i
VALIDASI METODE DAN PENETAPAN KADAR NIKOTIN DALAM
EKSTRAK TEMBAKAU ROKOK “MEREK X” DENGAN METODE
KROMATOGRAFI CAIR KINERJA TINGGI (KCKT) FASE TERBALIK
MENGGUNAKAN STANDAR INTERNAL ASETANILIDA
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat
Memperoleh Gelar Sarjana Farmasi (S.Farm)
Program Studi Farmasi
Oleh:
Is Sumitro
NIM : 098114127
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA
YOGJAKARTA
iv
HALAMAN PERSEMBAHAN
“Hidup seorang laki-laki jangan takut akan segala hal sebab ketakutan hanya akan
menghambat jalanmu, namun juga selalu berpegang pada prinsip yang benar karena
hidup hanyalah hidup jika bermanfaat bagi orang lain”
“Kebaikan belum tentu akan dimengerti orang lain, maka jangan menuntut orang juga
akan mengerti kebaikanmu namun selalulah berbuat baik dan bekerja keras sebab doa
orang tua dan Tuhan selalu menyertaimu”
(Mintju dan Effendi)
Kupersembahkan karyaku ini untuk kedua orang tuaku Mintju dan Effendi, Sahabatku, dan
v
PERNYATAAN KEASLIAN KARYA
Saya menyatakan dengan sesungguhnya bahwa skripsi yang saya susun ini
tidak memuat karya atau bagian dari pekerjaan orang lain, kecuali yang telah
disebutkan dalam kutipan dan daftar pustaka, sebagaimana layaknya sebuah karya
ilmiah.
Apabila dikemudian hari ditemukan adanya indikasi plagiarisme dalam
naskah yang saya susun ini, maka saya bersedia menanggung segala resiko dan sanksi
sesuai dengan peraturan perundang-undangan yang berlaku.
Yogyakarta, 10 Juli 2013
Penulis,
vi
LEMBAR PERNYATAAN PERSETUJUAN
PUBLIKASI KARYA ILMIAH UNTUK KEPENTINGAN AKADEMIS
Yang bertanda tangah di bawah ini, saya mahasiswa Universitas Sanata Dharma:
Nama
: Is Sumitro
Nomor Mahasiswa
: 098114127
Demi pengembangan ilmu penegtahuan, saya memberikan kepada perpustakaan
Sanata Dharma karya ilmiah yang berjudul:
“VALIDASI METODE DAN PENETAPAN KADAR EKSTRAK TEMBAKAN
DALAM ROKOK “MEREK X” DENGAN METODE KROMATOGRAFI
CAIR KINERJA TINGGI (KCKT) FASE TERBALIK MENGGUNAKAN
STANDAR INTERNAL ASETANILIDA”
Beserta perangkat yang diperlukan (bila ada). Dengan demikian saya memberikan
kepada Perpustakaan Universitas Sanata Dharma hak untuk menyimpan,
mengalihkan dalam bentuk media lain, mengelolanya dalam bentuk pangkalan data,
mendistribusikannya secara terbatas, dan mempublikasikannya di internet atau media
lain untuk kepentingan akademis tanpa perlu meminta izin dari saya maupun
memberikan royalti kepada saya selama tetap mencantumkan nama saya sebagai
penulis.
Dengan demikian pernyataan ini saya buat dengan sebenarnya.
Dibuat di Yogyakarta
Pada tanggal : 10 Juli 2013
Yang menyatakan
vii
PRAKATA
Puji dan syukur kepada Tuhan Yang Maha Esa atas cinta kasih, berkat, ijin
dan peryertaan-Nya yang begitu besar, sehingga penulis dapat menyelesaikan skripsi
yang berjudul “Penetapan Kadar Nikotin Dalam Rokok “MEREK X” Dengan Metode
Kromatografi Cair Kinerja Tinggi (KCKT) Fase Terbalik Menggunakan Standar
Internal Asetanilida” sebagai salah satu syarat yang harus dipenuhi demi memperoleh
gelar Sarjana Farmasi (S.Farm.) di Fakultas Farmasi Universitas Sanata Dharma
Yogyakarta.
Penulis menyadari bahwa penelitian dan penyusunan skripsi ini dapat
terselesaikan karena adanya masukan, kritikan, diskusi, arahan, saran, dan bimbingan
dari berbagai pihak. Oleh karena itu penulis mengucapkan terima kasih yang
sebesar-besarnya kepada :
1.
Ipang Djurnarko, M.Sc., Apt. Selaku Dekan Fakultas Farmasi Uninversitas Sanata
Dharma Yogyakarta atas teladan seorang pemimpin yang diberikan
2.
Dra. M.M. Yetty Tjandrawati, M.Si. selaku dosen pembimbing, dosen penguji,
dan pengganti orang tua saya yang telah meluangkan waktunya untuk
memberikan perhatian, bimbingan, masukan, motivasi, kritikan, dan saran selama
penulis berkuliah di Fakultas Farmasi Universitas Sanata Dharma dan selama
viii
3.
Jeffry Julianus, M.Si. selaku dosen penguji yang memberikan banyak kritik dan
saran yang membangun untuk skripsi ini.
4.
Lucia Wiwid Wijayanti, M,Si. selaku dosen penguji yang memberikan banyak
kritik dan saran yang membangun untuk skripsi ini.
5.
Christine Patramurti, M.Si., Apt. selaku dosen pembimbing di laboratorium dan
teman selama penelitian skripsi yang telah memberikan masukan, diskusi, saran,
dan dukungan moral kepada penulis selama penelitian skripsi ini.
6.
C.M.Ratna Rini Nastiti, M.Pharm., Apt. sebagai Kaprodi Fakultas Farmasi
Universitas Sanata Dharma Yogyakarta atas teladan kepemimpinan, masukan,
dan saran yang diberikan selama penulis berkuliah dan menyusun naskah.
7.
Rini Dwi Astuti, M.Sc., Apt. sebagai Kepala Laboratorium Fakultas Farmasi
Universitas Sanata Dharma Yogyakarta
8.
Prof. Dr. Sudibyo Martono, M.S., Apt. atas waktu yang diluangkan untuk
memberikan sedikit masukan diawal penelitian
9.
Bimo Adithya, Suparlan, dan Kunto dan segenap staf laboran yang senantiasa
siap membantu dan meluangkan waktunya dalam penyediaan bahan dan alat
selama penelitian.
10.
Semua dosen dan karyawan Fakultas Farmasi Universitas Sanata Dharma atas
ix
11.
Demas dan Eric sebagai rekan kerja dalam penelitian skripsi ini. Terima kasih
atas kesabaran, kepercayaan, kerjasama, persahabatan, canda dan semangat
selama ini.
12.
Lucia Shinta R, Sisilia Mirsya A, Metri S.K., Agnes Mutiara, Victor Purnama
Agung, dan Novia Sarwoningtyas sebagai teman seperjuangan dalam satu lantai
Laboratorium Analisis Instrumental.
13. Teman angkatan 2009 yang bersama-sama berjuang dan mengisi sebagian cerita
hidupku, terima kasih atas kebersamaan, diskusi, dan bantuan selama perkuliahan.
14. Semua pihak yang tidak dapat disebutkan satu persatu, atas segala bantuan,
semangat dan doa yang menyertai penulis dari awalnya penelitian hingga
diselesaikannya penulisan skripsi ini.
Penulis merasakan dan menyadari atas kekurangan dalam penyusunan
skripsi ini, karena keterbatasan wawasan dan kemampuan. Penulis dengan senang
hati membuka diri menerima kritik dan saran yang membangun dari semua pihak,
dengan segala kerendahan hati penulis mengharapkan skripsi ini memberikan
manfaat yang berarti bagi para pembaca. Akhir kata, penulis mempersembahkan
skripsi ini demi majunya ilmu pengetahuan farmasi.
Yogyakarta, 10 Juli 2013
Penulis
x
DAFTAR ISI
HALAMAN JUDUL……… i
HALAMAN PERSETUJUAN PEMBIMBING ……….
ii
HALAMAN PENGESAHAN……….
iii
HALAMAN PERSEMBAHAN………..
iv
PERNYATAAN KEASLIAN KARYA………..
v
LEMBAR PERNYATAAN PUBLIKASI………..
vi
xi
3.
Ekstraksi Padat-Cair…………..………
12
4.
Ekstraksi Cair-Cair………....
13
F. Spektrofotometri UV………... 13
G. Kromatografi Cair Kinerja Tinggi (KCKT)………. 16
1.
Definisi dan Instrumentasi……….…………
17
2.
Analisis Kualitatif dan Kuantitatif ……….………...
19
H. Validasi Metode Analisis………..…………..
19
1. Akurasi……….…………..
20
2. Presisi……….
21
3. Selektivitas atau Spesifisitas……….
22
4. Linearitas………..
23
5. Rentang……….
23
I. Landasan Teori……….
24
J. Hipotesis………...
25
BAB III METODOLOGI PENELITIAN………...
26
A. Jenis dan Rancangan Penelitian………
26
xii
1.
Variabel Bebas……….……..
26
2.
Variabel Tergantung……….….……. 26
3.
Variabel Pengacau Terkendali………..……….. 26
C. Definisi Operasional……….……….…… 27
D. Bahan Penelitian……….……….….. 27
E. Alat Penelitian……….……….. 27
F. Tata Cara Penelitian……….………. 28
1.
Pembuatan Fase Gerak……….…………..……. 28
2.
Pembuatan Larutan Baku Standar Internal Asetanilida………. 29
3.
Pembuatan Larutan Baku Nikotin………... 29
4.
Penetapan Panjang Gelombang Pengamatan……….. 30
5.
Pembuatan Kurva Baku……….……….……. 31
6.
Penyiapan Sampel……….……….…………. 31
7.
Pembuatan Ekstrak Tembakau Rokok “MEREK X”…….…………. 32
8.
Validasi Metode……….. 33
9.
Penetapan Kadar Nikotin Dalam Sampel Rokok “MEREK X” ….… 35
G. Analisis Hasil……….…… 36
xiii
F. Pembuatan Kurva Baku Nikotin……….…
52
G. Ekstraksi Nikotin pada Sampel Rokok “Merek X”………....
53
H. Optimasi Ekstraski Nikotin pada Sampel Rokok …………..…………
57
I. Ekstraksi dengan Waktu Optimum 30 menit……….……….... 60
J. Preparasi Sampel……….…..
61
K. Validasi Metode Analsis………..……….……….
61
L. Analisis Kualitatif Nikotin………..
67
M. Penetapan Kadar Nikotin dalam Ekstrak Etanol Fraksi Kloroform Tembakau
Sampel Rokok………...…… 69
BAB V KESIMPULAN DAN SARAN……….………
71
DAFTAR PUSTAKA……….………....
72
LAMPIRAN………..………..
74
xiv
DAFTAR TABEL
Tabel I. Karakteristik beberapa pelarut yang digunakan dalam KCKT….. 17
Tabel II. Nilai
recovery
yang diperbolehkan untuk setiap kadar analit…… 21
Tabel III. Kriteria penerimaan presisi untuk setiap kadar analit……… 22
Tabel IV. Hasil pengukuran AUC asetanilida dengan ekstraksi dan tanpa
ekstraksi………..………... 45
Tabel V. Jumlah nikotin pada kemasan rokok……….…. 47
Tabel VI. Hasil pengukuran AUC nikotin dengan 2 kali ekstraksi dan tanpa
ekstraksi……….…...…… 57
Tabel VII. Uji Normalitas………...…….………. 59
Tabel VIII. Uji T tidak berpasangan………... 60
Tabel IX. Hasil pengukuran AUC nikotin dan standar asetanilida pada ekstrak
tembakau rokok “MEREK X”……….... 61
Tabel X. Hasil perhitungan resolusi sampel………...…..
62
Tabel XI. Hasil persen perolehan kembali (%
recovery
) baku nikotin….. 64
Tabel XII. Hasil
intraday precision
……….
65
Tabel XIII. Hasil
interday precision
... 66
xv
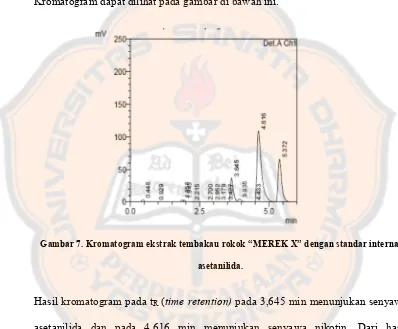
Gambar 7. Kromatogram ekstrak tembakau rokok “MEREK X” dengan standar
internal asetanilida……….... 41
Gambar 8. Kromatogram sampel rokok dan asetanilida………... 43
Gambar 9. Kromatogram asetanilida hasil ekstraksi dan tanpa ekstraksi.. 44
Gambar 10. Spektra λ maksimum nikotin 3 konsentrasi... 49
Gambar 11. Spektra λ maksimum asetanilida 3 konsentrasi……… 50
Gambar 12. Kromofor nikotin dan asetanilida………. 52
Gambar 13. Grafik hubungan antara konsentrasi nikotin dan asetanilida dengan
AUC………. 53
Gambar 14. Tingkat protonasi nikotin berdasarkan hubungan dengan pH… 55
Gambar 15. Kromatogram baku nikotin dengan ektraksi dan tanpa ekstraks. 57
Gambar 16. Kurva baku hubungan antara konsentrasi baku nikotin dengan
AUC………. 63
xvi
Gambar 18. Struktur nikotin……… 68
xvii
DAFTAR LAMPIRAN
Lampiran 1. Sertifikat analisis asetanilida……… 75
Lampiran 2. Sertifikat analisis nikotin………. 76
Lampiran 3. Kromatogram optimasi waktu 10 menit replikasi 1……... 77
Lampiran 4. Kromatogram optimasi waktu 10 menit replikasi 2………. 78
Lampiran 5. Kromatogram optimasi waktu 10 menit replikasi 3………. 79
Lampiran 6. Kromatogram optimasi waktu 20 menit replikasi 1………. 80
Lampiran 7 Kromatogram optimasi waktu 20 menit replikasi 2……….. 81
Lampiran 8. Kromatogram optimasi waktu 20 menit replikasi 3….….... 82
Lampiran 9. Kromatogram optimasi waktu 30 menit replikasi 1……... 83
Lampiran 10. Kromatogram optimasi waktu 30 menit replikasi 2….….. 84
Lampiran 11. Kromatogram optimasi waktu 30 menit replikasi 3……... 85
Lampiran 12. Kromatogram optimasi waktu 40 menit replikasi 1……... 86
Lampiran 13. Kromatogram optimasi waktu 40 menit replikasi 2….….. 87
Lampiran 14. Kromatogram optimasi waktu 40 menit replikasi 3….….. 88
Lampiran 15. Kromatogram penetapan kadar nikotin replikasi 1……… 89
xviii
Lampiran 17. Kromatogram penetapan kadar nikotin replikasi 3…….. 91
Lampiran 18. Kromatogram penetapan kadar nikotin replikasi 4…….. 92
xix
VALIDASI METODE DAN PENETAPAN KADAR NIKOTIN DALAM EKSTRAK TEMBAKAU ROKOK “MEREK X” DENGAN METODE KROMATOGRAFI CAIR KINERJA TINGGI (KCKT)
MENGGUNAKAN STANDAR INTERNAL ASETANILIDA
Is Sumitro 098114127
INTISARI
Telah dilakukan penelitian tentang validasi metode dan penetapan kadar nikotin dalam ekstrak tembakau rokok “Merek X” dengan metode kromatografi cair kinerja tinggi (KCKT) fase terbalik menggunakan standar internal asetanilida. Penelitian ini bertujuan untuk mengetahui validitas metode dan kadar nikotin yang terdapat dalam ekstrak tembakau rokok “MEREK X”.
Penelitian ini mengikuti jenis dan rancangan penelitian non eksperimental deskriptif. Sistem kromatografi cair kinerja tinggi (KCKT) menggunakan kolom fase diam oktil silika (C8), fase gerak metanol : ammonium asetat + TEA 0,1% (70 : 30), kecepatan alir 1 mL/menit, dan detector UV pada panjang gelombang 260 nm. Pada validasi KCKT fase terbalik memenuhi parameter selektivitas (Rs = 2,929), linearitas (r = 0,999893), akurasi dan presisi pada rentang kadar sampel 40-60 µg/mL.
Hasil penelitian menunjukan kadar rata-rata nikotin dalam ekstrak tembakau rokok “Merek X” adalah 0.57385 ± 0.007224 %b/b dengan nilai CV = 1,2588%. Nilai CV yang diperoleh memenuhi syarat presisi yang baik yaitu <2%.
xx
ABSTRACT
A study concerned the determination amount of nicotine in cigarettes
“BRAND X” by reversed phase high performance liquid chromatography with
standar internal acetanilide. This study aims to determine amount nicotine in tobacco
extract cigarettes “BRAND X”.
This research is conducted with a descriptive non-experimental plan and
design. The HPLC system used for quantitative analysis of nicotine consists of octyl
silica (C
8) as the stationary phase, mixture of methanol : ammonium acetate + TEA
0,1% (70:30) as mobile phase, and UV detector with λ max of 260 nm. The
parameters of method validation used in this research are selectivity (Rs = 2,929),
liniearity (r = 0,999), resulted good accuracy and precision (intraday and interday) in
range concentrations 40- 60 µg/mL.
The results of this research of average levels of nicotine contained in tobacco
extract cigarettes “BRAND X” is 0.57385 ± 0.007224 %w/w with value of CV =
1,2588%. Values of CV obtained qualified good precision is < 2%.
BAB I
PENGANTAR
A. Latar Belakang
Rokok merupakan produk yang banyak dikonsumsi masyarakat luas, data
WHO (World Health Organization) mencatat bahwa perokok aktif di Indonesia
mencapai jumlah 62,8 juta orang pada tahun 2011 (WHO, 2011). Kandungan
senyawa kimia dalam rokok yang menyebabkan ketergantungan adalah nikotin.
Nikotin memiliki Lethal Dose sebesar 40 sampai 60 mg (0,5-1,0 mg/kg) pada
manusia dewasa dan kosentrasi nikotin dalam darah lebih besar dari 5 mg/L akan
menyebabkan kematian (Clarke, 2003).
Masyarakat umum yang menjadi konsumen rokok biasanya mengetahui
kandungan nikotin dalam tiap bungkus rokok dengan melihat informasi yang
terdapat pada bungkusan rokok, dengan informasi kandungan nikotin dalam tiap
bungkus rokok ini dapat menjadi dasar patokan berapa banyak nikotin yang
terserap dalam tubuh saat merokok. Namun informasi dalam bungkus rokok
tentang kadar nikotin masih perlu diteliti kembali tentang kebenaran informasinya
yang diperlukan untuk penjaminan mutu produk rokok dari kadar nikotinnya.
Pencantuman kadar nikotin dalam rokok sesuai dengan peraturan
pemerintah no 109 tahun 2012 dimana terdapat pada pasal 10 disebutkan “setiap
orang yang memproduksi produk tembakau berupa Rokok harus melakukan
pengujian kandungan kadar nikotin dan tar perbatang untuk varian yang
mengimpor produk tembakau berupa rokok wajib mencantumkan informasi
kandungan kadar nikotin dan tar sesuai hasil pengujian sebagaimana dimaksud”
(Peraturan Pemerintah RI, 2012).
Dengan melakukan pengujian kadar nikotin dalam tiap batang rokok,
secara tidak lansung dapat membantu pemerintah dalam memastikan kadar
nikotin dalam rokok. Selain dari penjaminan mutu kadar nikotin dalam rokok,
konsumen rokok juga perlu untuk dipenuhi hak konsumennya terkait kebenaran
informasi nikotin dalam rokok.
Hak konsumen ini tercantum pada Undang-Undang Republik Indonesia
nomor 8 tahun 1999, dimana pasal 4 yang berbunyi “Hak konsumen adalah hak
atas informasi yang benar, jelas dan jujur mengenai kondisi dan jaminan barang
dan/atau jasa” ( Undang-Undang RI, 1999).
Rokok “Merek X” yang akan dianalisis dipilih berdasarkan kadar nikotin
yang tinggi dibanding rokok sejenis dan juga dari jumlah konsumen yang banyak.
Kadar nikotin yang tercantum pada label kemasan yang tinggi ini diharapkan
dapat mudah untuk mendapatkan hasil ekstraksi dan pengukuran yang baik terkait
kadar nikotin dalam rokok.
Rokok yang akan dianalisis kadar nikotinnya, nantinya akan diekstraksi
dan didapatkan ekstrak kental rokok. Untuk meningkatkan kadar nikotin dalam
ekstrak kental rokok tersebut maka dipilih metode ekstraksi yang dapat
menghasilkan ekstrak dengan kandungan nikotin yang maksimal. Metode yang
cair-cair. Dimana tahap pertama metode ekstraksi padat-cair dapat berfungsi
untuk mengekstraksi senyawa nikotin dengan maksimal yang menjadi acuan
adalah metode ektraksi dari jurnal “Determination of Nicotine From Tobacco by
LC-MS-MS” ( Vlase, Filip, Mindrutau dan Leucuta, 2005). Tahap selanjutnya
dilakukan metode ekstraksi cair-cair untuk melakukan clean up terhadap senyawa
ekstrak yang telah dihasilkan, sehingga diharapkan hasil kadar nikotin lebih
maksimal dan terpisah dari zat pengotornya yang menjadi acuan adalah metode
ekstraksi cair-cair dari penelitian “Penetapan Kadar Nikotin Dalam Ekstrak
Etanolik Daun Tembakau Vorstenlanden Bawah Naungan dan NA OOGST Secara
KCKT Fase Terbalik” (Dewi, 2012). Cairan penyari yang digunakan adalah etanol
karena dari sifat nikotin yang dapat larut dalam etanol.
Standar internal digunakan untuk mencegah kesalahan dalam pengukuran
karena proses metode yang cukup panjang dengan sampel uji yang cukup kecil
kosentrasinya (Basset,1994). Proses ektraksi pada penetapan kadar nikotin dalam
rokok “MEREK X” cukup panjang karena adanya proses clean up ekstrak yang
berulang-ulang sehingga mencegah hilangnya senyawa nikotin yang banyak
digunakan satandar internal. Pemilihan asetanilida sebagai standar internal
mengacu pada jurnal “Improved highly sensitive method for determination of
nicotine and cotinine in human plasma by high performance liquid
chromatography” ( Nakajima, Yamamoto, Kuroiwa, dan Yokoi, 2000).
Metode Kromatografi Cair Kinerja Tinggi (KCKT) dipilih untuk
menetapkan kadar nikotin dalam ekstrak tembakau pada rokok “MEREK X”,
hasil pemisahan yang baik, dan waktu relatif singkat. Detektor yang digunakan
adalah UV, karena nikotin memiliki struktur kromofor dan memiliki serapan
maksimum pada panjang gelombang tertentu (Cordell, 1981).
Penelitian ini merupakan tahap lanjutan dari serangkaian penelitian kadar
nikotin ekstrak tembakau dalam rokok “MEREK X” yang meliputi tahap
optimasi, validasi metode, dan penetapan kadar nikotin dalam sampel rokok
“Merek X”. Pada penelitian tentang optimasi metode KCKT fase terbalik
didapatkan metode KCKT yang optimal dengan menggunakan kolom fase diam
OktilSilika (C8) dan fase gerak Metanol : Ammonium asetat 10mM + TEA 0,1%
(70 : 30), kecepatan alir 1 mL/menit, detektor UV pada panjang gelombang 262
nm (Antonius, 2013). Metode analisis yang digunakan perlu divalidasi terlebih
dahulu agar hasil analisis yang dilakukan nantinya dapat dipercaya dan dapat
diterima. Parameter-paramater validasi yang digunakan, yaitu selektivitas,
linearitas, akurasi, presisi, dan rentang. Tahap akhir dilakukan penetapan kadar
nikotin dalam ekstrak tembakau rokok “MEREK X”.
1. Permasalahan
Permasalahan yang dapat dirumuskan berdasarkan latar belakang tersebut
antara lain:
a. Apakah metode kromatografi cair kinerja tinggi fase terbalik yang
menggunakan fase diam oktil silika (C8) dan fase gerak metanol : ammonium
penetapan kadar nikotin dalam ekstrak tembakau rokok “MEREK X” memenuhi
parameter-parameter validasi yaitu selektivitas, linearitas, akurasi, presisi, dan
rentang ?
b. Berapakah kadar nikotin dalam ekstrak tembakau rokok “MEREK X”?
2. Keaslian Penelitian
Berdasarkan penelusuran literatur yang telah dilakukan, penetapan kadar
nikotin yang pernah dilakukan adalah penetapan kadar nikotin dalam sampel
biologis menggunakan Kromatografi Cair Kinerja Tinggi (KCKT), kromatografi
gas, spektrofotometri massa, dan kromatografi cair MS (LC-MS) (Nakajima,
Yamamoto, Kuroiwa, Yokoi, 2000); penetapan kadar nikotin dalam
macam-macam merek rokok (Alali dan Massadeh, 2003); penetapan kadar nikotin dalam
tembakau dengan metode LC-MS-MS (Vlase, Filip, Mindrutau, dan Leucuta,
2005); validasi metode KCKT fase terbalik pada penetapan kadar nikotin dalam
ekstrak etanolik daun tembakau (Syenina, 2011); penetapan kadar nikotin dalam
ekstrak etanolik daun tembakau Vorstenlanden Bawah Naungan dan NA OOGST
secara KCKT Fase Terbalik (Dewi, 2012); optimasi komposisi dan kecepatan alir
fase gerak sistem KCKT fase terbalik pada penetapan kadar nikotin dalam rokok
“Merek X” menggunakan standar internal asetanilida (Antonius, 2013).
Validasi metode dan penetapan kadar nikotin ekstrak etanol pada rokok
“Merek X” dengan standar internal asetanilida metode Kromatografi Cair Kinerja
fase gerak Metanol : Ammonium asetat 10mM+ TEA 0,1% (70:30) belum pernah
dilakukan.
3. Manfaat Penelitian
a. Manfaat Metodologis. Hasil penelitian ini diharapkan dapat menjadi
alternatif metode dalam penentuan kadar nikotin dalam ekstrak tembakau rokok
“MEREK X” yaitu menggunakan metode kromatografi cair kinerja tinggi
(KCKT) fase terbalik dengan standar internal asetanilida.
b. Manfaat Praktis. Hasil penelitian ini dapat menambah informasi
tentang parameter validasi yaitu selektivitas, linearitas, akurasi, presisi, dan
rentang serta kadar nikotin dalam ekstrak tembakau rokok “MEREK X” dengan
metode kromatografi cair kinerja tinggi (KCKT) fase terbalik menggunakan
standar internal asetanilida.
B. Tujuan Penelitian
Tujuan dilakukan penelitian ini adalah untuk mengetahui :
a. Validitas metode KCKT fase terbalik yang menggunakan fase diam
oktil silika (C8) dan fase gerak metanol:ammonium asetat 10 mM + TEA 0,1%
(70:30) dengan kecepatan alir 1,0 mL/menit pada penetapan kadar nikotin dalam
ekstrak tembakau rokok “MEREK X” dengan melihat parameter validasi yaitu
selektivitas, linearitas, akurasi, presisi, dan rentang.
b. Kadar nikotin yang terdapat dalam ekstrak tembakau rokok “Merek
BAB II
PENELAAHAN PUSTAKA
A. Rokok
1. Pengertian Rokok
Rokok merupakan suatu produk yang dibungkus oleh kertas berbentuk
seperti silinder dengan panjang mendekati 90 mm, ketika dibakar dan dihisap asap
dari tembakau atau rokok tersebut maka mulailah terjadinya absorpsi dari nikotin
menuju tubuh (Stratton,2001). Terdapat sekitar empat ribu macam zat kimia
dalam rokok yang terdiri dari komponen gas (85%) dan sisanya merupakan
partikel. Diantara ribuan zat kimia tersebut setidaknya dua ratus senyawa
dinyatakan berbahaya bagi kesehatan.Beberapa zat kimia darisekitar empat ribu
zat tersebut ialah nikotin, gas karbon monoksida, nitrogen oksida, nitrogen
sianida, amoniak, benzaldehid, benzen, dan metanol. Racun utama pada rokok
adalah tar, nikotin, dan karbon monoksida (Ma’arif, 2012).
Ada dua jenis rokok yaitu rokok yang berfilter dan tidak berfilter. Filter
pada rokok terbuat dari bahan busa serabut sintesis yang berfungsi menyaring
nikotin. Rokok biasanya dijual dalam bungkusan berbentuk kotak atau kemasan
kertas yang dapat dimasukkan dengan mudah kedalam kantong. Sejak beberapa
tahun terakhir, bungkusan-bungkusan tersebut juga umumnya disertai pesan
kesehatan yang memperingatkan perokok akan bahaya kesehatan yang dapat
ditimbulkan dari merokok, misalnya akan ke penyakit paru-paru atau serangan
2. Bagian-Bagian Rokok
a. Cigarette paper
Kertas rokok (Cigarette paper) terbuat dari bahan kertas selulosa hasil
dari pengolahan serat kain contoh flax atau hemp, atau dari serat kayu. Kertas
rokok ini mampu untuk dilewati udara sehingga dapat memudahkan untuk proses
pembakaran tembakau (Geiss dan Kotzias, 2007).
b. Filter
Filter atau penyaring, umumnya terdapat pada kebanyakan rokok apalagi
pada rokok berfilter. Bagian rokok filter ini terbuat dari asetat selulosa atau tow.
Bagian filter ini mempunyai fungsi sebagai penjebak nikotin dan tar ketika asap
rokok dihisap melewati bagian filter. Fungsi kerja dari filter ini bergantung pada
bagian ventilasi filter dimana diatur oleh tipping paper, selanjutnya bagian ini
akan mengatur kemampuan udara melewati bagian filter juga bersamaan akan
menangkap senyawa nikotin, tar serta senyawa lain (Geiss dan Kotzias, 2007).
B. Tembakau
Tanaman tembakau (Nicotina tabaccum L.) termasuk dalam family
terong-terongan (Solanaceae) (Cahyono,1998).
a. Akar, tanaman tembakau merupakan tanaman berakar tunggang yang
tumbuh tegak ke pusat bumi. Akar tunggangnya dapat menembus tanah
bulu-bulu akar. Perakaran akan berkembang baik jika tanahnya gembur, mudah
menyerap air.
b.Batang, tanaman tembakau memiliki bentuk batang agak bulat, agak
lunak tetapi kuat, semakin keujung semakin kecil.Ruas-ruas batang mengalami
penebalan yang ditumbuhi daun.
c.Daun, tanaman tembakau memiliki tulang daun menyirip, bagian tepi
daun agak bergelombang dan licin. Lapisan atas daun terdiri atas lapisan palisade
parenkim dan spongy parenkim pada bagian bawah. Jumlah daun dalam satu
tanaman 28-32 helai (Hanum, 2008).
C. Nikotin
Nikotin merupakan golongan alkaloid yang diperoleh dari daun tanaman
temabakau (Nicotina tabacum L.).Senyawa ini tidak berwarna, mudah menguap,
sangat higroskopis, jika teroksidasi oleh udara atau cahaya akan berubah menjadi
warna coklat. Senyawa ini larut dalam etanol, eter , kloroform serta memiliki titik
didih sekitar 247oC, dengan indeks refraktif sebesar 1,5280. Nikotin dapat
diesktraksi dengan pelarut organic yang bersifat alkalis (Clarke, 2003).
Nikotin mengandung dua jenis gugus amin tersier yang bersifat basa
dengan pKa cincin piridin adalah 3,04 sedangkan pKa pada cincin pirolidin adalah
7,84. Nilai pKa pada cincin aromatik lebih rendah dikarenakan efek hibridisasi sp2
Hibridisasi sp2 digunakan bila suatu atom karbon membentuk ikatan
rangkap, ikatan rangkap menggambarkan satu ikatan sigma yang kuat dan satu
ikatan pi yang lemah. Ikatan pi akan membuat elektron lebih mudah bergerak
antar ikatan melalui ikatan ini dan juga membuat suatu molekul mempunyai
bentuk yang kaku (Fessenden dan Fessenden, 1986).
Gambar 1. Struktur kimia nikotin (Clarke, 1969 ).
D. Standar Internal
Standar internal merupakan suatu senyawa yang ditambahkan pada suatu
prosedur kerja analisis dalam penetapan kadar secara spektroskopi dan
kromatografi. Senyawa yang dilibatkan berupa sejumlah bahan
pembanding(standar internal) kepada senyawa yang akan diukur dengan
konsentrasi yang diketahui. Fungsi dari standar internal ini adalah untuk
mencegah kesalahan dalam pengukuran karena proses metode yang cukup
panjang dengan sampel uji yang cukup kecil konsentrasinya ( Basset, 1994 ).
Syarat-syarat yang diperlukan senyawa untuk menjadi standar internal
pada metode kromatografi cair kinerja tinggi ( KCKT ) adalah senyawa tersebut
harus dapat terelusi dari komponen lain yang terdapat pada ekstrak campuran
sampel dan dapat dibaca hasil kromatogramnya, serta tidak ada kandungan
kromatogramnya harus mendekati senyawa yang ingin dianalisis untuk
meminimalisir efek instrumental drift.Senyawa harus stabil secara kimia dan
fisika terhadap metode yang digunakan.Akurasi dan presisi yang baik didapatkan
dari peak kromatogram senyawa standar internal yang mendekati peak senyawa
analit.Senyawa standar internal harus dapat secara keseluruhan terpisah dari
senyawa analit saat dipisahkan secara kromatografi. Senyawa standar internal
harus memiliki kemiripan sifat kimia dan fisika dengan analit yang akan dianalisis
( Boyd, 2008 ).
E. Ekstraksi
1. Ekstraksi
Ekstrak adalah sediaan pekat yang diperoleh dengan mengekstraksi zat
aktif dari simplisia nabati atau simplisia hewani menggunakan pelarut yang
sesuai, kemudian semua atau hampir semua pelarut diuapkan dan massa atau
serbuk yang tersisa diperlakukan sedemikian hingga memenuhi syarat yang telah
ditetapkan ( Direktorat Jendral Pengawasan Obat dan Makanan,1995 ).
Ekstrak tumbuhan merupakan material yang diperoleh dengan cara
menyari sampel tumbuhan dengan pelarut tertentu. Terdapat beberapa jenis
ekstrak yaitu : ekstrak cair, ekstrak kental, dan ekstrak kering (Direktorat Jendral
Pengawasan Obat dan Makanan,2000).
Ekstrak diperoleh dengan cara ekstraksi. Ekstraksi adalah kegiatan
penarikan zat aktif yang dapat larut sehingga terpisah dari bahan yang tidak dapat
dapat dipermudah dengan mengetahui terlebih dahulu zat aktif yang dikandung
simplisia. Ekstraksi dipengaruhi oleh derajat kehalusan serbuk dan perbedaan
konsentrasi. Jika hanya dengan mencelupkan serbuk simplisia kedalam pelarut,
maka ekstraksi tidak akan sempurna karena terjadi kesetimbangan antara larutan
zat aktif di luar sel dan larutan zat aktif di dalam sel (Direktorat Jendral
Pengawasan Obat dan Makanan, 1986).
2. Cairan Penyari
Pemilihan cairan penyari harus mempertimbangkan banyak faktor.
Cairan penyari yang baik harus memenuhi kriteria berikut : murah dan mudah
diperoleh, stabil secara fisika dan kimia, bereaksi netral, dan tidak mudah
terbakar, selektif yaitu mudah menarik zat berkhasiat yang dikehendaki, tidak
mempengaruhi zat yang berkhasiat dan diperbolehkan oleh peraturan.
Etanol dipertimbangkan sebagai penyari karena lebih selektif, kapang
dan kuman sulit tumbuh dalam etanol 20%, tidak beracun, netral, absorpsinya
baik dan suhu yang digunakan untuk pemekatan lebih rendah. Etanol dapat
melarutkan alkaloid basa, minyak menguap, glikosida, kurkumin (Direktorat
Jendral Pengawasan Obat dan Makanan, 1986).
3. Ekstraksi padat-cair
Untuk ekstraksi padat-cair ini, prosedur yang paling sering dijumpai
adalah ekstraksi senyawa dari bentuk sediaan padat. Prosedur ini merupakan
prosedur yang sederhana karena melibatkan pemilihan pelarut atau gabungan
dianalisis dan hanya sedikit melarutkan senyawa lain yang akan mengganggu
analisis lebih lanjut, misalkan akan mengganggu pemisahan pada kromatografi.
Kebanyakan prosedur ini dilakukan dengan terlebih dahulu menggerus
matriks padat hingga diperoleh serbuk yang halus lalu dilanjutkan dengan
ekstraksi pelarut, penyaringan, atau sentrifugasi untuk menghilangkan partikulat
(Moldoveanu dan David, 2002).
4. Ekstraksi cair-cair ( liquid-liquid extraction, LLE)
Ekstraksicair-cair digunakan sebagai cara untuk praperlakuan sampel
atau clean-up sampel untuk memisahkan analit-analit dari komponen-komponen
matriks yang mungkin mengganggu pada saat kuantifikasi atau deteksi analit. Di
samping itu, ekstraksi pelarut juga digunakan untuk memekatkan analit yang ada
dalam sampel dengan jumlah kecil sehingga tidak memungkinkan atau
menyulitkan untuk deteksi atau kuantifikasinya
Analit-analit yang mudah terekstraksi dalampelarut organik adalah
molekul-molekul netral yang berikatan secara kovalen dengan substituen yang
bersifat nonpolar atau agak polar. Sementara itu, senyawa-senyawa polar dan juga
senyawa-senyawa yang mudah mengalami ionisasi akan tertahan dalam fase air
(Moldoveanu dan David, 2002).
F. Spektrofotometri UV
Spektrofotometri UV adalah teknik analisis spektroskopik yang
menggunakan sumber radiasi elektromagnetik ultraviolet (λ < 400 nm) dengan
Jika suatu molekul dikenakan radiasi elektromagnetik (REM) maka
molekul akan menyerap REM yang energinya sesuai. Interaksi antara molekul
dengan REM akan meningkatkan energi potensial elektron pada tingkat keadaan
tereksitasi. Transisi elektronik yang terjadi diantara tingkat energi dalam suatu
molekul yaitu transisi σ σ*, n π* dan π π*
Gambar 2. Diagram tingkat energi elektronik (Gandjar dan Rohman, 2007).
1.Transisi σ σ*
Energi yang diperlukan untuk transisi ini besarnya sesuai dengan energy
sinar yang frekuensinya terletak diantara UV vakum (>180 nm) sehingga kurang
begitu bermanfaat untuk analisis dengan cara spektrofotometri UV-Vis
2. Transisi n σ*
Jenis transisi ini terjadi pada senyawa organik jenuh yang mengandung
atom-atom yang memiliki elektron bukan ikatan (elektron n). Energi yang
diperlukan untuk transisi n menuju σ* lebih kecil dibanding transisi σ σ*
sehingga sinar yang diabsorpsi memiliki panjang gelombang lebih panjang
(150-250 nm) (Sastrohamidjojo, 2001).
3.Transisi n π* dan π π*
Jenis transisi ini molekul organik harus mempunyai gugus fungsional
yang tidak jenuh sehingga ikatan rangkap dalam gugus tersebut memberikan
Pelarut dapat mempengaruhi transisi n π* dan π π*, hal ini berkaitan
dengan adanya perbedaan kemapuan pelarut untuk mensolvasi antara keadaan
dasar dengan keadaan tereksitasi (Sastrohamidjojo, 2001).
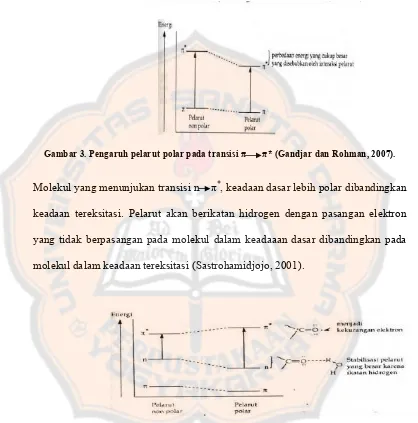
Gambar 3. Pengaruh pelarut polar pada transisi π π* (Gandjar dan Rohman, 2007).
Molekul yang menunjukan transisi n π*, keadaan dasar lebih polar dibandingkan
keadaan tereksitasi. Pelarut akan berikatan hidrogen dengan pasangan elektron
yang tidak berpasangan pada molekul dalam keadaaan dasar dibandingkan pada
molekul dalam keadaan tereksitasi (Sastrohamidjojo, 2001).
Gambar 4.Pengaruh pelarut polar pada transisi n π*(Gandjar dan Rohman, 2007).
Terjadinya eksitasi elektronik pada panjang gelombang yang memberikan
gelombang maksimum yang tetap dapat digunakan untuk identifikasi molekul
yang bersifat karakterisitik sebagai data, sehingga spectrum UV-Vis dapat untuk
tujuan anlisis kualtitaif dan kuantitatif (Mulja dan Suharman, 1995).
G. Kromatografi Cair Kinerja Tinggi (KCKT)
1. Definisi dan Instrumentasi
Kromatografi cair kinerja tinggi ( KCKT) atau biasa disebut juga dengan
HPLC ( High Performance Liquid Chromatography) merupakan teknik
pemisahan yang diterima secara luas untuk analisis sampel obat, baik dalam bulk
atau sediaaan farmasetik, serta dalam cairan biologis (Gandjar dan Rohman,
2007).
Gambar 5. Instrumentasi KCKT (Kazekevich and Lobrutto, 2007).
a.Wadah fase gerak dan fase gerak, alat KCKT yang baru dilengkapi
dengn satu atau lebih wadah gelas, yang mengandung 500 mL atau lebih fase
gerak. Sonikasi (penghilangan gas) biasanya dilakukan terlebih dahulu pada fase
gerak untuk menghilangkan gas yang mungkin terdapat didalamnya.Adanya gas
dapat menyebabkan flow rate yang tidak reprodusibel serta dapat mengganggu
Fase gerak atau eluen biasanya terdiri atas campuran pelarut yang dapat
bercampur dimana secara keseluruhan berperan dalam daya elusi dan resolusi.
Fase gerak yang sering digunakan adalah campuran metanol dan
asetonitril dengan air atau dengan larutan buffer. Untuk analit yang bersifat asam
atau basa lemah, peranan pH sangat penting karena jika pH fase gerak tidak diatur
maka analit akan mengalami ionisasi sehingga ikatan dengan fase diam akan
menjadi lemah jika dibandingkan dengan bentuk tidak terionisasi, spesies yang
terionisasi akan terelusi lebih cepat (Rohman dan Gandjar, 2007).
Pelarut yang digunakan dalam analisis menggunakan KCKT detektor UV
hendaknya memiliki UVcut-off yang jauh dari panjang gelombang serapan analit.
Hal ini karena pada panjang gelombang tersebut kepekaan detector UV sangat
lemah (Mulja dan Suharman, 1995).Karakteristik beberapa pelarut yang sering
digunakan pada analisis menggunakan KCKT disajikan pada tabel 1.
b.Pompa, dalam alat KCKT syarat pompa yang baik bagi pelarut fase
gerak, yakni: pompa harus inert terhadap fase gerak. Pompa yang digunakan
sebaiknya mampu memberikan tekanan sampai 350 sampai 500 bar dan mampu
mengalirkan fase gerak dengan kecepatan alir yang biasa digunakan yaitu 0.1-10
mL/min (Meyer, 2004 ).
c.Tempat penyuntikan sampel, sampel berupa cairan atau larutan
disuntikkan secara lansung ke tempat penyuntikan maka sampel akan dibawa fase
gerak yang mengalir dibawah tekanan menuju kolom (Gandjar dan Rohman,
2007).
d.Kolom, kolom merupakan bagian KCKT yang terdapat fase diam di
dalamnya. Oktadesilsilan (C18) dan oktil silika (C8) merupakan fase diam yang
paling banyak digunakan karena mampu memisahkan senyawa-senyawa dengan
kepolaran yang rendah, sedang, maupun tinggi. Oktil atau rantai alkil yang lebih
pendek lagi lebih sesuai untuk pelarut yang bersifat polar (Meyer, 2004).
e.Detektor, persyaratan detektor KCKT adalah sensitivitas yang tinggi,
rentang senstivitas (108 – 1015 analit/detik), kestabilan dan reprodusibilitas yang
baik memberikan respon yang linier terhadap konsentrasi analit, dapat bekerja
dari temperatur kamar sampai 400oC, tidak dipengaruhi oleh perubahan
temperatur dan kecepatan dari fase gerak, mudah didapat dan mudah
dioperasikan, selektif terhadap berbagai macam analit di dalam fase gerak, tidak
merusak sampel, dapat menghilangkan zone broadening dengan adanya pengaruh
2. Analisis Kualitatif dan Kuantitatif
a. analisis kualitatif, merupakan identifikasi terhadap analit yang terdapat
dalam ekstrak sampel. Analisis kualitatif KCKT umumnya menggunakan
komponen yaitu: waktu retensi.Waktu retensi analit diukur ketika kondisi dari
KCKT konstan, selanjutnya dibandingkan dengan waktu retensi baku, analit harus
memiliki variasi dengan waktu retensi baku yaitu (± 0,02-0,05 menit) (Snyder,
2010).
b. analisis kuantitatif, merupakan identifikasi terhadap jumlah kadar
analit dalam sampel atau ekstrak. Untuk KCKT kuantifikasi dapat dilakukan
dengan mengukur tinggi puncak atau dengan luas puncak.Tinggi puncak diukur
sebagai jarak dari garis dasar ke puncak maksimum.Sedangkan luas puncak
diukur sebagai hasil kali tinggi puncak dan lebar pada setengah tinggi (W1/2)
(Gandjar dan Rohman, 2007).
Kalibrasi menggunakan standar internal, senyawa baku dengan variasi
konsentrasi ditambahakan dengan jumlah baku standar internal yang konstan,
hasil ratio luas area peak kromatogram antara senyawa baku dan standar internal
digunakan sebagai kurva baku untuk pengukuran terhadap jumlah kadar analit
(Snyder, 2010).
H. Validasi Metode Analisis
Validasi metode analisis merupakan suatu proses untuk menilai suatu
Penilaian tersebut dapat dilihat dengan menggunakan parameter-parameter
tertentu yang berdasarkan percobaan di laboratorium (Harmita, 2004).
Validasi metode dilakukan berdasarkan tipe prosedur yang dianalisis.
Tipe prosedur yang umum dianalisis ada tiga macam, yaitu :
a) Kategori I : metode analitik untuk penentuan bahan baku obat atau bahan
aktif pada hasil akhir farmasetika.
b) Kategori II : metode analitik untuk penentuan campuran dalam bahan baku
atau komponen sisa pada produk akhir farmasetika.
c) Kategori III : metode analitik untuk penentuan performa karakteristik obat
(disolusi, pelepasan obat) (Harmita, 2004).
1. Akurasi
Akurasi merupakan suatu prosedur analisis untuk melihat ketelitian
metode analisis atau kesesuaian antara nilai yang diperoleh dari hasil analisis dan
nilai sebenarnya (Ermer dan Miller, 2005).
Akurasi dinyatakan sebagai persen perolehan kembali. Akurasi dapat
ditentukan dengan dua cara, yaitu metode simulasi (spiked-placebo recovery) dan
metode penambahan baku (standard addition method). Metode penambahan baku
dilakukan dengan cara menambahkan sejumlah baku standar ke dalam sampel.
Sebelumnya sampel telah dianalisis terlebih dahulu. Selisih kedua hasil yang
didapat dibandingkan dengan kadar sebenarnya baku standar yang ditambahkan
(Harmita, 2004).
Tabel tentang acuan nilai recovery untuk penetapan akurasi dapat dilihat
Tabel II. Nilai recovery yang diperbolehkan untuk setiap kadar analit (Gonzalez dan Herrador, 2007).
2. Presisi
Presisi merupakan prosedur analisis untuk melihat derajad kesesuaian
hasil uji individual beberapa penginjeksian suatu seri standard. Presisi diukur
sebagai simpangan baku atau simpangan baku relatif. Presisi dapat dilakukan pada
tiga tingkatan yang berbeda, yaitu keterulangan (repeatability), presisi antara
(intermediate precision), dan ketertiruan (reproducibility) (Gandjar dan Rohman,
2010).
Presisi terdiri dari dua komponen, yaitu keterulangan dan presisi antara
(intermediate precision).Keterulangan merupakan variasi yang dilakukan oleh
satu analis pada satu instrument. Keterulangan tidak dilakukan pada variasi
instrument atau sistem. Keterulangan dilakukan dengan cara menganalisis
beberapa replikasi sampel dengan menggunakan metode analisis. Kemudian
dihitung simpangan baku relatifnya (koefisien variasi) (Snyder, dkk., 2010).
Intermediate precision merupakan variasi yang terjadi pada saat di
berbeda.Sebelumnya hal ini dikenal dengan ketangguhan (ruggednes) (Bliesner,
2006).
Kriteria penerimaan diberikan jika metode analisis memberikan
simpangan baku relatif atau koefisien variasi sebesar 2% atau kurang. Akan tetapi
kriteria ini dapat berubah sesuai dengan konsentrasi analit yang diperiksa, jumlah
sampel, dan kondisi laboratorium (Harmita, 2004). Kiteria penerimaan presisi
dapat dilihat pada tabel dibawah ini.
Tabel III. Kriteria penerimaan presisi untuk setiap kadar analit (Gonzalez dan Herrador, 2007).
3. Selektifitas atau Spesifisitas
Selektivitas atau spesifisitas menggambarkan kemampuan suatu metode
analisis untuk mengukur analit yang diinginkan secara tepat dan spesifik pada
matriks sampel. Pada matriks sampel ada kemungkinan terdapat
komponen-komponen lainnya. Komponen-komponen-komponen lain yang mungkin terdapat di dalam
matriks sampel, yaitu pengotor, degradants, dan lain lain (Ermer dan Miller,
2005).
Spesifisitas suatu metode analisis dapat diketahui dengan cara melihat
satu cara untuk mengetahui spesifisitas metode analisis. Nilai resolusi yang
dianjurkan harus mendekati atau lebih dari 1,5 (Snyder, dkk., 2010).
4. Liniearitas
Linearitas menggambarkan kemampuan suatu metode analisis untuk
mendapatkan hasil uji yang secara langsung proporsional konsentrasi kurva baku
dengan analit di dalam sampel. Pengukuran linearitas dapat dilakukan langsung
pada analit atau dapat dilakukan pada sampel yang telah ditambah baku standar.
Linearitas dapat dilihat dengan dua cara, yaitu secara evaluasi lansung pada garis
persamaaan kurva baku dan secara statistika menggunakan regresi linear (Ermer
dan Miller, 2005).
Pengukuran linearitas dilakukan dengan cara membuat seri baku standar
terlebih dahulu. Seri baku yang dibuat biasanya memiliki rentang antara 50-150%
dari kadar analit di dalam sampel. Suatu metode analisis dikatakan linear apabila
memenuhi persyaratan nilai koefisien korelasi (r) ≥ 0,999. Pembuatan kurva baku
yang akan digunakan untuk perhitungan kadar zat sampel dapat dilakukan dengan
tiga macam teknik standar. Teknik standar tersebut, yaitu standar eksternal,
standar internal, dan standar adisi (Snyder, dkk., 2010).
5. Rentang
Rentang merupakan interval antara batas terendah dan tertinggi analit yang
telah memenuhi persyaratan keakuratan, keseksamaan, dan lineritas (Harmita,
2004). Rentang kerja dari suatu metode analisis didapatkan dari hasil karakteristik
validasi yang didapatkan pada bagian akurasi, presisi, dan lineritas (Ermer dan
I. Landasan Teori
Rokok merupakan produk yang terbuat dari bahan baku daun tembakau,
dalam tembakau tersebut banyak mengandung berbagai senyawa alkaloid salah
satunya adalah senyawa nikotin. Nikotin merupakan senyawa alkaloid yang
terdapat pada famili Solanaceae, dengan sifat senyawa basa yang terdapat pada
molekul nikotin yaitu pada cincin pirolidin dengan pKa 7,84 dan cincin piridin
dengan pKa 3,04. Kandungan nikotin dalam rokok perlu diteliti untuk penjaminan
mutu kandungan nikotin dan juga memenuhi hak konsumen untuk mendapat
informasi terkait kadar nikotin dalam rokok sesuai dengan peraturan pemerintah
nomor 109 tahun 2012 Pasal 10 Ayat 1.
Metode KCKT (Kromatografi Cair Kinerja Tinggi) fase terbalik yang
telah dioptimasi dapat memisahkan beberapa campuran senyawa pada ekstrak
tembakau, karena adanya perbedaan interaksi antara senyawa-senyawa tersebut
dengan fase diam oktil silica (C8) dan fase gerak metanol : ammonium asetat
10mM +TEA 0,1% (70 : 30). Metode ini harus divalidasi terlebih dahulu sebelum
dilakukan penetapan kadar agar hasil analisis yang didapatkan nantinya dapat
dipertanggungjawabkan, dapat dipercaya, dan dapat diterima berdasarkan
parameter-parameter validasi yang digunakan. Parameter-paramater yang
divalidasi yaitu Parameter-paramater validasi yang digunakan, meliputi
selektivitas yang ditentukan dengan resolusi, linearitas yang ditentukan dengan
koefisien korelasi (r), akurasi yang ditentukan dengan persen perolehan kembali
ditentukan dari kadar terendah sampai tertinggi sampel yang memenuhi parameter
linearitas, akurasi, dan presisi.
Penetapan kadar nikotin dalam sampel rokok “MEREK X” dilakukan
dengan membandingkan nilai AUC (Area Under Curve) antara sampel ekstrak
tembakau yang telah ditambahkan dengan standar internal asetanilida dengan
AUC standar baku nikotin yang juga telah ditambahkan dengan standar internal
asetanilida. Dengan menggunakan persamaan kurva baku nikotin dan asetanilida,
y = bx + a, dimana y adalah AUC dan x adalah kadar nikotin., maka AUC sampel
dimasukkan dalam persamaan, kemudian kadar dari sampel nikotin dalam ekstrak
tembakau rokok “MEREK X” dapat diketahui.
J. Hipotesis
a. Metode KCKT fase terbalik yang menggunakan fase diam oktil silika
(C8) dan fase gerak metanol:ammonium asetat 10 mM + TEA 0,1% (70:30)
dengan kecepatan alir 1,0 mL/menit pada penetapan kadar nikotin dalam ekstrak
tembakau rokok “MEREK X” memenuhi parameter-parameter validasi, meliputi
selektivitas yang ditentukan dengan resolusi, linearitas yang ditentukan dengan
koefisien korelasi (r), akurasi yang ditentukan dengan persen perolehan kembali
(recovery), presisi yang ditentukan dengan koefisien variasi, dan rentang yang
ditentukan dari kadar terendah sampai tertinggi sampel yang memenuhi parameter
linearitas, akurasi, dan presisi.
b. Ekstrak tembakau rokok “MEREK X” mengandung senyawa analit
BAB III
METODE PENELITIAN
A. Jenis dan Rancangan Penelitian
Penelitian ini merupakan jenis penelitian non eksperimental, karena tidak
dilakukan perlakuan atau manipulasi pada subjek uji yang digunakan dan
merupakan rancangan deskriptif karena hanya menggambarkan data yang
diperoleh.
B. Variabel Penelitian
1. Variabel bebas pada penelitian ini adalah sistem kromatografi cair kinerja
tinggi dengan fase diam oktil silika (C8) dan fase gerak methanol:ammonium
asetat 10 mM + TEA 0,1% (70:30) dengan kecepatan alir 1,0 mL/menit dan
ekstrak tembakau rokok “Merek X”.
2. Variabel tergantung pada penelitian ini adalah parameter validasi yaitu
selektivitas, linearitas, akurasi, presisi, dan rentang serta kadar nikotin yang
terdapat pada ekstrak tembakau rokok “Merek X”.
3. Variabel pengacau terkendali pada penelitian ini adalah
a. Kemurnian pelarut, sehingga digunakan pelarut pro analysis, yang
memiliki kemurnian tinggi.
b. Larutan baku nikotin yang bersifat mudah teroksidasi oleh udara dan
cahaya, diatasi dengan menggunakan aluminium foil untuk menutupi
C. Definisi Operasional
1. Sistem kromatografi cair kinerja tinggi (KCKT) yang digunakan dalam
penelitian ini menggunakan kolom fase diam oktilsilika (C8) dan komposisi
fase gerak metanol : ammonium asetat 10mM + TEA 0,1% (70 : 30).
2. Ekstrak tembakau rokok “Merek X”.
3. Validasi metode yang dilakukan pada penelitian ini meliputi pengukuran
terhadap parameter-parameter validasi yaitu selektivitas, linearitas, akurasi,
presisi, dan rentang.
4. Kadar nikotin dalam 1 gram ekstrak dinyatakan dalam satuan %b/b ± SD.
D. Bahan Penelitian
Bahan yang digunakan memiliki kualitas pro analysis kecuali
dinyatakan lain yaitu baku nikotin (E. Merck), asetanilida (E. Merck), ammonium
asetat (E. Merck), Metanol (E.Merck), kalium hidroksida (E. Merck) memiliki
kualitas teknis, kloroform (E.Merck) memiliki kualitas teknis, Etanol (E. Merck)
memiliki kualitas teknis, aquadest dan aquabidest. Sampel yang digunakan dalam
penelitian ini adalah ekstrak tembakau rokok “Merek X”
E. Alat Penelitian
Alat yang digunakan adalah spektrofotometer UV-Vis (merek optima
SP-300 Plus), seperangkat alat KCKT fase terbalik terdiri: pompa (merek Shimadzu
LC-10 AD No. C20293309457 J2) dengan sistem elusi gradien dan isokratik,
C8merek Shimadzu (spesifikasi ukuran diameter internal 4,6mm x 25 cm, ukuran
diameter partikel 5µm fully encapped residual silanol), seperangkat alat computer
(merek Dell Vostro 220), printer (merek HP D2566), alat ultrasonikator (Retsch
tipe T640 no 935922013), organic and anorganic solvent membrane filter
(Whatman) ukuran pori 0,45 m dengan diameter 47mm, alat sentrifugasi, alat
vortex, neraca analitik merek Ohaus, milipore, mikropipet, indicator PH, pompa
vakum dan seperangkat alat gelas.
F. Tata Cara Penelitian
1. Pembuatan Campuran Fase Gerak
a. Pembuatan Ammonium Asetat 10 mM dan TEA 0,1%
1. Pembuatan larutan ammonium asetat 10 mM. Menimbang seksama
kurang lebih 0,7708 g ammonium asetat (BM = 77,08), dilarutkan dengan
aquabidest pada labu takar 1000 mL hingga batas tanda. Didapatkan larutan
ammonium asetat 10 mM.
2. Pembuatan TEA 0,1% v/v.Mengambil sebanyak 1 mL trietilamin,
ditambahkan ke dalam larutan ammonium asetat, dilarutkan dengan aquabidest
pada labu takar 1000 mL hingga batas tanda. Didapatkan larutan ammonium
asetat 10 mM + TEA 0,1%.
b. Pembuatan Fase Gerak
Fase gerak yang digunakan yaitu campuran metanol : ammonium asetat
10mM + TEA 0,1% (70 : 30). Masing-masing larutan disaring menggunakan
larutan tea dan ammonium asetat, dibantu dengan pompa vakum dan
diawaudarakan selama 15 menit.Pencampuran fase gerak dilakukan secara manual
didalam wadah fase gerak.
2. Pembuatan Larutan Baku Standar Internal Asetanilida
a. Pembuatan larutan stok asetanilida. Menimbang seksama kurang
lebih 0,5 gram asetanilida, laruttkan dengan metanol dalam labu takar 10,0 mL
hingga tanda. Didapatkan larutan stok asetanilida 0,05 g/mL (50 mg/mL).
b. Pembuatan larutan intermediet asetanilida. Larutan asetanilida 2,5
mg/mL dibuat dengan cara mengambil 0,5 mL larutan stok asetanilida 50 mg/mL
ke dalam labu takar 10,0 mL, encerkan hingga tanda dengan metanol.
c. Pembuatan larutan intermediet kerja asetanilida. Larutan intermediet
kerja asetanilida 0,1 mg/mL dibuat dengan cara mengambil 0,2 mL larutan
intermediet asetanilida 2,5 mg/mL ke dalam labu takar 5,0 mL, encerkan hingga
tanda dengan methanol.
3. Pembuatan Larutan Baku Nikotin
a. Pembuatan larutan stok baku nikotin. Larutan stok dibuat dengan
cara mengambil 497 µL baku nikotin dan dimasukkan ke dalam labu takar 5,0
mL. Larutan diencerkan dengan metanol hingga tanda. Didapatkan larutan stok
b. Pembuatan larutan intermediet baku nikotin. Larutan intermediet
nikotin 10 mg/mL dibuat dengan cara mengambil 0,5 mL larutan stok nikotin 100
mg/mL ke dalam labu takar 5,0 mL, encerkan hingga tanda dengan metanol.
c. Pembuatan larutan intermediet kerja baku nikotin. Larutan
intermediet kerja nikotin 0,2 mg/mL dibuat dengan cara mengambil 0,2 mL
larutan intermediet asetanilida 10 mg/mL ke dala labu takar 10,0 mL, encerkan
hingga tanda dengan metanol.
d. Pembuatan seri larutan baku nikotin. Dibuat seri larutan baku dengan
konsentrasi 20, 40, 60, 80, dan 100 µg/mL dengan cara mengambil sebanyak 500,
600, 700, 800 dan 900 µL dari larutan intermediet kerja nikotin, dimasukkan ke
dalam labu takar 5,0 mL.
e. Pembuatan seri larutan baku nikotin dengan penambahan standar
internal asetanilida. Standar internal asetanilida 20 µg/mL dibuat dengan cara
mengambil sebanyak 500 µL dari larutan intermediet kerja asetanilida,
dimasukkan ke dalam labu takar 5,0 mL yang sebelumnya telah diisi dengan seri
larutan baku nikotin, encerkan hingga tanda dengan metanol.
4. Penetapan Panjang Gelombang Pengamatan
a. Penentuan panjang gelombang maksimum pengamatan nikotin.
Dilakukan screening larutan baku nikotin 20 µg/mL, 30 µg/mL, dan 40 µg/mL
pada daerah panjang gelombang 225-300 nm, menggunakan spektrofotometer
UV-Vis. Panjang gelombang maksimum pengamatan ditentukan berdasarkan
b. Penentuan panjang gelombang maksimum pengamatan asetanilida.
Dilakukan screening larutan baku asetanilida 1 µg/mL, 5 µg/mL, dan 10 µg/mL
pada panjang gelombang 225-300 nm, menggunakan spektrofotometer UV-Vis.
Panjang gelombang maksimum pengamatan ditentukan berdasarkan spektra
dengan serapan yang maksimal.
5. Pembuatan Kurva Baku Nikotin dengan Standar Internal Asetanilida
Pembuatan seri larutan baku nikotin dengan konsentrasi 20, 40, 60, 80,
dan 100 µg/mL, masing-masing larutan ditambahkan standar internal asetanilida
20 µg/mL, kemudian disaring dengan menggunakan milipore, lalu diawaudarakan
selama 15 menit. Selanjutnya masing-masing campuran larutan baku diinjeksikan
pada system kromatografi cair kinerja tinggi (KCKT) fase terbalik dengan fase
diam oktil silica (C8) dan fase gerak metanol : ammonium asetat 10mM + TEA
0,1% (70:30), dengan kecepatan alir 1,0 mL/menit. Dari hasil luas area masing
baku campuran baku, selnajutnya dibandingkan kemudian diplotkan terhadap
konsentrasi nikotin untuk memperoleh regresi linier dengan persamaan y = bx + a
6. Penyiapan Sampel
a. Pembuatan larutan KOH 10 M. Menimbang seksama lebih kurang
56,11 g (BM = 56,11), masukkan ke dalam labu takar 100,0 mL, kemudian
larutkan dengan aquades hingga tanda.
b. Pembuatan larutan KOH 0,1 M. Mengambil 2,0 mL KOH 10 M,
masukkan ke dalam labu takar 200,0 mL, kemudian encerkan dengan aquades
c. Pemilihan dan Pengambilan Sampel. Sampel yang dipilih adalah
rokok dengan “Merek X” yang diambil dari toko penjualan rokok “MEREK X”
Kabupaten Sleman, Yogyakarta dengan nomor batch sama. Selanjutnya dari 90
bungkus rokok diambil masing-masing 1 batang rokok lalu dipreparasi.
d. Preparasi sampel rokok. Diambil 90 batang rokok “MEREK X”
yang telah dibeli, dipotong tegak lurus bagian batang rokok. Bagian batang rokok
yang mengandung serbuk tembakau dan cengkeh dikeluarkan. Serbuk diaduk
kemudian diblender. Campuran serbuk hasil blender yang dihasilkan kemudian
diayak dengan ayakan nomor mesh 16, didapatkan campuran serbuk halus
tembakau yang lolos dari ayakan. Campuran serbuk halus tembakau ini siap untuk
diekstraksi lebih lanjut.
7. Pembuatan Ekstrak Tembakau Rokok “MEREK X”
a. Optimasi lama waktu ekstraksi. Serbuk rokok “MEREK X” yang
telah diayak ditimbang sebanyak 200 mg. Selanjutnya dimasukan ke dalam beker
gelas, ditambahkan etanol teknis sebanyak 20 mL, dan asetanilida 10mg/mL
sebanyak 20 µL. Selanjutnya beker gelas dipanaskan di atas waterbath selama
waktu optimasi yaitu : 10 menit, 20 menit, 30 menit, dan 40 menit dengan suhu ±
70oC. Setelah proses pemanasan, diambil sebanyak 5 mL ekstrak tembakau rokok
“MEREK X” untuk diuapkan. Lalu setelah proses penguapan selesai,
ditambahkan sejumlah 1 mL aquades, 3 mL kloroform dan 1 mL larutan KOH 0,1
M dalam ekstrak kental tembakau rokok “MEREK X”. Selanjutnya dimasukkan
selama 24 menit. Tahap selanjutnya diambil bagian fase kloroform, dan dilakukan
pengulangan dengan penambahan 3 mL kloroform lagi ke dalam ekstrak rokok
“MEREK X” yang telah diambil fase kloroformnya, dan di vortex selama 30 detik
dan disentrifugasi selama 24 menit dengan kecepatan 4000 rpm. Selanjutnya
diambil bagian kloroformnya. Bagian kloroform yang telah terkumpul dalam vial,
selanjutnya diuapkan hingga kering, sampai didapatkan ekstrak kental rokok.
ditambahkan 5,0 mL fase gerak, diawaudarakan selama lebih kurang 5 menit.
Diambil 1,0 mL larutan yang telah diawaudarakan, disaring dengan milipore dan
dimasukkan ke dalam vial KCKT, vial KCKT diawaudarakan selama lebih kurang
2 menit. Larutan siap diinjeksikan. Masing-masing waktu optimasi dilakukan 3
kali replikasi.
yang telah disaring dengan milipore dan diawaudarakan selama 15 menit
diinjeksikan pada sistem KCKT fase terbalik dengan fase diam oktil silika (C8)
dan fase gerak metanol:ammonium asetat 10 mM + TEA 0,1% (70:30) dengan
kecepatan alir 1,0 mL/menit. Dilakukan repetisi tiga kali. Resolusi dihitung
dengan memasukkan selisih waktu retensi dan lebar setengah tinggi peak nikotin
b. Pembuatan kurva baku dan penentuan linearitas. Dibuat seri larutan
baku nikotin dengan konsentrasi 20, 40, 60, 80, dan 100 µg/mL sebanyak 1 mL,
masing-masing larutan ditambahkan standar internal asetanilida 20 µg/mL
sebanyak 100 µL, kemudian disaring dengan menggunakan milipore kemudian
diawaudarakan selama 15 menit. Sebanyak 20 µL dari masing-masing larutan
diinjeksikan pada sistem kromatografi cair kinerja tinggi fase terbalik dengan fase
diam oktil silika (C8) dan fase gerak metanol:ammonium asetat 10 mM + TEA
0,1% (70:30) dengan kecepatan alir 1,0 mL/menit. Dari kromatogram akan
diperoleh luas area nikotin dan luas area asetanilida untuk masing-masing
konsentrasi. Luas area ini kemudian dibandingkan sehingga didapatkan
perbandingan luas area nikotin terhadap asetanilida. Perbandingan kedua luas area
ini kemudian diplotkan terhadap konsentrasi nikotin untuk memperoleh regresi
linear dengan persamaan y = bx + a dan nilai koefisien korelasi (r) yang akan
digunakan untuk menentukan parameter validasi linearitas
c. Penentuan persen kembali (recovery) dan penentuan koefisen variasi
adisi baku nikotin dalam sampel (presisi). Dibuat dua macam larutan yaitu larutan
sampel dan larutan sampel yang ditambahkan baku nikotin (adisi). Larutan sampel
dibuat dengan tiga tingkatan berdasarkan penimbangan sampel rokok. Larutan
sampel pertama untuk level rendah dibuat dengan cara menimbang sampel
sebanyak 125 mg, kemudian dilakukan ekstraksi sampel. Larutan sampel kedua
untuk level sedang dibuat dengan cara menimbang sampel sebanyak 150 mg,
kemudian dilakukan ekstraksi sampel.Larutan sampel ketiga untuk level tinggi
untuk diinjeksikan ke dalam sistem KCKT dengan cara mengambil 1,0 mL
ekstrak sampel, disaring dengan milipore dan dimasukkan ke dalam vial KCKT,
vial KCKT diawaudarakan selama lebih kurang 2 menit.Sampel siap
diinjeksikan.Dilakukan replikasi sebanyak tiga kali untuk tiap level. Larutan
sampel yang ditambahkan baku nikotin (adisi) dibuat dengan cara menambahkan
baku nikotin pada vial KCKT untuk setiap level, untuk level rendah ditambahkan
2,5 µg/mL, untuk level sedang ditambahkan 5 µg/mL, dan untuk level tinggi
ditambahkan 10 µg/mL, 20 µg/mL, dan 50µg/mL. Setiap level perlakuan
dilakukan replikasi tiga kali. Kadar baku nikotin yang ditambahkan dalam sampel
merupakan selisih nilai kadar sampel adisi dan kadar sampel. Kemudian dihitung
persen perolehan kembali (recovery), Standard Deviation (SD), dan koefisien
variasi (KV).
9. Penetapan Kadar Nikotin Dalam Sampel Rokok “MEREK X”
Sampel yang telah dipreparasi, diinjeksikan sebanyak 20 µL ke dalam
system KCKT yang telah dioptimasi sehingga didaptkan kromatogram sampel dan
dibaca AUC dari masing-masing replikasi. Masukkan hasil AUC ke persamaan
regresi linier baku nikotin dengan standar internal asetanilida dari hasil validasi
G. Analisis Hasil
1. Selektivitas
Selektivitas ditentukan dengan menghitung resolusi dari kromatogram
yang dihasilkan oleh ekstraksi sampel rokok. Menurut Synder dkk. (2010), syarat
resolusi yang baik yaitu dimana senyawa analit terpisah dari senyawa-senyawa
yang lain adalah ≥ 1,5.
Resolusi dihitung dengan rumus :
(1)
Dimana : Rs = resolusi
t2 = waktu retensi puncak kedua t1 = waktu retensi puncak pertama
0,5W(1) = lebar setengah tinggi puncak pertama 0,5W(2) = lebar setengah tinggi puncak kedua
2. Linearitas dan Rentang
Linearitas ditentukan dengan nilai koefisien korelasi (r), yang diperoleh
dari AUC baku nikotin yang diplotkan terhadap konsentrasi baku. Nilai r yang
dipersyaratkan adalah ≥ 0,999 (Snyder, dkk, 1997). Sedangkan rentang diperoleh
dari kadar terendah hingga tertinggi sampel yang memberikan akurasi, presisi, dan
linearitas yang baik.
3. Akurasi
Menurut Harmita (2004), akurasi ditentukan dengan persen perolehan
kembali (recovery), yang dapat dihitung dengan rumus :