The catalytic properties of ribozymes depend on the
sophisticated structures of the respective ribozyme–substrate complexes. Although it has been suggested that ribozyme-mediated cleavage of RNA occurs via a rather strictly defined mechanism, recent findings have clearly demonstrated the diversity of reaction mechanisms.
Addresses
*Department of Chemistry and Biotechnology, Graduate School of Engineering, The University of Tokyo, Hongo, Tokyo 113-8656, Japan
†National Institute for Advanced Interdisciplinary Research, AIST, MITI,
Tsukuba Science City 305-8562, Japan
‡Polish Academy of Science, Centre of Molecular and Macromolecular
Studies, Department of Bioorganic Chemistry, Sienkiewicza 112, 90-363 Lodz, Poland
Correspondence: Kazunari Taira; e-mail: [email protected] Current Opinion in Biotechnology2000, 11:354–362
0958-1669/00/$ — see front matter
© 2000 Elsevier Science Ltd. All rights reserved. Abbreviations
GS ground state HDV hepatitis delta virus TS transition state
Introduction
Catalytic RNAs are called ribozymes and they include hammerhead, hairpin and hepatitis delta virus (HDV) ribozymes, group I and II introns, the RNA subunit of RNase P, and ribosomal RNA [1–5]. Among these ribozymes, the first two ribozymes to be discovered, by Altman and Cech, respectively, were the RNA subunit of RNase P and a group I intron [1–3]. Within the following five years, small ribozymes, such as hammerhead, hairpin and HDV ribozymes, were discovered in studies of the replication, via a rolling-circle mechanism, of certain viroids, satellite RNAs and an RNA virus [1,4]. These ribozymes catalyze RNA cleavage/ligation reactions. In the case of the ribozyme-mediated cleavage of RNA, first a nucleophile attacks the phosphorus atom at the scissile phosphate with resultant formation of a pentavalent phos-phorus species, which is either a transition state or a short-lived intermediate, then a leaving oxygen departs from the phosphorus. The nucleophile can either be an internal 2′-oxygen in small ribozymes, such as hammer-head, hairpin, and HDV ribozymes, or an external nucleophile in larger ribozymes, such as the group I intron and the RNA subunit of RNase P.
Ribozymes are considered to be fossil molecules that origi-nated in a hypothetical prebiotic RNA world. A full understanding of their mechanisms of action should enhance our understanding of the evolution of primitive organisms [1–5]. It has long been assumed that all ribozymes
must be metalloenzymes that require divalent metal ions for catalysis and that the cleavage mechanism is unique to ribozymes but common to all types of ribozyme. Recent advances, however, have revealed examples of metal-ion-independent cleavage by hairpin ribozymes and it has become clear that multiple mechanisms for cleavage by ribozymes must exist, depending upon the architecture of each ribozyme. It has also become clear that the architecture of HDV ribozyme–substrate complexes can influence the pKaof the ring nitrogens of cytosine and adenine, with the resultant perturbed ring-nitrogens being directly involved in acid/base catalysis [6,7•,8••]. Nevertheless, there is no doubt
that RNA catalysts lacking functional substituents under physiological conditions exploit metal ions for an appropri-ate structure and for catalytic function and that the majority of ribozymes use metal ions as cofactors. The extensive potential of RNA molecules to act as catalysts and the sequential events that occur in reactions catalyzed by ribozymes have been of great interest, and the role of the catalytic metal ions have been the focus of much experi-mentation and discussion [9–15,16•,17–19]. Among various
ribozymes studied in the past, we will focus on a few ribozymes whose enzymatic mechanism became unveiled over the past year at the atomic level.
Reactions catalyzed by the
Tetrahymena
ribozyme: a double-metal-ion mechanism
The reaction mechanism of ribozymes that catalyze the cleavage of RNA phosphodiester linkages, in particular that of large ribozymes such as group I introns and the cat-alytic RNA subunit of RNase P, involves external nucleophiles. By contrast, small ribozymes, such as ham-merheads, hairpins and HDV ribozymes, use an internal nucleophile, namely, the 2′-oxygen of a ribose moiety at the cleavage site, with the resultant formation of a 3′ -ter-minal 2′-O,3′-O-cyclic phosphate. Despite the large size of the group I ribozyme of Tetrahymenawith its sophisticated higher-order structure, the extensive efforts made over the past decade have revealed the details of the mechanism of the Tetrahymena ribozyme-mediated cleavage of RNAs. Detailed kinetic and thermodynamic analyses of wild-type and mutated forms of this ribozyme have helped to define the reaction mechanism at the atomic level [1,20,21,22••].
Chemical modification at the atomic level has generally involved introduction of a sulfur atom to replace an oxygen atom that has the potential to interact with a catalytically important metal ion. The reduction in the cleavage rate in the presence of Mg2+after such a modification (the so-called thio
effect) and the subsequent observation of cleavage with the phosphorothioate-modified substrate or ribozyme at the same rate as that of cleavage with the normal RNA components in the presence of Mn2+(the so-called manganese rescue effect)
Differences among mechanisms of ribozyme-catalyzed reactions
Masaki Warashina
*
†
, Yasuomi Takagi
†
, Wojciech J Stec
‡
and
have been taken as evidence that supports the direct coordi-nation of the oxygen atom in question with a metal ion. This phenomenon can be explained by the hard–soft acid base (HSAB) rule. According to this rule, a ‘hard acid’, such as Mg2+, prefers to bind to a ‘hard base’ oxygen atom rather than
to a ‘soft base’ sulfur atom. By contrast, a ‘soft acid’, such as Cd2+, prefers to bind to a ‘soft base’ sulfur atom. Mn2+is
soft-er than Mg2+and, thus, can bind to a soft sulfur atom (as well
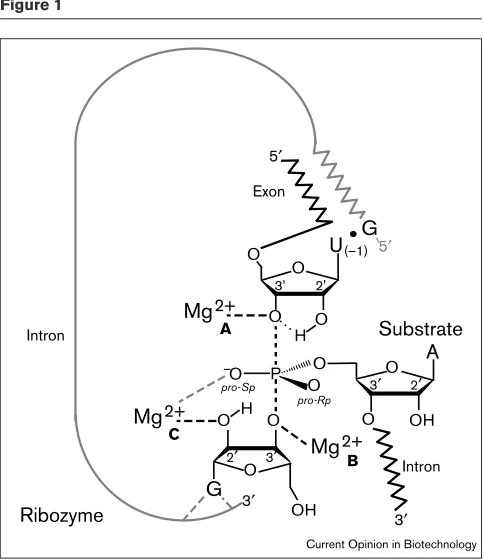
as to a hard oxygen atom) and is believed to be the origin of the manganese rescue effect. Analysis of both the thio effect and the manganese rescue effect has contributed significant-ly to our understanding of the mechanism of action of the Tetrahymenaribozyme. Such analysis has revealed the three metal-binding sites that are shown in Figure 1 [22••]. Thus, it
is generally accepted that the Tetrahymenaribozyme is a met-alloenzyme that operates via the double-metal-ion mechanism of catalysis, in which Mg2+at the B site enhances
the deprotonation of the 3′-OH of the guanosine nucleophile and Mg2+at the A site stabilizes the leaving 3′-bridging
oxy-gen of U(–1) in the transition state (TS). A third Mg2+ is
postulated to bind at the C site to interact directly with the 2′-OH of the guanosine nucleophile.
Although the above mechanism is, indeed, generally accept-ed, a linear free-energy analysis of the cleavage of oligonucleotide substrates with a series of 2′-substituents at U(–1) suggested that the effect on the catalytic rate of the
inductive effects. Specifically, the weaker electron-with-drawing 2′-OH group enhances the chemical cleavage step to a greater extent than did the stronger electron-withdrawing 2′-F atom of the corresponding 2′-deoxy-2′-fluoro derivative. Therefore, the possibility of a symmetrical TS, in which the 2′-OH of U(–1) might or might not interact with a metal ion (as observed at site C), was recently examined [22••].
Despite the absence of lone-pair electrons at the 2′-NH3+
group that might interact with a metal ion, the higher reac-tivity of the substrate with a 2′-deoxy-2′-NH3+ group than
that of the substrate with a 2′-OH group at U(–1) suggested that interaction of a metal ion with the 2′-OH of U(–1) might not be important for catalysis by the Tetrahymenaribozyme. The higher reactivity of the derivative suggests that donation of a hydrogen bond from the neighboring 2′-group to the 3′-bridging oxygen might allow specific stabilization of the TS relative to the ground state (GS), thereby facilitating the chemical cleavage step. The network of interactions between the 2′-OH of U(–1), the 2′-OH of A207 and the exo-cyclic amino group of G22 have been referred to as a catalytic triad. However, the observation that the chemical cleavage step with the substrate with a 2′-NH3+moiety is faster than
that with the 2′-OH group, despite the absence of lone-pair electrons at the 2′-NH3+group that can accept a hydrogen
bond from A207, suggests another possibility for the arrange-ment of active-site groups within this network [22••].
Even though the ribozyme-mediated chemical cleavage step for the substrate with a 2′-OH group is significantly (103-fold) faster than that with 2′-H at U(–1), with
metal-binding site A being occupied by a Mn2+, the rate constants
for reactions with 3′-deoxy-3′-thio-modified substrates are similar irrespective of whether there is a 2′-OH group or 2′-H at U(–1). Furthermore, in the presence of Mg2+, with
metal-binding site A unoccupied by a metal ion, the rate constants for reactions with 3′-deoxy-3′-thio-modified sub-strates are similar with a 2′-OH group or with 2′-H at U(–1), indicating that the 2′-OH group at U(–1) does not con-tribute significantly to the chemical cleavage of the phosphorus–sulfur bond with a 3′-mercapto leaving group. Since sulfur is a weaker acceptor of a hydrogen-bond than oxygen and, furthermore, since sulfur is a significantly bet-ter leaving group than oxygen, the 3′-mercapto leaving group suppresses the catalytic advantage provided by a hydrogen bond from the 2′-OH in the native TS [22••].
Analysis of the thio effect and rescue experiments have contributed significantly to elucidation of the mechanism of action of the large group I ribozyme of Tetrahymena. All the available data appear to support the refined double-metal-ion mechanism of catalysis that is shown in Figure 1.
Thio effects and rescue experiments in
reactions catalyzed by hammerhead ribozymes
Metal-ion rescue experiments have also provided consid-erable information about the sites at which functional metal ions bind in reactions catalyzed by hammerhead The transition state during the splicing reaction mediated by a
Tetrahymenagroup I intron. Replacement of a specific oxygen atom with a sulfur atom followed by metal-dependent cleavage, revealed three important metal-ion binding sites (A, Band C).
O O O
–O O O
O OH O O
OH
G
O O
O H
H
pro-Rp pro-Sp P
Mg2+
U(–1)
A
Exon
Intron 5′
3′
G
5′
3′ Intron
Mg2+
Ribozyme
Substrate
A
Mg2+
C
B 2' 3'
3′ 2′
2′ 3′
ribozymes [9,23,24,25•,26••]. In such experiments, analogs
of hammerhead ribozymes with phosphorothioate modifi-cations were synthesized, with a sulfur atom replacing either a bridging or a non-bridging oxygen at the internu-cleotide phosphorus atom at a preselected position. In some cases, such substitution resulted in the loss of activi-ty in the presence of Mg2+, which is generally accepted as
the natural metal cofactor. Addition of even trace amounts of either Mn2+or Cd2+, however, restored catalysis. These
results supported the hypothesis that the cleavage process requires direct coordination of a Mg2+with the phosphate
oxygen and in the case of phosphorothioate analogs involvement of interaction of a thiophilic metal ion with the sulfur atom. Specifically, in the presence of Mg2+or
Ca2+ions, the rate of the ribozyme-catalyzed cleavage of a
phosphorothioate substrate, in which one of the non-bridg-ing oxygens (the pro-Rp oxygen atom) had been replaced by sulfur, was nearly four orders of magnitude lower than that of the reaction with the natural substrate (thio effect). However, the rates of the ribozyme-catalyzed reactions with the natural substrate and the phosphorothioate sub-strate were enhanced by Cd2+(Cd2+rescue). Such results
have generally been considered to provide evidence of the direct interaction of the sulfur atom at the Rp position of the cleavage site with the added Cd2+; however, that
con-clusion has been controversial. Results from our laboratory have indicated that the added Cd2+binds at the P9 site and
that, contrary to the conclusion reached by other investiga-tors, Cd2+does not interact with the sulfur atom at the Rp
position of the scissile phosphate in the GS or in the TS [26••]. Our intepretation is based on the analysis shown
in Figure 2, as discussed below.
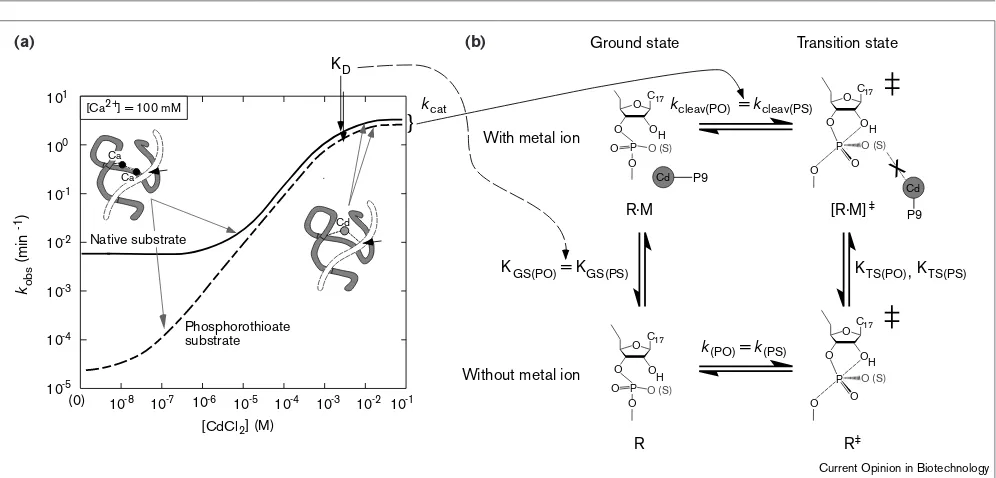
We investigated the effects of Cd2+against a background
of Ca2+. In the case of cleavage of unmodified and
phos-phorothioate-modified substrates, each curve of the logarithm of activity versus the logarithm of the concen-tration of Cd2+can be divided into three regions. In the
absence of Cd2+, the ribozyme–substrate complex
remains in a less active state in the presence of back-ground Ca2+. Upon addition of Cd2+, Cd2+ replaces
pre-existing Ca2+within the ribozyme–substrate complex
and, in proportion to the extent of such replacement, the activity of the ribozyme increases linearly until the rele-vant Ca2+ has been completely replaced by Cd2+. After
the completion of the replacement of Ca2+ by Cd2+(at
Cd2+ concentrations greater than the apparent K D of
~3 mM; Figure 2, left), the ribozyme–substrate complex enters the activated state with bound Cd2+.
From the titration curves in Figure 2, an estimate can be made of respective kinetic and thermodynamic parameters in the thermodynamic cycle, as follows. As the pre-existing Ca2+within the ribozyme–substrate complex was replaced Figure 2
10-5
10-3
10-1
101
(0) 10-6 10-3
100
10-2
10-4
10-8 10-7 10-5 10-4 10-2 10-1
[CdCl2] (M)
[Ca2+] = 100 mM
Cd
KD
Ca
Ca
kcleav(PO) = kcleav(PS)
O
Cd O
O (S)
OC17
O O
H P
With metal ion
Without metal ion KGS(PO) = KGS(PS)
Cd P9
OC
O O
O P O
H
O (S)
17
OC
O O
O P O O (S)H
17
KTS(PO), KTS(PS)
k(PO) = k(PS)
Ground state Transition state
kcat
}
kobs
(min
-1)
Phosphorothioate substrate
[R·M]‡
R·M
R R‡
O O
O (S)
OC17
O O
H P
Native substrate
P9
X
Current Opinion in Biotechnology
(a) (b)
Evidence for the identical affinity of a Cd2+for the native substrate and a phosphorothioate substrate. (a)Titration with Cd2+of the ribozyme-catalyzed cleavage of each substrate (native substrate, solid line; phosphorothioate substrate, dashed line) with 100 mM Ca2+as background (50 mM MES-Na [pH 6], 37°C). The black arrows indicate the apparent KDfor each ribozyme–substrate complex (left).
(b)Thermodynamic cycles for the rescue of cleavage of the native and/or the phosphorothioate substrate by addition of Cd2+. Binding of a Cd2+ to
the Rp position of the scissile phosphate in the scheme proposed by other groups [24,25•] is unlikely. ‘Without metal ion’ means that the
and phosphorothioate (PS) substrates, the binding affinity of the added Cd2+to each respective complex in the GS should
be the same: KGS(PO)= KGS(PS). The rate of the cleavage in
the activated complex [R•M]‡is 1 min–1at saturating Cd2+
and is the same for both the natural and phosphorothioate substrates: kcleav(PO)=kcleav(PS). Furthermore, the rate of nonenzymatic cleavage of the P–O bond in the two sub-strates is similar: k(PO)=k(PS).
The application of thermodynamics and TS theory permits the calculation of KTS(PO)and KTS(PS). Because three out
of four parameters were the same for the natural substrate and the phosphorothioate substrate — KGS(PO)= KGS(PS), kcleav(PO)=kcleav(PS)and k(PO)=k(PS)— the remaining para-meter should also be the same for the two substrates: KTS(PO)= KTS(PS). It is apparent, therefore, that the
affini-ty of Cd2+for the ribozyme–substrate complex is the same
for both complexes not only in the GS, KGS(PO)= KGS(PS),
but also in the TS, KTS(PO)= KTS(PS), irrespective of
whether the cleavage site includes a regular phosphate or a modified phosphorothioate moiety.
We must conclude that Cd2+does not interact directly with
the sulfur atom at the Rp position of the scissile phosphate either but binds to the P9/G10.1 motif to restore the cat-alytic activity of the cleavage site [25•,26••]. Consequently,
observations of the thio effect and manganese rescue effect themselves may not provide decisive arguments in support of the hypothesis that direct coordination of a metal ion with the pro-Rp oxygen at the cleavage site is a precondi-tion for hammerhead ribozyme-catalyzed reacprecondi-tions [9,26••].
Even a single substitution of sulfur for oxygen has serious steric effects, rather than the expected kinetic thio effect, in reactions that are catalyzed by RNase P [27•]. With a
3′-deoxy-3′-S-phosphorothiolate-modified precursor tRNA, which can be processed by RNase P, the cleavage site shifts from the expected phosphorothiolate-modified site to the adjacent unmodified phosphodiester linkage in the 5′ direction. The bulkiness of the sulfur atom might prevent the correct positioning of the scissile bond at the active site.
Hepatitus delta virus ribozyme-catalyzed
reactions
Recent reports by three groups have revolutionized our understanding of the mechanism of HDV ribozyme-cat-alyzed reactions [6,7•,8••]. For the cleavage of
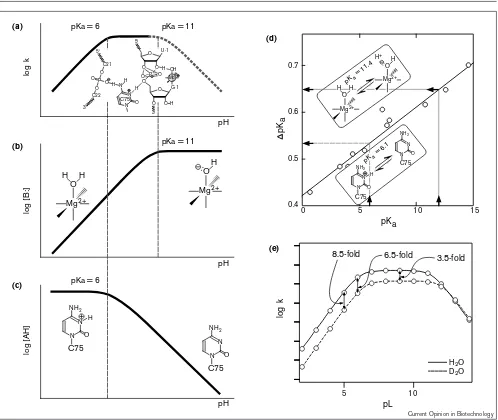
phosphodiester bonds, the nucleophile must be deproto-nated and the leaving group must be protodeproto-nated or stabilized by a metal ion. As shown in Figure 1, the devel-oping negative charges in the TS in the Tetrahymena ribozyme-catalyzed reaction are stabilized by direct inter-actions with metal ions. The novel finding with respect to the mechanism of catalysis by the HDV ribozyme is that the pKa of C75 is perturbed to neutrality in the ribozyme–substrate complex and, more importantly, C75 acts as a general acid catalyst in combination with a metal
this phenomenon provided the first direct demonstration that a nucleoside can act as an acid–base catalyst in RNA. As a result, as shown in Figure 3a, the curve showing the dependence on pH of the self-cleavage of the precursor HDV genomic ribozyme has a slope of unity at pH values below 7 (the activity increases linearly as pH increases with a slope of +1) and then, at higher pH values, the observed rate constant is insensitive to pH.
The slope of unity at low pH is consistent with an increase, with pH, in the concentration of the metal hydroxide, which acts as the general base upon deprotonation (Figure 3b), and a constant amount of the functional pro-tonated form of C75, which acts as the general acid (Figure 3c). The slope of zero from pH 7 to pH 9 indicates that the concentration of the metal hydroxide [B:] increas-es, whereas the concentration of the protonated C75 [AH] decreases by the same amount [8••]. In agreement with
this interpretation, when C75 was replaced by uracil, the resultant C75→U mutant, which was unable to assist in the transfer of a proton, did indeed lack catalytic activity. The activity of the C75→U mutant could be restored, however, by the addition of imidazole whose protonated form (imi-dazolium ion) is known to act as a good proton donor [7•,8••]. Another mutant, C75→A, in which the ring
nitro-gen N1 at A75 has a slightly lower pKa than the
corresponding ring nitrogen N3 of C75, supported self-cleavage activity, albeit with reduced efficiency. The observed pKa of the C75→A mutant was slightly lower
than that of the wild-type C75 ribozyme, supporting the interpretation that a nucleic acid base acts as a catalyst in HDV ribozyme-catalyzed reactions as shown in Figure 3.
Further convincing evidence for this model comes from the observation that, in the absence of divalent metal ions (in the absence of base B; Figure 3b) and in the presence of a high concentration of monovalent cations (~2 M NaCl and 1 mM EDTA), the logarithm of the observed rate con-stant decreased with pH with a slope of –1 (as shown in Figure 3c) with only general acid catalysis operative (in the absence of the metal hydroxide as a general base). This type of profile clearly demonstrates that the observed pKa
for self-cleavage results from the general acid rather than from the general base. It is noteworthy that [Co(NH3)6]3+
which is known to be a substitutionally inert transition metal complex (kex< 10–10s–1even at acidic pH)
inhibit-ed the Mg2+-catalyzed reaction in a competitive fashion,
suggesting that [Co(NH3)6]3+might bind to the same site
as the functional Mg2+with outer-sphere coordination but
does not ionize. The similar rate constants in the presence of Ca2+and of Mg2+are also consistent with the action of a
hydrated metal ion as a Brönsted base rather than a Lewis acid in the HDV ribozyme.
There was a significant, apparent D2O solvent isotope
constants, kcat(H2O):kcat(D2O), being as high as 10; see [8••]). Moreover, because, firstly, the observed pK
a for
self-cleavage is that for the general acid and secondly, the overall rate-limiting step appears to be the cleavage of the bond between the phosphorus and the 5′-leaving oxygen (see below and Figure 4), the transferred proton in the TS must be that of C75. As the concentration of
the protonated C75 in H2O can be higher than that in
D2O at a fixed pH, the difference in pKa(∆pKa), which
equals log(KaH2O/KaD2O), must be taken into account
when the D2O solvent isotope effect is calculated [9,28,29]. It is possible to estimate the relative concen-trations of the active species in H2O and in D2O because
a linear relationship exists between them, as shown in Figure 3
log k
5 10
H2O D2O
pL
0 5 10 15
0.7
0.6
0.5
0.4
pKa
pK
a
N
N O
NH2
C75 Mg2+
O H
:
H+
Mg2+
O H pKH
a = 11.4
N
N O
NH2
H
C75 Mg2+
O H H
pKa = 11
pH
log [B:]
(b)
Mg2+ O
H
:
pKa = 6
pH
log [AH]
(c)
N N O NH2
C75 N
N O NH2
H
C75
pH
log k
pKa = 11 pKa = 6
(a)
O O P O
C22 5'
3' C21
N
H H
O N
O
O U-1
O O
O
O
O O H
G-1 5'
3' Mg OH H O P
H N
C75
O
(d)
(e)
pKa = 6.1
8.5-fold 6.5-fold 3.5-fold
Current Opinion in Biotechnology
Kinetic analysis of HDV ribozyme-catalyzed reactions. (a)A schematic replotting of the pH-rate profile of HDV ribozyme-catalyzed reactions [8••]. This profile has pKavalues of 6 and 11. The solid line and the dotted line represent the experimentally obtained curve and the theoretical curve, respectively. This profile can be explained by a combination of the dependence on pH of the concentration of the active base [B:], as shown in (b), and of the concentration of the active acid [AH], as shown in (c). (b)The logarithm of the concentration of a general base catalyst in an active form is plotted against pH. The pKais 11. (c) The logarithm of the concentration of a general acid catalyst in an active form is plotted against pH. The pKais 6. (d)Isotope effects on the acidities of phenols and alcohols as a function of their acid strength, pKa (for details, see [9,28,29]). (e)Theoretical curves for the activity of the
HDV ribozyme in H2O and D2O. The pKavalues in H2O and in D2O for C75 of 6.1 and 6.5, respectively, were taken from the experimental data [8••]. Using the pKain H2O for a magnesium-bound water molecule as being 11.4, we calculated the corresponding pKavalues in D2O, from the linear plot in (d), to be 12 [9]. Theoretical curves were plotted on the assumption that the value of the intrinsic isotope effect was 2. The following equation was used to plot the graph of pL versus log(rate): logkobs= logkmax– log (1 + 10[pKa(base) – pL]) – log (1 + 10[pL – pKa(acid)]) – logα. Where kmaxis the rate constant in the case of all acid and base catalysts in active forms, and αis the coefficient of the intrinsic isotope effect, that is, kmax(H2O)/kmax(L2O). In the pH profile, α= 1; in the pD profile,
the pKaof C75 is 6.1 and, thus, the estimated ∆pKaturns out to be 0.53, as indicated by the dashed arrow in Figure 3d. Indeed, in the pD-logk profile, the pKa is
shifted upward by ~0.4±0.1 pH units, consistent with the estimated value and with proton transfer by a ring nitrogen [8••]. If we take the pK
a of C75 as 6.1 in H2O
and 6.5 in D2O, and the pKa values for
magnesium-bound water to be 11.4 and 12.0, respectively, and if we assume that the value of the intrinsic D2O solvent iso-tope effect is two, we can generate theoretical curves for HDV-catalyzed reactions in H2O and D2O, as shown in
Figure 3e. The agreement of our theoretical curves with the observed curves [8••] confirms that proton transfer
does indeed occur from protonated C75 in the P–(5′–O) bond-cleaving TS. This conclusion is different from the case of hammerhead ribozymes because, in that case, the observed isotope effect (k
cat(H2O)/kcat(D2O)= 4.4; see
[9,28,29]) can be fully explained by the difference in rel-ative concentrations of the active species in H2O and in
D2O, with an intrinsic D2O solvent isotope effect being
zero [9,28,29].
Thus, in the case of HDV ribozyme-catalyzed reactions, all the available data support the reaction mechanism shown in Figure 3a. The donation of a proton is favored by this model, which involves an acid with an optimal pKaof 7.
The ability of the HDV ribozyme to perform general acid–base catalysis effectively under physiological condi-tions appears to be unique among known ribozymes.
Importance of acid catalysis in reactions
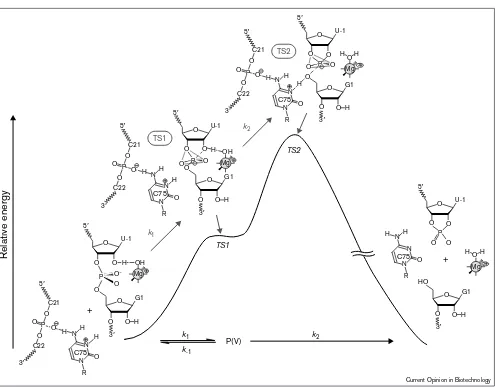
catalyzed by hammerhead and HDV ribozymes
Nonenzymatic hydrolysis, as well as small-ribozyme-medi-ated hydrolysis, of RNAs occurs via nucleophilic attack by the 2′-oxygen that generates the pentacoordinate interme-diate or TS (via the first putative TS [TS1]). Such attack is followed by the cleavage of the bond between the phos-phorus and the 5′-oxygen leaving group (via the second putative TS [TS2]; Figure 4). Among the two putative TSs, TS2 is the overall rate-limiting TS (attack by the 2′-OH on the phosphorus atom is easier than cleavage of the P–O[5′] bond and, thus, TS2 always has higher energy than TS1). This conclusion was confirmed in experiments with an RNA analog with a 5′-mercapto leaving group. If the formation of the intermediate were the rate-limiting step (i.e. if TS1 were to be a higher-energy state than TS2), the phosphorothiolate substrate should be hydrolyzed at a rate similar to the rate of the hydrolysis of the natural RNA, because the 5′-bridging phosphorothio-late linkage would not be expected to enhance the attack by the 2′-oxygen. In contrast, if the decomposition of the intermediate were the rate-limiting step (i.e. if TS2 were a higher-energy state than TS1), we would expect that the phosphorothiolate substrate would be hydrolyzed much more rapidly than the natural RNA because the pKaof a
thiol is more than 5 units lower than that of the corre-sponding alcohol. Several groups have confirmed that the
reactive than the natural RNA in nonenzymatic hydrolytic reactions [9,10] and, thus, TS2 is indeed always a higher-energy state than TS1 in the cleavage of natural RNAs.
In the case of the hammerhead-ribozyme-catalyzed reac-tions, the RNA substrate with a 5′-mercapto leaving group is extremely reactive and handling is difficult [30,31]. In our laboratory, however, we recently succeeded in measuring rate constants of 5′-phosphorothiolate-modified and natural substrates under controlled conditions (measurements were performed under conditions such that curves of pH versus log[rate] had a slope of unity for both substrates, and all undesired side reactions could also be avoided; Y Takagi and K Taira, unpublished data). As the 5′ -phosphorothio-late analog of the substrate was hydrolyzed by hammerhead ribozymes at significantly higher rates than those measured with the natural RNA substrate, in agreement with our pre-vious conclusion [31], TS2 must also be a higher-energy state than TS1 in these ribozyme-catalyzed reactions. Because the overall transition-state structure in the hydrol-ysis of RNA is TS2, regardless of whether the reaction proceeds via a concerted one-step mechanism or via a two-step mechanism with a stable pentacoordinate intermediate, it is important that any enzyme that catalyzes the hydrolysis of RNA should stabilize TS2 by donating a proton to the 5′-leaving oxygen or by ensuring coordination of a metal ion to the leaving oxygen.
The above scenario should also be applicable to HDV-ribozyme-catalyzed reactions and, thus, the removal of the possibility of acid catalysis in the C75→U mutant sup-pressed the enzymatic activity. By contrast, self-cleavage of the HDV ribozyme in the absence of divalent cations, but in the presence of high concentrations of monovalent cations, suggests that, at high concentrations, monovalent cations can replace divalent cations in the tertiary folding of the RNA. Thus, divalent cations are not absolutely essential for the folding or cleavage activity of the HDV ribozyme, although a functional Mg2+does participate in
the cleavage process under physiological conditions.
Metal ion involvement in hammerhead
ribozyme-catalyzed reactions
Base catalysis mediated by Mg2+-hydroxide was postulated as
the mechanism of the chemical cleavage step in reactions cat-alyzed by a hammerhead ribozyme on the basis of the original profiles of pH versus the rates of various metal ion-catalyzed reactions of hammerhead ribozymes [32]. The single-metal-ion mechanism, in which Mg2+-hydroxide acts as a general
base and abstracts a proton from the 2′-OH internal nucle-ophile [32], is supported by the finding that no metal ion was found close to the 5′-leaving oxygen in the crystallographic structure of a freeze-trapped conformational intermediate of a hammerhead ribozyme [33,34]. In the more recently deter-mined crystal structure of a modified hammerhead ribozyme [35], however, Co2+was located close to the 5′oxygen atom of
structure, molecular dynamic simulations led to the proposal of a new and alternative single-metal-ion mechanism. In the proposed mechanism, one Mg2+ coordinates directly and
simultaneously to the pro-Rp oxygen and the adjacent oxygen of the attacking hydroxyl group at the cleavage site, acting as a Lewis acid to enhance the attack by the 2′-OH moiety on the phosphorus atom. In addition, it was also proposed that one of the outer-sphere water molecules that surround the metal ion is located such that it might act as a general acid to donate a proton to the 5′-leaving group [36]. As mentioned above, however, three independent groups recently found no evidence for the direct coordination of a metal ion to the pro-Rp oxygen, at least in the GS [25•,26••,37]. It was also noted
that the absence of such an interaction could explain the absence of a thio effect in the hammerhead-catalyzed cleav-age of a phosphorodithioate linkcleav-age [37]. Our analysis, depicted in Figure 2, also did not support such an interaction in the TS [26••].
In contradiction, several reports have now appeared that pro-vide experimental epro-vidence for a double-metal-ion mechanism of catalysis in hammerhead-catalyzed reactions. Studies on solvent isotope effects and kinetic analysis of a modified substrate with a 5′-mercapto leaving group at the cleavage site have lent strong support to the double-metal-ion mechanism of catalysis [28,31]. Furthermore, hammerhead-ribozyme-mediated cleavage has been ana-lyzed as a function of the concentration of La3+ions in the
presence of a fixed concentration of Mg2+ions. In this way,
the role of lanthanum could be monitored and the metal ions could be identified that were directly involved in the cleav-age reaction [38,39]. The resultant bell-shaped curve that originated from activation by the 5′-oxygen-bound La3+and
deactivation by the 2′-oxygen-bound La3+, shown in
Figure 5, was used to support the proposed double-metal-ion mechanism of catalysis. Other studies, however, demonstrat-ed that binding of a metal ion (the P9 metal ion) to the Figure 4
Current Opinion in Biotechnology
pro-Rp oxygen (P9 oxygen) of the phosphate moiety of nucleotide A9 and to N7 of nucleotide G10.1 is critical for efficient catalysis, despite the considerable distance (~20 Å) between the P9 metal ion and the phosphorus of the scissile phosphodiester group in the GS. In fact, it was demonstrat-ed that an adddemonstrat-ed Cd2+binds first to the pro-Rp phosphoryl
P9 oxygen and not to the pro-Rp phosphoryl oxygen at the cleavage site [25•,26••]. Therefore, it was difficult to exclude
completely the possibility that La3+ions might have replaced
the P9 metal ion and, as a result, might have created condi-tions represented by the bell-shaped curve. In order to clarify this situation, we examined a modified hammerhead ribozyme that included an N7-deazaguanine residue instead of G10.1. Because the data from our experiments with the 7-deaza-ribozyme were free from potential artifacts that might have resulted from the binding of a La3+to N7 at G10.1, the
bell-shaped curve obtained for 7-deaza-ribozyme upon the addition of La3+to a reaction mixture that contained Mg2+,
as reproduced in Figure 4, strongly supports the proposal that a double-metal-ion mechanism operates in the cleavage reac-tions catalyzed by hammerhead ribozymes [40•]. Recent ab initiomolecular orbital calculations also support the dou-ble-metal-ion mechanism [41].
Conclusions: diversity among the mechanisms
of ribozyme-catalyzed reactions
It has been well established that the Tetrahymenaribozyme is a metalloenzyme that operates according to the double-metal-ion mechanism of catalysis. In general, as indicated above, other ribozymes also require metal ions for catalysis. Metal ions are almost certainly needed to induce the folding of each ribozyme into its active conformation, and the folded ribozyme might recruit metal ions at correct positions for direct participation in the chemical cleavage process, as in the case of the Tetrahymena ribozyme. Alternatively, the folded RNA structure might itself play a role in the cleavage
this latter ribozyme, the folded RNA perturbs the pKaof C75
and, as a result, C75 can perform effective general-acid catal-ysis under physiological conditions. Evidence that the folded RNA structure itself functions in the cleavage mechanism comes from studies of hairpin ribozymes. Three independent groups have demonstrated that the hairpin ribozyme is almost fully functional when [Co(NH3)6]3+ replaces Mg2+ [42–44].
The studies of cleavage induced by the exchange-inert [Co(NH3)6]3+complex have been extended to include
non-metallic compounds, and hairpin ribozyme-mediated cleavage has been induced both by aminoglycoside antibi-otics and by polyamines [45]. The mechanism of catalysis of the simplest and most extensively studied hammerhead ribozyme remains controversial. The activities of hammer-head ribozymes under physiological conditions apparently depend on Mg2+. Nevertheless, high concentrations of
mono-valent metal ions (1–4 M monomono-valent cations, such as Li+,
Na+, and NH
4+) can perform the functions of divalent metal
ions [46]. Furthermore, the cleavage activity of hammerhead ribozymes in the absence of divalent cations suggests that divalent cations are not absolutely essential for folding or cleavage in hammerhead ribozyme-catalyzed reactions, as is also the case for the HDV ribozyme. It remains to be deter-mined whether such high concentrations of monovalent metal ions might stabilize the substantial negative charge that develops on the 5′-oxygen atom of the leaving group in ham-merhead ribozyme-catalyzed reactions.
In conclusion, the evidence that has accumulated over the past two years clearly demonstrates the diversity of ribozyme-catalyzed reactions. Nevertheless, acid-base catalysis appears to operate in every case even if many details remain to be clarified.
Acknowledgement
M Warashina and Y Takagi contributed equally to the work. WJ Stec thanks MITI for the financial support that paid for his three-month stay at the Laboratory of Molecular Biology of the National Institute of Advanced Interdisciplinary Research in Tsukuba.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest ••of outstanding interest
1. Gesteland RF, Cech TR, Atkins JF (Eds): The RNA World, edn 3. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999.
2. Cech TR, Zaug AJ, Grabowski PJ: In vitrosplicing of the ribosomal RNA precursor of Tetrahymena: involvement of a guanosine nucleotide in the excision of the intervening sequence.Cell1981, 27:487-496.
3. Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S: The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell1983, 35:849-857.
4. Symons RH: Small catalytic RNAs.Annu Rev Biochem 1992, 61:641-671.
5. Yarus M: Boundaries for an RNA world.Curr Opin Chem Biol1999, 3:260-267.
6. Ferré-D’Amaré AR, Zhou K, Doudna J: Crystal structure of a hepatitis delta virus ribozyme.Nature1998, 395:567-574. A bell-shaped titration curve for cleavage by hammerhead
ribozymes. Both the wild-type ribozyme and the ribozyme with a 7-deaza group at G10.1 generated the same bell-shaped titration curve (for details, see [38,39,40•]).
O O O
P O O O
O–
C
Mg2+
Mg2+
La3+
Mg2+ O O
O
P O O O
O–
C
La3+
La3+
O O O
P O O O
O–
C
LaCl3 (µM)
Activity of ribozyme
7. Perrotta AT, Shih I, Been MD: Imidazole rescue of a cytosine
• mutation in a self-cleaving ribozyme.Science1999, 286:123-126. A chemical group within HDV RNA is shown, for the first time, to be able to act as a true catalyst in a ribozyme reaction.
8. Nakano S, Chadalavada DM, Bevilacqua PC: General acid-base
•• catalysis in the mechanism of a hepatitis delta virus ribozyme. Science2000, 287:1493-1497.
This paper provides a complete analysis of HDV ribozyme-catalyzed reac-tions. The most important finding is that C75 acts as a general acid catalyst rather than as a general base catalyst. The importance of the acid catalyst for stabilization of the leaving group is also emphasized.
9. Zhou DM, Taira K: The hydrolysis of RNA: from theoretical calculations to the hammerhead ribozyme-mediated cleavage of RNA.Chem Rev 1998, 98:991-1026.
10. Kuimelis RG, McLaughlin LW: Mechanism of ribozyme-mediated cleavage.Chem Rev1998, 98:1027-1044.
11. Stage-Zimmermann TK, Uhlenbeck OC: Hammerhead ribozyme kinetics.RNA1998, 4:875-889.
12. Doudna JA: Ribozymes: the hammerhead swings into action.Curr Biol1998, 8:R495-R497.
13. Walter NG, Burke JM: The hairpin ribozyme: structure, assembly and catalysis.Curr Opin Chem Biol1998, 2:24-30.
14. Carola C, Eckstein F: Nucleic acid enzymes.Curr Opin Chem Biol 1999, 3:274-283.
15. Li Y, Breaker RB: Deoxyribozymes: new players in the ancient game of biocatalysis.Curr Opin Struct Biol 1999, 9:315-323.
16. Lilley DMJ: Structure, folding and catalysis of the small nucleolytic
• ribozymes.Curr Opin Struct Biol1999, 9:330-338.
The importance of RNA folding in ribozyme-mediated reactions is summarized. 17. Scott WG: RNA structure, metal ions, and catalysis.Curr Opin
Chem Biol1999, 3:703-709.
18. Strobel SA: A chemogenetic approach to RNA function/structure analysis.Curr Opin Struct Biol1999, 9:346-352.
19. Westhof E: Chemical diversity in RNA cleavage.Science1999, 286:61-62.
20. Weinstein LB, Jones BC, Cosstick R, Cech TR: A second catalytic metal ion in group I ribozyme.Nature1997, 388:805-808.
21. Shan S, Yoshida A, Sun S, Piccirilli JA, Herschlag D: Three metal ions at the active site of the Tetrahymenagroup I ribozyme.Proc Natl Acad Sci USA1999, 96:12299-12304.
22. Yoshida A, Shan S, Herschlag A, Piccirilli A: The role of the cleavage
•• site 2′′-hydroxyl in the Tetrahymenagroup I ribozyme reaction. Chem Biol2000, 7:85-96.
Replacement of the leaving oxygen and the 2′-OH by a sulfur atom and an amino group, respectively, and detailed kinetic analysis are used to analyze a role of the 2′-OH group in catalysis.
23. Warashina M, Zhou DM, Kuwabara T, Taira K: Ribozyme structure and function.In Comprehensive Natural Products Chemistry, vol 6. Edited by Söll D, Nishimura S, Moore PB. Oxford: Elsevier Science Ltd.; 1999:235-268.
24. Scott EC, Uhlenbeck OC: A re-investigation of the thio effect at the hammerhead cleavage site.Nucleic Acids Res1999, 27:479-484.
25. Wang S, Karbstein K, Peracchi A, Beigelman L, Herschlag D:
• Identification of the hammerhead ribozyme metal ion-binding site responsible for rescue of the deleterious effect of a cleavage site phosphorothioate.Biochemistry1999, 38:14363-14378.
This paper provides evidence that a metal ion does not bind to the pro-Rp oxygen of the scissile phosphate bond in the ground state.
26. Yoshinari K, Taira K: A further investigation and reappraisal of the
•• thio effect in the cleavage reaction catalyzed by a hammerhead ribozyme.Nucleic Acids Res2000, 28:1730-1742.
Evidence is provided that, contrary to the conclusion reached by other inves-tigators, a metal ion does not interact with the sulfur atom at the Rp position of the scissile phosphate either in the ground state or in the transition state.
27. Warnecke JM, Sontheimer EJ, Piccirilli JA, Hartmann RK: Active site
• constraints in the hydrolysis reaction catalyzed by bacterial RNase P: analysis of precursor tRNAs with a single 3′′ -S-phosphorothiolate internucleotide linkage.Nucleic Acids Res 2000, 28:720-727.
A good example of the large side effects of phosphorothiolate substitution at the cleavage site.
28. Sawata S, Komiyama M, Taira K: Kinetic evidence based on solvent isotope effects for the nonexistence of a proton-transfer process in reactions catalyzed by a hammerhead ribozyme: implication to the double-metal-ion mechanism of catalysis.J Am Chem Soc 1995, 117:2357-2358.
29. Kumar PKR, Zhou DM, Yoshinari K, Taira K: Mechanistic studies on hammerhead ribozymes. In Catalytic RNA, Nucleic Acids and Molecular Biology,vol 10. Edited by Eckstein F, Lilley DMJ. Berlin: Springer-Verlag; 1996:217-230.
30. Zhou DM, Usman N, Wincott FE, Matulic-Adamic J, Orita M, Zhang LH, Komiyama M, Kumar PKR, Taira K: Evidence for the rate-limiting departure of the 5′′-oxygen in nonenzymatic and hammerhead ribozyme-catalyzed reactions. J Am Chem Soc 1996, 118:5862-5866.
31. Zhou DM, Zhang LH, Taira K: Explanation by the double-metal-ion mechanism of catalysis for the differential metal ion effects on the cleavage rates of 5′′-oxy and 5′′-thio substrates by a hammerhead ribozyme.Proc Natl Acad Sci USA 1997, 94:14343-14348.
32. Dahm SC, Derrick WB, Uhlenbeck OC: Evidence for the role of solvated metal hydroxide in the hammerhead cleavage mechanism. Biochemistry1993, 32:13040-13045.
33. Scott WG, Murray JB, Arnold JRP, Stoddard BL, Klug A: Capturing the structure of a catalytic RNA intermediate: the hammerhead ribozyme.Science1996, 274:2065-2069.
34. Wedekind JE, McKay DB: Crystallographic structures of the hammerhead ribozyme: relationship to ribozyme folding and catalysis.Annu Rev Biophys Biomol Struct1998, 27:475-502.
35. Murray JB, Terwey DP, Karpeisky A, Usman N, Beigelman L, Scott WG: The structural basis of hammerhead ribozyme self-cleavage. Cell1998, 92:665-673.
36. Torres RA, Bruice TC: The mechanism of phosphodiester hydrolysis: near in-line attack conformations in the hammerhead ribozyme.J Am Chem Soc2000, 122:781-791.
37. Murray JB, Scott WG: Does a single metal ion bridge the A-9 and scissile phosphate groups in the catalytically active hammerhead ribozyme structure?J Mol Biol2000, 296:33-41.
38. Pontius BW, Lott WB, von Hippel PH: Observations on catalysis by hammerhead ribozymes are consistent with a two-divalent-metal-ion mechanism. Proc Natl Acad Sci USA1997, 94:2290-2294.
39. Lott WB, Pontius BW, von Hippel PH: A two-metal-ion mechanism operates in the hammerhead ribozyme-mediated cleavage of an RNA substrate.Proc Natl Acad Sci USA1998, 95:542-547.
40. Nakamatsu Y, Warashina M, Tanaka Y, Yoshinari K, Taira K: Significant
• activity of a modified ribozyme with N7-deazaguanine at G10.1: implication to the double-metal-ion mechanismof catalysis in reactions catalyzed by hammerhead ribozymes.Genes Cells 2000, 5:in press.
A detailed bell-shaped curve for the activation of cleavage by the added La3+
ions is used to explain the double-metal-ion mechanism.
41. Lyne PD, Karplus M: Determination of the pKaof the 2′′-hydroxyl
group of a phosphorylated ribose: implications for the mechanism of hammerhead ribozyme catalysis.J Am Chem Soc2000, 122:166-167.
42. Hampel A, Cowan JA: A unique mechanism for RNA catalysis: the role of metal cofactors in hairpin ribozyme cleavage.Chem Biol 1997, 4:513-517.
43. Nesbitt S, Hegg LA, Fedor MJ: An unusual pH-independent and metal-ion-independent mechanism for hairpin ribozyme catalysis. Chem Biol1997, 4:619-630.
44 Young KJ, Gill F, Grasby JA: Metal ions play a passive role in the hairpin ribozyme-catalysed reaction.Nucleic Acids Res1997, 25:3760-3766.
45. Earnshaw DJ, Gait MJ: Hairpin ribozyme cleavage catalyzed by aminoglycoside antibiotics and the polyamine spermine in the absence of metal ions.Nucleic Acids Res1998, 26:5551-5561.
46. Murray JB, Seyhan AA, Walter NG, Burk JM, Scott WG: The hammerhead, hairpin and VS ribozymes are catalytically proficient in monovalent cations alone. Chem Biol1998, 5:587-595.
![Figure 5mechanism [16•,37], as in the case of the HDV ribozyme. Inthis latter ribozyme, the folded RNA perturbs the pKa of C75and, as a result, C75 can perform effective general-acid catal-](https://thumb-ap.123doks.com/thumbv2/123dok/1036524.929302/8.595.57.301.92.265/figure-mechanism-ribozyme-inthis-ribozyme-perturbs-effective-general.webp)
