Informasi Dokumen
- Penulis:
- Vincilia Indriyani
- Pengajar:
- Drs. Mulyono, Apt.
- Rita Suhadi, M. Si., Apt.
- Christine Patramurti, M. Si., Apt.
- C. M Ratna Rini Nastiti, S. Si., Apt.
- Yosef Wijoyo, M. Si., Apt.
- Sekolah: Universitas Sanata Dharma
- Mata Pelajaran: Ilmu Farmasi
- Topik: Perbandingan Bioavailabilitas Antara Tablet Biogesic Dan Tablet Pamol Dengan Tablet Parasetamol Generik Pada Kelinci Putih Jantan
- Tipe: Skripsi
- Tahun: 2007
- Kota: Yogyakarta
Ringkasan Dokumen
I. PENGANTAR
Bagian ini menjelaskan latar belakang penelitian mengenai bioavailabilitas obat generik dan obat bermerek dagang. Penelitian ini penting karena obat yang beredar di masyarakat harus terjamin mutu, khasiat, dan keamanannya. Penelitian ini bertujuan untuk membandingkan bioavailabilitas tablet Biogesic dan Pamol dengan tablet parasetamol generik pada kelinci putih jantan. Hal ini relevan dengan tujuan pendidikan di bidang farmasi, di mana mahasiswa diharapkan memahami perbedaan antara obat generik dan bermerek serta pentingnya penelitian farmakokinetika.
1.1. Latar Belakang
Latar belakang penelitian ini menyoroti tingginya permintaan masyarakat akan obat, yang mendorong perkembangan industri farmasi. Penelitian ini mencoba menjawab pertanyaan apakah tablet Biogesic dan Pamol memiliki bioavailabilitas yang sama dengan tablet parasetamol generik, sehingga memberikan wawasan tentang kualitas dan efektivitas obat yang beredar.
1.2. Tujuan Penelitian
Tujuan umum dari penelitian ini adalah untuk mengetahui bioavailabilitas tablet parasetamol generik, Biogesic, dan Pamol. Tujuan khususnya adalah untuk mengetahui perbedaan bioavailabilitas yang bermakna antara kedua jenis tablet tersebut. Hal ini penting untuk membantu mahasiswa memahami bagaimana penelitian dapat memberikan informasi yang berguna dalam pengembangan obat.
II. PENELAAHAN PUSTAKA
Bagian ini mencakup teori-teori dasar tentang nasib obat di dalam tubuh, fase farmakokinetika, dan bioekivalensi. Pemahaman tentang fase-fase ini sangat penting dalam pendidikan farmasi, karena mahasiswa perlu memahami bagaimana obat berinteraksi dalam tubuh dan bagaimana bioavailabilitas dapat mempengaruhi efektivitas obat.
2.1. Nasib Obat di Dalam Tubuh
Dalam bagian ini, dijelaskan tentang fase farmasetik, farmakokinetika, dan farmakodinamika. Pemahaman ini penting bagi mahasiswa farmasi untuk menganalisis bagaimana obat diserap, didistribusikan, dimetabolisme, dan diekskresi dalam tubuh, yang merupakan dasar dari studi farmakokinetika.
2.2. Fase Farmakokinetika
Fase farmakokinetika mencakup absorpsi, distribusi, metabolisme, dan ekskresi obat. Mahasiswa perlu memahami proses ini untuk dapat mengevaluasi efektivitas dan keamanan obat dalam terapi klinis. Pengetahuan ini juga membantu dalam merancang penelitian yang relevan dalam bidang farmasi.
III. METODOLOGI PENELITIAN
Metodologi penelitian ini menjelaskan jenis dan rancangan penelitian, serta prosedur yang digunakan untuk mengumpulkan dan menganalisis data. Ini merupakan bagian penting dari pendidikan farmasi, di mana mahasiswa belajar tentang desain penelitian yang valid dan analisis statistik yang tepat.
3.1. Jenis dan Rancangan Penelitian
Penelitian ini merupakan penelitian eksperimental murni dengan rancangan silang. Mahasiswa diajarkan bagaimana memilih desain penelitian yang sesuai untuk menjawab pertanyaan penelitian serta cara mengontrol variabel yang mungkin mempengaruhi hasil.
3.2. Analisis Hasil
Data yang diperoleh dari penelitian ini dianalisis menggunakan ANOVA dengan tingkat kepercayaan 90%. Pemahaman tentang analisis statistik sangat penting bagi mahasiswa untuk dapat mengevaluasi dan menginterpretasi hasil penelitian secara tepat.
IV. HASIL DAN PEMBAHASAN
Bagian ini menyajikan hasil penelitian dan membahas perbandingan bioavailabilitas antara tablet Biogesic, Pamol, dan tablet parasetamol generik. Diskusi ini penting untuk memahami implikasi klinis dari hasil penelitian dan bagaimana hasil tersebut dapat diterapkan dalam praktik farmasi.
4.1. Uji Sifat Fisik Tablet
Hasil uji sifat fisik tablet memberikan informasi penting mengenai kualitas produk obat. Mahasiswa perlu memahami bagaimana sifat fisik dapat mempengaruhi bioavailabilitas dan efektivitas obat, yang merupakan bagian dari pengetahuan dasar dalam farmasi.
4.2. Perbandingan Bioavailabilitas
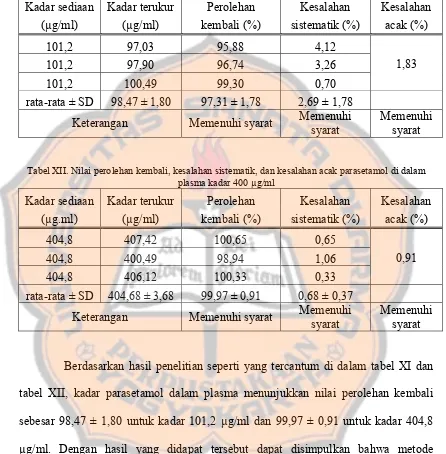
Hasil penelitian menunjukkan perbedaan bioavailabilitas yang signifikan antara tablet Biogesic dan Pamol dibandingkan dengan tablet parasetamol generik. Diskusi ini memberikan wawasan tentang pentingnya pemilihan produk obat yang tepat dalam praktik klinis.
V. KESIMPULAN DAN SARAN
Kesimpulan dari penelitian ini menegaskan bahwa tablet Biogesic bioekivalen dengan tablet generik, sedangkan tablet Pamol bioinekivalen. Ini menunjukkan pentingnya penelitian bioavailabilitas dalam menentukan pilihan obat yang tepat. Saran untuk penelitian selanjutnya juga memberikan arah bagi pengembangan ilmu pengetahuan di bidang farmasi.
5.1. Kesimpulan
Kesimpulan dari penelitian ini menunjukkan bahwa pemahaman tentang bioavailabilitas dapat membantu dalam pengambilan keputusan klinis terkait penggunaan obat. Mahasiswa diharapkan dapat menerapkan pengetahuan ini dalam praktik mereka di masa depan.
5.2. Saran
Saran untuk penelitian selanjutnya mencakup perlunya penelitian lebih lanjut untuk mengeksplorasi faktor-faktor yang mempengaruhi bioavailabilitas. Ini memberikan kesempatan bagi mahasiswa untuk berkontribusi dalam penelitian dan pengembangan ilmu farmasi.