PASIEN DIABETES MELITUS TIPE 2
TESIS
Disusun untuk kualifikasi mencapai derajat Magister Kesehatan pada Program Studi Magister Kedokteran Keluarga
minat utama Ilmu Biomedik
Oleh:
Didik Supriyadi Kusumo Budoyo S500809124
PROGRAM PASCASARJANA
UNIVERSITAS SEBELAS MARET SURAKARTA 2014
i
PASIEN DIABETES MELITUS TIPE 2
TESIS
Disusun untuk kualifikasi mencapai derajat Magister Kesehatan pada Program Studi Magister Kedokteran Keluarga
minat utama Ilmu Biomedik
Oleh:
Didik Supriyadi Kusumo Budoyo S500809124
PROGRAM PASCASARJANA
UNIVERSITAS SEBELAS MARET SURAKARTA 2014
ii
PENGARUH SIMVASTATIN TERHADAP KADAR TISSUE FACTOR DAN PLASMINOGEN ACTIVATOR INHIBITOR-1 PADA
PASIEN DIABETES MELITUS TIPE 2
TESIS
Oleh:
Didik Supriyadi Kusumo Budoyo S500809124
Komisi Pembimbing
Nama Tanda Tangan Tanggal
Pembimbing I Dr. dr. Sugiarto, SpPD, FINASIM
NIP.196205221989011001 ………... ……… Pembimbing II Dr. dr. Hari Wujoso, SpF, MM
NIP.196210221995031001 ………... ………
Telah dinyatakan memenuhi syarat pada tanggal 14 Agustus 2014
Ketua Program Studi Magister Kedokteran Keluarga Program Pascasarjana UNS
Dr. dr. Hari Wujoso, SpF, MM NIP. 196210221995031001
iii
PENGARUH SIMVASTATIN TERHADAP KADAR TISSUE FACTOR DAN PLASMINOGEN ACTIVATOR INHIBITOR-1
PADA PASIEN DIABETES MELITUS TIPE 2
TESIS
Oleh:
Didik Supriyadi Kusumo Budoyo S500809124
Tim Penguji:
Jabatan Nama Tanda Tangan
Ketua Prof. Dr. Muchsin Doewes, dr, P.Fark, MARS
NIP. 194805311976031001 ………
Sekretaris dr. Suradi Maryono, SpPD-KHOM, FINASIM
NIP. 194708121973107001 ... Anggota Penguji Dr. dr. Sugiarto, SpPD, FINASIM
NIP. 196205221989011001 ... Dr. dr. Hari Wujoso, SpF, MM
NIP. 196210221995031001 ...
Mengetahui,
iv
PERNYATAAN
Nama : Didik Supriyadi Kusumo Budoyo NIM : S 500809124
Menyatakan dengan sesungguhnya bahwa tesis dengan judul “Pengaruh
Simvastatin terhadap Kadar Tissue Factor dan Plasminogen Activator Inhibitor-1 pada Pasien DM Tipe 2” adalah betul-betul karya sendiri. Hal-hal yang bukan karya
saya dalam tesis tersebut diberi tanda sitasi dan ditunjukkan dalam daftar pustaka. Apabila dikemudian hari terbukti pernyataan saya tidak benar maka saya bersedia menerima sangsi akademik berupa pencabutan tesis dan gelar yang saya peroleh dari tesis tersebut.
Surakarta, 24 Juli 2014 Yang membuat pernyataan
Didik Supriyadi Kusumo Budoyo
v
KATA PENGANTAR
Puji syukur Alhamdulillahirabbil'alamin penulis panjatkan ke hadirat Allah SWT atas segala limpahan kasih sayang, rahmat dan hidayahNya sehingga penyusunan tesis yang berjudul Pengaruh Simvastatin terhadap Kadar Tissue Factor dan Plasminogen Activator Inhibitor-1 pada Pasien DM tipe 2 ini dapat terselesaikan. Penelitian ini untuk memenuhi sebagian persyaratan dalam menyelesaikan Program Pendidikan Dokter Spesialis I bidang Ilmu Penyakit Dalam di Fakultas Kedokteran Universitas Sebelas Maret Surakarta.
Pada kesempatan ini penulis mengucapkan terima kasih yang tulus dan penghargaan yang tinggi kepada:
1. Prof. Dr. Ravik Karsidi, M.S, selaku Rektor Universitas Sebelas Maret Surakarta yang telah memberikan kemudahan penulis dalam melaksanakan pendidikan Pasca Sarjana Program Studi Magister Kedokteran Keluarga minat utama Biomedik.
2. Prof. Dr. Ir. Ahmad Yunus, M.S, sebagai Direktur Program Pasca Sarjana Universitas Sebelas Maret Surakarta beserta staf atas kebijakannya yang mendukung dalam penulisan penelitian tesis ini.
3. Dr. Hari Wujoso, dr. SpF. MM, sebagai Ketua Program Studi Magister Kedokteran Keluarga minat utama Ilmu Biomedik sekaligus sebagai pembimbing II yang telah memberikan dorongan dan arahan kepada penulis untuk pelaksanaan serta penulisan tesis ini.
4. Prof. Dr. Muchsin Doewes, dr. PFark, MARS, sebagai Sekretaris Program Studi Magister Kedokteran Keluarga minat utama Ilmu Biomedik sekaligus sebagai Ketua Tim Penguji yang telah memberikan dorongan, masukan dan kritik membangun kepada penulis untuk memperbaiki penulisan tesis ini.
5. R. Basoeki Soetardjo, drg. MMR, sebagai Direktur RSUD Dr. Moewardi beserta seluruh jajaran staf direksi yang telah berkenan dan mengijinkan untuk menjalani program pendidikan PPDS I Ilmu Penyakit Dalam.
vi
6. Dr. dr. Sugiarto, SpPD, FINASIM, selaku pembimbing I yang telah membimbing dan memberi pengarahan dalam penyusunan tesis ini, serta memberi kemudahan dalam menjalani pendidikan PPDS I Ilmu Penyakit Dalam.
7. Prof. Dr. H. Zainal Arifin Adnan, dr. SpPD-KR, FINASIM, selaku Dekan Fakultas Kedokteran Universitas Sebelas Maret Surakarta yang telah memberikan kemudahan dan dukungan kepada penulis selama menjalani pendidikan PPDS I Ilmu Penyakit Dalam.
8. Prof. Dr. HA. Guntur Hermawan, dr. SpPD-KPTI, FINASIM, selaku Kepala Bagian Ilmu Penyakit Dalam FK UNS/RSUD Dr Moewardi yang telah memberikan ijin dan bimbingan sehingga tugas penulisan tesis ini terwujud. 9. Prof. Dr. HM. Bambang Purwanto, dr. SpPD-KGH, FINASIM, selaku Ketua
Program Studi PPDS I Ilmu Penyakit Dalam yang telah mendidik dan memberikan kemudahan penulis dalam melaksanakan pendidikan.
10.dr. Suradi Maryono, SpPD-KHOM, FINASIM, yang telah memberikan ide, kesempatan, bimbingan dan arahan dalam penyusunan tesis ini, serta memberikan kemudahan dalam menjalani pendidikan PPDS I Ilmu Penyakit Dalam.
11.Drs. Sumardi, MM, selaku pembimbing statistik yang telah sabar membimbing dan membrikan pengarahan dalam penyusunan tesis.
12.Seluruh staf pengajar Ilmu Penyakit Dalam FK UNS/RSUD Dr Moewardi Surakarta. Prof. Dr. HA. Guntur Hermawan, dr. SpPD-KPTI, FINASIM; Prof. Dr. Zainal Arifin Adnan, dr. SpPD-KR, FINASIM; Prof. Dr. Djoko Hardiman, dr. SpPD-KEMD, FINASIM; Prof. Dr. HM. Bambang Purwanto, dr. SpPD-KGH, FINASIM; Suradi Maryono, dr. SpPD-KHOM, FINASIM; Sumarmi Soewoto, dr. SpPD-KGER, FINASIM; Tatar Sumandjar, dr. SpPD-KPTI, FINASIM; Tantoro Harmono, dr. SpPD-KGEH, FINASIM; Tri Yuli Pramana, dr. SpPD-KGEH, FINASIM; P. Kusnanto, dr. SpPD-KGEH, FINASIM; Dr. Sugiarto, dr. SpPD, FINASIM; Supriyanto Kartodarsono, dr. SpPD-KEMD, FINASIM; Supriyanto Muktiatmojo, dr. SpPD, FINASIM; Dhani Redhono, dr, SpPD-KPTI, FINASIM; Wachid Putranto, dr. SpPD, FINASIM; Arifin, dr. SpPD, FINASIM; Fatichati B,
vii
dr. SpPD; Agung Susanto, dr. SpPD; Arief Nurudin, dr. SpPD; Agus Joko Susanto, dr. SpPD; Yulyani W, dr. SpPD; Sri Marwanta, dr. SpPD, MKES; Aritantri, dr. SpPD; Bayu Basuki Wijaya, dr. SpPD, MKES; Eva Niamuzisilawati, dr. SpPD, MKES; Evi Nurhayatun, dr. SpPD. MKES; R. Satrio, dr. SpPD. MKES yang telah memberi dorongan, bimbingan dan bantuan dalam segala bentuk sehingga penulis bisa menyelesaikan penyusunan tesis ini.
13.Seluruh teman sejawat Residen Penyakit Dalam yang telah memberikan dukungan dan bantuan kepada penulis baik dalam penelitian ini maupun selama menjalani pendidikan.
14.Perawat Poli Interna yang telah memberikan bantuan selama pengambilan sampel dalam penelitian ini.
15.Laboratorium Klinik Prodia yang telah membantu dalam pengambilan dan pengelolaan sampel penelitian .
16.Istri, anak-anak, orang tua, mertua, saudara dan keponakan yang telah memberikan dorongan baik moril maupun meteriil selama menjalani pendidikan PPDS I Ilmu Penyakit Dalam.
17.Semua pihak yang tidak dapat penulis sebutkan satu-persatu yang telah membantu dalam menjalani maupun dalam penelitian ini.
Penyusun menyadari bahwa dalam penyusunan dan penulisan tesis ini banyak terdapat kekurangan, untuk itu penyusun mohon maaf dan sangat mengharapkan saran dan kritik dalam rangka perbaikan penulisan penelitian tesis ini.
Surakarta, Juli 2014 Penyusun
viii
Didik Supriyadi. 2014. Pengaruh simvastatin terhadap kadar tissue factor dan plasminogen activator inhibitor-1 pada pasien DM tipe 2. TESIS. Pembimbing I: Dr. Sugiarto, dr. SpPD, FINASIM, Pembimbing II: Dr. Hari Wujoso, dr. SpF. MM. Program Studi Kedokteran Keluarga, Program Pascasarjana, Universitas Sebelas Maret Surakarta.
ABSTRAK
Latar Belakang
Penyakit kardiovaskuler bertanggung jawab sekitar 70% kasus kematian, terutama penyakit jantung koroner akibat aterosklerosis dini, yang merupakan penyebab utama morbiditas dan mortalitas penderita DM tipe 2. Trombosis merupakan tahap krusial perkembangan dan progresivitas aterosklerosis serta kejadian kardiovaskuler sehubungan dengan aterosklerosis. Tissue factor (TF) merupakan pemicu koagulasi yang paling kuat sedangkan plasminogen activator inhibitor-1 (PAI-1) merupakan inhibitor fibrinolisis. Pada DM tipe 2 terjadi peningkatan kadar TF dan kadar PAI-1. Simvastatin menunjukkan sifat antitrombosis dengan menekan TF, serta meningkatkan profibrinolisis dengan menekan PAI-1. Tujuan Penelitian
Penelitian ini bertujuan untuk mengetahui pengaruh simvastatin terhadap kadar TF dan PAI-1 pada pasien DM tipe 2.
Metode Penelitian
Metode yang digunakan adalah randomized double blind controlled trial, melibatkan 24 pasien, 12 pasien kelompok kontrol diberikan plasebo dan 12 pasien kelompok perlakuan diberikan simvastatin 20 mg/hari. Penelitian berlangsung selama 6 minggu. Analisis statistik dengan SPSS 17 for windows, signifikan bila p<0,05. Hasil Penelitian
Hasil penelitian menunjukkan, pada kelompok perlakuan didapatkan penurunan kadar TF (pre vs post: 54,69±37,99 pM vs 31,62±1,58 pM; p=0,02) tetapi tidak didapatkan perubahan kadar PAI-1 (pre vs post: 1,05±1,27 U/ml vs 1,29±1,08 U/ml; p=0,07). Pada kelompok kontrol tidak didapatkan perbedaan kadar TF dan PAI-1 sebelum maupun sesudah perlakuan (TF pre vs post: 32,23±3,28 pM vs 34,34 ±7,86 pM, p=0,29; PAI-1 pre vs post: 1,08±1,19 U/ml vs 2,1±2,67 U/ml, p=0,13). Kesimpulan
Simvastatin menurunkan kadar TF tetapi tidak berpengaruh terhadap kadar PAI-1 pada pasien DM tipe 2.
Kata kunci: simvastatin, tissue factor, plasminogen activator inhibitor-1, DM tipe 2
ix
Didik Supriyadi. 2014. The effect of simvastatin on the level of tissue factor and plasminogen activator inhibitor-1 in patients with type 2 DM. THESIS. Supervisor I: Dr. Sugiarto, dr. SpPD, FINASIM, Supervisor II: Dr. Hari Wujoso, dr. SpF. MM. Program Study of Medical Family, Post-graduate Program of Sebelas Maret University Surakarta.
ABSTRACT
Background
Cardiovascular disease is responsible for about 70% cases of death, especially coronary heart disease due to premature atherosclerosis, which is a major cause of morbidity and mortality of patients with type 2 DM. Thrombosis is a crucial stage of development and progression of atherosclerosis and cardiovascular events in relation to atherosclerosis. Tissue factor (TF) is the most powerful trigger of coagulation, while plasminogen activator inhibitor-1 (PAI-1) is an inhibitor of fibrinolysis. The level of TF and PAI-1 increased in type 2 DM. Simvastatin showed antithrombotic properties by pressing TF, as well as improving profibrinolysis by pressing PAI-1. Objectives
This study aims to determine the effect of simvastatin on the level of TF and PAI-1 in patients with type 2 DM.
Methods
The method used was a double blind randomized controlled trial, involving 24 pateints, 12 patients were given placebo in the control group and 12 pateints were given simvastatin 20 mg/day. The study lasted for 6 weeks. Statitical analysis with SPSS 1 for windows, significant if p<0.05.
Results
The results showes that the treatment group obtained a decrease in the level of TF (pre vs post: 54,69±37,99 pM vs 31,62±1,58 pM; p=0,02) but did not change the level of PAI-1 (pre vs post: 1,05±1,27 U/ml vs 1,29±1,08 U/ml; p=0,07). In the control group was not found differences in the level of TF and PAI-1 before and after treatment (TF pre vs post: 32,23±3,28 pM vs 34,34±7,86 pM, p=0,29; PAI-1 pre vs post: 1,08±1,19 U/ml vs 2,1±2,67 U/ml, p=0,13).
Conclusions
Simvastatin decreased TF level but had no effect on PAI-1 level in patients with type 2 DM.
Key words: simvastatin, tissue factor, plasminogen activator inhibitor-1, type 2 DM
x DAFTAR ISI
HALAMAN JUDUL………... i
HALAMAN PENGESAHAN PEMBIMBING………... ii
HALAMAN PENGESAHAN PENGUJI……… iii
PERNYATAAN………... iv
KATA PENGANTAR………. v
ABSTRAK………... viii
DAFTAR ISI………... x
DAFTAR GAMBAR……….. xiv
DAFTAR TABEL………... xv
DAFTAR SINGKATAN……… xvi
BAB I. PENDAHULUAN……….. 1
A. Latar Belakang………... 1
B. Rumusan Masalah………. 3
C. Tujuan Penelitian………... 3
1. Tujuan umum………... 3
2. Tujuan khusus………... 3
D. Manfaat Penelitian……….... 4
1. Manfaat teoritis………. 4
xi
2. Manfaat terapan………... 4
BAB II. TINJAUAN PUSTAKA... 5
A. Kajian Teori………... 5
1. Diabetes Melitus... 5
a. Patogenesis komplikasi diabetes melitus... 6
a.1. Sumber ROS pada DM tipe 2... 7
a.2. Mekanisme ROS mengaktivasi empat mekanisme komplikasi DM... 10
b. Implikasi klinis disfungsi endotel... 13
c. Diabetes melitus, disfungsi endotel dan protrombosis... 14
2. Tissue Factor (TF)………. 17
a. Struktur protein TF... 17
b. Ekspresi TF... 19
3. Plasminogen Activator Inhibitor – 1 (PAI-1)... 23
a. Struktur protein PAI-1………... 23
b. Ekspresi PAI-1……… 24
4. Statin... 26
a. Struktur, sumber dan sifat statin... 26
b. Farmakokinetik dan farmakodinamik statin... 26
c. Efek pleiotrofik statin... 30
d. Efek samping statin... 34
xii
B. Penelitian Relevan………. 36
BAB III. KERANGKA KONSEPTUAL DAN HIPOTESIS………. 39
A. Kerangka Konseptual... 39
B. Hipotesis Penelitian………... 41
BAB IV. METODE PENELITIAN………. 42
A. Tempat dan Waktu……… 42
B. Jenis Penelitian……….. 42
C. Populasi, Sampel dan Teknik Sampling………... 42
1. Populasi sasaran………... 42
2. Populasi sumber………... 42
3. Sampel... 43
4. Teknik pengambilan sampel... 45
D. Identifikasi dan Definisi Opersional Variabel... 45
1. Variabel tergantung... 45
2. Variabel bebas... 46
3. Variabel perancu... 46
4. Definisi operasional variabel... 46
E. Cara Kerja... 47
F. Teknik Analisis Data... 52
G. Alur Penelitian………... 53
xiii
BAB V. HASIL PENELITIAN... 54
A. Karakteristik Subyek Penelitian... 54
B. Pengujian Variabel Utama... 64
C. Efek Samping... 71
BAB VI. PEMBAHASAN... 72
A. Hasil Utama... 72
1. Pengaruh Simvastatin terhadap Kadar TF... 73
2. Pengaruh Simvastatin terhadap Kadar PAI-1... 76
B. Keterbatasan Penelitian... 81
BAB VII. PENUTUP... 83
A. Kesimpulan... 83
B. Saran... 83
DAFTAR PUSTAKA... 84
LAMPIRAN... 91
xiv
DAFTAR GAMBAR
Gambar 2.1 Faktor risiko kardiovaskuler dan disfungsi endotel... 6
Gambar 2.2 Mekanisme terjadi komplikasi DM... 7
Gambar 2.3 Rantai elektron transport di mitokondria... 9
Gambar 2.4 Produksi ROS pada resistensi insulin... 10
Gambar 2.5 Mekanisme ROS mengaktivasi empat mekanisme komplikasi DM... 11
Gambar 2.6 Hubungan hiperglikemia dengan inflamasi... 13
Gambar 2.7 Hubungan inflamasi dengan sistem koagulasi dan fibrinolisis... 15
Gambar 2.8. Imunopatogenesis... 17
Gambar 2.9 Induksi ekspresi dan aktivitas TF... 21
Gambar 2.10 Mekanisme sinyal yang terlibat pada regulasi TF... 22
Gambar 2.11 Efek statin terhadap aktivitas small G-protein... 32
Gambar 2.12 Efek pleiotrofik statin... 33
Gambar 2.13 Sifat antioksidan statin... 34
Gambar 3.1 Kerangka konsep penelitian... 39
Gambar 4.1 Alur penelitian... 53
Gambar 5.1 Perubahan kadar TF sebelum (pre) dan sesudah (post) pada kelompok kontrol dan kelompok perlakuan simvastatin... 67
Gambar 5.2 Perubahan PAI-1 sebelum (pre) dan sesudah (post) pada kelompok kontrol dan kelompok perlakuan simvastatin... 68
xv
DAFTAR TABEL
Tabel 2.1 Karakteristik statin... 30 Tabel 4.1 Definisi operasional variabel...
Tabel 4.2 Pengenceran reagen... 50 Tabel 4.3 Assay mix... 50 Tabel 5.1 Perbandingan variabel karakteristik umur, GDP pre, GDP
post, delta GDP, HbA1c dan BMI kelompok kontrol dan
kelompok perlakuan... 58 Tabel 5.2 Perbandingan karakteristik jenis kelamin, lama sakit,
olahraga, insulin, OAD, hipertensi dan dislipidemia pada
kelompok kontrol dan kelompok perlakuan... 63 Tabel 5.3 Perbandingan Kadar TF dan PAI-1 Sebelum dan Sesudah
Perlakuan pada Kelompok Kontrol………... 65 Tabel 5.4 Perbandingan kadar TF dan PAI-1 sebelum dan sesudah
perlakuan pada kelompok perlakuan... 66
Tabel 5.5 Perbandingan delta TF dan delta PAI-1 pada kelompok
kontrol dan kelompok perlakuan... 70
xvi
DAFTAR SINGKATAN
ADP : Adenosine Diphosphate
AGEs : Advanced Glycation End Products ANG-II : Angiotensin-II
APC : Antigen Processing and Presenting Cell AT-III : Antithrombin-III
CSF : Colony Stimulating Factor
DAG : diacylglycerol
DAMP : Damage Associated Molecular Pattern DM : Diabetes Mellitus
ERK : Extracellular-Signal Regulated Kinase FADH : Flavine Adenine Dinucleotide
FDP : Fibrin Degradation Product FFA : Free Fatty Acid
GAPDH : Glyceraldehide-3-Phosphate Dehydrogenase GLUT-1 : Glucosa Transporter-1
HMG-CoA : 3-Hydroxy-3-Methylglutaryl-Coenzym A ICAM-1 : Intercelluler Adhesion Molecule-1
IFN- : Interferon-
xvii IL-1 : Interleukine-1
JNK : c-Jun Terminal Kinase LRE : LPS Responsive Region
MAPK : Mitogen Activated Protein Kinase MHC II : Major Histocompatibility Complex II mTOR : Mammalian Target of Rapamycin
NA : Nicotinic Acid
NAD : Nicotinamide Adenine Dinucleotide
NADH : Reduced Nicotinamide Adenine Dinucleotide NADPH : Nicotinamide Adenine Dinucleotide Phosphate NF-ĸ : Nuclear Factor-ĸ
NO : Nitric oxide
OATP-2 : Organic Anion Transporter-2 PAI-1 : Plasminogen Activator Inhibitor – 1 PARP : Poly ADP-Ribose Polymerase PDI : Protein Disulfide Isomerase
PERKENI : Perkumpulan Endokrinologi Indonesia PGI-2 : Prostacyclin
PI-3K : Phosphatidil Inositol-3 Kinase PKC : Protein Kinase C
PKC- : Protein Kinase C type
xviii RCL : Reactive Centere Loop ROS : Reactive Oxigen Species SOD : Superoxide Dismutase
sTF : Extracelluler Soluble Form TF TCA cycle : Tricarboxylic Acid Cycle
TF : Tissue Factor
TFPI : Tissue Factor Pathway Inhibitor TGT : Toleransi Glukosa Terganggu TGF- : Tumour Growth Factor- Th1 : T helper type 1
TLR-9 : Toll Like Receptor-9 TNF : Tumour Necrosing Factor t-PA : Tissue Plasminogen Activator
TXA : Thromboxane
u-PA : Urokinase Plasminogen Activator VCAM-1 : Vascular Cell Adhesion Molecule-1 VSMC : Vascular Smooth Muscle Cell vWF : Von Willibrand’s Factor
1 BAB I PENDAHULUAN
A. Latar Belakang Masalah
Diabetes mellitus (DM) dapat digambarkan sebagai kelainan metabolik dengan multipel etiologi yang ditandai adanya hiperglikemia kronik dengan gangguan metabolisme karbohidrat, lemak dan protein akibat defek sekresi insulin, aksi insulin maupun kombinasi keduanya. Prevalensi diabetes di dunia, usia dewasa (antara 20-79 tahun) diperkirakan 6,4%, yaitu sekitar 285 juta penduduk pada tahun 2010 dan diprediksi meningkat menjadi 7,7%, yaitu sekitar 439 juta penduduk pada tahun 2030 (Balasubramaniam et al., 2012). Biro Pusat Statistik memperkirakan pada tahun 2030 nanti akan ada 194 juta penduduk Indonesia yang berusia diatas 20 tahun, dengan asumsi prevalensi DM pada urban (14,7%) dan rural (7,2%), maka diperkirakan terdapat 12 juta penderita diabetes di daerah urban dan 8,1 juta di daerah rural (PERKENI, 2011).
Angka harapan hidup penderita DM menurun hampir delapan tahun disebabkan karena meningkatnya mortalitas (Mohan et al., 2010). Penyakit kardiovaskuler bertanggung jawab sekitar 70% kasus kematian, terutama penyakit jantung koroner akibat aterosklerosis dini, yang merupakan penyebab utama morbiditas dan mortalitas penderita DM tipe 2 (Balasubramaniam et al., 2012). DM tipe 2 berhubungan dengan kejadian aterosklerosis yang dipercepat, kerusakan endotel dan tingginya kecenderungan terjadi komplikasi trombosis seperti penyakit
pembuluh darah perifer, kejadian kardiovaskuler dan stroke (El-Hagracy et al., 2010). Trombosis merupakan tahap krusial perkembangan dan progresivitas aterosklerosis serta kejadian kardiovaskuler sehubungan dengan aterosklerosis. Pemicu trombosis ada dua, yaitu akibat rupturnya plak aterosklerosis sehingga protein prokoagulan terpapar dengan darah yang akan memicu koagulasi darah, dan akibat kontak antara darah dengan endotel yang rusak (Krysiak et al., 2010; El-Hagracy et al., 2010).
Tissue factor merupakan pemicu kaskade koagulasi yang paling kuat dan
didapatkan peningkatan kadarnya pada DM dan sindrom koroner akut (Meerarani et al., 2007) sedangkan PAI-1 merupakan inhibitor fisiologis utama untuk t-PA (tissue
plasminogen activator) dan u-PA (urokinase plasminogen activator) sehingga
menghambat fibrinolisis (Ludwig et al., 2005). Pada DM terjadi peningkatan kadar TF (Zoccai et al., 2003; Alzahrani dan Ajjan, 2010; El-Hagracy et al., 2010) dan peningkatan kadar PAI-1 (Zoccai et al., 2003; Creager et al., 2003; Schneider dan Sobel, 2005; Dunn dan Grant, 2005; Virella dan Virella, 2005; Ludwig et al., 2005; Alzahrani dan Ajjan, 2010; Krysak et al., 2010).
Statin, suatu HMG-CoA (3-hydroxy-3-methylglutaryl-Coenzym A) reduktase inhibitor mempunyai berbagai efek terhadap hemostasis dan fibrinolisis. Statin menunjukkan sifat antitrombosis dengan menekan ekspresi dan aktivitas TF, serta meningkatkan faktor profibrinolisis t-PA dengan menekan sintesis PAI-1 (Mason, 2003).
Penelitian in-vitro mendapatkan hasil yang meyakinkan bahwa simvastatin mampu menurunkan kadar TF dan PAI-1 (Krysak et al., 2003), tetapi penelitian
klinis dengan simvastatin terhadap PAI-1 mendapatkan hasil yang bervariasi, serta belum ada penelitian yang mengukur efek simvastatin terhadap kadar TF dan PAI-1 sekaligus dalam satu penelitian meskipun TF merupakan pemicu utama kaskade koagulasi sedangkan PAI-1 merupakan inhibitor kuat proses fibrinolisis dimana keduanya berperan sinergistik dalam proses trombosis. Berdasarkan kesenjangan tersebut diatas maka disusunlah penelitian ini untuk mengetahui pengaruh simvastatin terhadap kadar TF dan PAI-1 pada pasien DM tipe 2.
B. Rumusan Masalah
1. Apakah simvastatin berpengaruh terhadap kadar TF pada pasien DM tipe 2. 2. Apakah simvastatin berpengaruh terhadap kadar PAI-1 pada pasien DM tipe 2.
C. Tujuan Penelitian 1. Tujuan umum:
Penelitian ini bertujuan untuk mengetahui pengaruh simvastatin terhadap faktor-faktor protrombosis pada pasien DM tipe 2
2. Tujuan khusus:
a. Mengetahui pengaruh simvastatin terhadap kadar TF pada pasien DM tipe 2. b. Mengetahui pangaruh simvastatin terhadap kadar PAI-1 pada pasien DM tipe 2.
D. Manfaat Penelitian 1. Manfaat teoritis:
Mengetahui pengaruh simvastatin terhadap faktor-faktor protrombosis pada pasien DM tipe 2.
2. Manfaat terapan:
Mengetahui pengaruh simvastatin terhadap kadar TF dan PAI-1 pada pasien DM tipe 2, yang mana keduanya bekerja sinergistik terhadap kejadian trombosis yang berperan penting dalam perkembangan progresivitas aterosklerosis dan kejadian klinik sehubungan rupturnya plak aterosklerosis. Bila didapatkan penurunan kadar TF dan PAI-1 pada penelitian ini, maka simvastatin dapat digunakan untuk menekan kejadian penyakit kardiovaskuler yang merupakan penyebab utama morbiditas dan mortalitas pada DM tipe 2.
5 BAB II
TINJAUAN PUSTAKA
A. Kajian Teori 1. Diabetes melitus
Diabetes melitus merupakan suatu kelompok penyakit metabolik dengan karakteristik hiperglikemia kronik dengan gangguan metabolisme karbohidrat, lemak dan protein yang terjadi karena kelainan sekresi insulin, kerja insulin atau kedua-duanya (Bennet dan Knowler, 2006; ADA, 2010). DM tipe 2 merupakan tipe terbanyak, yaitu sekitar 90% total pasien diabetes (Ludwig et al., 2005; Bennet dan Knowler, 2006).
Pandangan tradisional tentang aterosklerosis sebagai akibat patologis deposisi lipid didalam dinding arteri, telah di re-definisi dengan teori yang lebih kompleks dimana disfungsi endotel sebagai pemeran utama (Mannarino dan Pirro, 2008). Disfungsi endotel berperan dalam patogenesis dan manifestasi klinis aterosklerosis, telah dibuktikan berhubungan dengan DM tipe 2 dan resistensi insulin pada penelitian ekperimental dan klinis (Van der Oever et al., 2010; Tabit et al., 2010; Balasubramaniam et al., 2012; Bambang, 2012).
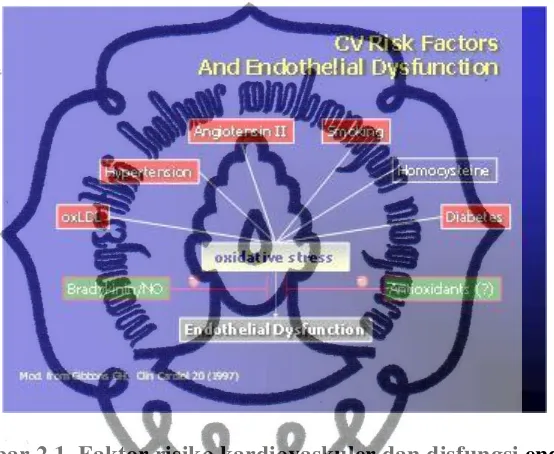
Diabetes melitus tipe 2, tidak hanya didapatkan hiperglikemia saja tetapi juga disertai dislipidemia, resistensi insulin, hipertensi dan obesitas, yang kesemuanya merupakan gambaran sindroma metabolik (Skrha, 2007). Beberapa faktor risiko seperti oxLDL, hipertensi, angiotensin II, merokok, homosistein dan DM dapat
merangsang enzim NADPH (nicotinamide adenine dinucleotide phosphate) oksidase pada mitokondria sehingga akan terjadi stres oksidatif akibat peningkatan ROS (reactive oxygen species) yang akan menyebabkan disfungsi endotel seperti tampak
pada gambar 2.1 (Bambang, 2012).
Gambar 2.1. Faktor risiko kardiovaskuler dan disfungsi endotel. (dikutip dari Bambang, 2012 modifikasi dari Gibbons, 1997).
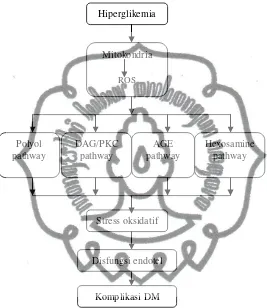
a. Patogenesis komplikasi DM
Empat mekanisme terjadinya komplikasi pada DM; polyol pathway, AGEs pathway, PKC pathway dan hexosamine pathway, bukan merupakan proses yang berjalan sendiri-sendiri akan tetapi suatu kesatuan proses dengan faktor pemicu yang sama yaitu ROS (Brownlee, 2005; Skrha, 2007; Brownlee et al., 2008; Van den Oeven et al., 2010). Produksi ROS yang berlebihan akan menyebabkan ketidakseimbangan antara ROS (oksidan) dengan scavenger system (antioksidan) dan
berlanjut dengan terpicunya empat mekanisme komplikasi DM seperti tampak pada gambar 2.2 (Skrha, 2007; Van den Oeven et al., 2010).
Gambar 2.2. Mekanisme terjadi komplikasi DM. (dikutip dari Van den Oeven et al., 2010).
a.1. Sumber ROS pada DM tipe 2
Peningkatan masukan glukosa ke sel endotel melalui GLUT-1 (glucosa transporter-1) akan menyebabkan meningkatnya metabolisme glukosa sehingga
terjadi hiperaktivasi rantai transport elektron di mitokondria sehingga terjadi Hiperglikemia
Hexosamine pathway AGE
pathway DAG/PKC
pathway Polyol
pathway
Stress oksidatif Mitokondria
ROS
overproduksi ROS (Brownlee, 2005; Skrha, 2007; Brownlee et al., 2008; Van den Oeven et al., 2010; Tabit et al., 2010). Pada kondisi fisiologis, produksi ROS melalui rantai respirasi ini hanya sekitar 5%, yaitu terbentuk O2- (superoksida) (Beltowski,
2005).
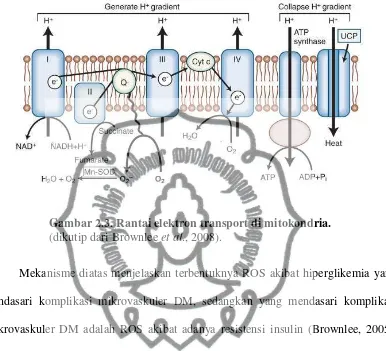
Rantai transport elekron di mitokondria secara sederhana digambarkan sebagai berikut. Ketika glukosa dimetabolisme melalui siklus Kreb (TCA cycle; tricarboxylic acid cycle) akan dihasilkan donor elektron dalam bentuk NADH
(reduced nicotinamide adenine dinucleotide) dan FADH2 (flavine adenine
dinucleotide). Elektron dari NADH masuk ke kompleks I sedangkan FADH2 ke
kompleks II, kemudian berturut-turut ke coenzim Q, kompleks III, sitokrom C, kompleks IV dan terakhir ke molekul O2 yang akan direduksi menjadi air. Rangkaian
reaksi tersebut merupakan pompa proton yang akan menyebabkan terjadinya perbedaan gradien. Perbedaan gradien tersebut menimbulkan energi yang akan memutar ATP sintase sehingga terbentuk ATP (Mayes dan Botham, 2003; Brownlee, 2005; Brownlee et al., 2008).
Pada kondisi hiperglikemia, akan terbentuk lebih banyak NADH dan FADH2
akibat peningkatan metabolisme glukosa melalui siklus Kreb, yang pada titik batas tertentu akan terjadi blokade transport elektron di kompleks III sehingga terjadi penumpukan elektron di coenzim Q. Elektron ini akan direaksikan dengan molekul O2 sehingga terbentuk O2- (superoksida). Bila produksi O2- melebihi kemampuan
SOD (superoxide dismutase) maka akan terbentuk ROS seperti tampak pada gambar
Gambar 2.3. Rantai elektron transport di mitokondria. (dikutip dari Brownlee et al., 2008).
Mekanisme diatas menjelaskan terbentuknya ROS akibat hiperglikemia yang mendasari komplikasi mikrovaskuler DM, sedangkan yang mendasari komplikasi makrovaskuler DM adalah ROS akibat adanya resistensi insulin (Brownlee, 2005). Resistensi insulin menyebabkan peningkatan pelepasan FFA (free fatty acid) dari jaringan lemak. Pada sel endotel makrovaskuler, tidak pada mikrovaskuler, peningkatan FFA akan menyebabkan peningkatan oksidasi FFA di mitokondria. Karena oksidasi asam lemak dan oksidasi asetil CoA yang berasal dari FFA menghasilkan donor elektron yang sama dengan oksidasi glukosa, yaitu NADH dan FADH2 maka peningkatan oksidasi FFA akan menyebabkan peningkatan produksi
Gambar 2.4. Produksi ROS pada resistensi insulin. (dikutip dari Brownlee et al., 2008).
a.2. Mekanisme ROS mengaktivasi empat mekanisme komplikasi DM
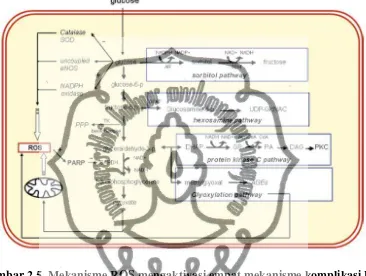
Peningkatan produksi ROS, yaitu O2- (superoxide) pada rantai transport
elektron di mitokondria merupakan kunci aktivasi empat mekanisme komplikasi DM. Produksi O2- akibat hiperglikemia akan menurunkan aktivitas GAPDH
(glyceraldehide-3-phosphate dehydrogenase) sebesar 66% akibat ribosilasi poli ADP
pada GAPDH oleh enzim PARP (poly ADP-ribose polymerase). Enzim PARP ini aktif karena rusaknya DNA akibat ROS (Van den Oever et al., 2010).
Enzim PARP merupakan enzim yang bertugas untuk memperbaiki kerusakan DNA dan akan aktif bila ada kerusakan struktur DNA. Ketika teraktivasi, akan memecah NAD (nicotinamide adenine dinucleotide) menjadi NA (nicotinic acid) dan ADP-ribose (adenosine diphosphate). PARP kemudian memicu polimerisasi ADP-ribose yang akan terakumulasi pada GAPDH sehingga mengganggu aktivitas enzim
ini dalam glikolisis dan berakibat terakumulasinya metabolit glikolisis seperti tampak pada gambar 2.5 (Brownlee et al., 2008).
Gambar 2.5. Mekanisme ROS mengaktivasi empat mekanisme komplikasi DM. (dikutip dari Schalkwijk dan Stehouwer, 2005).
Akumulasi glyceraldehide-3-phosphate akan mengaktivasi AGEs pathway dan PKC pathway karena prekursornya yaitu methylglyoxal (prekursor AGEs) dan diacylglycerol (DAG, prekursor PKC), terbentuk dari glyceraldehide-3-phosphate.
Metabolit yang lebih atas lagi yaitu fructose-6-phosphate akan mengaktivasi hexosamine pathway dan metabolit tertinggi yaitu glukosa akan mengaktivasi polyol
uncouple eNOS serta akan menekan aktivitas enzim katalase dan SOD (superoxide
dismutase). AGEs pathway juga berperan meningkatkan ROS (Schalkwijk dan
Stehouwer, 2005; Van den Oever et al., 2010). Tampak jelas terjadi lingkaran setan produksi ROS pada diabetes melitus.
Reactive oxygen species pada konsentrasi rendah dapat berfungsi sebagai
signal molekul yang berperan pada aktivitas seluler seperti pertumbuhan sel dan respon adaptasi. Pada konsentrasi tinggi akan menyebabkan stres oksidatif, celluler injury dan apoptosis. ROS dapat mempengaruhi banyak jalur signal seluler seperti
G-protein, protein kinase, ion channel dan faktor transkripsi. Pada akhirnya, ROS yang
timbul akibat hiperglikemia dapat menginduksi aktivasi dan disfungsi endotel dengan berbagai mekanisme seperti peroksidasi membran lipid, aktivasi NF-ĸβ dan menurunkan aktivitas NO (Van den Oever et al., 2010).
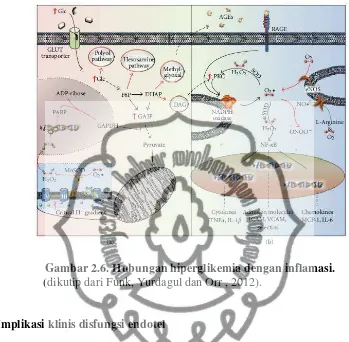
Hiperglikemia mengakibatkan disfungsi metabolik melalui peningkatan produksi superoksida pada mitokondria yang akan mengaktivasi enzim PARP sehingga terjadi penumpukan metabolit glikolisis seperti DAG, metylglyoxal, hexosamine dan polyol pathway (a). Stres oksidatif akibat hiperglikemia akan diperkuat lagi oleh kelebihan produksi DAG dan penurunan NADH+ / reduced gluthatione (GSH) yang akan mengaktivasi reseptor AGE (RAGE). Stres oksidatif
akan menurunkan kemampuan mediator protektif (bioavailabilitas NO) dan akan meningkatkan aktivasi NF-ĸβ sehingga terjadi peningkatan produksi sitokin proinflamasi (b), seperti tampak pada gambar 2.6 (Funk, Yurdagul dan Orr , 2012).
Gambar 2.6. Hubungan hiperglikemia dengan inflamasi. (dikutip dari Funk, Yurdagul dan Orr , 2012).
b. Implikasi klinis disfungsi endotel
Endotel berfungsi mempertahankan homeostasis vaskuler melalui kompleks interaksi multipel antar sel pada lumen dan dinding pembuluh darah. Pertama, endotel mengatur tonus vaskuler dengan menyeimbangkan antara vasodilator dan vasokonstrikor. Kedua, endotel mengontrol blood fluidity and coagulation dengan memproduksi faktor-faktor yang mengatur aktivitas platelet, kaskade koagulasi dan fibrinolisis. Ketiga, endotel mempunyai kemampuan memproduksi sitokin dan molekul adhesi yang mengatur proses inflamasi (Widlansky et al., 2003).
setelah stimulasi, sehingga terjadi perburukan fungsi organ (Van den Oever et al., 2010; Balasubramanian et al., 2012).
Disfungsi endotel akan memicu endotel untuk mengekspresikan sitokin proinflamasi, yaitu TNF-α, IL-1β, IL-6 dan TGF-β1. Bila proses ini tidak terkontrol dalam jangka waktu yang lama akan mengakibatkan aterosklerosis dan komplikasi pada target organ, yaitu ginjal, jantung, pembuluh darah koroner dan serebral (Van den Oever et al., 2010; Bambang, 2012).
c. Diabetes melitus, disfungsi endotel dan protrombosis
Endotel memproduksi molekul protrombosis seperti TF, PAI-1, thromboxane dan vWF (von Willibrand’s factor) dalam kondisi seimbang dengan produksi molekul antitrombosis seperti NO, heparin, tPA dan trombomodulin. Pada DM, keseimbangan tersebut bergeser ke kondisi protrombosis dan antifibrinolisis. Hal ini terjadi akibat menurunnya sinyal melalui PI-3K pathway tetapi tidak terjadi gangguan sinyal yang melalui MAPK pathway yang merupakan ciri khas resistensi insulin pada DM tipe 2. Terjadi juga peningkatan aktivitas NADPH oksidase sehingga produksi superoksida (O2-) meningkat (Balasubramaniam et al., 2012). ROS akan menurunkan
NO dan mengaktivasi NFĸβ sehingga mengaktivasi transkripsi gen untuk produksi
VCAM-1, e-selectin, ICAM, IL-1, IL-6, IL-8, TF, PAI dan iNOS (Van den Oever et al., 2010; Funk, Yurdagul dan Orr, 2012).
Hubungan antara inflamasi dengan koagulasi sangat kompleks, inflamasi akan menggeser keseimbangan hemostasis kearah koagulasi dan jauh dari antikoagulan
sebaliknya koagulasi tidak hanya terbentuk fibrin dan aktivasi trombosit tetapi juga mengakibatkan pengaktifan sel endotel vaskuler yang berperan untuk aktivasi lekosit (Guntur, 2008; Suradi, 2011).
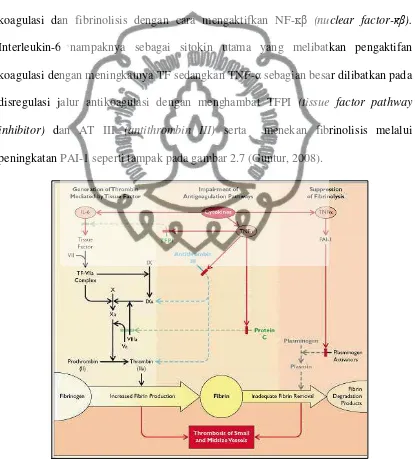
Proses inflamasi secara langsung berhubungan dengan aktivasi sistem koagulasi dan fibrinolisis dengan cara mengaktifkan NF-ĸβ (nuclear factor-ĸ ). Interleukin-6 nampaknya sebagai sitokin utama yang melibatkan pengaktifan koagulasi dengan meningkatnya TF sedangkan TNF-α sebagian besar dilibatkan pada disregulasi jalur antikoagulasi dengan menghambat TFPI (tissue factor pathway inhibitor) dan AT III (antithrombin III) serta menekan fibrinolisis melalui
peningkatan PAI-1 seperti tampak pada gambar 2.7 (Guntur, 2008).
Balasubramaniam et al. (2012) mendukung penjelasan diatas dan menyatakan bahwa protrombosis pada DM akibat adanya disregulasi pada sistem koagulasi dan fibrinolisis, yaitu meningkatnya aktivitas sistem koagulasi yang ditandai dengan peningkatan kadar TF, F VII, trombin, tingginya kadar IL-6 dan fibrinogen, tetapi disisi lain terjadi penurunan aktivitas fibrinolisis akibat peningkatan PAI-1 (Balasubramaniam et al., 2012).
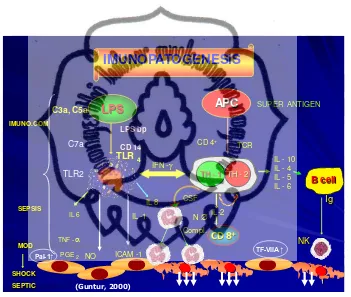
Penjelasan singkat hubungan DM dengan protrombosis dapat dijelaskan dengan imunopatogenesis (Guntur, 2000) sebagai berikut; hiperglikemia bertindak sebagai DAMP (damage associated molecular pattern) akan ditangkap oleh APC (antigen processing and presenting cell) melalui TLR 9 (toll like receptor) dan akan
mempresentasikannya melalui MHC II (major histocompatibility complex II) yang akan menggeser keseimbangan kearah Th1 yang akan memproduksi CSF (colony
stimulating factor) dan IFN- (interferon- ). CSF akan mengaktifkan netrofil,
sedangkan IFN- akan mengaktivasi makrofag mengeluarkan sitokin proinflamasi seperti TNF-α, IL-1, IL-6 dan IL-8. Kondisi inilah yang disebut sebagai low grade inflamation pada DM yang akan menyebabkan disfungsi endotel sehingga terjadi
peningkatan produksi TF dan PAI-1 oleh endotel seperti tampak pada gambar 2.8 (Guntur, 2000).
Diabetes melitus tidak hanya menyebabkan perubahan kuantitas faktor-faktor yang berpengaruh pada koagulasi dan fibrinolisis tetapi juga menyebabkan perubahan kualitas struktur jendalan (clot/trombus) yang terbentuk. Percobaan dengan plasma-purified fibrinogen 150 pasien DM tipe 2 dibandingkan 50 kontrol sehat, ditemukan
jendalan/clot pasien DM lebih padat, ukuran pori-pori lebih kecil, benang fibrin lebih tebal dan percabangan lebih banyak. Hal ini karena terjadi modifikasi post translation pada fibrinogen. Akibatnya jendalan/clot pada DM lebih sulit dilisiskan dibanding kontrol (Alzahrani dan Ajjan, 2010).
1
Tissue factor disebut juga thromboplastine atau factor III; merupakan
asam amino rantai tunggal, diklasifikasikan sebagai CD 142 (reseptor sitokin klas II) dengan 219 asam amino ekstraseluler N-terminus, 23 asam amino transmembran dan 21 asam amino intraseluler C-terminus (Tremoli et al., 1999; Steffel et al., 2006; Monroe, 2010; Breintenstein et al., 2010; Chu, 2011). Regio ekstraseluler mengandung binding domain FVII/VIIa. Extracelluler soluble form (sTF) dapat dilepaskan dari sel endotel sebagai respon terhadap sitokin proinflamasi. Domain intraseluler dapat mengalami serine phosphorylation yang dapat merubah fungsinya, sebagai contoh domain sitoplasmik dapat menekan ekspresi TF akibat tertekannya fosforilasi Erk1/2 ( Chu, 2011).
Gen yang bertanggung jawab untuk produksi TF terletak pada kromosom 1p21-p22, tersusun atas enam exon dan lima intron dengan panjang sekitar 12 kb (kilo basa). Exon pertama untuk bagian promoter (signal peptide), kedua sampai kelima untuk domain ekstraseluler sedangkan exon keenam untuk domain transmembran dan sitoplasmik (Tremoli et al., 1999; Monroe, 2010). Bagian promoter memungkinkan gen ini diatur oleh rangsangan (seperti pada monosit, makrofag, sel endotel) ataupun produksi terus menerus (seperti pada fibroblast, sel epitel). Lima Sp1 untuk produksi basal TF di banyak sel dan dua enhancer didapatkan pada bagian promoter. Proksimal enhancer untuk induksi oleh growth factor dan phorbol ester dan distal enhancer untuk induksi oleh LRE (LPS responsive region) yang terdiri dari dua AP-1 yang berikatan dengan c-fos/c-jun heterodimer dan
satu ĸβ yang akan dikenali oleh c-rel/p65 heterodimer, termasuk famili faktor
NF-ĸ /rel transcription (Tremoli et al., 1999).
Ekspresi TF secara terus-menerus didapatkan pada beberapa sel seperti fibroblas, otot polos dan sel epitel, tetapi pada penelitian kultur sel didapatkan hasil bahwa bagian prmoter TF dapat diinduksi oleh LPS, IL-1β dan TNF-α (Maly et al., 2007). Sitokin proinflamasi ini akan menginduksi fosforilasi IĸBα sehingga terjadi translokasi NF-ĸβ ke nucleus, berikatan dengan urutan gen DNA tertentu sehingga terjadi aktivasi trasnkripsi gen TF (Tremoli et al., 1999).
b. Ekspresi TF
Tissue factor biasanya dalam bentuk inaktif (encrypted) dan akan teraktivasi
menjadi bentuk aktif (crypted) bila ada kerusakan vaskuler (vascular injury) akibat paparan PDI (protein disulfide isomerase) dengan PS (phosphatidylserine). Inflamasi (LPS, ILs, TNF-α, CRP, C pneumoniae), IFN, MCP-1, ICAM, p-selectin, CD40/40L, PDGF, oxLDL, Lp(a), angiotensin II, plasmin, complement anaphylatoxin C5a, antiphospholipid antibody, AGEs dan hipoksia akan mengakibatkan upregulasi
aktivasi TF (Chu, 2011). Secara umum, ekspresi TF diperantarai oleh aktivasi kinase sinyal intraseluler seperti PKC, MAPK (Erk, p38) dan komponen sinyal yang lain seperti faktor transkripsi AP-1, NF-ĸβ, Erg-1 (Tremoli et al., 1999; Steffel et al., 2006; Breintenstein et al., 2010; Chu, 2011).
Downregulasi ekspresi TF bila ada paparan HMG-CoA reduktase inhibitor, cyclooxygenase inhibitor, paclitaxel, phosphatidylcholine, nikotinamide, NO, soluble
guanylate cyclase, hydroxyurea, etil piruvat, DMSO (dimethyl sulfoxide), ACE
agonist, pentoksifilin, indobufen, phenolics/resveratrol derivative, amiodarone, metformin, peningkatan sinyal cAMP, dan PI-3K/Akt/PKB. Short hairpin RNA, miR-19, hairpin ribozyme dan antisense ODN menekan translasi dan ekspresi TF mRNA
(Chu, 2011).
Sel endotel, pada kondisi fisiologis, hanya sedikit sekali mengekspresikan TF. Akan tetapi sitokin seperti TNF-α, IL-1β, CD40 ligand, biogenik amin seperti histamin, serotonin dan mediator seperti trombin, oxLDL serta VEGF dapat menginduksi ekspresi TF bila berikatan dengan reseptornya seperti tampak pada gambar 2.9. Stimulasi ini akan mengaktivasi MAPK (mitogen activated protein kinase) p38, ERK (extracellular-signal regulated kinase) dan JNK (c-jun terminal
kinase) (Steffel et al., 2006; Breintenstein et al., 2010). TNF-α, histamin dan trombin akan mengaktivasi melalui MAPK p38, ERK dan JNK, sedangkan VEGF mengaktivasi melalui MAPK p38 dan ERK. TNF-α dan VEGF juga diketahui mengaktivasi ekspresi TF melalui PKC (Steffel et al., 2006) dan trombin juga mengaktivasi melalui Rho-kinase pathway (Breintenstein et al., 2010). Sinyal transduksi tersebut akan mengaktivasi gen TF pada bagian promoter dengan mengaktivasi faktor transkripsi seperti AP-1, NF-ĸβ dan EGR-1, sehingga terjadi upregulasi TF mRNA (Steffel et al., 2006). Dalam hal aktivasi melalui NF-ĸβ, aktivasi MAPK akan mengakibatkan degradasi protein inhibitor Iĸβ sehingga terjadi
translokasi NF-ĸβ ke nukleus (Breintenstein et al., 2010).
Gambar 2.9. Induksi ekspresi dan aktivitas TF. (dikutip dari Steffel et al., 2006).
Regulasi negatif ekspresi TF dilakukan oleh PI-3K pathway, berbeda dengan MAPK dan PKC yang mengaktivasi ekspresi TF. Stimulasi sel endotel dengan TNF-α, trombin ataupun VEGF akan menginhibisi PI-3K disatu sisi tetapi justru
mengaktivasi MAPK disisi lain, sehingga terjadi peningkatan ekspresi TF. Telah diketahui keterlibatan downstream target PI-3K seperti Akt dan GSK-3β (glycogen synthase kinase-3 ), dimana Akt menghambat aktivasi MAPK sedangkan GSK-3β
mengatur pada tingkat transkripsi gen. Downstream target PI-3K yang lain seperti mTOR (mammalian target of rapamycin) dan p70S6 menghambat pada tingkat translasi TF (Breintenstein et al., 2010).
Gambar 2.10. Mekanisme sinyal yang terlibat pada regulasi TF. (dikutip dari Breintenstein et al., 2010).
Tissue factor tidak hanya ditemukan pada sel vaskuler saja tetapi juga
ditemukan dalam darah, yang disebut circulating atau blood-borne TF (Steffel et al., 2006). Sumber blood-borne TF adalah monosit, eosinofil, platelet, MPs (microparticles) dan asTF (alternative splicing TF). Monosit merupakan sumber
utama blood-borne TF (Bogdanov dan Osterud, 2010; Breintenstein et al., 2010). Vaidyula et al telah membuktikan pada sukarelawan sehat bahwa kombinasi hiperglikemia dan hiperinsulinemia pada kadar seperti DM tipe 2 akan meningkatkan ekspresi TF pada monosit dan meningkatkan interaksi antara monosit dengan platelet. Lebih lanjut juga melaporkan pada pasien DM tipe 2, terjadi peningkatan basal blood-borne TF dan TF mRNA pada monosit (Bogdanov dan Osterud, 2010).
Mediator proinflamasi yang berasal dari Th1 (T helper type 1) seperti IFN- (interferon- ) dan TNF-α akan menginduksi ekspresi TF pada monosit. Transformasi makrofag menjadi sel busa (foam cell) juga akan meningkatkan ekspresi TF. Sedangkan mediator yang berasal dari Th2 seperti IL-4, IL-10 dan IL-13 mencegah Th1 menginduksi ekspresi TF (Breintenstein et al., 2010).
3. Plasminogen Activator Inhibitor – 1 (PAI-1) a. Struktur protein PAI-1
Plasminogen activator inhibitor-1 adalah glikoprotein rantai tunggal dengan
berat molekul ± 50 kDa yang merupakan anggota famili serpin (serine proteinase inhibitor) (Aso, 2007). Bentuk matur yang disekresi terdiri dari 379 asam amino dan
mengandung ± 13% karbohidrat. Pusat reaksi inhibisi PAI-1 terletak pada reactive centere loop (RCL) yang mengandung Arg346 –Met 347 pada carboxy terminus sebagai pseudosubstrat target protease serin (Binder et al., 2002; Hajjar, 2010). Serpin ini aktivitasnya tidak stabil, supaya aktivitasnya stabil maka akan membentuk komplek dengan vitronectin yang merupakan komponen plasma dan matrik periseluler (Hajjar, 2010).
sequence, bertanggung jawab untuk stabilitas mRNA. Urutan dengan akhiran 5’ yang
lebih pendek, mengandung TATA box (transcription initiation site) dan regulatory element, sehingga bagian ini disebut bagian promoter (Binder et al., 2002; Aso,
2007).
b. Ekspresi PAI-1
Ekspresi PAI-1 dapat ditingkatkan pada tingkat transkripsi oleh banyak faktor seperti growth factor dan sitokin (TGF-β1, IL-1, FGF, VEGF), hormon (glukokortikoid, insulin), mediator inflamasi (TNF-α, LPS), metabolit glukosa dan lipid (glukosa, FFA, triglycerol, VLDL), faktor yang mengatur tonus vaskuler (angiotensin II), bahan kimia (phorbol ester), dan faktor lingkungan/fisik (ROS, hipoksia, stres, luka) (Huang dan Lee, 2005). Ekspresi PAI-1 ditekan oleh forskolin dan endothelial growth factor ketika ada heparin (Hajjar, 2010).
Sejumlah elemen pengatur ekspresi gen PAI-1 telah ditemukan pada bagian promoter, diantaranya dua elemen Sp1 (pada -42 dan -73) yang memperantarai respon terhadap glukosa, HIF (hypoxia responsive element, pada -194), VLDL elemen (pada -672/-657), SMAD 3 dan 4 (pada -280, -580, -730) yang memperantarai respon terhadap TGF-β. CCAAT enhancer (pada -226) yang memperantarai upregulasi oleh IL-1 dan IL-6. TNF-α meningkatkan PAI-1 melalui NF-ĸ binding site yang terletak pada -14,7 kb. Polimorfisme 4G/5G (pada -653 ) juga berperan pada
ekspresi PAI-1 meskipun masih diperdebatkan (Kruithof, 2008).
Jalur sinyal transduksi yang berperan dalam respon PAI-1 terhadap inflamasi telah diketahui, yaitu MAPK pathway dan NFĸβ. Stimulus ekstraseluler seperti sitokin proinflamasi, growth factor, TLR (toll like receptor) ligand dan ligand dari G-protein receptor akan mengaktivasi kaskade fosforilasi, yaitu MAP3K akan
mengaktivasi MAP2K yang selanjutnya mengaktivasi MAP kinase melalui MAPK p38, ERK dan c-jun-N-terminal kinase (JNK), yang akan mengaktivasi faktor transkripsi. IL-1 dan LPS mengaktivasi melalui NFĸβ, aktivasi Iĸ-kinase akan memfosforilasi Iĸβ, selanjutnya terjadi pelepasan NFĸβ ke nukleus dibagian
promoter. MAPK dan NFĸβ saling berinteraksi pada beberapa titik seperti MAP3K2, MAPγKγ dan MAPγK7 (TAK1) sehingga mampu memfosforilasi Iĸβ dan
menginduksi pelepasan NFĸβ (Kruithof, 2008).
Plasminogen activator inhibitor-1 plasma berada tiga bentuk molekul yang
berbeda, yaitu aktif, inaktif (cleaved) dan laten. Bentuk aktif berada dalam sirkulasi dengan half life 10 menit, untuk segera dirubah menjadi bentuk laten. PAI-1 menunjukkan variasi mengikuti irama sirkadian dengan konsentrasi puncak pada pagi hari dan segera menurun kadarnya pada siang hari (Aso, 2007). Bentuk aktif mampu berikatan dengan plasminogen activator (PA), baik itu t-PA (tissue type plasminogen activator) ataupun u-PA (urokinase type plasminogen activator) sehingga menjadi
4. Statin
a. Struktur, sumber dan sifat statin
Statin adalah obat yang paling efektif dan paling dapat ditoleransi dengan baik untuk mengatasi peningkatan LDL-C (low-density lipoprotein cholesterol). Obat ini merupakan kompetitif inhibitor HMG-CoA (3-hydroxy-3-methylglutaryl coenzim A) reduktase, enzim yang bertanggung jawab merubah HMG-CoA menjadi mevalonat yang merupakan langkah penentu kecepatan biosintesis kolesterol (Tamargo et al., 2007; Goodman dan Gillman, 2008; Sadowitz et al., 2010). Statin mempunyai struktur mirip HMG-CoA yang dapat menghambat HMG-CoA reduktase dengan cara mengikat sisi aktif enzim tersebut sehingga tidak dapat berikatan dengan substrat aslinya (Tamargo et al., 2007; Yanez et al., 2008). Hal ini akan berakibat HMG-CoA tidak dapat dirubah menjadi mevalonat dan akan terjadi penurunan sintesis kolesterol terutama di hepatosit (Yanez et al., 2008).
Statin berdasarkan sumbernya dibagi menjadi dua, yaitu statin produk alamiah yang berasal dari metabolit jamur, dan produk sintetik. Produk alamiah disebut juga statin generasi pertama memiliki decaline ring/hexahydronaphtalene ring, seperti lovastatin, pravastatin dan simvastatin, sedangkan produk sintetik disebut juga generasi kedua memiliki fluorophenyl group, seperti fluvastatin, atorvastatin dan rosuvastatin (Beltowski, 2005; Tamargo et al., 2007). Produk sintetik mempunyai efek yang lebih kuat, tetapi juga memiliki efek samping yang lebih buruk yaitu rhabdomyolysis (Fenton et al., 2005). Statin generasi ketiga yang merupakan produk
menyebabkan rhabdomyolysis yang fatal (Beltowski, 2005; Fenton et al., 2005), dilaporkan 31 pasien meninggal karena penyakit ginjal akut akibat rhabdomyolysis (Stancu dan Sima, 2001).
Struktur statin dapat dibagi menjadi tiga bagian, yaitu analog HMG-CoA, struktur hydrophobic ring yang berikatan dengan HMG-CoA redukatase dan side ring group yang menentukan solubilitas statin (Sadowitz et al., 2010). Atorvastatin,
fluvastatin, lovastatin, pitavastatin, cerivastatin dan simvastatin adalah lipofilik statin, sedangkan pravastatin dan rosuvastatin adalah hidrofilik statin (Tamargo et al., 2007; Sadowitz et al., 2010). Lipofilik statin dapat dengan mudah menembus membran sel di semua organ, akumulasinya di hepatosit karena difusi pasif, sedangkan akumulasi hidrofilik statin di liver melalui carrier-mediated uptake. Distribusi lipofilik statin jauh lebih luas dibandingkan hidrofilik statin sehingga efek pleiotrofik lipofilik statin lebih banyak dibandingkan hidrofilik statin (Sadowitz et al., 2010).
b. Farmakokinetik dan farmakodinamik statin
masih dalam bentuk inactive lactone sehingga perlu dirubah menjadi bentuk active -hydroxy acid di liver (Goodman dan Gillman, 2008; Stancu dan Sima, 2001).
Seluruh statin yang diserap akan melalui metabolisme pertama di liver, tetapi mekanisme masuk ke liver berbeda-beda. Atorvastatin, pravastatin dan rosuvastatin melalui OATP-2 (organic anion transporter-2), sedangkan bentuk lipofilik lactone dari simvastatin dan lovastatin dengan cara difusi (Goodman dan Gillman, 2008). Liver merupakan organ target statin, persentase dosis statin yang berada di liver sebagai berikut; fluvastatin dan lovastatin > 70%, simvastatin > 80%, pravastatin > 46%, sedangkan atorvastatin dan cerivastatin belum ada data (Stancu dan Sima, 2001). Akibat dari metabolisme pertama di liver menyebabkan bioavailabilitas statin bervariasi antara 5-30% dari dosis yang diberikan. Di plasma, semua statin dan metabolitnya berikatan dengan protein > 95%, kecuali pravastatin dan metabolitnya yang hanya 50% berikatan denga protein plasma (Goodman dan Gillman, 2008).
Konsentrasi statin di plasma setelah pemberian oral mencapai puncak setelah 1-4 jam. Waktu paruh komponen induk 1-4 jam, kecuali atorvastatin dan rosuvastatin yang mencapai 20 jam, yang berperan semakin kuat efek menurunkan kolesterolnya. Biotransformasi statin terjadi di liver dan lebih dari 70% diekskresi melalui liver yang selanjutnya dibuang melalui feses (Goodman dan Gillman, 2008). Rute ekskresi utama melalui empedu setelah mengalami biotransformasi di liver, sebagian kecil di ekskresi melalui ginjal sehingga konsentrasinya akan lebih tinggi pada pasien penyakit ginjal dan perlu dosis yang lebih kecil pada pasien dengan penyakit liver. Kontraindikasi semua statin untuk diberikan pada wanita hamil karena bersifat
teratogenik, tetapi statin tidak mempengaruhi steroidogenesis di adrenal dan gonadal (Knopp, 1999). Karakteristik statin disajikan pada tabel 2.4 berikut ini.
Tabel 2.1. Karakteristik Statin (Knopp, 1999)
Karakteristik Lovastatin Pravastatin Simvastatin Atorvastatin Fluvastatin Dosis Max
2008). Statin juga dapat menurunkan kadar LDL dengan cara menurunkan produksi VLDL di hepar sehingga prekursor LDL (VLDL dan IDL) akan menurun. Mekanisme ini merupakan penyebab penurunan trigliseride akibat statin dan bertanggung jawab pada penurunan sekitar 25% LDL kolesterol pada pasien familial hiperkolesterolemia homozigot yang diterapi dengan 80 mg atorvastatin atau simvastatin (Goodman dan Gillman, 2008).
c. Efek pleiotrofik statin
Statin, selain mempunyai kemampuan menurunkan kadar LDL kolesterol tetapi juga mempunyai efek-efek yang lain. Efek statin selain menurunkan kadar kolesterol seringkali disebut sebagai efek pleiotrofik, yang diambil dari bahasa Yunani; pleio berarti banyak, dan tropos berarti sifat (Kotyla, 2010; Yanez et al., 2008). Efek ini terjadi segera setelah dimulai terapi dan seringkali mendahului efek penurunan kolesterol (Kotyla, 2010).
Mekanisme efek pleiotrofik statin berhubungan dengan inhibisi sintesis
isoprenoid intermediates jalur mevalonat seperti isopentenyl adenosine,
farnesylpyrophosphate dan geranyl-geranyl pyrophosphate. Intermediate ini
berfungsi sebagai pengait protein ke lipid di membran sel (lipid anchors) untuk modifikasi paska translasi sejumlah protein yang terlibat dalam jalur transduksi sinyal intraseluler termasuk heterotrimeric G proteins dan small guanosine-triphosphate (GTP)-binding protein, seperti Ras, Rho dan Rac1 (Tamargo et al., 2007). Small
molecular weight G-protein tersebut terlibat dalam proliferasi sel, diferensiasi,
apoptosis, migrasi, kontraksi dan pengaturan trankripsi gen (McFarlane et al., 2002). Pengaitan (anchoring) small G-protein ke membran sel membutuhkan
phrenylation; Ras membutuhkan farnesylation sedangkan Rho membutuhkan
geranylgeranylation. Small G-protein berada di sitoplasma dalam bentuk inaktif
berikatan dengan GDP (guanosine diphosphate), untuk menjadi aktif membutuhkan phrenylation sehingga GDP menjadi GTP (guanosine triphosphate), kemudian terjadi
translokasi ke membran sel yang akan menimbulkan aktivitas biologisnya (McFarlane et al., 2002). Statin akan menghambat proses phrenylation dengan menghambat pembentukan farnesylation dan geranylgeranylation small G-protein dengan cara menghambat konversi HMG-CoA menjadi mevalonat sehingga tidak terbentuk substrat untuk proses phrenylation seperti tampak pada gambar 2.11 (McFarlane et al., 2002; Paul dan Gahtan, 2003, Wolfrum et al., 2003; Tamargo et al., 2007; Yanez et al., 2008; Kotyla, 2010; Sadowitz et al., 2010).
Isoprenoids penting untuk mempertahankan fluiditas membran, pertumbuhan
dan proliferasi sel, ekspresi gen, assembly cytoskeletal dan motilitas sel, pengambilan lipid dan protein, nuclear transport dan pertahanan host. Efek pleiotrofik statin meliputi memperbaiki disfungsi endotel, modulasi fungsi autonom, stabilisasi plak, antioksidan, antiinflamasi, antitrombotik dan kardioprotektif (Tamargo et al., 2007).
Ras berhubungan dengan migrasi dan proliferasi VSMC serta penumpukan fatty streaks. Berbagai penelitian membuktikan, inhibisi aktivasi Ras akan
Ras terletak diatas (upstream) jalur MAPK sehingga inhibisi Ras maka akan terjadi inhibisi MAPK (Paul dan Gahtan, 2003).
Gambar 2.11. Efek statin terhadap aktivitas small G-protein. (dikutip dari McFarlane et al., 2002).
Rho mempunyai aktivitas biologis yang sangat luas, meliputi pengaturan actin cytoskeleton, migrasi seluler, perkembangan neuronal, morfogenesis, transkripsi gen
dan stabilitas mRNA serta divisi dan adhesi sel. Juga berperan pada struktur dan fungsi vaskuler. Secara singkat, efek Rho terhadap VSMC dan sel endotel adalah proaterogenik (Sadowitz et al., 2010).
Rac mengaktifkan NADPH oksidase pada SMC dan endotel, merupakan sumber utama ROS pada dinding vaskuler. Peningkatan produksi ROS akan berakibat terjadinya disfungsi endotel dan perkembangan aterosklerosis. Wassmann et al melaporkan bahwa atorvastatin menurunkan produksi ROS yang dipicu oleh angiotensin II dan EGF (endothelial growth factor). Lebih lanjut dijelaskan bahwa atorvastatin tersebut menurunkan Rac di membran dan meningkatkan Rac di
sitoplasmik sehingga terjadi penurunan aktivitas NADPH oksidase (Paul dan Gahtan, 2003).
Kureishi et al melaporkan bahwa statin dapat mengaktivasi Akt yang penting dalam metabolisme dan apoptosis. Akt merupakan bagian jalur PI-3K, aktivasi Akt oleh statin akan menghambat apoptosis dan meningkatkan produksi eNOS pada sel endotel (Wolfrum et al., 2003). Aktivasi PI-3K akan merubah keseimbangan kearah antiapoptosis (Bcl-2) sehingga tidak terjadi aktivasi caspase-9 (Wolfrum et al., 2003). Aktivasi Akt mengakibatkan peningkatan ekspresi GLUT-4 (glucose transporter-4) yang akan mengatasi resistensi insulin dan peningkatan produksi eNOS dengan cara melepas ikatan eNOS-caveolin dan mengikatkan eNOS dengan calmodulin (McFarlane et al., 2002). Efek pleiotrofik tersebut dapat dilihat pada gambar 2.12.
Statin mempunyai kemampuan antioksidan sehingga mampu menghambat aktivitas IKK (inhibitor ĸ kinase) dan NF-ĸβ. Akibatnya NF-ĸβ tetap akan terikat dengan IKβ (inhibitor ĸ ) sehingga tidak bisa mengaktivasi target gen dan tidak terjadi produksi sitokin seperti tampak pada gambar 2.13 (Guntur, 2008).
Gambar 2.13. Sifat antioksidan statin. (dikutip dari Guntur, 2008).
d. Efek samping statin
Statin secara umum dapat ditoleransi dengan baik. Efek samping yang paling penting adalah toksisitas ke liver dan otot. Miopati dapat terjadi bila inhibitor sitokrom P-450 atau inhibitor metabolisme statin yang lain diberikan bersamaan dengan statin sehingga terjadi peningkatan kadar statin dalam darah (Stancu dan Sima, 2001). Lovastatin, simvastatin, atorvastatin, dimetabolisme oleh sitokrom P-450 3A4; fluvastatin oleh sitokrom P-P-450 2C9; sedangkan pravastatin tidak melalui sitokrom P-450 tatapi melalui proses sulfation. Obat yang menghambat sitokrom P-450 akan meningkatkan kadar statin sehingga efek samping juga akan meningkat,
sedangkan obat yang menginduksi sitokrom P-450 akan menurunkan kadarnya sehingga menurunkan aktivitas biologisnya (Knopp, 1999).
Hepatotoksisitas terjadi kurang dari 1% pasien yang diberikan statin dosis tinggi dan lebih jarang lagi pada dosis rendah (Knopp, 1999). Hepatotoksisitas berat sangat jarang terjadi, hanya satu kasus persejuta orang pemakai statin pertahun (Goodman dan Gillman, 2008). Bahkan kebanyakan hepatologis sudah tidak menganggap statin menyebabkan hepatotoksis yang signifikan. Para ahli tersebut menyimpulkan peningkatan aminotransferase sehubungan terapi statin bukan merupakan bukti kerusakan liver (Bader, 2010).
Efek samping yang paling signifikan terkait pemakaian statin adalah miopati. Insiden miopati sangat rendah (0,01%) tetapi resiko miopati dan rhabdomyolysis meningkat sesuai dengan peningkatan kadar statin di plasma. Faktor yang menghambat katabolisme statin seperti usia lanjut (>80 tahun), gangguan hepar dan renal, periode perioperatif dan hipotiroidisme akan meningkatkan resiko tersebut. Obat-obat seperti fibrat terutama gemfibrozil, siklosporin, digoxin, warfarin, antibiotik golongan makrolide, mibefradil dan antijamur golongan azole juga meningkatkan resiko miopati (Goodman dan Gillman, 2008).
menyusui sebaiknya dihindari karena keamanannya belum jelas (Goodman dan Gillman, 2008).
B. Penelitian Relevan
Krysiak et al., 2003, melakukan meta-analisis terhadap pemakaian statin dan melaporkan ada empat penelitian dengan simvastatin. (1) Simvastatin 20-40 mg selama 12 bulan pada 30 subyek dengan coronary arterial disease (CAD) dengan kadar kolesterol ≥ 4,0 mmol/L didapatkan tidak didapatkan penurunan signifikan
PAI-1 dan tPA. (2) Simvastatin 20-40 mg selama 2 tahun pada 111 subyek dengan kadar kolesterol total ≥ γ,5 mmol/L dan resiko CAD tinggi justru didapatkan
peningkatan signifikan kadar PAI-1. (3) Simvastatin 20 mg selama 8 minggu pada 16 subyek post menopause dengan hiperkolesterol dan CAD didapatkan penurunan tidak signifikan PAI-1, terjadi penurunan signifikan setelah pemberian terapi pengganti hormon. (4) Simvastatin 20-40 mg selama 14 minggu pada 13 subyek CAD dan kadar LDL > 130 mg/dL didapatkan penurunan tidak signifikan kadar PAI-1. Beragamnya metode penelitian yang dipakai menentukan hasil penelitian seperti pemilihan subyek, dosis, lama pemberian, saat pengambilan sampel dan faktor-faktor seperti kadar trigliseride, ox-LDL, glukosa darah, resistensi insulin serta obesitas (Krysial et al., 2003).
Penelitian pada 63 subyek hiperkolesterol selama 4 dan 12 minggu menggunakan simvastatin 20 mg/hari dan fluvastatin 40 mg/hari didapatkan kecenderungan peningkatan kadar fibrinogen mulai 4 minggu pada kedua kelompok
dan setelah 12 minggu terjadi peningkatan signifikan fibrinogen pada kelompok simvastatin meskipun terdapat penurunan bermakna LDL dan kolesterol total pada kedua kelompok. Efek terhadap fibrinogen tidak berkorelasi dengan penurunan profil lipid dan jenis kelamin. Pada penelitian ini dieksklusi pemakaian antilipidemia, antikoagulan, antitrombotik, ACE inhibitor, CCB dan NSAID dalam 3 bulan sebelum penelitian (Okopien et al., 2004).
Perlakuan dengan simvastatin 20 mg/hari sampai 12 bulan pada 26 subyek DM tipe 2 didapatkan penurunan signifikan fragmen protrombin (F1+2) dan PAI-1 setelah 6 minggu. Kadar A1c, glukosa puasa dan insulin tidak dipengaruhi oleh simvastatin. Terdapat korelasi positif antara PAI-1 dengan trigliseride dan LDL. Pada penelitian ini kadar A1c antara 7-10%, tidak membedakan jenis kelamin, obat antihipertensi tetap dilanjutkan tetapi pemakaian antilipidemia dan kontrasepsi oral dieksklusi (Ludwig et al., 2005).
Penelitian pada 125 subyek dengan risiko tinggi kardiovaskuler non DM selama 12 minggu dengan pioglitazone 30 mg/hari, simvastatin 20 mg/hari dan kombinasi pioglitazone-simvastatin didapatkan penurunan signifikan PAI-1 pada kelompok pioglitazone dan kombinasi pioglitazon-simvastatin. Tidak terdapat penurunan pada kelompok simvastatin. Penelitian ini mengeksklusi pemakaian antilipidemia dalam 1 bulan sebelum penelitian sedangkan antihipertensi dan antitrombotik tidak diekslusi (Hanefeld et al., 2007).
signifikan kadar PAI-1 dan fibrinogen mulai minggu keempat. Penelitian ini mengeksklusi pemakaian antilipidemia, antihipertensi (ACE inhibitor dan CCB), steroid, NSAID dan pemakaian kontrasepsi oral serta terapi pengganti hormon. Simvastatin diminum malam hari sebelum tidur dan pengambilan sampel di pagi hari antara jam 8-9 pagi (Krysiak et al., 2010).
Penelitian ini mengkonfirmasi hasil penelitian-penelitian sebelumnya yang secara klinis hasilnya masih bervariasi tetapi dengan memperbaiki metodologi penelitian seperti membatasi usia yang tidak terlalu muda/tua, memilih subyek laki-laki sehingga tidak dipengaruhi kadar hormonal, simvastatin diminum pada jam 19.00-22.00, kadar A1c > 7%, tidak konsumsi antilipidemia/antitrombotik minimal 1 bulan sebelum penelitian dan pengambilan sampel di pagi hari antara jam 08.00-09.00. Persamaan dengan penelitian sebelumnya yaitu obat antidiabetes (OAD/insulin), antihipertensi tetap dilanjutkan tetapi dikendalikan dengan randomisasi.
Pemakaian simvastatin karena obat ini murah, mudah didapat, bersifat lipofilik sehingga mendukung efek pleiotrofiknya serta merupakan produk alamiah (metabolit jamur) sehingga lebih aman karena efek sampingnya lebih kecil dibanding produk sintetis. ADA 2009 membagi simvastatin menjadi dua berdasar dosisnya, yaitu terapi standar (20 mg) dan terapi agresif (40 mg). Pada penelitian ini dipilih simvastatin 20 mg berdasarkan faktor keamanan karena semakin tinggi dosis statin maka efek sampingnya semakin kuat dan dengan dosis tersebut diyakini sudah memberikan efek seperti yang diharapkan berdasarkan penelitian sebelumnya.
39 BAB III
KERANGKA KONSEP DAN HIPOTESIS
A. Kerangka Konsep
Gambar 3.1. Kerangka konsep penelitian Keterangan:
1. : meningkat (DM) 2. : menurun (simvastatin) 3. : menghambat (simvastatin)
DM tipe 2
AGE pathway
ROS
NF-ĸβ
Low grade inflammtion
Disfungsi endotel
PKC pathway Hexosamine pathway
Polyol pathway
Simvastatin
TF PAI-1