8 BAB II
TINJAUAN PUSTAKA 2.1 Uraian Bahan
2.1.1 Parasetamol
2.1.1.1 Sifat Fisika dan Kimia Rumus struktur
Gambar 2.1 Parasetamol (Sweetman, 2009).
Menurut Dijen BKAK., (2014) dan Moffat, dkk., (2011), uraian tentang parasetamol adalah sebagai berikut:
Rumus molekul : C8H9NO2 Berat Molekul : 151,16
Nama IUPAC : N-(4-Hydroxyphenyl)acetamide
Sinonim : Acetaminophen dan N-acetyl-p-aminophenol
Pemerian : Serbuk hablur, kristal putih, tidak berbau, sedikit pahit. Kelarutan : Larut dalam air mendidih; natrium hidroksida, etanol,
methanol, dimetilformamida, etilene diklorida, aseton dan etil asetat; kloroform; kurang larut dalam eter; air dingin. Titik lebur : 169 – 172 0C
pKa : 9,5
2.1.1.2 Farmakologi
9
metabolit aktif dari acetanilide dan fenasetin. Parasetamol dan fenasetin memiliki efek analgetik dan antipiretik. Efek antiinflamasi yang dimilikinya lemah. Parasetamol memiliki kemampuan yang lemah dalam menghambat biosintesis prostaglandin (Goodman Gilmann, dkk., 2001).
Parasetamol dimetbolisme oleh enzim mikrosomal hati. Parasetamol cepat dan hampir semua diabsorbsi dalam saluran cerna. Konsentrasi meningkat dalam plasma setelah 30-60 menit dan waktu paruh kira - kira 2 jam, hanya 20-50% konsentrasi terikat yang menyebabkan intoksikasi akut. Setelah dosisis terapi diberikan 90-100% akan terdapat urin pada hari pertama. Setelah konjugasi hati dengan asam glukoronat (35%), sistein (60%), asam sulfat (3%); sejumlah kecil akan dihidroksilasi dan diasetilasi (Goodman Gilman, dkk., 2001).
Parasetamol diberikan secara oral atau sebagai supositoria rektal untuk nyeri ringan hingga sedang dan untuk demam. Hal ini juga dapat diberikan melalui infus intravena untuk pengobatan jangka pendek nyeri sedang, terutama setelah operasi, dan demam. Parasetamol sering digunakan sebagai analgesik antipiretik pilihan (Sweetman, 2009).
Parasetamol mudah diserap dari saluran pencernaan yang saluran dengan konsentrasi puncak plasma terjadi sekitar 10 sampai 60 menit setelah dosis oral. Eliminasi paruh parasetamol bervariasi dari sekitar 1 sampai 3 jam. Parasetamol dimetabolisme terutama di hati dan diekskresikan dalam urin terutama sebagai glukuronida dan sulfat konjugat (Sweetman, 2009).
10
didetoksifikasi melalui konjugasi dengan glutation tapi mungkin menumpuk setelah overdosis parasetamol dan menyebabkan kerusakan jaringan (Sweetman, 2009).
Efek samping dari parasetamol jarang terjadi dan biasanya ringan, meskipun reaksi hematologi termasuk trombositopenia, leukopenia, pansitopenia, neutropenia, dan agranulositosis telah dilaporkan. Kulit ruam dan reaksi hipersensitivitas lainnya sering terjadi. Hipotensi telah dilaporkan jarang dengan penggunaan parenteral (Sweetman, 2009).
Overdosis dengan parasetamol dapat menyebabkan kerusakan hati yang parah hingga nekrosis. Asetilasi gugus amino dari para-aminofenol akan menurunkan toksisitasnya, pada dosis terapi relatif aman tetapi pada dosis yang lebih besar dan pemakaian jangka panjang dapat menyebabkan methemoglobin dan kerusakan hati. Hepatotoksisitas untuk anak setelah pemberian dosis tunggal 10-15g (150-250 mg/kg berat badan) sedangkan dosis 20-25 g akan berakibat fatal. Pemberian acetylcystein intravena dan oral dengan dosis 140 mg/kg BB untuk pasien yang memiliki gagal hati fulminan telah terbukti mengurangi morbiditas dan mortalitas (Sweetman, 2009; Goodman Gilman, dkk., 2001). 2.1.1.3 Analisis Kualitatif dan Kuantitatif
11
kalium dikromat (100g/L); perlahan – lahan akan terbentuk larutan violet hingga kemerahan (WHO, 1986).
Analisis kualitatif parasetamol juga dapat dilakukan dengan metode spektroskopi inframerah. Hal ini diamati dengan memperhatikan puncak utamanya pada beberapa bilangan gelombang 1506, 1657, 1565, 1263, 1227, 1612 cm-1 (Moffat, dkk., 2011). Selain itu, identifikasi juga dapat dilakukan dengan metode kromatografi lapis tipis. Dalam hal ini fase diam yang digunakan adalah lempeng KLT silika gel F254 berukuran 3x10 cm, fase gerak kloroform: etil asetat (6:4). Deteksi bercak dilakukan dengan pengamatan di bawah lampu UV 254 nm dan 365 nm serta dengan direaksikan dengan FeCl3. Bercak yang muncul dihitung nilai Rf dan dibandingkan antara Rf bercak sampel dan Rf baku parasetamol (Gandjar dan Rohman, 2008).
Metode spektrofotometri UV-Vis dapat digunakan untuk analisis kualitatif parasetamol. Bentuk spektra senyawa dapat digunakan sebagai informasi untuk analisis kualitatif. Karena transisi yang diperbolehkan untuk suatu molekul dengan struktur kimia berbeda tidak sama sehingga spektra absorpsinya juga berbeda (Gandjar dan Rohman, 2008).
Penetapan kadar parasetamol dapat dilakukan secara: titrimetri dengan metode diazotasi; spektrofotometri UV-Vis; dan teknik kromatografi. Analisis kuantitatif Parasetamol secara titrimetri dengan metode nitrimetri (diazotasi) dan juga titrasi dengan N,N-dibromodimetilhidantoin (Gandjar dan Rohman, 2�08).
12
dan metilen biru , pengamatan titik akhir titrasi tercapai jika muncul warna biru segera pada kertas kanji iodida. Metode titrimetri dapat juga dilakukan dengan N,N-dibromodimetilhidantoin (Gandjar dan Rohman, 2�08).
Penetapan kadar parasetamol secara spektrofotometri UV dapat dilakukan karena parasetamol mempunyai gugus kromofor yang mampu menyerap sinar UV. Metode analisis dengan spektrofotometri ultraviolet dilakukan pada panjang gelombang 245 nm (A 1%, 1 cm dalam larutan asam= 668a) dan 257 nm (A 1%, 1 cm dalam larutan alkali = 715a). Dua metode spektrofotometri UV yakni dengan spektra derivatif dan berdasarkan pada metode Vierordt’s telah digunakan untuk analisis parasetamol. Spektrofotometri visible meggunakan metode Bratton-Marshall, metode ammonium molibdat, dan metode natrium 1,2-naftalkuinon-4-sulfonat (Moffat, dkk., 2011; Ditjen BKAK. 2014; Sudjadi dan Rohman, 2�12).
Metode spektrofluorometri dengan batas deteksi yang rendah telah diusulkan untuk penetapan kadar parasetamol. Metode kromatografi juga digunakan untuk penetapan kadar parasetamol khususnya parasetamol yg bercampur dengan bahan obat lain. Metode kromatografi yang digunakan adalah Metode kromatografi lapis tipis (KLT-densitometri); kromatografi cair kinerja tinggi; dan elektroforesis kapiler (Moffat, dkk., 2011; Ditjen BKAK. 2014; Sudjadi dan Rohman, 2�12).
2.1.2 Fenilpropanolamin HCl 2.1.2.1 Sifat Fisika dan Kimia
Rumus Struktur
13
Menurut Moffat, dkk., (2011), uraian tentang fenilpropanolamin hidroklorida adalah sebagai berikut:
Rumus Molekul: C9H13NO. HCl Berat Molekul : 187.7
Titik lebur : 191 – 196 0C pH dan pKa : 4.5 – 6 dan 9.4
Pemerian : Serbuk hablur; putih; kristal putih higga putih kuning gading; tidak berbau; rasa sedikit pahit.
Kelarutan : larut dalam air ; etanol; praktis tidak larut dalam kloroform dan eter.
2.1.2.2 Farmakologi
Fenilpropanolamin adalah simpatomimetik terutama tidak langsung bertindak dengan tindakan yang sama dengan efedrin tapi kurang aktif sebagai stimulan SSP. Fenilpropanolamin telah diberikan secara oral sebagai hidroklorida untuk pengobatan hidung tersumbat (dekongestan). Sering digunakan dalam sediaan kombinasi untuk mengobati batuk dan demam. Fenilpropanolamin telah digunakan untuk menekan nafsu makan dalam pengelolaan obesitas, tetapi penggunaan ini tidak lagi dianjurkan (Sweetman, 2009).
Puncak konsentrasi rata-rata dalam plasma sekitar 0,08 mg / L tercapai sekitar 2 jam. Kematian akibat overdosis fenilpropanolamin, konsentrasi jaringan yang menyebabkan kematian: darah 48 mg/L, otak 86 mg/g, hati 460 mg/g. Waktu Paruh sekitar 4 jam (Moffat, dkk., 2011).
14
penglihatan kabur, pusing, gelisah, agitasi, tremor, kebingungan, dan hipersensitivitas reaksi. Reaksi lain termasuk krisis hipertensi dengan hipertensi ensefalopati, kejang, aritmia, psikosis, dan nekrosis tubular akut. Selanjutnya, setelah studi kasus-kontrol yang besar di Amerika Serikat yang menemukan peningkatan risiko stroke perdarahan yang berhubungan dengan penggunaan sediaan yang mengandung fenilpropanolamin (khususnya pada wanita yang menggunakan fenilpropanolamin sebagai penekan nafsu makan) (Sweetman, 2009).
2.1.2.3 Analisa Kualitatif dan Kuantitatif
Analisa Kualitatif fenilpropanolamin HCl yang dilakukan oleh Naid (2011) dilakuka melalui reaksi pembentukan warna. Hal ini dilakukan dengan cara melarutkan sampel dengan metanol, disaring, diuapkan kemudian ditambahkan dengan larutan FeCl3. Hasil positif ditunjukkan dengan terbentuknya endapan biru keunguan. Identifikasi juga dapat dilakukan dengan metode spektroskopi inframerah dimana zat yang telah didispersikan dalam kalium bromida P menunjukkan maksimum pada bilangan gelombang yang sama seperti pada fenilpropanolamin hidroklorida BPFI (Ditjen BKAK., 2014). Melalui Metode spektroskopi inframerah puncak utama akan terlihat pada bilangan gelombang 700, 746, 1030, 1500, 1055, 1590 cm-1 (Moffat, dkk., 2011).
15
Fenilpropanolamin HCl dapat ditetapkan kadarnya dengan beberapa cara yaitu titrasi bebas air (Gandjar dan Rohman, 2008), kromatografi cair kinerja tinggi seperti yang telah dilakukan oleh Dowse. dkk., (1982); kromatografi lapis tipis; kromatografi gas; spektrofotometri ultraviolet pada panjang gelombang 251 nm ,257 nm; 262 nm (A 1%, 1 cm dalam larutan asam = 11,7a) tidak dapat dianalisis dalam larutan alkali (Ditjen BKAK. 2014; Moffat, dkk., 2011).
Penetapan kadar fenilpropanolamin hidroklorida dapat dilakukan dengan metode titrasi argentometri. Argentometri merupakan metode umum untuk menetapkan kadar halogenida dan senyawa-senyawa lain yang membentuk endapan dengan perak nitrat (AgNO3) pada suasana tertentu. Larutan baku sekunder yang digunakan adalah AgNO3 (Gandjar dan Rohman, 2008).
Terdapat beberapa metode titrasi argentometri, yaitu : Metode Metode Guy Lussac (cara kekeruhan); Metode Mohr (pembentukan endapan berwarna pada titik akhir); Metode Fajans (adsorpsi indikator pada endapan); Metode Volhard (terbentuknya kompleks berwarna yang larut pada titik akhir) (Gandjar dan Rohman, 2008).
2.3 Spektrofotometri UV-Vis 2.3.1 Penyerapan Radiasi
16
Sinar ultraviolet dan sinar tampak memberikan energi yang cukup untuk terjadinya transisi elektronik. Keadaan energi yang paling rendah disebut dengan keadaan dasar (ground state). Transisi elektronik akan meningkatkan energi molekuler dari keadaan dasar ke satu atau lebih tingkat energi tereksitasi. Jika suatu molekul sederhana dikenakan radiasi elektromagnetik maka molekul tersebut akan menyerap radiasi elektromagnetik yang energinya sesuai. Interaksi ini akan meningkatkan energi potensial elektron pada tingkat keadaan tereksitasi. Penyerapan sinar UV dan sinar tampak pada umumnya dihasilkan oleh eksitasi elektron- elektron ikatan (Gandjar dan Rohman, 2008).
Eksitasi menyebabkan terbentuknya pita spektrum. Penyerapan radiasi dibatasi oleh sejumlah gugus fungsional yang disebut kromofor. Elektron yang terlibat pada penyerapan radiasi UV-Vis ini ada tiga, yaitu elektron sigma, phi , dan elektron bukan ikatan atau non bonding electron (Gandjar dan Rohman, 2008).
Terdapat berbagai faktor yang mengatur pengukuran serapan UV-Vis yakni: adanya gugus- gugus penyerap (kromofor), pengaruh pelarut, pengaruh suhu, ion-ion anorganik, dan pengaruh PH. Kromofor merupakan semua gugus atau atom dalam senyawa organik yang mampu menyerap sinar ultraviolet dan sinar tampak. Kromofor yang paling banyak ditemukan dalam molekul obat adalah cincin benzene (Gandjar dan Rohman, 2012).
17
lebih besar (pergeseran batokromik) dan kadang –kadang disertai dengan peningkatan intensitas (efek hiperkromik ) (Gandjar dan Rohman, 2012).
2.3.2 Hukum Lambert-Beer
Menurut Hukum Lambert, serapan berbanding lurus terhadap ketebalan sel yang disinari. Sedangkan menurut Beer, serapan berbanding lurus dengan konsentrasi. Kedua pernyataan ini dapat dijadikan satu dalam Hukum Lambert-Beer, sehingga diperoleh bahwa serapan berbanding lurus terhadap konsentrasi dan ketebalan sel, yang dapat ditulis dengan persamaan :
A= a.b.c (g/liter) atau A= ε. b. c (mol/liter)
Dimana: A = serapan a = absorptivitas b = ketebalan sel c = konsentrasi
ε = absorptivitas molar (Gandjar dan Rohman, 2012).
Hukum Lambert-Beer menjadi dasar aspek kuantitatif spektrofotometri dimana konsentrasi dapat dihitung berdasarkan rumus di atas. Absorptivitas merupakan suatu tetapan dan spesifik untuk setiap molekul pada panjang gelombang dan pelarut tertentu. Penggunaan utama spektrofotometri ultraviolet adalah dalam analisis kuantitatif. Apabila dalam alur spektrofotometer terdapat senyawa yang mengabsorpsi radiasi, akan terjadi pengurangan kekuatan radiasi yang mencapai detektor (Gandjar dan Rohman, 2012).
18
fotometer sebagai alat pengukur intensitas cahaya yang ditransmisikan atau yang diabsorpsi (Gandjar dan Rohman, 2012).
2.3.3 Komponen spektrofotometer Ultraviolet
Spektrofotometer yang sesuai untuk pengukuran di daerah spektrum ultraviolet dan sinar tampak terdiri atas suatu system optik dengan kemampuan mengahasilkan sinar monokromatis. Komponen – komponennya meliputi:
1. Sumber Sinar
Senyawa- senyawa yang menyerap di spektrum daerah ultraviolet digunakan lampu deuterium. Sedangkan untuk sinar tampak digunakan lampu tungsten. Lampu tungsten mengemisikan sinar pada panjang gelombang 35� -2��� nm, sedangkan lampu deuterium pada panjang gelombang 2��-37� nm (Gandjar dan Rohman, 2012).
2. Monokromator
Kebanyakan pengukuran kuantitatif sinar harus bersifat monokromatik yakni sinar dengan satu panjang gelombang tertentu. Hal ini dicapai dengan melewatkan sinar polikromatik melalui suatu monokromator. Terdapat dua jenis monokromator dalam spektrofotometer modern; yaitu prisma dan kisi difraksi (Gandjar dan Rohman, 2012).
3. Detektor
19 2.4 Spektrofotometri Derivatif
Konsep derivatif telah diperkenalkan pertama kali pada tahun 195�, dimana terlihat memberikan banyak keuntungan. Aplikasi utama spektrofotometri derivatif UV – Vis adalah untuk identifikasi kualitatif dan analisis senyawa dalam sampel. Metode spektrofotometri derivatif sangat cocok untuk analisis pita absorpsi yang tumpang tindih (Nurhidayati, 2��7).
Spektrofotometri derivatif merupakan suatu metode manipulatif terhadap
spektra pada spektrofotometri UV-Vis. Metode spektrofotometri derivatif atau metode kurva turunan adalah salah satu metode spektrofotometri yang dapat digunakan untuk analisis campuran beberapa zat secara langsung, tanpa harus melakukan pemisahan terlebih dahulu walaupun dengan panjang gelombang yang berdekatan. Pada spektrofotometri konvensional (derivat kenol), spektrum serapan
20
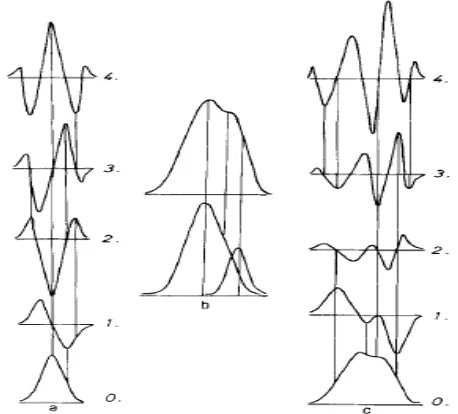
Gambar 2.3 Spektrum derivat pertama sampai derivat keempat (Talsky, 1994).
2.4.1 Metode Spektrofotometri Derivatif
Metode spektrofotometri derivatif yang sering digunakan dalam anlisa kuantitatif antara lain: metode zero crossing; metode peak to peak; dan metode tangen (Talsky, 1994). Metode zero-crossing adalah prosedur yang paling umum untuk menentukan campuran biner yang spektranya saling tumpang tindih dapat dilihat pada Gambar 2.4.
Gambar 2.4 Aplikasi sederhana teknik zero crossing (O’Haver, 1979)
21
gelombang zero-crossing lebih dari satu, maka yang dipilih untuk dijadikan panjang gelombang analisis adalah panjang gelombang zero-crossing yang serapan pasangannya dan campurannya persis sama, karena pada panjang gelombang tersebut dapat secara selektif mengukur serapan senyawa pasangannya dan memiliki serapan yang paling besar. Pada serapan yang paling besar, serapannya lebih stabil sehingga kesalahan analisis dapat diperkecil (Nurhidayati, 2��7).
Metode yang lain adalah derivatif quotient spectra atau rasio spektra derivatif. Metode ini berdasarkan pada pembagian spektrum campuran menjadi spektrum standard setiap analisis dan mengarahkan quotient untuk mendapatkan spektrum yang tidak tergantung pada konsentrasi analit yang digunakan sebagai pembagi. Bila dibandingkan dengan metode zero-crossing, pengukuran menggunakan rasio spektra derivatif lebih mudah dan sinyal analit lebih tinggi. Di sampingitu, adanya maksimum dan minimum pada rasio spektra derivatif memberikan kemungkinan untuk penentuan kadar komponen-komponen tersebut bila terdapat komponen aktif dan eksipien lain yang mempengaruhi penetapan kadar (Nurhidayati, 2��7).
2.4.2 Penggunaan Spektrofotometri Derivatif
22
zat anorganik, analisis makanan, analisis komponen biologi misalnya protein dan asam amino, analisis kandungan dalam sediaan farmasetika, serta analisis stabilitas obat-obatan (Ojeda dan Rojas, 2013; Talsky, 1994).
2.4.3 Komponen Spektrofotometer Derivatif
Komponen-komponen pada spektrofotometer UV-Vis biasa sama dengan komponen pada spektrofotometer derivatif. Alat spektrofotometer harus dilengkapi dengan peralatan sedemikian rupa untuk dapat menghasilkan spektrum derivatif (Ditjen POM., 1995). Biasanya spektrofotometer telah mempunyai software untuk mengolah data yang dapat dioperasikan malalui komputer yang telah terhubung dengan spektrofotometer (Moffat, dkk., 2011).
Spektrum derivatif dihasilkan oleh spektrofotometer yang dirancang untuk
melakukan transformasi elektronik. Derivatif dA/dλ didekati dengan
meningkatnya rasio ΔA/Δλ, di mana ΔA adalah perubahan serapan terhadap
perbedaan Δλ. Makin kecil Δλ, ΔA/Δλ makin mendekati derivatif sesungguhnya
dA/dλ. Mode derivatif pertama dan kedua adalah fitur standar microprocessor
spektrofotometer UV-Vis dan beberapa instrumen dilengkapi dengan mode derivatif ketiga, keempat, kelima sampai dengan ketujuh. Perangkat lunak komputer juga tersedia untuk mengolah data spektra UV-Vis sampai dengan derivat kesembilan (O’Haver, 1979).
2.5 Validasi Metode Analisis
23
dilakukan untuk menjamin bahwa setiap pengukuran serupa yang dilakukan di masa yang akan datang akan menghasilkan nilai terhitung (calculated value) yang cukup dekat atau sama dengan nilai sebenarnya dari jumlah analit yang terdapat dalam sampel. Adapun karakteristik dalam validasi yaitu akurasi/kecermatan, presisi/keseksamaan, spesifisitas, batas deteksi, batas kuantitasi, linieritas, rentang, kekasaran dan ketahanan (robutness) (Gandjar dan Rohman, 2012).
2.5.1 Akurasi (Kecermatan)
Akurasi adalah kedekatan antara nilai hasil uji yang diperoleh melalui metode analitik dengan nilai sebenarnya. Untuk pengujian senyawa obat akurasi diperoleh dengan membandingkan hasil pengukuran dengan bahan rujukan standar (stadar reference material, SRM). Akurasi dinyatakan dalam persen perolehan kembali (% recovery). Akurasi dapat ditentukan dengan tiga cara yaitu: (1) membandingkan hasil analisis dengan CRM (certified reference material) dari organisasi standar internasional; (2) spiked – placebo recovery; dan (3) standard addition method (Gandjar dan Rohman, 2012).
Placebo recovery atau metode simulasi, analit murni ditambahkan (spiked)
24 2.5.2 Presisi (Keseksamaan)
Presisi dari suatu metode analisis adalah derajat kesesuaian di antara masing-masing hasil uji, jika prosedur analisis diterapkan berulang kali pada sejumlah cuplikan yang diambil dari satu sampel homogen. Presisi dinyatakan sebagai deviasi standar atau deviasi standar relatif (koefisien variasi). Presisi dapat diartikan pula sebagai derajat keterulangan dari prosedur analisis pada kondisi kerja normal (Satiadarma, dkk., 2004).
Presisi ditentukan dengan menggunakan sejumlah alikot secukupnya dari satu sampel homogen, agar dapat dihitung secara statistik perkiraan deviasi stadar atau deviasi standar yang sahih. Pada uji tersebut setiap cuplikan mendapatkan perlakuan analisis yang sama, lengkap dan mandiri, mulai dari persiapannya sampai dengan didapatkan hasil akhirnya (Satiadarma, dkk., 2004).
Sesuai dengan ICH, presisi dilakukan pada 3 tingkatan yang berbeda yaitu: keterulangan (repeatibility), presisi antara (intermediate precision), dan ketertiruan (reproducibility). Keterulangan yakni presisi pada kondisi percobaan yang sama (berulang) baik orangnya, peralatannya, tempatnya maupun waktunya. Presisi seringkali diekspresikan dengan SD atau standar deviasi relatif (RSD) dari serangkaian data. Nilai RSD dirumuskan dengan:
RSD =1�� X SD
X
� ; yang mana X� merupakan rata – rata data, dan SD adalah standar deviasi serangkaian data. Sementara nilai SD dihitung dengan rumus:
SD =�Σ(x−X�)2
(N−1�) ; yang mana: X adalah nilai dari masing – masing pengukuran;
�� adalah rata – rata dari pengukuran; N adalah banyaknya data. Biasanya replikasi
25 2.5.3 Spesifisitas
Spesifisitas adalah kemampuan untuk mengukur analit yang dituju secara tepat dan spesifik dengan adanya komponen lain dalam matriks sampel seperti ketidakmurnian, produk degradatif dan komponen matriks. Secara umum, spesifisitas dapat ditunjukkan oleh pendekatan secara langsung maupun tidak langsung. Pendekatan langsung dapat ditunjukkan oleh minimalnya gangguan oleh senyawa lain terhadap hasil analisis misalnya mendapatkan hasil yang sama dengan atau tanpa senyawa pengganggu, resolusi kromatografik yang bagus dan kemurnian puncak (peak purity). Pendekatan tidak langsung adalah lewat pengamatan karakteristik akurasi dari metode tersebut. Bila akurasi metode telah dapat diterima (acceptable) dan valid, maka metode tersebut otomatis telah masuk kriteria sebagai metode yang spesifik (Ermer dan McB Miller, 2005).
2.5.4 Batas Deteksi dan Batas Kuantitasi
Limit deteksi dari suatu metode analisis adalah nilai parameter uji batas yaitu konsentrasi analit terendah yang masih dapat dideteksi. Limit kuantitasi adalah konsentrasi analit terendah dalam sampel yang dapat ditentukan dengan presisi dan akurasi yang dapat diterima pada kondisi eksperimen yang ditentukan. LOD dan LOQ dapat dihitung dengan rumus:
LOD= 3,3(SD/ S) LOQ= 10( SD/S) Dimana: SD : standard deviasi
S : kemiringan (slope) (Gandjar dan Rohman, 2012; Satiadarma, dkk., 2004).
2.5.5 Linearitas
26
diberikan. Linieritas dapat ditentukan secara langsung dengan pengukuran sampel (analit) yang ditambahkan baku pada sekurang-kurangnya lima titik konsentrasi yang mencakup seluruh rentang konsentrasi kerja (Ermer dan McB Miller, 2005).
Linearitas paling baik dievaluasi dengan pengamatan visual terhadap suatu plot yang menyatakan hubungan antara fungsi konsentrasi analit dengan signal yang diukur (absorbansi, luas puncak, tinggi puncak, area di bawah kurva dsb). Pada uji linearitas, paling tidak 6 konsentrasi yang berbeda digunakan pada uji. Pada keadaan normal, linearitas diperoleh ketika nilai koefisien determinasi (r2) ≥ 0,997 dan yang kurang diterima ketika r2 < 0,997 (Gandjar dan Rohman, 2012). 2.5.6 Rentang
Rentang adalah konsentrasi terendah dan tertinggi yang mana suatu metode analitik menunjukkan akurasi, presisi dan linieritas yang mencukupi. Rentang suatu prosedur dapat divalidasi lewat pembuktian bahwa prosedur analitik tersebut mampu memberikan presisi, akurasi dan linieritas yang dapat diterima ketika digunakan untuk menganalisis sampel (Gandjar dan Rohman, 2012).