
Dia Mem
U
TODE PENETAPAN KADAR ASAM TRAN AGEN PENDERIVATO-FTALALDEHID S
PEKTROFOTOMETRI ULTRAVIOLET
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S. Farm.)
Program Studi Farmasi
Oleh:
Natalia Windari Rahardjo NIM : 088114052
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
2012
ANEKSAMAT SECARA
VALIDASI METO DENGAN AG
SPE
Dia Mem
U
i
TODE PENETAPAN KADAR ASAM TRAN AGEN PENDERIVATO-FTALALDEHID S
PEKTROFOTOMETRI ULTRAVIOLET
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S. Farm.)
Program Studi Farmasi
Oleh:
Natalia Windari Rahardjo NIM : 088114052
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
2012
ANEKSAMAT SECARA
ii
Persetujuan Pembimbing
VALIDASI METODE PENETAPAN KADAR ASAM TRANEKSAMAT
DENGAN AGEN PENDERIVATO-FTALALDEHID SECARA
SPEKTROFOTOMETRI ULTRAVIOLET
Skripsi yang diajukan oleh: Natalia Windari Rahardjo
NIM : 088114052
iv
HALAMAN PERSEMBAHAN
Tr u st i n th e LOR D ; an d do good; so sh al t th ou dw el l i n th e l an d, an d ver i l y th ou sh al t be fed. (P sal m 3 7 :3 )
We l ea r n w i sd om f r om f a i l u r e m u ch m or e th a n su ccess. We of ten d i scover w h a t w e w i l l d o, b y f i n d i n g ou t w h a t w e w i l l n ot d o.
Sam u el Sm i l es
K arya ini kupersembahkan untuk:
Papa, M ama, A dikku atas doa, perhatian dan semangat yang
diberikan padaku.
Om K iong Bing dan keluarganya yang telah banyak membantuku
dalam penyelesaian studi ini.
Penolong dan penyemangatku F ranky
v
PERNYATAAN KEASLIAN KARYA
Saya menyatakan dengan sesungguhnya bahwa skripsi yang saya tulis ini
tidak memuat karya atau bagian karya orang lain, kecuali yang telah disebutkan
dalam kutipan dan daftar pustaka, sebagaimana layaknya karya ilmiah.
Apabila di kemudian hari ditemukan indikasi plagiarisme dalam naskah
ini, maka saya bersedia menanggung segala sanksi sesuai peraturan
perundang-undangan yang berlaku.
Yogyakarta, Februari 2012
Penulis
vi
LEMBAR PERNYATAAN PERSETUJUAN PUBLIKASI KARYA ILMIAH UNTUK KEPENTINGAN AKADEMIS
Yang bertanda tangan di bawah ini, saya mahasiswa Universitas Sanata Dharma:
Nama : Natalia Windari Rahardjo
Nomor mahasiswa : 088114052
Demi pengembangan ilmu pengetahuan, saya memberikan kepada Perpustakaan Universitas Sanata Dharma karya ilmiah saya yang berjudul:
VALIDASI METODE PENETAPAN KADAR ASAM TRANEKSAMAT
DENGAN AGEN PENDERIVATO-FTALALDEHID SECARA
SPEKTROFOTOMETRI ULTRAVIOLET
beserta perangkat yang diperlukan (bila ada). Dengan demikian saya memberikan kepada Perpustakaan Universitas Sanata Dharma hak untuk menyimpan, mengalihkan dalam bentuk media lain, mengelolanya dalam bentuk pangkalan data, mendistribusikan secara terbatas, dan mempublikasiknnya di Internet atau media lain untuk kepentingan akademis tanpa perlu meminta ijin dari saya maupun memberikan royalti kepada saya selama tetap mencantumkan nama saya sebagai penulis.
Demikian pernyataan ini yang saya buat dengan sebenarnya.
Dibuat di Yogyakarta Pada tanggal: 3 April 2012 Yang menyatakan
vii
PRAKATA
Puji dan syukur penulis panjatkan ke hadirat Tuhan Yang Maha Esa atas
segala anugerah dan penyertaan-Nya kepada penulis selama menyelesaikan
penelitian dan penyusunan naskah skripsi ini.
Skripsi berjudul “Validasi Metode Penetapan Kadar Asam Traneksamat
dengan Agen Penderivat O-Ftaladehid secara Spektrofotometri Ultraviolet” ini
disusun untuk memenuhi salah satu syarat memperoleh gelar Sarjana Farmasi
(S.Farm) Program Studi Ilmu Farmasi Universitas Sanata Dharma.
Keberhasilan dalam penulisan skripsi ini juga tidak terlepas dari bantuan
dan dukungan berbagai pihak yang telah memberikan saran, kritik, dan dukungan
kepada penulis, maka dari itu penulis mengucapkan terima kasih yang
setulus-tulusnya kepada:
1. Ipang Djunarko, M.Sc., Apt. selaku Dekan Fakultas Farmasi Universitas
Sanata Dharma Yogyakarta.
2. Prof. Dr. Sudibyo Martono, M.S., Apt. selaku dosen pembimbing yang
dengan penuh kesabaran memberikan pengarahan, masukan, dukungan,
kritik dan saran baik selama penelitian maupun penyusunan skripsi ini.
3. Jeffry Julianus, M.Si. selaku dosen penguji yang telah memberikan
masukan, kritik dan saran yang bermanfaat untuk skripsi ini.
4. Prof. Dr. Sri Noegrohati, Apt. selaku dosen penguji yang telah
viii
5. Lucia Wiwid Wijayanti M.Si. selaku dosen pembimbing akademik yang
telah memberikan semangat, masukan dan dukungan yang bermanfaat
kepada penulis dalam menjalankan studi di Fakultas Farmasi USD.
6. Christine Patramurti, M.Si., Apt. selaku dosen yang telah banyak
memberikan kritik, saran dan dukungan yang bermanfaat dalam
penyusunan skripsi ini.
7. PT. Ifars-Solo yang telah bersedia memberikan baku asam traneksamat
yang berguna dalam skripsi.
8. Anggun Aji Mukti, Fitriana Susanti dan Agnes Anania yang telah banyak
memberikan bantuan baik reagen o-ftalaldehid, diskusi, maupun
semangat dalam penyelesaian skripsi ini.
9. Segenap dosen dan karyawan atas ilmu dan pengalaman yang berharga
sehingga berguna dalam proses penyusunanan skripsi ini.
10. Pak Parlan dan Mas Bimo selaku laboran Laboratorium Kimia Organik
dan Laboratorium Kimia Analisis Instrumental, Fakultas Farmasi
Universitas Sanata Dharma Yogyakarta atas bantuan dan kerjasamanya
selama penelitian.
11. Pak Bambang dan Mas Devi selaku laboran Laboratorium Analisis
Makanan, Fakultas Farmasi Universitas Gadjah Mada Yogyakarta yang
telah banyak membantu selama proses penelitian di laboratorium.
12. Papa, Mama dan Novi, yang tak pernah bosan memberikan semangat,
ix
13. Om Kiong Bing dan keluarganya. Terimakasih atas segala bantuan yang
telah diberikan kepada penulis sehingga studi ini dapat selesai.
14. Teman seperjuanganku, Franky Limawan. Terima kasih atas
kebersamaan, kekompakan, semangat, pengalaman baik maupun buruk
selama melakukan penelitian ini. Semoga pengalaman berharga yang
telah kita lewati dapat berguna bagi masa depan kita kelak.
15. Yanuar, Jefta, Pius, Yuni, Elisa, Melisa, Usi, Satya, Edward, Widi dan
Cynthia. Terimakasih atas semua bantuan dan semangat yang diberikan
sehingga skripsi ini dapat selesai.
16. Teman-teman kosku Mba Icha, Bobo, Novia, Feny, Dewi, Elen, Novi,
Felis, Puji dan Anin yang selalu memberikan semangat dalam
penyelesaian skripsi ini.
17. Teman-teman FST A dan B 2008, terimakasih atas kebersamaan yang
telah kita lalui di fakultas farmasi ini. Semoga kebersamaan ini dapat
terus berlanjut.
18. Semua pihak lainnya yang tidak dapat disebutkan satu per satu yang telah
membantu dalam penyelesaian skripsi ini.
Penulis menyadari bahwa masih banyak kekurangan dalam penyusunan
skripsi ini, oleh karena itu penulis sangat mengharapkan kritik dan saran yang
membangun. Semoga skripsi ini dapat bermanfaat bagi setiap pembacanya.
Yogyakarta, Februari 2012
x
DAFTAR ISI
Halaman
HALAMAN JUDUL...i
HALAMAN PERSETUJUAN PEMBIMBING ...ii
HALAMAN PENGESAHAN...iii
HALAMAN PERSEMBAHAN ...iv
PERNYATAAN KEASLIAN KARYA ... v
PERSETUJUAN PUBLIKASI KARYA ILMIAH...vi
PRAKATA...vii
DAFTAR ISI...x
DAFTAR TABEL...xiv
DAFTAR GAMBAR ...xv
DAFTAR LAMPIRAN ...xvii
INTISARI...xx
ABSTRACT...xxi
BAB I PENGANTAR ...1
A. Latar Belakang ... 1
xi
2. Keaslian Penelitian...5
3. Manfaat Penelitian ...5
B. Tujuan Penelitian...6
BAB II PENELAAHAN PUSTAKA ... 7
A. Asam Traneksamat ...7
B.O-Ftalaldehid (OPA) ...8
C. Derivatisasi ...10
D. Spektrofotometri UV...13
E. Teknik Perhitungan Kuantitatif ...18
F. Kesalahan dalan Analisis ...19
G. Validasi Metode Analisis ...21
1. Spesifisitas ... 22
2. Linieritas ...23
3. Batas Deteksi (LOD) dan Batas Kuantitasi (LOQ)...23
4. Ketepatan (accuracy) ...24
5. Ketelitian (precision) ...24
H. Landasan Teori ... 25
xii
BAB III METODE PENELITIAN ...29
A. Jenis dan Rancangan Penelitian ...29
B. Variabel Penelitian ...29
C. Definisi Operasional ...30
D. Bahan...30
E. Alat ...31
F. Tata Cara Penelitian ...31
1. Pembuatan Dapar Borat pH 8 ...31
2. Pembuatan Larutan OPA...31
3. Pembuatan Larutan Stok Asam Traneksamat ...32
4. Pembuatan Larutan Intermediet Baku Asam Traneksamat...32
5. Pembuatan Larutan Seri Baku dan Kurva Baku Asam Traneksamat ...32
6.RecoveryBaku ...33
7. Akurasi dan Presisi Menggunakan Standar Adisi...33
8. Analisis Hasil ...34
9. Penetapan Kadar Asam Traneksamat dalam Sampel Kapsul ...35
BAB IV HASIL DAN PEMBAHASAN ...37
xiii
1. Larutan Asam traneksamat...37
2. Larutan Dapar Borat...37
3. Larutan OPA ...38
4. Larutan Baku Asam traneksamat ... 41
B. Pembuatan Kurva Baku Asam Traneksamat ...44
C. Validasi Metode...47
1. Spesifisitas ... 47
2. Linieritas ...52
3. Batas Deteksi (LOD) dan Batas Kuantitasi (LOQ)...52
4. Akurasi ...56
5. Presisi ...58
D. Penetapan Kadar Asam Traneksamat dalam Kapsul...59
BAB V KESIMPULAN DAN SARAN ...64
A. Kesimpulan ...64
B. Saran ...64
DAFTAR PUSTAKA ...65
LAMPIRAN ...69
xiv
DAFTAR TABEL
Tabel I. Kategori pengujian parameter validasi...22
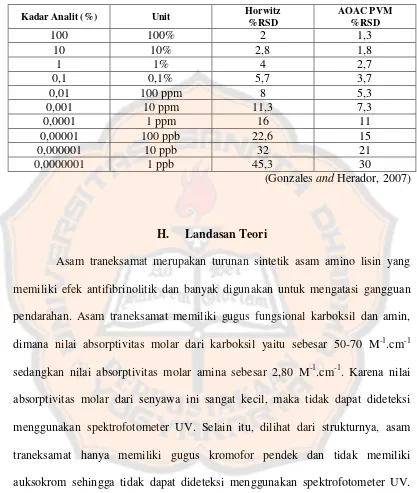
Tabel II. Kriteria penerimaanrecoveryberdasarkan kadar analit...24
Tabel III. Kriteria penerimaan RSD dari ketentuan Horwitz dan dari ketentuan AOAC Peer Verified Methods (PVM) berdasarkan kadar analit...25
Tabel IV. Data replikasi kurva baku asam traneksamat...46
Tabel V. Hasil penetapan kadar asam traneksamat dalam sampelbatch1...61
xv
DAFTAR GAMBAR
Gambar 1. Struktur asam traneksamat ...7
Gambar 2. Strukturo-ftalaldehid ...8
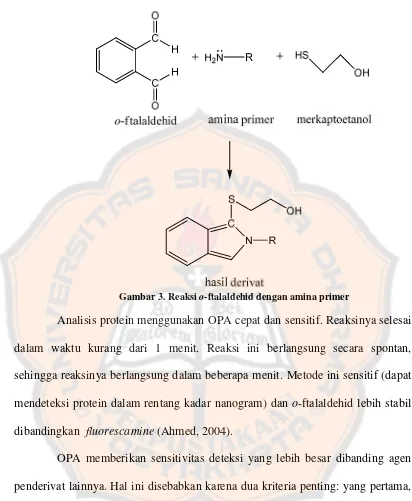
Gambar 3. Reaksio-ftalaldehid dengan amina primer ...9
Gambar 4. Diagram Tingkat Energi Elektronik...14
Gambar 5. SpektrofotometerSingle-beam...17
Gambar 6. SpektrofotometerDouble-beam...17
Gambar 7. Reaksi Cannizzaro pada OPA ...40
Gambar 8. Usulan reaksi fotodegradasi OPA ...41
Gambar 9. Spektra absorbansi OPA dalam dapar borat pH 8...42
Gambar 10. Penurunan absorbansi hasil derivatisasi asam traneksamat dengan OPA ...43
Gambar 11. Reaksi antara asam traneksamat dengan OPA dengan adanya merkaptoetanol menghasilkan senyawa hasil derivatisasi... ...44
Gambar 12. Gugus kromofor dan auksokrom dari senyawa hasil derivatisasi ...44
xvi
Gambar 14. Spektra hasil derivatisasi asam traneksamat dengan OPA
setelah 4 menit reaksi ...48
Gambar 15. Spektra hasil degradasi produk derivatisasi asam
traneksamat dengan OPA setelah 35 menit reaksi ...49
Gambar 16. Reaksi degradasi produk derivatisasi asam traneksamat
dengan OPA ...49
Gambar 17. Spektra hasil derivatisasi asam traneksamat dengan OPA
setelah 4 menit reaksi dan tingkatoverlappingyang terjadi...50
Gambar 18. Spektra larutan asam traneksamat dalam sampel kapsul
tanpa derivatisasi menggunakan OPA ...51
Gambar 19. Spektra larutan asam traneksamat 30 µg/mL (dari sampel
kapsul) dengan OPA setelah 4 menit reaksi... 51
xvii
DAFTAR LAMPIRAN
Lampiran 1. Sertifikat analisis asam traneksamat ...70
Lampiran 2. Hasil scanning panjang gelombang larutan OPA dalam
dapar borat pH 8 segera setelah dibuat ...71
Lampiran 3. Hasil scanning panjang gelombang larutan OPA dalam
dapar borat pH 8 setelah 1 jam dibuat ...72
Lampiran 4. Hasil scanning panjang gelombang larutan OPA dalam
dapar borat pH 8 setelah 2 jam dibuat ...73
Lampiran 5. Hasil scanning panjang gelombang larutan OPA dalam
dapar borat pH 8 setelah 3 jam dibuat ...74
Lampiran 6. Hasil scanning panjang gelombang larutan OPA dalam
dapar borat pH 8 setelah 4,5 jam dibuat ...75
Lampiran 7. Hasil scanning panjang gelombang hasil derivatisasi asam traneksamat dan OPA setelah 4 menit reaksi ...76
Lampiran 8. Hasil scanning panjang gelombang hasil degradasi dari
hasil derivatisasi antara asam traneksamat dan OPA setelah
35 menit reaksi...77
Lampiran 9. Spektra penurunan absorbansi hasil derivatisasi asam
traneksamat dengan OPA selama 1 jam ...78
xviii
Lampiran 11. Perhitungan persamaan kurva baku asam traneksamat ...81
Lampiran 12. Rangelinieritas kurva baku asam traneksamat...83
Lampiran 13. Pembuatan kurva baku asam traneksamat untuk
menghitung LOD dan LOQ ...87
Lampiran 14. Perhitungan LOD dan LOQ teoritis...90
Lampiran 15. Perhitungan LOD dan LOQ praktis ...92
Lampiran 16. Perhitungan akurasi dan presisi larutan baku asam
traneksamat ...94
Lampiran 17. Spektra larutan asam traneksamat dalam serbuk kapsul
tanpa derivatisasi menggunakan OPA ...97
Lampiran 18. Spektra hasil derivatisasi asam traneksamat dari sampel
kapsul...98
Lampiran 19. Penetapan kadar asam traneksamat dalam kapsulbatch1 ...99
Lampiran 20. Perhitungan akurasi dan presisi larutan baku asam
traneksamat dalam sampel serbuk kapsulbatch1...104
Lampiran 21. Penetapan kadar asam traneksamat dalam kapsulbatch2 ...109
Lampiran 22. Perhitungan akurasi dan presisi larutan baku asam
traneksamat dalam sampel serbuk kapsulbatch2...113
Lampiran 23. Penetapan kadar asam traneksamat dalam sampel serbuk
xix
Lampiran 24. Penetapan kadar asam traneksamat dalam sampel serbuk
kapsulbatch2 ...119
Lampiran 25. Perhitungan standar deviasi slope dan intersep dari kurva baku untuk LOQ dan standar deviasi slope dan intersep
dari kurva adisi padabatch1 ...120
Lampiran 26. Perhitungan standar deviasi slope dan intersep dari kurva baku untuk LOQ dan standar deviasi slope dan intersep
dari kurva adisi padabatch2 ...124
Lampiran 27. Uji signifikansi slope pada kurva LOQ dengan slope pada
kurva adisibatch1 danbatch2 ...128
Lampiran 28. Uji signifikansi intersep pada kurva LOQ dengan intersep
xx
INTISARI
Asam traneksamat merupakan senyawa dengan amina primer yang hanya memiliki gugus kromofor pendek dan tidak memiliki auksokrom, sehingga tidak dapat ditetapkan kadarnya secara langsung menggunakan spektrofotometer ultraviolet (UV) ataupun visible (Vis). Untuk dapat menetapkan kadarnya, perlu dilakukan derivatisasi menggunakan agen penderivat tertentu dan menghasilkan senyawa turunan asam traneksamat yang memiliki kromofor dan auksokrom yang cukup untuk dapat ditetapkan kadarnya menggunakan spektrofotometer UV. Oleh karena itu, dalam penelitian ini dilakukan pengembangan metode spektrofotometri UV dengan derivatisasi menggunakan agen penderivato-ftalaldehid (OPA) untuk menetapkan kadar asam traneksamat dalam kapsul asam traneksamat®.
Penelitian ini merupakan penelitian non eksperimental dengan rancangan penelitian deskriptif. Pada penelitian ini dilakukan pembuatan kurva baku menggunakan regresi linier antara kadar asam traneksamat dengan absorbansi hasil derivatisasi asam traneksamat dengan OPA. Untuk menentukan validitas metode, digunakan parameter spesifisitas, linearitas, batas deteksi, batas kuantitasi, ketepatan, dan ketelitian.
Hasil penelitian menunjukkan bahwa nilai koefisien korelasi (r) dari kurva baku hasil derivatisasi asam traneksamat dengan OPA sebesar 0,9998, rentang nilai recovery-nya sebesar 98,44-101,63%, rentang nilai CV sebesar 0,190-1,261%, batas deteksinya 0,227 µg/mL dan batas kuantitasinya 0,758 µg/mL pada pengukuran di panjang gelombang 334 nm. Maka dapat disimpulkan bahwa metode ini memiliki validitas yang baik untuk spesifisitas, linearitas, batas deteksi, batas kuantitasi, ketepatan, dan ketelitiannya.
xxi
ABSTRACT
Tranexamic acid is a compound with primary amine which have short chromophore and have not auxochrome, so can not be directly determined by ultraviolet (UV) spectrophotometry method nor visible (Vis) spectrophotometry. To determine that compound, it needs derivatization with a specific derivatizing agent to produce the derivate of tranexamic acid which have enough chromophore and auxochrome to determined by ultraviolet (UV) spectrophotometry method. This research is to develop spectrophotometry method with derivatization by
o-phthalaldehyde (OPA) to determine the level of tranexamic acid concentration in pharmaceutical product (asam traneksamat®capsule).
This research is a descriptive non experimental research. In this study, done by making a standard curve of derivate of tranexamic acid by using a linear regression between concentration of tranexamic acid against the derivate of tranexamic acid absorbance. To determine the validity of the method, parameters such as selectivity, linearity, limit of detection, limit of quantitation, accuracy, and precision were determined.
The result of correlation coefficient (r) of standard curve of derivate of tranexamic acid was 0.9998, range of recovery values were 98.44-101.63%, range of CV values were 0.190-1.261%, limit of detection was 0.227 µg/mL and limit of quantitation was 0.758 µg/mL, which measured in 334 nm. Therefore, it can be concluded that the method has good validity for specificity, linearity, limit of detection, limit of quantitation, accuracy, and precision.
1
BAB I PENGANTAR
A. Latar Belakang
Asam traneksamat merupakan senyawa yang digunakan untuk
pengobatan jangka pendek pada orang yang memiliki gangguan pendarahan
(hemofilia) untuk mencegah dan mengurangi pendarahan ketika pencabutan gigi.
Senyawa ini juga digunakan pada orang dengan kondisi risiko pendarahan tinggi
untuk mengontrol pendarahan pada saat-saat seperti setelah operasi atau cidera,
selama mimisan berat, atau selama menstruasi berat. Asam traneksamat bekerja
dengan membantu pembekuan darah secara normal untuk mencegah dan
menghentikan pendarahan berkepanjangan. Senyawa ini termasuk dalam kelas
obat yang disebut anti-fibrinolitik (Anonim, 2011).
Saat ini asam traneksamat telah tersedia dalam beberapa bentuk sediaan
obat salah satunya yaitu bentuk kapsul. Untuk menjaga keamanan dan khasiat dari
suatu sediaan obat, diperlukan suatu metode yang valid dan cepat dalam
menetapkan kadar asam traneksamat, sehingga kualitas dan mutu sediaan kapsul
tersebut terjamin.
Dilihat dari struktur molekulnya, asam traneksamat merupakan senyawa
amin primer dan merupakan turunan dari asam amino lisin (DunnandGoa, 1999).
Senyawa ini tidak memiliki kromofor dan auksokrom yang cukup sehingga tidak
dapat ditetapkan kadarnya secara langsung menggunakan spektrofotometri UV
senyawa turunan asam traneksamat dengan perpanjangan kromofor dan
bertambahnya auksokrom untuk dapat dideteksi menggunakan spektrofotometri
UV sehingga dapat ditetapkan kadarnya dengan lebih spesifik dan sensitif.
Beberapa penelitian yang telah dilakukan sebelumnya yaitu analisis
kuantitatif asam traneksamat dalam serum manusia menggunakan kromatografi
cair detektor electrospray ionisasi spektrofotometri masa (Delyle, Abe, Batisse, Tremey, Fischler, Devillier et al., 2010), determinasi asam traneksamat menggunakan kromatografi cair spektrofotometri massa tandem (Chang, Yin,
andChow, 2004), determinasi asam traneksamat dalam tablet dengan HPLC dan detektor ELS (Evaporative Light Scattering) (Patil, Rane, Sangshetti,andShinde,
2010), metode spektrofotometri untuk estimasi simultan ethamsylate dan asam traneksamat dalam sediaan tablet kombinasi (Issarani, Vankar, andNayak, 2010). Namun demikian, beberapa instrumen tersebut di atas yang digunakan dalam
penelitian asam traneksamat tidak dimiliki oleh kebanyakan laboratorium analisis
di Indonesia karena terlalu canggih.
Penetapan kadar asam traneksamat dapat dilakukan dengan metode
konvensional titrasi. Dilihat dari strukturnya, asam traneksamat memiliki sifat
asam yang terletak pada gugus karboksilatnya, sehingga dapat ditetapkan
kadarnya menggunakan metode titrasi alkalimetri. Analisis asam traneksamat
dapat dilakukan secara titrasi alkalimetri dengan deteksi menggunakan
potensiometri (Anonim, 2005a). Namun, titrasi konvensional hanya dapat
dilakukan pada senyawa tunggal dengan kadar besar seperti pada bahan baku atau
memiliki selektifitas dan sensitifitas yang lebih rendah dibandingkan dengan
metode spektrofotometri UV.
Berdasarkan data tersebut, masih jarang ditemukan adanya penetapan
kadar asam traneksamat menggunakan spektrofotometri padahal metode
spektrofotometri memiliki kelebihan mudah dan cepat dalam penggunaannya,
memiliki selektivitas dan sensitivitas yang cukup baik untuk penetapan kadar
senyawa tunggal dalam suatu sediaan serta merupakan metode dengan instrumen
yang umum digunakan pada laboratorium di Indonesia. Untuk dapat dianalisis
menggunakan spektrofotometri, asam traneksamat harus diderivatisasi
menggunakan agen penderivat sehingga menghasilkan senyawa yang memiliki
kromofor dan auksokrom.
Salah satu agen penderivat yang dapat digunakan untuk derivatisasi asam
traneksamat yaituo-ftaladehid (OPA) (Danielson, Gallaghe,andBao, 2000). OPA
banyak digunakan untuk derivatisasi asam amino karena reaksinya cepat dan
hanya bereaksi dengan amina primer dalam medium larutan basa (pH 9-11) dan
dengan adanya merkaptan (seperti 2-mercaptoethanol) untuk membentuk derivat indol yang berfluoresensi (Coppex, 2000). OPA dipilih karena memiliki kelebihan
lain seperti, OPA memiliki sensitivitas yang tinggi bila dibandingkan dengan agen
penderivat lain yang umum digunakan seperti fluoresamin dan ninhidrin.
Fluoresamin mudah terhidrolisis dalam air, derivat fluoresen yang dihasilkan
fluoresamin sangat tidak stabil dalam pelarut air dan sensitivitasnya 2-5 kali lebih
rendah dari pada hasil derivatisasi dengan OPA. Reaksi antara amina primer (α
dibandingkan dengan OPA (Coppex, 2000). OPA memiliki gugus aldehid yang
dapat bereaksi dengan gugus amina primer pada asam traneksamat dan
menghasilkan senyawa hasil derivatisasi yang memiliki kromofor dan auksokrom
sehingga dapat dideteksi menggunakan spektrofotometri UV.
Penelitian ini merupakan penelitian gabungan dari optimasi metode
dengan agen penderivat o-ftalaldehid secara spektrofotometri UV, sehingga setelah mendapatkan metode yang optimal, perlu dilakukan validasi metode agar
metode tersebut dapat digunakan untuk penetapan kadar asam traneksamat dalam
sediaan kapsul asam traneksamat®. Validasi metode ini dilakukan untuk
memenuhi parameter spesifisitas, presisi, akurasi dan linieritas sehingga ada
jaminan bahwa metode yang digunakan ini dapat dipercaya. Setelah parameter
validasi metode pengukuran asam traneksamat tersebut terpenuhi, maka
didapatkan metode yang valid dan reprodusibel dalam penetapan kadar asam
traneksamat dalam sediaan kapsul asam traneksamat®.
1. Perumusan Masalah
Berdasarkan latar belakang di atas, maka dapat disusun permasalahan
sebagai berikut:
1. Apakah penetapan kadar asam traneksamat dengan agen penderivat
OPA secara spektrofotometri UV memenuhi parameter spesifisitas,
linieritas, batas deteksi, batas kuantitasi, akurasi dan presisi?
2. Apakah metode spektrofotometri ultraviolet (UV) yang telah
tervalidasi dapat diaplikasikan untuk penetapan kadar asam
2. Keaslian Penelitian
Berdasarkan data-data penelitian yang telah dilakukan sebelumnya,
masih jarang ditemukan metode untuk menetapkan kadar asam traneksamat
menggunakan spektrofotometri UV di Indonesia. Sejauh peneliti mengetahui,
determinasi asam traneksamat dalam tablet pernah dilakukan menggunakan
metode HPLC dan detektor ELS (Patil et al., 2010). Determinasi asam traneksamat dalam serum manusia pernah dilakukan dengan HPLC menggunakan
derivatisasi pre-kolom fenil isotiosianat (Matsubayashi, Kojima, andTachizawa,
1988) dan menggunakan kromatografi cair yang dikombinasikan dengan deteksi
spektrofotometri massa ionisasielectrospray(Delyleet al., 2010).
Analisis asam traneksamat menggunakan metode spektrofotometri yang
pernah dilakukan sebelumnya yaitu estimasi asam traneksamat dan ethamsilat
secara berkesinambungan dalam tablet kombinasi (Issarani et al., 2010). Selain
itu, determinasi asam traneksamat dalam sediaan hidrogel dengan metode
spektrofotometri juga pernah dilakukan menggunakan penderivat
naphthalene-2,3-dicarboxaldehyde/cyanide(Duangrat, Wongsri,andPongpaibul, 2007).
3. Manfaat Penelitian
Hasil penelitian ini diharapkan memiliki manfaat sebagai berikut:
a. Manfaat teoritis. Penelitian ini diharapkan dapat menambah informasi
di dunia kefarmasian, khususnya di bidang industri obat asam traneksamat
mengenai metode spektrofotometri UV dengan agen penderivat OPA untuk
menetapkan kadar asam traneksamat dalam sediaan kapsul asam traneksamat®
b. Manfaat metodologis. Penelitian ini diharapkan dapat memberikan
sumbangan ilmiah mengenai metode alternatif untuk penetapan kadar asam
traneksamat, yaitu menggunakan agen penderivat OPA dengan metode
spektrofotometri UV.
c. Manfaat praktis. Penelitian ini diharapkan dapat menyediakan metode
penetapan kadar asam traneksamat yang valid dan dapat dimanfaatkan oleh pihak
industri dalam penjaminan kualitas produknya.
B. Tujuan Penelitian
Berdasarkan latar belakang dan permasalahan yang ada, maka penelitian
ini bertujuan untuk:
1. Mengetahui spesifisitas, linieritas, batas deteksi, batas kuantitasi,
akurasi dan presisi metode penetapan kadar asam traneksamat dengan
agen penderivat OPA secara spektrofotometri UV.
2. Mengetahui bahwa metode spektrofotometri UV yang telah tervalidasi
untuk menetapkan kadar asam traneksamat menggunakan agen
penderivat OPA dapat diaplikasikan pada sediaan kapsul asam
7
BAB II
PENELAAHAN PUSTAKA
A. Asam Traneksamat
Gambar 1. Struktur asam traneksamat
Asam traneksamat seperti yang ditunjukkan oleh gambar 1 memiliki
rumus molekul C8H15NO2dengan berat molekul 157,21 g/mol dan titik lebur
386-392° C. Kelarutan asam traneksamat dalam air sekitar 1g/6 mL, sangat sedikit
kelarutannya dalam alkohol, eter, praktis tidak larut dalam kebanyakan pelarut
organik lain dan tidak higroskopis (Anonim, 2001). Asam traneksamat (trans -4-(Aminomethyl) cyclohexanecarboxilic acid) merupakan turunan sintetik asam amino lisin yang memiliki efek antifibrinolitik dengan cara memblok sisi ikatan
lisin dengan plasminogen dan banyak digunakan untuk mengatasi gangguan
pendarahan (DunnandGoa, 1999).
Asam traneksamat memiliki gugus fungsional karboksil dan amina,
dimana nilai absorptivitas molar dari karboksil yaitu sebesar 50-70 M-1.cm-1
dengan panjang gelombang 200-210 nm sedangkan nilai absorptivitas molar
amina sebesar 2,80 M-1.cm-1 dengan panjang gelombang 195 nm (Willard,
masih ditemukan banyak gangguan yang berasal dari pelarut yang dapat dideteksi
juga pada panjang gelombang sekitar 210 nm. Selain itu, nilai absorptivitas molar
senyawa ini sangat kecil, maka akan kesulitan untuk dideteksi menggunakan
spektrofotometer UV.
B. O-Ftalaldehid (OPA)
Gambar 2. Strukturo-ftalaldehid
OPA dengan rumus molekul C8H6O2 seperti yang ditunjukkan gambar 2
memiliki bentuk kristal atau serbuk berwarna kuning. Bobot molekul OPA
sebesar 134,14 g/mol, OPA larut dalam metanol dan dietil eter (Anonim, 2005b).
OPA banyak digunakan untuk derivatisasi asam amino karena reaksinya cepat dan
hanya bereaksi dengan amina primer dalam medium larutan basa (pH 9-11) dan
dengan adanya merkaptan (seperti 2-mercaptoethanol) untuk membentuk derivat
indol yang berfluoresensi (Gambar 3). Reaksi derivatisasi terjadi pada suhu ruang
selama 2 menit dalam campuran dapar borat (pH 6-8 untuk amina primer) dan
metanol. Reaksi derivatisasi menggunakan OPA pada amina primer sempurna
dalam waktu 5 menit pada suhu ruangan, namun hasil derivatnya yang berupa
Gambar 3. Reaksio-ftalaldehid dengan amina primer
Analisis protein menggunakan OPA cepat dan sensitif. Reaksinya selesai
dalam waktu kurang dari 1 menit. Reaksi ini berlangsung secara spontan,
sehingga reaksinya berlangsung dalam beberapa menit. Metode ini sensitif (dapat
mendeteksi protein dalam rentang kadar nanogram) dan o-ftalaldehid lebih stabil dibandingkan fluorescamine(Ahmed, 2004).
OPA memberikan sensitivitas deteksi yang lebih besar dibanding agen
penderivat lainnya. Hal ini disebabkan karena dua kriteria penting: yang pertama,
OPA tidak berfluoresensi sendiri sehingga tidak akan mengganggu deteksinya
(Toyo’oka, 1999). Kedua, reaksinya terjadi dengan cepat pada suhu kamar
sehingga meminimalkan penggunaan waktu yang lama (Blackburn, 1989).
Kelebihan OPA dibandingkan ninhidrin yaitu selain sensitivitasnya
tinggi (Lindroth, Hamberger, and Sandberg, 1985). Reaksi derivatisasi menggunakan OPA dapat diaplikasikan untuk derivatisasi pre- atau post- kolom.
Pada pre-kolom, derivatisasi dapat dicapai secara manual atau otomatis dengan
mengontrol waktu reaksi dan interval waktu sebelum injeksi. Metode tersebut
memberikan sensitivitas yang tinggi dan reprodusibilitas analisis. OPA juga
digunakan pada analisis post-kolom karena waktu reaksi yang singkat dan sifat
fluorogenik (tidak berfluoresen) (Coppex, 2000).
C. Derivatisasi
Dalam suatu analisis, kemungkinan banyak terdapat zat-zat yang
memberikan absorbansi maksimal pada panjang gelombang 200-210 nm,
umumnya merupakan bahan-bahan yang digunakan sebagai pelarut, khususnya
yang mempunyai ikatan hidrogen. Proses derivatisasi dilakukan untuk mengatasi
keadaan tersebut dengan cara:
1. Mereaksikan zat yang dianalisis dengan zat tertentu sehingga terjadi pergeseran
panjang gelombang maksimal ke arah pergeseran merah.
2. Mereaksikan zat yang dianalisis dengan zat tertentu sehingga menghasilkan
senyawa yang berfluoresensi.
Perlu diperhatikan bahwa zat penderivat harus memberikan reaksi yang
cepat dan stabil serta meningkatkan sensitivitas pengukuran (Mulja dan
Suharman, 1995). Selain OPA, agen penderivat yang dapat digunakan untuk
1. Ninhidrin
Ninhidrin hanya bereaksi dengan amina primer (terutama α-asam amino
kecuali untuk sistein). Hasil derivatisasinya memiliki sensitivitas yang rendah,
waktu reaksi yang lama dan biaya instrument yang tinggi.
2. Fluoresamin
Fluoresamin mudah terhidrolisis dalam air, derivat fluoresen yang
dihasilkan fluoresamin sangat tidak stabil dalam pelarut air dan sensitivitasnya
2-5 kali lebih rendah dari pada hasil derivatisasi dengan OPA.
3. 1-Fluoro-2,4-dinitrobenzene(FDNB)
FDNB bereaksi dengan gugus amina primer dan sekunder menghasilkan
2,4-dinitrophenyl (DNP). Meskipun produk derivat yang dihasilkan stabil, namun FDNB bersifat toksik, sehingga penanganannya menggunakan
protective gloves.
4. 4-Fluoro-3-nitrobenzotrifluoride(FNBT)
FNBT bereaksi dengan amina primer dan tidak dapat bereaksi dengan
amina sekunder. FNBT dapat bereaksi dengan poliamina menghasilkan N-2’
-nitro-4-trifluoromethylphenyl polyamine(NTP-polyamine). 5. 2,4,6-Trinitrobenzene-1-sulfonic Acid(TNBS)
TNBS bereaksi dengan gugus amina primer dan peptida pada larutan
dengan pH 8 dan suhu ruang tanpa menghasilkan reaksi samping yang tidak
Agen penderivat yang dapat digunakan untuk menderivatisasi gugus
karboksil menurut Toyo’oka(1999) yaitu:
1. Reagen alkil halida
Contoh reagen alkil halida yaitu phenacyl bromide (PHB), p-bromophenacyl bromide (BPB), α-bromo-2’-acetonaphtone (BAN) dan
panacyl bromide (PB). Reagen alkil halida bereaksi dengan asam karboksilat dalam asetonitril di bawah kondisi sejuk dan reaksinya dikatalisis oleh crown
etherdan ion potasium atau basa organik seperti trietilamin dan etilamin. 2. Amina aromatis
Contoh reagen amina aromatis yaitu p-methoxyaniline, p-chloroaniline
dan 1-naphthylamine yang dapat langsung bereaksi dengan asam klorida pada gugus karboksil. Derivat amida dapat dibentuk dengan amina aromatis dengan
mengkonversikan asam karboksilat menjadi asam klorida. Senyawa yang biasa
digunakan untuk membentuk asam klorida yaitu triethylphosphin atau oxalyl chloridedanthionyl chloride.
3. Hidrazin
2-Nitrophenylhydrazides (NPH) bereaksi dengan rantai asam amino pendek dan panjang dan juga rantai asam karboksilat lurus dan bercabang
dengan adanya 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
4. Hidroksil amina
Hidroksil amina dapat digunakan untuk derivatisasi sederhana dan cepat
pada ester asam lemak hingga asam hidroksamik, yang menyerap dengan kuat
pada panjang gelombang rendah daerah UV (206 atau 213 nm).
D. Spektrofotometri UV
Spektrofotometri UV adalah salah satu teknik analisis spektroskopik
yang menggunakan radiasi elektromagnetik ultraviolet dekat (190-380 nm)
dengan menggunakan alat spektrofotometer. Pada analisis menggunakan
spektrofotometri UV, dilakukan pembacaan absorbansi (penyerapan) atau
transmitansi (penerusan) radiasi elektromagnetik oleh suatu molekul. Hasil
pembacaan absorbansi disebut sebagai absorban (A) dan tidak memiliki satuan,
sedangkan hasil pembacaan transmitansi disebut transmitan dan memiliki satuan
%T (Mulja dan Suharman, 1995).
Absorbansi cahaya oleh molekul dalam daerah spektra ultraviolet dan
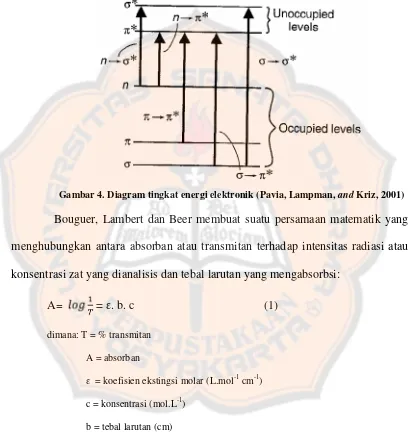
visible tergantung dari struktur elektronik molekul (Sastrohamidjojo, 2001). Apabila suatu molekul dikenai radiasi elektromagnetik (REM) maka akan terjadi
eksitasi ke tingkat energi yang lebih tinggi yang dikenal sebagai orbital elektron
antiikatan kalau ada kesesuaian dengan energi untuk eksitasi. Ada empat tipe
transisi elektronik yang mungkin terjadi yaitu σ → σ* , π → π*, n → π*, n → σ*.
Eksitasi elektron (σ → σ*) membutuhkan energi yang terbesar dan terjadi pada
daerah ultraviolet jauh yang diberikan oleh ikatan tunggal, misalnya alkana.
terjadi pada daerah ultraviolet jauh. Eksitasi elektron (n→ σ*) terjadi pada gugus
karbonil yang terjadi pada ultraviolet jauh (Mulja dan Suharman, 1995). Diagram
tingkat energi elektronik dapat dilihat pada gambar 4.
Gambar 4. Diagram tingkat energi elektronik (Pavia, Lampman,andKriz, 2001)
Bouguer, Lambert dan Beer membuat suatu persamaan matematik yang
menghubungkan antara absorban atau transmitan terhadap intensitas radiasi atau
konsentrasi zat yang dianalisis dan tebal larutan yang mengabsorbsi:
A= = ε. b. c (1)
dimana: T = % transmitan
A = absorban
ε = koefisien ekstingsi molar (L.mol-1cm-1)
c = konsentrasi (mol.L-1)
b = tebal larutan (cm)
Hubungan antara nilai % dengan absorptivitas molar (ε) adalah
sebagai berikut:
Nilai ε didefinisikan sebagai daya serap molar atau koefisien ekstingsi
molar. Nilai ε adalah karakteristik untuk molekul atau ion penyerap dalampelarut
tertentu, pada panjang gelombang tertentu dan tidak bergantung pada konsentrasi
dan panjang gelombang lintasan radiasi (Sastrohamidjojo, 2001). Secara umum,
dapat dikatakan bahwa nilai ε sangat mempengaruhi puncak spektra yang
dihasilkan oleh suatu zat. Rincian nilai ε terhadap puncak spektra adalah: 1-10:
sangat lemah; 10-102: lemah; 102-103: sedang; 103-104: kuat; 104-105: sangat kuat
(Mulja dan Suharman, 1995).
Radiasi dari spektrofotometer, diserap oleh molekul melalui adanya
eksitasi elektron pada ikatan antar atom penyusun senyawa, sehingga awan
elektron mengalami redistribusi. Radiasi yang digunakan biasanya berada pada
daerah panjang gelombang >200 nm untuk menghindari gangguan dari
pembacaan absorban pelarut. Semakin lemah ikatan antar atom maka dibutuhkan
lebih sedikit energi radiasi untuk mengeksitasi elektronnya. Pada daerah panjang
gelombang >200 nm, energi yang dibutuhkan tidak cukup untuk mengeksitasi
elektron σ ke σ*, sehingga diperlukan ikatan antar atom dengan ikatan yang lebih
lemah, yaitu ikatan dengan elektron π yang dengan mudah tereksitasi menjadi π*
pada panjang gelombang >200 nm. Adanya ikatan rangkap terkonjugasi (ikatan
rangkap yang diselingi ikatan tunggal), dapat meningkatkan intensitas absorpsi
yang terjadi dan juga meningkatkan panjang gelombang pengukuran (Watson,
2003).
Pemilihan pelarut yang digunakan dalam spektroskopi UV merupakan
mengabsorbsi radiasi UV pada daerah yang sama dengan analitnya. Biasanya,
pelarut yang dipilih adalah yang tidak memiliki sistem terkonjugasi. Air, etanol
95% dan n-heksan merupakan pelarut yang banyak digunakan. Zat-zat tersebut
tidak terlihat dalam daerah spektra ultraviolet dimana puncak absorbsi analit
biasanya muncul (Paviaet al., 2001).
Analisis kuantitatif zat tunggal pada spektrofotometri menggunakan
pengukuran absorbansi senyawa pada panjang gelombang maksimum, yaitu
panjang gelombang dimana terjadi eksitasi elektronik yang memberikan
absorbansi yang maksimum (Mulja dan Suharman, 1995). Beberapa alasan
digunakannya panjang gelombang maksimum dalam suatu analisis kuantitatif
adalah sebagai berikut:
- Pada panjang gelombang maksimum diperoleh kepekaan analisis yang
maksimal, karena pada panjang gelombang tersebut perubahan absorbansi
untuk setiap satuan konsentrasi adalah yang paling besar.
- Di sekitar panjang gelombang maksimum, bentuk kurva absorbansi datar dan
pada kondisi tersebut hukum Lambert-Beer akan terpenuhi.
- Jika dilakukan pengukuran ulang akan memberikan kesalahan yang kecil
ketika digunakan panjang gelombang maksimum (Gandjar dan Rohman, 2007).
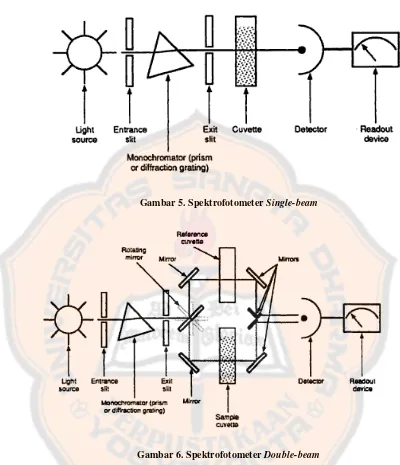
Pada umumnya konfigurasi dasar spektrofotometer UV berupa susunan
Gambar 5. SpektrofotometerSingle-beam
Gambar 6. SpektrofotometerDouble-beam
Spektrofotometer single beam, memiliki kelebihan antara lain sistemnya lebih sederhana dan murah dibandingkan spektrofotometer double beam, tetapi keterbatasannya adalah tidak dapat mengkoreksi perubahan respon serapan akibat
turbiditas sampel atau perbedaan intensitas cahaya baik dari sumber radiasi
maupun dari pengaruh luar. Keterbatasan ini merupakan alasan dikembangkannya
cahaya, fluktuasi pada kelistrikan instrumen, dan serapan blangko.
Keterbatasannya adalah sistemdouble beamlebih rumit dan harganya lebih mahal daripada spektrofotometersingle beam(Haven, Tetrault,andSchenken, 1994).
E. Teknik Perhitungan Kuantitatif
Pada banyak industri farmasi, produk obat dapat diproduksi dengan
berbagai variasi konsentrasi. Untuk mengembangakan dan melakukan validasi
suatu metode, dapat digunakan 3 macam teknik perhitungan kuantitatif:
1. Single-Point Calibration
Suatu metode analisis dapat dikembangkan dan divalidasi dengan
menggunakan satu konsentrasi standar yang digunakan untuk menguji semua
level konsentrasi analit. Metode ini dipilih karena sederhana dan mudah
dilakukan untuk menguji kadar analit. Walaupun demikian, metode ini
membutuhkan cara ekstraksi dan pengenceran yang berbeda untuk tiap
konsentrasi analit agar dapat dihasilkan final solution dengan konsentrasi
yang mendekati konsentrasi standar.
2. Multiple-Point Calibration
Metode lain yang dapat digunakan untuk kuantifikasi kadar analit adalah
dengan membuat berbagai konsentrasi standar yang mencakup seluruh
konsentrasi analit. Plot standar yang dihasilkan, selanjutnya digunakan untuk
menghitung konsentrasi analit dalam sampel. Namun, metode ini hanya valid
apabila semua konsentrasi analit tercakup dalam plot standar yang dibuat.
berbeda-beda untuk tiap konsentrasi analit. Keterbatasannya adalah
pembuatan konsentrasi standar yang bervariasi, dapat meningkatkan
kemungkinan terjadinya kesalahan penimbangan atau pengenceran dalam
pembuatan larutan standar.
3. One Standard Calibration for Each Strength
Metode yang menjadi alternatif terakhir untuk mengembangkan dan
memvalidasi metode adalah dengan membuat satu konsentrasi standar untuk
tiap konsentrasi analit. Metode ini ditempuh apabila tidak diperoleh respon
analit yang linier pada rentang konsentrasi standar yang dibuat (Chan, Lam,
Lee,andZang, 2004).
F. Kesalahan dalam Analisis
Istilah kesalahan didasarkan pada perbedaan antara hasil pengukuran
(nilai perhitungan) dengan nilai sebenarnya. Nilai sebenarnya dari suatu kuantitas
yang diukur merupakan sesuatu yang tidak pernah kita ketahui secara pasti,
namun nilai sebenarnya (true value) diterima jika nilai tersebut mempunyai ketidakpastian yang paling kecil diantara nilai-nilai lain dari suatu pengukuran
kuantitas (Gandjar dan Rohman, 2007). Menurut Gandjar dan Rohman (2007),
ada tiga macam kesalahan dalam analisis kimia yaitu: 1). Kesalahan gamblang
(gross error), 2). Kesalahan acak (random error), dan 3). Kesalahan sistematik
(systematic error).
Kesalahan gamblang merupakan kesalahan yang sudah jelas karena
mengabaikan percobaan yang telah kita lakukan dan memulainya dari awal lagi
secara menyeluruh. Contoh kesalahan gamblang adalah sampel tumpah; pereaksi
yang akan digunakan tercemar; larutan yang dipersiapkan salah; dan alat yang
akan digunakan rusak.
Kesalahan acak merupakan kesalahan yang nilainya tidak dapat
diramalkan dan tidak ada aturan yang mengaturnya serta nilainya berfluktuasi.
Kesalahan acak merupakan jenis kesalahan yang selalu terjadi dalam analisis
sebagai akibat adanya sedikit variasi yang tidak dapat ditentukan (dikontrol)
dalam pelaksanaan prosedur analisis.
Kesalahan sistematik merupakan kesalahan yang mempunyai nilai
definitif (nilai tertentu). Hasil analisis yang mengandung kesalahan ini dapat
mengarah ke arah yang lebih kecil atau ke arah yang lebih besar dari rata-rata.
Kesalahan sistematik bersifat ajeg (konstan) dan berhubungan dengan ketelitian
(akurasi) hasil analisis. Kesalahan jenis ini mengakibatkan penyimpangan tertentu
dari rata-rata (mean). Beberapa faktor yang mempengaruhi kesalahan sistematik
antara lain:
a. Kesalahan personil dan operasi
Kesalahan ini disebabkan oleh cara pelaksanaan analisis dari analis (personil)
dan bukan karena metode.
b. Kesalahan alat dan pereaksi
Kesalahan ini dapat disebabkan oleh pereaksi yang kurang murni, alat yang
kurang valid atau pemakaian alat yang kurang tepat walaupun alatnya sendiri
c. Kesalahan metode
Kesalahan metode dapat disebabkan kesalahan pengambilan sampel dan
kesalahan akibat reaksi kimia yang tidak sempurna (Gandjar dan Rohman,
2007).
Untuk memperkecil kesalahan sistematik dapat dilakukan beberapa cara,
antara lain:
1. Kalibrasi (peneraan) alat yang dipakai
Cara ini dimaksudkan untuk memperkecil kesalahan alat.
2. Dilakukan penetapan blanko
Di sini dilakukan pekerjaan seperti pada percobaan sebenarnya, tetapi dengan
tidak menggunakan sampel yang diselidiki (Gandjar dan Rohman, 2007).
G. Validasi Metode Analisis
Validasi metode analisis adalah suatu tindakan penilaian terhadap
parameter tertentu, berdasarkan percobaan laboratorium, untuk membuktikan
bahwa parameter tersebut memenuhi persyaratan untuk penggunaannya (Harmita,
2004). Parameter-parameter yang harus dipertimbangkan dalam validasi metode
analisis antara lain spesifisitas, linearitas, akurasi, presisi, LOD dan LOQ.
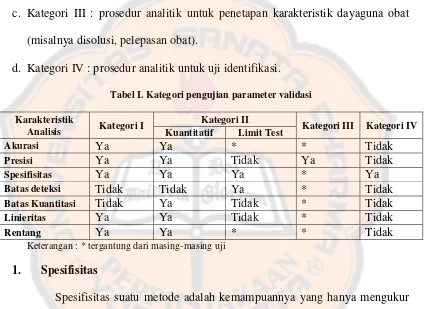
Perbedaan prosedur pengujian suatu analit membutuhkan parameter
validasi yang berbeda. Kategori pengujian yang paling umum yang data
a. Kategori I : prosedur analitik untuk penetapan kadar komponen utama dalam
bahan baku atau bahan aktif (termasuk pengawet) dalam produk akhir
farmasetik.
b. Kategori II : prosedur analitik untuk penetapan ketidakmurnian dalam bahan
baku atau senyawa degradasi pada produk akhir farmasetik.
c. Kategori III : prosedur analitik untuk penetapan karakteristik dayaguna obat
(misalnya disolusi, pelepasan obat).
d. Kategori IV : prosedur analitik untuk uji identifikasi.
Tabel I. Kategori pengujian parameter validasi Karakteristik
Presisi Ya Ya Tidak Ya Tidak
Spesifisitas Ya Ya Ya * Ya
Batas deteksi Tidak Tidak Ya * Tidak
Batas Kuantitasi Tidak Ya Tidak * Tidak
Linieritas Ya Ya Tidak * Tidak
Rentang Ya Ya * * Tidak
Keterangan : * tergantung dari masing-masing uji
1. Spesifisitas
Spesifisitas suatu metode adalah kemampuannya yang hanya mengukur
zat tertentu saja secara cermat dan seksama dengan adanya komponen lain yang
mungkin ada dalam matriks sampel. Spesifisitas metode ditentukan dengan
membandingkan hasil analisis sampel yang mengandung cemaran, hasil urai,
senyawa sejenis, senyawa asing lainnya, atau pembawa plasebo dengan hasil
2. Linieritas
Linearitas adalah kemampuan metode analisis (pada rentang tertentu)
untuk menghasilkan respon yang proporsional terhadap konsentrasi analit di
dalam sampel. Syarat suatu metode dikatakan memiliki linearitas yang baik
adalah apabila nilai koefisien korelasi (r)-nya ≥ 0,999, terutama untuk penetapan
kadar senyawa utama (Snyder, Kirkland,andGlajch, 1997).
3. Batas Deteksi (limit of detection / LOD) dan Batas Kuantitasi (limit of quantitation / LOQ)
Batas deteksi adalah konsentrasi terkecil analit dalam sampel yang dapat
dideteksi dan masih memberikan respon signifikan dibandingkan dengan respon
blanko. Batas deteksi dinyatakan sebagai perbandingan signal-to-noise (S/N) antara hasil uji sampel dengan analit yang diketahui konsentrasinya dan blangko.
Rasio signal-to-noise untuk batas deteksi sekurang-kurangnya 3:1 (Snyder et al., 1997). Penentuan batas deteksi juga dapat didasarkan pada perhitungan tiga kali
nilai standar deviasi blangko dibagi dengan nilai slope kurva baku (Anonim, 2005c).
Batas kuantitasi merupakan konsentrasi terkecil analit dalam sampel
yang masih dapat memenuhi kriteria akurasi dan presisi (Harmita, 2004). Batas
kuantitasi dapat dihitung berdasarkan perhitungan sepuluh kali nilai standar
4. Ketepatan (accuracy)
Ketepatan (accuracy) adalah ukuran yang menunjukkan kedekatan hasil analisis dengan kadar analit yang sebenarnya. Akurasi dinyatakan sebagai persen
perolehan kembali (recovery) analit yang ditambahkan. Ada tiga cara dalam menentukan akurasi yaitu: 1). Dengan membandingkan dengan reference
standard; 2). Menghitung recovery analit yang ditambahkan ke dalam blangko matriks dan 3). Standar adisi analit (Snyderet al., 1997). Kriteria rentangrecovery
yang dapat diterima ditentukan berdasarkan kadar analit, seperti yang tertera pada
tabel II.
Tabel II. Kriteria penerimaanrecoveryberdasarkan kadar analit Kadar Analit (%) Unit Recovery range(%)
100 100% 98-102
(AOACcit., GonzalesandHerrador, 2007)
5. Ketelitian (precision)
Ketelitian adalah ukuran yang menunjukkan derajat kesesuaian antara
hasil uji individual yang diperoleh dari pengambilan sampel berulang yang telah
dihomogenkan dengan suatu metode analisis (Snyder et al., 1997). Presisi umumnya dinyatakan dengan koefisien variasi (CV) atau standar deviasi relatif
Tabel III. Kriteria penerimaan RSD dari ketentuan Horwitz dan dari ketentuan AOACPeer
Verified Methods(PVM) berdasarkan kadar analit
Kadar Analit (%) Unit Horwitz %RSD
0,00001 100 ppb 22,6 15
0,000001 10 ppb 32 21
0,0000001 1 ppb 45,3 30
(GonzalesandHerador, 2007)
H. Landasan Teori
Asam traneksamat merupakan turunan sintetik asam amino lisin yang
memiliki efek antifibrinolitik dan banyak digunakan untuk mengatasi gangguan
pendarahan. Asam traneksamat memiliki gugus fungsional karboksil dan amin,
dimana nilai absorptivitas molar dari karboksil yaitu sebesar 50-70 M-1.cm-1
sedangkan nilai absorptivitas molar amina sebesar 2,80 M-1.cm-1. Karena nilai
absorptivitas molar dari senyawa ini sangat kecil, maka tidak dapat dideteksi
menggunakan spektrofotometer UV. Selain itu, dilihat dari strukturnya, asam
traneksamat hanya memiliki gugus kromofor pendek dan tidak memiliki
auksokrom sehingga tidak dapat dideteksi menggunakan spektrofotometer UV.
Derivatisasi terhadap asam traneksamat dilakukan agar didapatkan senyawa yang
memiliki gugus kromofor dan auksokrom sehingga dapat dideteksi menggunakan
OPA merupakan agen penderivat yang memiliki gugus aldehid yang akan
bereaksi dengan amina primer pada asam traneksamat. Senyawa hasil derivatisasi
ini akan memiliki gugus kromofor dan auksokrom sehingga dapat ditetapkan
kadarnya menggunakan spektrofotometer UV dengan lebih spesifik dan sensitif.
OPA dipilih karena reaksinya cepat dan hasilnya sensitif.
Penetapan kadar asam traneksamat dapat dilakukan dengan metode
konvensional titrasi. Dilihat dari strukturnya, asam traneksamat memiliki sifat
asam yang terletak pada gugus karboksilatnya, sehingga dapat ditetapkan
kadarnya menggunakan metode titrasi alkalimetri. Namun, titrasi konvensional
hanya dapat dilakukan pada senyawa tunggal dengan kadar besar seperti pada
bahan baku atau sediaan dengan zat aktif tunggal. Hal ini disebabkan karena titrasi
konvensional memiliki selektivitas dan sensitivitas yang lebih rendah
dibandingkan dengan metode spektrofotometri UV.
Metode spektrofotometri UV digunakan karena cukup selektif dan
sensitif untuk menetapkan kadar senyawa hasil derivatisasi asam traneksamat
dengan OPA. Selain itu, prosesnya cepat dan alatnya mudah digunakan sehingga
spektrofotometri UV banyak digunakan untuk menetapkan kadar senyawa tunggal
dalam jumlah besar dalam suatu sediaan. Senyawa hasil derivatisasi yang lebih
sensitif ini diharapkan memiliki linieritas yang baik antara kadar dengan
absorbansinya, memiliki batas deteksi dan batas kuantitasi yang lebih kecil serta
memberikan nilai presisi dan akurasi yang memenuhi kriteria.
Penelitian ini bertujuan untuk mengetahui apakah penetapan kadar asam
dapat memenuhi parameter validasi seperti spesifisitas, linieritas, batas deteksi,
batas kuantitasi, presisi dan akurasi yang baik. Spesifisitas dapat diketahui dari
perbandingan absorbansi masing-masing senyawa, baik senyawa hasil derivatisasi
dan OPA maupun asam traneksamat dan hasil derivat asam traneksamat dengan
OPA akan memiliki panjang gelombang yang berbeda pada rekaman spektra
UV-nya. Linieritas dianalisis berdasarkan nilai r ≥ 0,999, batas deteksi dan batas
kuantitasi berdasarkan perhitungan antara standar deviasi blangko dan slopekurva baku, akurasi dianalisis berdasarkan % recovery untuk analit 100% adalah
98-102% (AOAC cit., Gonzales and Herrador, 2007) dan presisi dianalisis berdasarkan CV (Coefficient of Varience) ≤ 1,3% (AOAC cit., Gonzales and
Herrador, 2007). Penentuan % recovery yang harus memenuhi syarat tersebut ditujukan untuk mengurangi kesalahan sistematik sedangkan penentuan CV yang
harus memenuhi syarat yang ditetapkan tersebut ditujukan untuk mengurangi
kesalahan acak.
I. Hipotesis
Berdasarkan landasan teori di atas, dapat disusun hipotesis sebagai
berikut:
1. Penetapan kadar asam traneksamat menggunakan agen penderivat OPA
dengan metode spektrofotometri UV memiliki validitas yang baik untuk
parameter spesifisitas, linearitas, batas deteksi, batas kuantitasi, akurasi,
2. Metode spektrofotometri ultraviolet (UV) yang telah tervalidasi dapat
diaplikasikan untuk menetapkan kadar asam traneksamat dalam sediaan
29
BAB III
METODE PENELITIAN
A. Jenis dan Rancangan Penelitian
Penelitian ini merupakan jenis penelitian non eksperimental dengan
rancangan penelitian deskriptif. Jenis penelitian non eksperimental karena subjek
penelitian tidak diberikan perlakuan, yaitu pH, operating time (OT) dan panjang gelombang pengukuran tetap. Rancangan penelitian bersifat deskriptif karena
peneliti hanya mendeskripsikan keadaan yang ada.
B. Variabel Penelitian 1. Variabel bebas
Variabel bebas dalam penelitian ini adalah seri kadar baku asam
traneksamat.
2. Variabel tergantung
Variabel tergantung dalam penelitian ini adalah spesifisitas, linearitas,
batas deteksi, batas kuantitasi,akurasi, dan presisi.
3. Variabel pengacau terkendali
Variabel pengacau terkendali dalam penelitian ini adalah cahaya, suhu
reaksi, pengotor pada alat, dan kemurnian pelarut yang digunakan. OPA bersifat
fotosensitif sehingga mudah terdegradasi jika terpapar cahaya. Untuk mencegah
hal tersebut maka larutan OPA yang telah dibuat dimasukkan dalam gelas Beaker
pendingin (2-8ºC). Alat yang digunakan dicuci dengan menggunakan asam
pencuci dan metanol. Pelarut yang digunakan adalah pelarut dengan derajat pro
analysisyang memiliki tingkat kemurnian yang tinggi.
C. Definisi Operasional
1. Baku asam traneksamat yang divalidasi adalah baku asam traneksamat yang
diperoleh dari P.T. Ifars - Solo (Certificate of Analysis terlampir di Lampiran 1).
2. Larutan dapar yang digunakan merupakan larutan dapar borat dengan pH 8
yang merupakan hasil optimasi Limawan (2012).
3. Spektrofotometri yang digunakan adalah seperangkat alat spektrofotometer
UV merek Hitachi U-2900.
4. Absorbansi yang diukur merupakan absorbansi senyawa hasil derivatisasi
asam traneksamat menggunakan agen penderivat OPA.
5. Parameter validasi metode yang digunakan adalah spesifisitas, linearitas,
batas deteksi, batas kuantitasi, akurasi, dan presisi.
D. Bahan
Bahan-bahan yang digunakan dalam penelitian ini meliputi baku asam
traneksamat (P.T. Ifars) dengan kemurnian 99,8% secara titrasi, o-ftalaldehid
(p.a., Nacalai), merkaptoetanol, metanol, asam borat, NaOH, KCl, (p.a., E. Merck), kapsul asam traneksamat®, kertas saring, kapas merk Selection, aquades
E. Alat
Alat-alat yang digunakan dalam penelitian ini meliputi Spektrofotometer
UV-Vis merk Hitachi U-2900, kuvet UV, pH meter merk Hanna HI 83141, neraca
merk Precisa A 125 SCS dengan kepekaan 0,1 mg (4 digit di belakang koma,
satuan g), neraca analitik merk Boeco Germany dengan kepekaan 0,1 mg (4 digit
di belakang koma, satuan g), mikropipet skala 100-1000 µL merk Socorex,
mikropipet skala 10-200 µL merk Gilson pipetman dan seperangkat alat gelas
yang lazim digunakan di laboratorium analisis.
F. Tatacara Penelitian 1. Pembuatan dapar borat pH 8
Ditimbang 1,55 g H3BO3 dan 1,85 g KCl, kemudian dilarutkan dalam
aquades sampai 250 mL. Larutan tersebut diambil sebanyak 125 mL, kemudian
ditambahkan dengan 9,75 mL larutan NaOH 0,1 M dan aquades sampai volume
250 mL. pH larutan diukur, kemudian pH-nya ditepatkan menjadi 8 dengan
menambahkan larutan H3BO3atau larutan NaOH (PerrinandDempsey, 1974).
2. Pembuatan larutan OPA
Ditimbang lebih kurang saksama 100,0 mg OPA, kemudian dilarutkan
dalam 2 mL metanol. Ditambah 100 µL merkaptoetanol dan dapar borat pH 8
hingga volume tepat 200 mL, kemudian dicampur hingga homogen. Larutan
disimpan dalam lemari pendingin dan ditempat gelap (± 1 jam). Larutan OPA
3. Pembuatan larutan stok asam traneksamat (2 mg/mL)
Ditimbang lebih kurang saksama 100,2 mg baku asam traneksamat,
kemudian dilarutkan dengan aquades. Larutan dimasukkan ke dalam labu takar
50,0 mL dan ditambahkan aquades hingga tanda.
Keterangan: Tingkat kemurnian baku asam traneksamat 99,8% (b/b) dengan teknik titrasi,
sehingga untuk memperoleh asam traneksamat 100 mg, maka bobot baku asam traneksamat yang
ditimbang sebesar 100,2 mg.
4. Pembuatan larutan intermediet baku asam traneksamat (1 mg/mL)
Larutan stok asam traneksamat 2 mg/mL dipipet 5 mL dan dimasukkan
dalam labu ukur 10,0 mL. Aquades ditambahkan hingga tanda.
5. Pembuatan larutan seri baku dan kurva baku asam traneksamat
Dipipet 50; 100; 150; 200; dan 250 µL dari larutan intermediet asam
traneksamat 1 mg/mL, kemudian dimasukkan dalam labu takar 5,0 mL yang telah
dibungkus kertas aluminium. Larutan OPA pH 8 yang telah didinginkan pada
suhu 2-8ºC selama ± 1 jam ditambahkan hingga volume tepat 5,0 mL kemudian
digojog, sehingga diperoleh kadar seri baku sebesar 10; 20; 30; 40; dan 50 µg/mL.
Absorbansinya diukur pada panjang gelombang 334 nm menggunakan
spektrofotometer UV setelah didiamkan di tempat gelap pada suhu lemari
pendingin selama 4 menit (terhitung setelah 45 detik penambahan larutan OPA
pH 8). Kurva regresi linear antara kadar asam traneksamat dengan absorbansi
senyawa hasil derivatisasi dibuat, kemudian ditentukan persamaan garis regresi
linear dan tentukan nilai koefisien korelasinya. Syarat suatu metode dikatakan
memiliki linearitas yang baik adalah apabila nilai koefisien korelasi (r)-nya ≥
6. Recoverybaku
Larutan intermediet asam traneksamat 1000 µg/mL dipipet sebanyak 50;
150; dan 250 µL dan dimasukkan ke dalam labu ukur 5,0 mL yang telah
dibungkus dengan kertas aluminium. Larutan OPA pH 8 yang telah didinginkan
pada suhu lemari pendingin selama 1 jam ditambahkan pada masing–masing labu
hingga volume tepat 5,0 mL. Labu digojog dan didiamkan di tempat gelap pada
suhu lemari pendingin selam 4 menit (terhitung setelah 45 detik penambahan
larutan OPA pH 8). Replikasi masing-masing konsentrasi dilakukan sebanyak 5
kali sehingga diperoleh 15 data. Absorbansi yang diperoleh dicatat. Kadar asam
traneksamat dihitung dengan memasukkan nilai absorbansi ke persamaan kurva
baku yang diperoleh.
7. Akurasi dan presisi menggunakan standar adisi
Ditimbang lebih kurang seksama 117,6 mg (untuk batch 1) dan 121, 7
mg (untuk batch2) serbuk kapsul yang telah dihomogenkan dalam mortir. Untuk akurasi dan presisi kadar rendah: ditambahkan 10,0 mL larutan baku asam
traneksamat (10 mg/mL). Untuk akurasi dan presisi kadar sedang: ditambahkan
20,0 mL larutan baku asam traneksamat (10 mg/mL). Untuk akurasi dan presisi
kadar tinggi: ditambahkan 30,0 mL larutan baku asam traneksamat (10 mg/mL).
Masing-masing dimasukkan dalam gelas beker dan dilarutkan dengan aquades,
kemudian dimasukkan dalam labu ukur 100,0 mL dan ditambah aquades hingga
volume tepat 100,0 mL. Campuran tersebut digojog hingga homogen. Penyarian
asam traneksamat dilakukan menggunakan corong, kapas dan kertas saring
mL. Larutan OPA pH 8 ditambahkan hingga volume tepat 10,0 mL.
Absorbansinya diukur pada λ 334 nm setelah didiamkan di tempat dingin dan
gelap selama 4 menit. Replikasi dilakukan sebanyak 3 kali untuk setiap kadar
(rendah, sedang dan tinggi) sehingga didapatkan 9 data untuk setiapbatch.
8. Analisis Hasil
Validasi metode analisis yang digunakan dalam penetapan kadar asam
traneksamat pada penelitian ini dapat ditentukan berdasarkan parameter berikut:
a. Spesifisitas
Pada metode ini, spektra panjang gelombang maksimum yang dihasilkan
oleh campuran larutan OPA dengan pelarut dibandingkan dengan spektra panjang
gelombang maksimum yang dihasilkan oleh campuran larutan OPA dengan asam
traneksamat. Jika panjang gelombang maksimum (selective UV-wavelength) dari pola spektra berbeda, dapat dikatakan bahwa metode tersebut spesifik (Ermerand
Miller, 2005). Spesifisitas metode ini juga dapat dilihat dari besarnyaoverlapping
yang terjadi antara produk derivatisasi dengan larutan OPA.
b. Linearitas kurva baku
Linearitas dilihat dari nilai koefisien korelasi (r) dari hasil pengukuran
seri larutan baku asam traneksamat. Suatu metode dikatakan memiliki linearitas
c. Batas deteksi (LOD) dan batas kuantitasi (LOQ)
Absorbansi larutan baku konsentrasi terkecil setelah diderivatisasi
dengan OPA diukur minimal 3 kali. Nilai LOD diperoleh pada 3 x SD blangko
dibagi slope kurva baku dan nilai LOQ diperoleh pada 10 x SD blangko dibagi
slopekurva baku (Anonim, 2005c).
d. Ketelitian (precision)
Ketelitian metode analisis dinyatakan dengan koefisien variasi (CV) yang
dihitung dengan cara berikut:
CV =. x 100%
Kriteria presisi yang diterima untuk kadar zat analit 100% adalah CV ≤
1,3% (AOACcit., GonzalesandHerrador, 2007).
e. Ketepatan (accuracy)
Akurasi metode analisis dinyatakan dengan % perolehan kembali
(recovery) yang dihitung dengan cara berikut:
%recovery= x 100%
Kriteria akurasi yang diterima untuk kadar zat analit 100% adalah
98-102% (AOACcit., GonzalesandHerrador, 2007).
9. Penetapan kadar asam traneksamat dalam sampel kapsul
Uji keseragaman bobot dilakukan dengan cara: ditimbang 20 kapsul.
Ditimbang lagi kapsul satu persatu. Isi semua kapsul dikeluarkan, ditimbang
isi kapsul. Perbedaan dalam persen bobot isi tiap kapsul terhadap bobot rata-rata
tiap isi kapsul tidak boleh lebih dari ± 7,5% untuk kapsul yang memiliki bobot
rata-rata lebih dari 120 mg (Direktorat Jendral Pengawasan Obat dan Makanan RI,
1995).
Dua puluh kapsul asam traneksamat® yang telah dilakukan uji
keseragaman bobot kapsul dihomogenkan dalam mortir. Kemudian ditimbang
saksama serbuk kapsul 119,5 mg untuk batch1 dan 121,1 mg untukbatch2 yang setara dengan 100 mg asam traneksamat (sesuai leaflet yang tertulis). Serbuk
kapsul dilarutkan dalam aquades hingga volume tepat 100,0 mL dan digojog
hingga homogen. Penyarian dilakukan 3 kali menggunakan kapas dan kertas
saring sehingga didapatkan filtrat yang jernih. Filtrat dipipet 300 µL, kemudian
dimasukkan dalam labu ukur 10 mL. Larutan OPA pH 8 ditambahkan hingga
volume tepat 10,0 mL. Diamkan ditempat gelap dan dingin selama 4 menit
(setelah 45 detik penambahan larutan OPA). Absorbansinya diukur menggunakan
spektrofotometer UV. Replikasi dilakukan sebanyak 5 kali untuk setiap batch
sehingga didapatkan 5 data absorbansi untuk setiap batch. Kadar terukur asam traneksamat dihitung dengan cara memasukkan absorbansi yang didapat ke dalam
persamaan regresi linier kurva baku dan ditentukan kadar rata-rata asam
traneksamat untuk setiap batch. Menurut Supplement II Japanese Pharmacopeia
kapsul asam traneksamat mengandung tidak kurang dari 95,0% dan tidak lebih
37
BAB IV
HASIL DAN PEMBAHASAN A. Pembuatan Larutan 1. Larutan Asam Traneksamat
Asam traneksamat baku yang digunakan merupakan senyawa
berbentuk serbuk yang sangat mudah larut dalam air dan memiliki tingkat
kemurnian 99,8% secara titrasi (CoA pada Lampiran 1.). Pada pembuatan
larutan asam traneksamat digunakan pelarut aquades, yaitu air yang
mengalami penyulingan untuk menjamin kemurniannya dan tidak
mengandung bahan-bahan lain yang mungkin dapat mengganggu analisis.
Aquades juga dipilih karena memenuhi kriteria yang baik untuk analisis
secara spektrofotometri ini, diantaranya tidak memberikan serapan pada
daerah yang sama dengan analit, tidak berinteraksi dengan analit, tidak
berwarna dan memiliki kemurnian yang cukup tinggi jika digunakan untuk
keperluan analisis.
2. Larutan Dapar Borat
Larutan dapar borat terdiri dari campuran asam borat (H3BO3) dan
kalium klorida (KCl) serta larutan natrium hidroksida (NaOH) untuk
mengatur agar larutan berada pada kondisi pH tertentu, dalam penelitian
ini dibuat larutan dapar borat dengan pH 8 yang merupakan hasil optimasi
(Limawan, 2012). Dapar borat ini berfungsi untuk memberikan suasana