PERBANDINGAN BIOAVAILABILITAS ANTARA TABLET BIOGESIC® DAN TABLET PAMOL® DENGAN TABLET PARASETAMOL GENERIK
PADA KELINCI PUTIH JANTAN
SKRIPSI
Diajukan Untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Ilmu Farmasi
Oleh:
Vincilia Indriyani
NIM : 038114008
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
PERBANDINGAN BIOAVAILABILITAS ANTARA TABLET BIOGESIC® DAN TABLET PAMOL® DENGAN TABLET PARASETAMOL GENERIK
PADA KELINCI PUTIH JANTAN
SKRIPSI
Diajukan Untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Ilmu Farmasi
Oleh:
Vincilia Indriyani
NIM : 038114008
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
2007
HALAMAN PERSEMBAHAN
PRAKATA
Puji syukur kepada Tuhan Yang Maha Besar atas kasih, kuasa, mujizat, dan
penyertaan-Nya yang sempurna sehingga penulis dapat menyelesaikan skripsi yang
berjudul “Perbandingan Bioavailabilitas antara Tablet Biogesic® dan Tablet Pamol® dengan Tablet Parasetamol Generik pada Kelinci Putih Jantan”. Skripsi ini ditulis
untuk memenuhi salah satu syarat memperoleh gelar Sarjana Farmasi (S.Farm.) pada
Program Studi Farmasi di Universitas Sanata Dharma.
Penulisan skripsi ini tidak mungkin terwujud tanpa bimbingan, bantuan,
dukungan, dan doa dari berbagai pihak, maka pada kesempatan ini penulis hendak
mengucapkan terima kasih kepada:
1. Orang tua tercinta dan adikku, Willy, atas doa, pengertiannya, dan dukungan
semangatnya yang tak pernah berhenti hingga hari ini.
2. Rita Suhadi, M. Si., Apt. selaku dekan Fakultas Farmasi Universitas Sanata
Dharma dan terima kasih atas pinjaman bukunya yang sangat membantu
penyelesaian skripsi ini.
3. Drs. Mulyono, Apt. selaku dosen pembimbing yang telah meluangkan waktu dan
tenaga, juga atas masukan, saran, pengajaran, dukungan, dan semangat yang
selalu menginspirasi dalam penyusunan skripsi ini.
4. Christine Patramurti, M. Si., Apt. selaku dosen penguji yang telah berkenan
menguji dan memberikan masukan serta saran atas penulisan skripsi ini.
5. C. M Ratna Rini Nastiti, S. Si., Apt. selaku dosen penguji yang telah berkenan
menguji, memberikan bimbingan, masukan dan saran yang sangat berarti dalam
penyusunan skripsi ini.
6. Yosef Wijoyo, M. Si., Apt. selaku dosen yang telah berkenan memberikan
masukan, dukungan dan dorongan semangat dalam pengerjaan skripsi ini.
7. Clara ”Jephi”ana Sri Widyarini, sahabat, teman praktikum, dan rekan kerja, atas
persahabatan yang indah, kerja sama yang luar biasa, canda tawa, pemikiran,
pengetahuan, semangat, motivasi, dan doa hingga kita bisa menyelesaikan kuliah
kita, PKM dan skripsi kita bersama.
8. Mas Heru, Mas Parjiman, Mas Kayat, Mas Yuwono, Pak Mus, Pak Mukmin, Mas
Wagiran, dan segenap karyawan yang telah membantu dan menyemangati.
9. Teman-teman seperjuangan di laboratorium : Fany, Essy, Surya, Galih, dan
Angga atas dukungan semangat dan canda tawanya selama ini.
10.Kesukaan Bapa’s crew yaitu Ci Anita, Ci V’ri, Ci Esme, Ci Vina, Astri, Juwi, Ine,
Indri, Dian, dan Christina, serta anak sel-ku Agnes, Heni, dan Jenny, atas doa,
perhatian, dan kebersamaan persekutuan kita yang indah.
11.Ko Andrey, Koko Can, dan Fang-fang, sahabat-sahabat terbaik, yang selalu ada
menemani hari-hariku, yang selalu mempercayaiku, mendukung dan
mendoakanku.
12.Bu Ina dan Ko Dian, teman sekaligus guru musikku yang selalu memberikan
semangat dan memaklumi kesibukanku dalam pengerjaan skripsi ini.
13.Adhy, Tirza, Eta, Arnie, dan teman-teman kuliah khususnya angkatan 2003
kelompok praktikum A, terima kasih atas semangat dan segala kebersamaan kita
selama ini.
14.Semua pihak yang telah banyak membantu.
Atas segala bantuan yang telah diberikan selama ini, penulis mengucapkan
banyak terima kasih. Penulis menyadari sepenuhnya penulisan skripsi ini tidak
terlepas dari keterbatasan dan kekurangan penulis. Oleh karena itu, diharapkan kritik
dan saran yang membangun demi penyempurnaan skripsi ini. Besar harapan penulis
bahwa skripsi ini dapat bermanfaat bagi perbendaharaan dan perkembangan ilmu
pengetahuan.
Penulis
INTISARI
Obat yang beredar di masyarakat dapat dibagi menjadi obat generik dan obat dagang. Perbandingan kedua produk obat tersebut dapat ditinjau dari penelitian farmakokinetika. Penelitian ini dimaksudkan untuk membandingkan bioavailabilitas obat dagang terhadap obat generik pada kelinci putih jantan.
Penelitian ini merupakan penelitian eksperimental murni dengan rancangan eksperimental silang. Konsentrasi parasetamol dalam plasma kelinci ditentukan dengan metode kolorimetri berdasarkan metode Chafetz et al. (1971) yang telah dimodifikasi. Data kemudian diubah menjadi parameter-parameter bioavailabilitas dan dianalisis dengan ANOVA mengggunakan taraf kepercayaan 90%.
Hasil penelitian yaitu tmaks (menit) untuk tablet parasetamol generik = 24,233
± 1,193; tablet Biogesic® = 28,000 ± 4,371; tablet Pamol® = 58,467 ± 1,976. Cmaks (μg
/ml) untuk tablet parasetamol generik = 193,927 ± 38,345; tablet Biogesic® = 162,870 ± 34,831; tablet Pamol® = 156,647 ± 42,072. AUC(0-∞) (μg.menit/ml) untuk tablet
parasetamol generik = 22896,410 ± 3731,193; tablet Biogesic® = 22198,470 ± 698,045; tablet Pamol® = 25525,490 ± 7181,70. Hasil ini menunjukkan ada perbedaan tidak bermakna nilai AUC(0-∞) dan nilai Cmaks antara tablet Biogesic® dan tablet
Pamol® dengan tablet parasetamol generik. Namun, terdapat perbedaan bermakna nilai tmaks tablet Pamol® terhadap tablet parasetamol generik. Jadi, dapat disimpulkan
tablet Biogesic® bioekivalen dengan tablet generik, sedangkan tablet Pamol® bioinekivalen dengan tablet generik.
Kata kunci utama : obat generik, obat dagang, parasetamol, bioavailabilitas, bioekivalen.
ABSTRACT
Drugs can be divided into two groups, are generic drugs and brand-name drugs. The comparison of them could be found out by pharmacokinetic research. This research was aimed to compare the bioavailability of brand-name drugs to generic drugs on male white rabbits.
The research was pure cross experimental research. Paracetamol concentrations in rabbits’ plasma were determined by a colorimetric method based on modified-Chafetz et al. method (1971). The data were presented as bioavailability parameters, and were analyzed using ANOVA with 90% confidence interval.
The results showed that tmax (min) for generic paracetamol tablets = 24,233 ±
1,193; Biogesic® tablets = 28,000 ± 4,371; Pamol® tablets = 58,467 ± 1,976. Cmax (μg
/ml) for generic paracetamol tablets = 193,927 ± 38,345; Biogesic® tablets = 162,870 ± 34,831; Pamol® tablets = 156,647 ± 42,072. AUC(0-∞) (μg.min/ml) for generic
paracetamol tablets = 22896,410 ± 3731,193; Biogesic® tablets = 22198,470 ± 698,045; Pamol® tablets = 25525,490 ± 7181,70. There were insignificant differences of AUC(0-∞) and Cmaks between Biogesic® and generic tablets, and between Pamol®
and generic tablets. However, significant difference of tmaks was found out between
Pamol® and generic tablets. Therefore, we conclude that Biogesic® and generic tablets were bioequivalent, but Pamol® and generic tablets were bioinequivalent.
Keywords : generic drugs, brand-name drugs, paracetamol, bioavailability, bioequivalent.
DAFTAR ISI
HALAMAN JUDUL ... ii
HALAMAN PERSETUJUAN PEMBIMBING ... iii
HALAMAN PENGESAHAN ... iv
HALAMAN PERSEMBAHAN ... v
PRAKATA ... vi
PERNYATAAN KEASLIAN KARYA ... ix
INTISARI ... x
ABSTRACT ... xi
DAFTAR ISI ... xii
DAFTAR TABEL ... xvi
DARTAR GAMBAR ... xviii
DAFTAR LAMPIRAN ... xx
BAB I PENGANTAR ... 1
A. Latar Belakang ... 1
1. Perumusan masalah ... 2
2. Keaslian Penelitian... 2
3. Manfaat ... 2
B. Tujuan ... 3
1. Tujuan Umum ... 3
2. Tujuan Khusus ... 3
BAB II PENELAAHAN PUSTAKA ... 4
A. Nasib Obat di Dalam Tubuh ... 4
B. Fase Farmakokinetika ... 5
1. Absorpsi dan Bioavailabilitas ... 6
2. Distribusi ... 23
3. Biotransformasi atau metabolisme ... 23
4. Ekskresi ... 24
C. Bioekivalensi ... 25
1. Definisi ... 25
2. Studi Bioavailabilitas dan Bioekivalensi ... 26
3. Korelasi in vitro dan in vivo ... 27
D. Dasar-Dasar Farmakokinetika ... 28
1. Definisi ... 28
2. Model Farmakokinetika ... 28
3. Parameter Farmakokinetika ... 29
4. Strategi Penelitian Farmakokinetika ... 35
E. Desain Cross Over ... 37
F. Parasetamol ... 37
G. Darah ... 41
H. Kolorimetri ... 43
1. Definisi ... 43
2. Kriteria Analisis Kolorimetri ... 43
3. Metode Kolorimetri untuk Parasetamol ... 44
I. Keterangan Empiris ... 47
BAB III METODOLOGI PENELITIAN ... 48
A. Jenis dan Rancangan Penelitian ... 48
B. Variabel dan Definisi Operasional ... 48
1. Variabel Penelitian ... 48
2. Definisi Operasional ... 50
C. Bahan Penelitian ... 51
D. Alat Penelitian ... 51
E. Tata Cara Penelitian ... 51
1. Uji Pendahuluan Tablet Parasetamol ... 51
2. Pembuatan Larutan ... 53
3. Pengambilan Plasma Darah ... 55
4. Validasi Metode Analisis ... 55
5. Orientasi Dosis ... 58
6. Metode Bioanalitik Parasetamol dalam Plasma Darah ... 59
F. Analisis Hasil ... 61
1. Nilai Perolehan Kembali (Recovery), Kesalahan Sistematik, dan Kesalahan Acak ... 61
2. Pengolahan Data dengan program STRIPE ... 62
3. Analisis Data secara statistik ... 62
BAB IV HASIL DAN PEMBAHASAN ... 64
A. Uji Sifat Fisik Tablet Parasetamol ... 64
1. Uji Keseragaman Bobot ... 64
2. Uji Kekerasan ... 66
3. Uji Kerapuhan ... 66
4. Uji Waktu Hancur ... 67
5. Uji Disolusi ... 68
B. Pengambilan Plasma Darah Kelinci ... 72
C. Validasi Metode Analisis ... 73
1. Penentuan Operating Time (OT) ... 78
2. Penentuan Panjang Gelombang Maksimum Parasetamol ... 80
3. Pembuatan Kurva Baku ... 81
4. Penentuan Nilai Perolehan kembali, Kesalahan Sistematik, dan Kesalahan Acak ... 83
D. Orientasi Dosis dan Orientasi Waktu Pengambilan Cuplikan ... 85
E. Perbandingan Bioavailabilitas ... 87
1. Nilai tmaks ... 89
2. Cmaks ... 90
3. AUC(0-∞) ... 91
BAB V KESIMPULAN DAN SARAN ... 101
A. Kesimpulan ... 101
B. Saran ... 102
DAFTAR PUSTAKA ... 103
LAMPIRAN ... 108
BIOGRAFI PENULIS ... 142
DAFTAR TABEL
Tabel I Desain Operasional Penelitian ... 59
Tabel II Parameter-parameter Farmakokinetika beserta satuannya ... 62
Tabel III Hasil Rata-rata Uji Keseragaman Bobot Tablet ... 65
Tabel IV Hasil Rata-rata Uji Kekerasan Tablet ... 66
Tabel V Hasil Uji Kerapuhan Tablet ... 67
Tabel VI Hasil Uji Waktu Hancur Tablet ... 68
Tabel VII Data Persamaan Kurva Baku Disolusi ... 69
Tabel VIII Hasil Uji Disolusi ... 70
Tabel IX Nilai Faktor Kemiripan (f2) ... 72
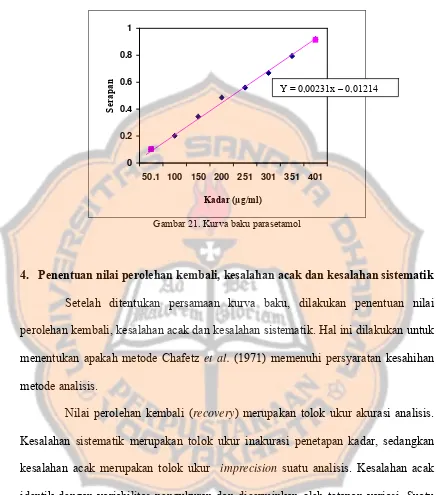
Tabel X Data Persamaan Kurva Baku ... 82
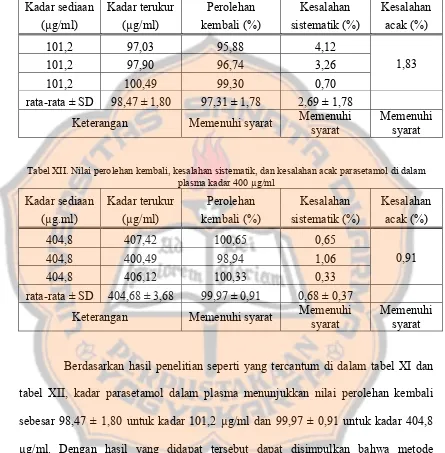
Tabel XI Nilai Perolehan Kembali, Kesalahan Sistematik, dan Kesalahan4 Acak Parasetamol di Dalam Plasma Kadar 100 µg/ml ... 84
Tabel XII Nilai Perolehan Kembali, Kesalahan Sistematik, dan Kesalahan Acak Parasetamol di Dalam Plasma Kadar 400 µg/ml ... 84
Tabel XIII Nilai Rata-Rata Parameter-Parameter Bioavailabilitas ... 87
Tabel XIV Hasil Analisis Statistik untuk tmaks ... 89
Tabel XV Hasil Analisis Statistik untuk ln Cmaks ... 90
Tabel XVI Hasil Analisis Statistik untuk AUC(0-∞) ... 92
Tabel XVII Nilai Rata-rata Geometrik Parameter-parameter Bioavailabilitas 98 Tabel XVIII Hasil Uji Keseragaman Bobot ... 108
Tabel XIX Hasil Uji Kekerasan ... 109
Tabel XX Hasil Uji Disolusi Tablet Parasetamol Generik ... 112
Tabel XXI Hasil Uji Disolusi Tablet Biogesic® ... 113
Tabel XXII Hasil Uji Disolusi Tablet Pamol® ... 114
Tabel XXIIII Perhitungan Faktor Kemiripan ... 115
Tabel XXIV Konversi Perhitungan Dosis Antar Jenis Hewan ... 118
Tabel XXV Data Tablet Parasetamol Generik 1 ... 124
Tabel XXVI Data Tablet Parasetamol Generik 2 ... 125
Tabel XXVII Data Tablet Parasetamol Generik 3 ... 126
Tabel XXVIII Data Tablet Biogesic® 1 ... 127
Tabel XXIX Data Tablet Biogesic® 2 ... 128
Tabel XXX Data Tablet Biogesic® 3 ... 129
Tabel XXXI Data Tablet Pamol®1 ... 130
Tabel XXXII Data Tablet Pamol® 2 ... 131
Tabel XXXIII Data Tablet Pamol® 3 ... 132
Tabel XXXIV Nilai Rata-rata Aritmatika Parameter-parameter Farmakokinetika ... 133
DAFTAR GAMBAR
Gambar 1 Proses Obat dalam Tubuh hingga Menimbulkan Efek ... 5
Gambar 2. Proses Farmakokinetika Obat di dalam Tubuh ... 6
Gambar 3 Proses Perjalanan Absorpsi Obat ... 7
Gambar 4 Struktur Parasetamol ... 38
Gambar 5 Metabolisme Parasetamol ... 41
Gambar 6 Reaksi Parasetamol dengan Asam Nitrat ... 45
Gambar 7 Reaksi Hidrolisis Parasetamol menjadi p-aminofenol ... 45
Gambar 8 Reaksi Pembentukan Warna ... 46
Gambar 9 Kurva Baku Disolusi ... 69
Gambar 10 Kurva Nilai Rata-rata Kumulatif Uji Disolusi ± SD ... 71
Gambar 11 Reaksi antara Asam Klorida dengan Natrium Nitrit Sehingga Membentuk ion Nitrosonium ... 74
Gambar 12 Reaksi antara Parasetamol dengan Ion Nitrosonium Membentuk 2-nitro-4-asetamidofenol ... 75
Gambar 13 Reaksi antara Asam Nitrit dengan Asam Sulfamat ... 76
Gambar 14 Reaksi antara 2-nitro-4 asetamidofenol dalam Suasana Basa Menghasilkan ion 2-nitro-4 asetamidofenolat ... 76
Gambar 15 Reaksi Menstabilkan Diri Ion 2-nitro-4 asetamidofenolat ... 77
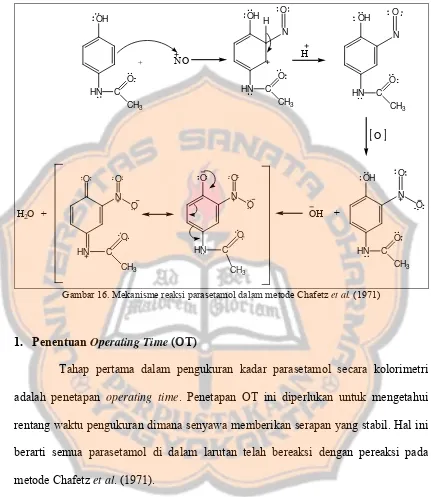
Gambar 16 Mekanisme Reaksi Parasetamol dalam Metode Chafetz et al. ... 78
Gambar 17 Pengukuran Operating Time (OT) Larutan Parasetamol dalam Plasma Kadar 100 μg/ml ... 79
Gambar 18 Pengukuran Operating Time (OT) Larutan Parasetamol dalam Plasma Kadar 400 μg/ml ... 79
Gambar 19 Pengukuran Panjang Gelombang Maksimum Larutan Parasetamol 100 μg/ml ... 80
Gambar 20 Pengukuran Panjang Gelombang Maksimum Larutan Parasetamol 400 μg/ml ... 81
Gambar 21 Kurva Baku Parasetamol ... 83
Gambar 22 Kurva Rata-rata Kadar Parasetamol dalam Plasma versus Waktu 93
Gambar 23 Kurva Rata-rata ln Kadar Parasetamol dalam Plasma versus Waktu 93 Gambar 24 Kurva Hasil Uji Disolusi Tablet Parasetamol Generik ... 112
Gambar 25 Kurva Hasil Uji Disolusi Tablet Biogesic® ... 113
Gambar 26 Kurva Hasil Uji Disolusi Tablet Pamol® ... 114
Gambar 27 OT Larutan Parasetamol Kadar 100µg/ml ... 120
Gambar 28 OT Larutan Parasetamol Kadar 400µg/ml ... 120
Gambar 29 λmaks Larutan Parasetamol Kadar 100µg/ml ... 121
Gambar 30 λmaks Larutan Parasetamol Kadar 400µg/ml ... 121
Gambar 31 Kurva Baku Parasetamol ... 122
Gambar 32 Sertifikat Analisis Parasetamol ... 123
Gambar 33 Kurva Hubungan Kadar Tablet Parasetamol Generik vs Waktu .. 134
Gambar 34 Kurva Hubungan Kadar Tablet Biogesic®vs Waktu ... 134
Gambar 35 Kurva Hubungan Kadar Tablet Pamol®vs Waktu ... 134
Gambar 36 Kurva Hubungan ln Kadar Tablet Parasetamol Generik vs Waktu 135
Gambar 37 Kurva Hubungan ln Kadar Tablet Biogesic® vs Waktu ... 135
Gambar 38 Kurva Hubungan ln Kadar Tablet Pamol® vs Waktu ... 135
DAFTAR LAMPIRAN
Lampiran 1 Hasil Uji Keseragaman Bobot ... 108
Lampiran 2 Hasil Uji Kekerasan ... 109
Lampiran 3 Contoh Cara Perhitungan Data Disolusi ... 110
Lampiran 4 Hasil Uji Disolusi ... 112
Lampiran 5 Perhitungan Pembuatan Larutan Parasetamol untuk Kurva Baku 116 Lampiran 6 Contoh Perhitungan Pembuatan Larutan Obat ... 117
Lampiran 7 Tabel Konversi Perhitungan Dosis Antar Jenis Hewan dan Perhitungan Orientasi Dosis ... 118
Lampiran 8 Contoh Perhitungan Volume Pemberian Larutan Parasetamol Pada Hewan Uji ... 119
Lampiran 9 Hasil Scanning Penentuan Operating time dan Panjang Gelombang Maksimum Parasetamol ... 120
Lampiran 10 Hasil Scanning Kurva Baku ... 122
Lampiran 11 Sertifikat Analisis Parasetamol ... 123
Lampiran 12 Hasil Pengolahan Data Dengan Program STRIPE Untuk Tablet Parasetamol Generik ... 125
Lampiran 13 Hasil Pengolahan Data Dengan Program STRIPE Untuk Tablet Biogesic® ... 127
Lampiran 14 Hasil Pengolahan Data Dengan Program STRIPE Untuk Tablet Pamol® ... 130
Lampiran 15 Nilai Rata-rata Parameter-parameter Farmakokinetika ... 133
Lampiran 16 Contoh Perhitungan Nilai Rata-rata Geometrik Parameter Bioavailabilitas ... 133
Lampiran 17 Kurva Kadar Parasetamol Dalam Plasma (Cp vs t) ... 134
Lampiran 18 Kurva ln Kadar Parasetamol Dalam Plasma (ln Cp vs t) ... 135
Lampiran 19 Hasil Pengolahan Data Secara Statistik Dengan Program SPSS 136
BAB I PENGANTAR
A. Latar Belakang
Obat tidak pernah lepas dari kehidupan masyarakat. Dewasa ini, semakin
tinggi permintaan masyarakat akan obat. Obat telah menjadi kebutuhan dalam
masyarakat. Hal tersebut memicu berkembangnya industri-industri obat sehingga obat
yang beredar di masyarakat menjadi sangat beragam. Obat dalam masyarakat dapat
dibedakan menjadi dua, yaitu obat generik dan obat bermerek dagang (trade mark). Dalam kehidupan sehari-hari, masyarakat menggunakan kedua jenis obat tersebut.
Oleh sebab itu, obat yang beredar di dalam masyarakat harus terjamin mutu, khasiat,
dan keamanannya.
Obat generik merupakan obat jadi yang menggunakan nama zat aktif yang
terkandung di dalamnya sesuai dengan yang ditetapkan dalam Farmakope Indonesia
atau buku resmi lainnya. Obat bermerek dagang merupakan obat jadi dengan nama
dagang yang dilindungi hukum yaitu merek terdaftar. Banyaknya obat jadi dengan zat
aktif yang sama yang beredar di masyarakat baik obat generik maupun obat bermerek
dagang, menimbulkan pertanyaan apakah obat-obat tersebut adalah sama.
Perbandingan kedua jenis obat tersebut dapat ditinjau dari segi farmakokinetika.
Dalam penelitian ini, dilakukan analisis farmakokinetika yaitu dengan
melakukan studi bioavailabilitas dan bioekivalensi dengan cara membandingkan
kecepatan dan jumlah obat yang diabsorpsi dalam tubuh. Penelitian ini menggunakan
tablet Biogesic® dan tablet Pamol® selaku obat bermerek dagang yang mengandung
senyawa aktif tunggal parasetamol dengan tablet parasetamol generik sebagai
pembandingnya. Penelitian ini dilakukan pada hewan uji yaitu kelinci putih jantan.
Parasetamol dipilih sebagai obat yang akan diteliti dikarenakan parasetamol sangat
lazim digunakan dalam masyarakat baik sebagai obat flu, demam, sakit kepala, nyeri
haid, dan sebagainya.
1. Perumusan masalah
Berdasarkan latar belakang yang dipaparkan maka dapat dirumuskan
permasalahan yaitu apakah tablet Biogesic® dan tablet Pamol® memiliki
bioavailabilitas yang sama dengan tablet parasetamol generik?
2. Keaslian penelitian
Sejauh yang diketahui penulis, belum pernah dilakukan penelitian mengenai
perbandingan bioavailabilitas antara tablet Biogesic® dan tablet Pamol® dengan tablet
parasetamol generik pada kelinci putih jantan di lingkungan penelitian Universitas
Sanata Dharma dan Universitas Gadjah Mada Yogyakarta.
3. Manfaat penelitian
Penelitian mengenai perbandingan bioavailabilitas tablet parasetamol ini
memberikan informasi akan perbandingan bioavailabilitas tablet obat bermerek
dagang dan tablet obat generik yang mengandung parasetamol pada hewan uji.
B. Tujuan
Tujuan dari penelitian ini yaitu:
1. Tujuan umum :
Untuk mengetahui bioavailabilitas tablet parasetamol generik, tablet
Biogesic®, dan tablet Pamol®.
2. Tujuan khusus:
Untuk mengetahui ada tidaknya perbedaan bioavailabilitas yang bermakna
antara tablet Biogesic® dan tablet Pamol® terhadap tablet parasetamol generik, pada
BAB II
PENELAAHAN PUSTAKA
Berkaitan dengan penelitian yang berjudul “Perbandingan Bioavailabilitas
Antara Tablet Biogesic® dan Tablet Pamol dengan Tablet Parasetamol Generik pada ® Kelinci Putih Jantan”, maka dilakukan studi pustaka yang akan mendukung analisis
profil bioavailabilitas yang dihasilkan dari penelitian ini. Studi pustaka yang
dilakukan meliputi penjelasan mengenai nasib obat di dalam tubuh, fase
farmakokinetika, bioekivalensi, dasar-dasar farmakokinetika, desain cross over,
parasetamol, darah, dan kolorimetri.
A. Nasib Obat di Dalam Tubuh
Proses yang terjadi pada selang antara pemberian obat hingga timbul efek
dibagi menjadi 3 fase yaitu fase farmasetik, fase farmakokinetika, dan fase
farmakodinamika. Fase farmasetik meliputi hancurnya bentuk sediaan obat dan
melarutnya bahan obat. Fase farmakokinetika termasuk proses-proses yang
berlangsung pada pengambilan suatu bahan obat ke dalam organisme (absorpsi,
distribusi), yang disebut juga proses invasi dan proses eliminasi yaitu proses-proses
yang menyebabkan penurunan konsentrasi obat (biotransformasi, ekskresi). Fase
farmakodinamika merupakan interaksi obat-reseptor dan juga proses-proses yang
Disintegrasi dari bentuk sediaan
Disolusi Obat
EFEK
pemberian
Fase farmasetik
Obat tersedia untuk diabsorpsi (availabilitas farmasetik)
Obat tersedia untuk aksi (availabilitas farmakologis)
Fase farmakokinetika
Fase farmakodinamika
Dosis formulasi obat
Absorpsi, distribusi, metabolisme,
ekskresi
Interaksi obat-reseptor
Gambar 1. Proses obat dalam tubuh hingga menimbulkan efek (Bowman and Rand, 1990)
B. Fase Farmakokinetika
Untuk obat-obat yang diberikan secara ekstravaskuler diperlukan suatu
proses absorpsi. Tempat aksi obat biasanya bukan di dalam darah sehingga obat yang
berada dalam sirkulasi sistemik harus menembus jaringan untuk dapat memberi efek.
Perpindahan obat ini disebut proses distribusi. Eliminasi adalah proses pengeluaran
obat, baik bentuk utuh maupun metabolitnya dari dalam tubuh, dapat melalui ginjal
Tempat Aksi “Reseptor” terikat bebas
Jaringan
terikat bebas
Absorpsi Ekskresi
Sirkulasi sistemik
obat bebas
obat terikat metabolit
Biotransformasi
Distribusi
Gambar 2. Proses farmakokinetika obat di dalam tubuh (Wilkinson, 2001)
1. Absorpsi dan Bioavailabilitas
a. Definisi absorpsi dan bioavailabilitas
Absorpsi menjelaskan mengenai perpindahan obat dari tempat pemberian ke
dalam sirkulasi sistemik (darah). Tetapi secara klinik, yang lebih penting adalah
bioavailabilitas (Wilkinson, 2001). Bioavailabilitas (ketersediaan hayati)
merupakan persentase dan kecepatan zat aktif dalam suatu produk obat yang
mencapai / tersedia dalam sirkulasi sistemik dalam bentuk utuh / aktif setelah
pemberian produk obat tersebut. Bioavailabilitas dapat diukur dari kadarnya dalam
disintegrasi
Tablet Granul atau deagregasi
agregat
Suspensi partikel halus di cairan gastrointestinal
Larutan obat dalam cairan gastrointestinal
disolusi disolusi disolusi
absorpsi
Obat dalam darah, cairan tubuh, dan jaringan
Gambar 3. Proses perjalanan absorpsi tablet (Proudfoot, 1990)
Produk obat umumnya mengalami absorpsi sistemik melalui suatu rangkaian
proses, yang meliputi disintegrasi produk obat yang diikuti pelepasan obat,
pelarutan obat dalam media aqueous, dan absorpsi melewati membran sel menuju
sirkulasi sistemik. Di dalam proses disintegrasi obat, pelarutan, dan absorpsi,
kecepatan obat mencapai sistem sirkulasi ditentukan oleh tahap yang paling lambat
(rate limiting step) (Shargel, Wu-Pong and Yu, 2005).
b. Mekanisme transpor obat
Setelah molekul obat dalam bentuk larutan maka obat harus berdifusi dari
cairan gastrointestinal ke membran kemudian berada dalam sirkulasi sistemik
dalam bentuk utuh (Mayersohn, 2002). Membran biologis tersusun dari protein dan
membran biologis. Kebanyakan dari obat menembus membran dengan mekanisme
yang disebut difusi pasif (Proudfoot, 1990). Difusi pasif menunjukkan perpindahan
komponen dari fase aqueous melewati suatu membran dimana membran tersebut
bersifat pasif, tenaga penggerak perpindahan tersebut hanya merupakan gradien
konsentrasi komponen (Mayersohn, 2002).
Mekanisme difusi pasif dapat ditunjukkan secara matematis dengan hukum
Fick :
(1)
( )
dt=
D
Δ
⎟
⎟
⎠
⎞
⎜⎜
⎝
⎛
−
→ m b g m/aq m m b g dQX
C
C
R
A
b( )
dQdtb g b→ = kecepatan obat berada di darah (b) setelah berdifusi dari cairan
saluran cerna (g)
Dm = koefisien difusi obat melewati membran
Am = luas permukaan membran yang tersedia untuk proses difusi obat = koefisien partisi obat antara membran dan cairan aqueous pada
saluran cerna Rm/aq
-C = gradien konsentrasi antara konsentrasi obat di cairan saluran cerna
(C Cg b
g) dengan konsentrasi obat di dalam darah pada tempat absorpsi
(C ) b
ΔXm = ketebalan dari membran
Pada kondisi dan obat tertentu maka nilai Dm, Am, Rm/aq, dan ΔXm adalah konstan
maka dapat digantikan sebagai koefisien permeabilitas (P).
(2)
( )
=P(
g −Cb)
Volume dimana obat terdistribusi dalam darah jauh lebih besar
dibandingkan volume cairan saluran cerna dan karena sirkulasi darah melewati
saluran gastrointestinal cepat dan terus menerus membawa obat yang terabsorpsi,
maka nilai C >> Cg b. Kondisi ini yang disebut kondisi sink (Mayersohn, 2002).
(3)
( )
dt g b gdQ
C . b
→ ≅P
c. Faktor-faktor yang Mempengaruhi Absorpsi dan Bioavailabilitas Obat
Faktor-faktor yang dapat mempengaruhi bioavailabilitas suatu obat seperti
tercantum di bawah ini.
1). Faktor mekanis
Faktor-faktor yang termasuk di dalamnya yaitu :
a). Rute dan metode pemberian
Ketika obat diberikan ke dalam tubuh, obat harus dapat menembus
membran hingga dapat masuk ke dalam sirkulasi sistemik. Contoh rute
dan metode pemberian mempengaruhi bioavailabilitas : ada beberapa obat
yang tidak terabsorpsi jika diberikan secara oral, ada obat yang bila
diberikan secara oral akan mengalami first-pass effect yang berlebihan
sehingga hanya sebagian kecil dari obat yang dapat masuk ke dalam
sirkulasi sistemik dan akan menghasilkan AUC kecil, sehingga obat perlu
Dosis dan aturan dosis b).
Dosis dan aturan dosis berkaitan dengan konsentrasi terapeutik yang dapat
dicapai suatu obat di dalam plasma, yang berarti berhubungan dengan
Cmaks dan AUC yang dihasilkan (mempengaruhi bioavailabilitas obat)
(Shargel et al., 2005).
c). Efek dari bentuk sediaan.
Faktor dari bentuk sediaan yang dapat mempengaruhi bioavailabilitas
yaitu:
(1). Faktor fisikakimia bahan dalam obat meliputi sebagai berikut.
Faktor yang mempengaruhi kelarutan
Absorpsi obat tergantung seberapa cepat obat larut dalam cairan
gastrointestinal, sehingga faktor yang mempengaruhi kecepatan disolusi
obat akan mempengaruhi bioavailabilitas obat. Kecepatan disolusi obat
ditentukan dari persamaan Noyes dan Whitney (Proudfoot, 1990) :
(4)
)
C
(
D
dm
=
C
−
s h
A dt
dm/dt = kecepatan disolusi partikel obat
D = koefisien difusi larutan obat di cairan gastrointestinal
A = luas permukaan efektif dari partikel obat
h = ketebalan lapisan difusi sekitar partikel obat
Cs = kelarutan jenuh obat di lapisan difusi
Faktor-faktor yang mempengaruhi kelarutan obat antara lain :
(a). Bentuk kristal
Polymorphism. Banyak obat dapat berada dalam lebih dari satu bentuk kristal. Polimorfi bentuk metastable memiliki kelarutan
dalam aqueous dan kecepatan disolusi yang lebih besar dibandingkan polimorfi bentuk stable.
Amorphous state. Obat dalam bentuk amorf biasanya lebih mudah
larut dan lebih cepat terdisolusi daripada obat dalam bentuk kristal
sehingga akan mempengaruhi bioavailabilitas.
Solvates. Obat bergabung dengan molekul dari pelarut dan membentuk bentuk kristal yang disebut solvates. Secara umum,
semakin banyak solvasi maka semakin rendah kelarutan dan
kecepatan disolusi obat sehingga dapat mempengaruhi
bioavailabilitas obat (Proudfoot, 1990; Wagner, 1975).
(b). Asam bebas, basa bebas, bentuk garam, nilai pKa
Bentuk garam akan lebih cepat larut di larutan aqueous
dibandingkan asam atau basa lemah (Wagner, 1975). Jumlah obat
asam lemah dan basa lemah yang terionisasi dalam larutan di cairan
lambung dan di darah dapat dihitung dengan persamaan
[ ]
[ ]
pH-pKa HAA log
-=
untuk obat asam lemah (5)
[ ]
[ ]
pKa-pH BBH log =
+
untuk obat basa lemah (6)
pH = keasaman media
pKa = keasaman senyawa
-] = fraksi terion dari senyawa yang bersifat asam lemah [A
[HA] = fraksi tak terion (molekul) dari senyawa yang bersifat asam
lemah
+
[BH ] = fraksi terion dari senyawa yang bersifat basa lemah
[B] = fraksi tak terion (molekul) dari senyawa yang bersifat basa
lemah
(c). Kompleksasi, larutan solid, dan eutetics
Bioavailabilitas tergantung dengan konsentrasi efektif obat.
Kompleksasi merupakan interaksi fisikakimia yang dapat terjadi
antara bahan-bahan di dalam bentuk sediaan atau di dalam cairan
gastrointestinal sehingga akan mempengaruhi konsentrasi efektif
obat di dalam cairan gastrointestinal (Proudfoot, 1990). Larutan
solid dan eutectics menghasilkan efek bervariasi pada kecepatan
disolusi karena dapat meningkatkan atau menurunkan kelarutan obat
(d). Surfaktan
Surfaktan dapat menghasilkan efek bervariasi pada proses disolusi
dan absorpsi. Surfaktan dapat menurunkan tegangan permukaan
sehingga meningkatkan kecepatan disolusi (Wagner, 1975).
Faktor yang mempengaruhi transpor obat
Faktor utama yang mempengaruhi obat dalam proses absorpsi obat
menembus membran adalah koefisien partisi, banyaknya ionisasi dalam
cairan biologis yang ditentukan dari nilai pKa, pH cairan medium obat
terlarut, dan berat molekul atau volume (Mayersohn, 2002).
(a). Koefisien partisi
Membran biologis merupakan lapisan lipid sehingga obat yang larut
dalam lemak (lipofil) lebih dapat menembus membran. Ko/w adalah
rasio kelarutan obat di dalam minyak (oil) dengan kelarutan obat di
dalam air (water). Hal ini berarti obat-obat yang memiliki nilai Ko/w
lebih besar akan lebih banyak yang dapat menembus membran
biologis dan dapat diabsorpsi. Peningkatan nilai Ko/w akan
meningkatkan kecepatan absorpsi (Mayersohn, 2002)
(b). Nilai pKa, pH, keberadaan muatan
akan terionisasi pada cairan biologis. Arti pentingnya ionisasi dalam
proses absorpsi obat didasarkan pada observasi dimana obat dalam
bentuk non ion memiliki nilai Ko/w lebih besar dibandingkan obat
dalam bentuk ion. Hal ini berarti membran bersifat permeabel
terhadap bentuk non ion dari obat asam lemah dan basa lemah
(Mayersohn, 2002; Proudfoot, 1990).
(c). Molal volume, difusivitas
Difusivitas berkaitan dengan berat molekular. Bentuk misel akan
berdifusi lebih lambat dari fase aqueous bulk menuju ke lapisan
difusi dan berdifusi lebih lambat dalam melewati lapisan difusi
dibandingkan molekul obat monomerik (Wagner, 1975).
(d). Stagnant water layers / aqueous diffusion layer
Proses pelarutan obat diawali dengan pelarutan obat pada permukaan
partikel padat yang membentuk larutan jenuh di sekeliling partikel
yang disebut stagnant water layers (Shargel et al., 2005). Obat harus
berdifusi melewati stagnant water layers yang bersifat aqueous, isi
cairan gastrointestinal dan lapisan membran, maka hal ini dapat
(2). Faktor farmasetik dan pembuatan obat
Faktor-faktor yang termasuk di dalamnya yang mungkin menyebabkan
adanya perbedaan pada parameter-parameter bioavailabilitas adalah
sebagai berikut.
(a). Ukuran partikel dan luas permukaan area
Peningkatan luas permukaan area obat untuk kontak dengan cairan
gastrointestinal akan meningkatkan kecepatan disolusi. Secara
umum, semakin kecil ukuran partikel obat, semakin besar luas
permukaan area dan semakin besar kecepatan disolusi, yang akan
meningkatkan bioavailabilitas (Proudfoot, 1990; Wagner, 1975).
(b). Static electrification dari obat padat
Banyak proses farmasetik seperti blending, pencampuran, coating,
dan sebagainya dapat menghasilkan static electrification dari bahan
padat. Hal ini dapat menyebabkan terjadinya agregasi dan obat tidak
bercampur. Agregasi dapat menurunkan luas permukaan efektif
sehingga dapat menurunkan kecepatan disolusi (Wagner, 1975).
(c). Jenis bentuk sediaan
Jenis bentuk sediaan mempengaruhi langkah-langkah obat dari
pemberian hingga terlarut dalam cairan gastrointestinal. Semakin
bentuk larutan di cairan gastrointestinal, maka makin banyak
penghalang absorpsi obat dan akan mempengaruhi bioavailabilitas
obat. Bioavailabilitas obat larutan aqueous > suspensi aqueous >
kapsul > tablet tidak bersalut > tablet bersalut (Proudfoot, 1990).
(d). Jenis dan jumlah bahan tambahan (eksipien) seperti bahan pengisi,
bahan pelicin, bahan pengikat, garam netral, garam asam atau garam
basa, dan lain-lain
Eksipien dianggap bahan yang inert, yang tidak memiliki pengaruh
terhadap aksi terapeutik dan tidak mengubah aksi biologik dari obat
yang terkandung di dalam bentuk sediaan. Namun, disadari bahwa
eksipien dapat mempengaruhi kecepatan dan jumlah obat yang
terabsorpsi dengan cara membentuk kompleks obat-eksipien yang
tidak larut seperti tetrasiklin dengan dikalsium fosfat. Selain itu,
perubahan eksipien dapat mempengaruhi bioavailabilitas (Proudfoot,
1990). Diluen yang tidak larut air akan memberikan kecepatan
disolusi yang lebih rendah dibandingkan bila digunakan diluen yang
larut air. Kemungkinannya karena kecepatan deagregasi obat
menurun dan obat menjadi lebih bersifat hidrofobik. Garam netral
dapat mempengaruhi disolusi karena air dapat lebih mudah masuk
sehingga mempercepat hancurnya tablet dan larutnya tablet
(e). Ukuran granul dan distribusi ukurannya
Dalam proses pembuatan tablet, proses granulasi merupakan proses
pengikatan campuran dan mempengaruhi sifat alir. Setelah granul
dibentuk menjadi tablet maka tablet akan mempertahankan
integritasnya. Ukuran granul dan distribusi ukurannya penting
karena mempengaruhi hancurnya tablet menjadi granul yang
kemudian hancur menjadi partikel-partikel kecil, sehingga akan
mempengaruhi ukuran partikel yang mempengaruhi luas permukaan
dan akan menentukan bioavailabilitas obat (Wagner, 1975).
(f). Jenis dan jumlah bahan penghancur dan metode mencampurnya
Bahan penghancur biasanya merupakan bahan yang akan
mengembang apabila ada air yang kemudian akan menekan tablet
untuk hancur. Proses disintegrasi tablet dalam cairan aqueous pada
saluran gastrointestinal merupakan salah satu rate limiting step yang
menentukan bioavailabilitas obat (Wagner, 1975).
(g).Waktu pencampuran
Pada proses pencampuran, diperlukan waktu optimum pencampuran
sehingga bahan-bahan tercampur sempurna, namun setelah melewati
tersebut tidak tercampur dengan baik sehingga akan mempengaruhi
konsentrasi obat dalam tubuh (Wagner, 1975).
(h). Tekanan kompresi
Tekanan kompresi menentukan waktu hancur tablet dan kecepatan
disolusi obat dari bentuk tablet (Wagner, 1975).
(i). Efek matrik
Untuk obat-obat yang lepas lambat maka terjadi efek matrik. Ketika
obat diberikan secara oral, maka pada fase aqueous, air akan masuk
ke dalam matrik yang terbuat dari polimer sintetik yang tidak
terabsorpsi pada saluran gastrointestinal, kemudian obat akan
terlepas dari matrik secara perlahan-lahan (Wagner, 1975).
(j). Jenis dan jumlah surfaktan
Surfaktan yang dimaksud dapat berupa agen pengemulsi, agen
pelarut, pensuspensi, penstabil, atau sebagai wetting agent. Surfaktan
dapat meningkatkan, menurunkan atau menunjukkan tidak adanya
efek pada proses transpor obat menembus membran. Surfaktan dapat
menurunkan tegangan permukaan antara obat dengan media disolusi
sehingga meningkatkan kecepatan disolusi. Selain itu, surfaktan
mempengaruhi enzim pemetabolisme obat atau ikatan obat dengan
reseptor. Surfaktan dapat mengganggu integritas dan fungsi
membran, surfaktan juga dapat mengubah waktu pengosongan
lambung (Wagner, 1975; Proudfoot, 1990).
(k). Bentuk dan geometri
Bentuk dan geometri akan mempengaruhi kecepatan disolusi obat.
Hal ini berhubungan dengan luas permukaan area efektif dan bentuk
sediaan (Wagner, 1975).
(l). Kondisi lingkungan selama pembuatan
Kelembaban selama pembuatan dapat mempengaruhi potensi dari
bentuk sediaan yang dibuat misalkan aspirin karena kondisi lembab
akan terhidrolisis sehingga mempengaruhi bentuk sediaan yang
dibuat (Wagner, 1975).
(m). Kondisi penyimpanan dan lama penyimpanan
Kondisi dan lama penyimpanan akan mempengaruhi stabilitas obat.
Stabilitas akan mempengaruhi waktu hancur dan kecepatan disolusi
2). Faktor Fisiologi
Faktor fisiologik mempengaruhi pelepasan, disolusi obat dari bentuk
sediaan, absorpsi pada saluran pencernaan dan dapat mempengaruhi profil
bioavailabilitas obat. Faktor-faktor tersebut yaitu :
a. Motilitas usus dan waktu transit obat dalam usus
Usus merupakan tempat utama terjadinya absorpsi obat sehingga semakin
besar kecepatan transit usus maka semakin kecil waktu tinggal obat di dalam
usus berarti makin kecil waktu obat kontak dengan tempat absorpsi sehingga
jumlah obat yang terabsorpsi menjadi kecil (Proudfoot 1990).
b. Kecepatan pengosongan lambung
Kebanyakan obat diabsorpsi di usus halus sehingga penurunan kecepatan obat
dalam bentuk larutan meninggalkan lambung, akan menurunkan kecepatan
absorpsi obat dan menunda onset efek terapeutik dari obat. Selain itu, ada
obat-obat yang akan mengalami degradasi akibat pH lambung dan aktivitas
enzim dalam cairan lambung jika terjadi penundaan pengosongan dalam
lambung sehingga akan menurunkan konsentrasi efektif obat dan
mempengaruhi bioavailabilitas. Salah satu faktor yang meningkatkan
kecepatan pengosongan lambung adalah rasa lapar Proudfoot, 1990).
c. Tempat absorpsi dan area permukaan yang efektif untuk absorpsi obat
villi dan mikrovilli pada usus halus sehingga kebanyakan obat akan
terabsorpsi maksimum di dalam usus halus yang berarti akan menghasilkan
kecepatan dan jumlah obat terabsorpsi yang maksimum (menentukan
bioavailabilitas). Glycocalyx merupakan lapisan pada mikrovilli. Absorpsi obat dari lumen usus halus untuk mencapai pembuluh darah harus melewati
beberapa barrier. Larutan obat untuk mencapai mikrovilli harus berdifusi menembus unstirred layer, lapisan mukus dan glycocalyx (Proudfoot, 1990).
d. Nilai pH cairan gastrointestinal, konsentrasi elektrolit
Keasaman (pH) cairan di saluran gastrointestinal bervariasi, pH cairan
lambung antara 1-3,5; pH cairan usus halus antara 5-8 (pH 5-6 di duodenum
dan sekitar pH 8 di ileum), pH cairan usus besar sekitar 8. Nilai pH cairan
gastrointestinal akan mempengaruhi absorpsi obat. Nilai pH cairan
gastrointestinal dapat menentukan absorpsi dalam berbagai cara karena
kebanyakan obat merupakan asam lemah atau basa lemah, kelarutan
komponen-komponen tersebut dalam air dipengaruhi pH, dan kecepatan
disolusi dari bentuk sediaan terutama tablet dan kapsul juga dipengaruhi pH.
Bagian obat yang terionisasi lebih larut dalam air daripada bagian obat yang
tak terionisasi (Mayersohn, 2002; Shargel et al., 2005).
e. Stabilitas obat pada saluran gastrointestinal
mengalami degradasi dan mengalami metabolisme di saluran gastrointestinal,
akibatnya fraksi obat yang terabsorpsi menjadi lebih kecil sehingga
menurunkan bioavailabilitas obat (Proudfoot, 1990; Wagner, 1975).
f. Metabolisme hepatik
Hati merupakan tempat utama terjadinya metabolisme. First-pass effect
merupakan fenomena dimana sebagian obat sebelum mencapai sirkulasi
sistemik mengalami metabolisme di hati sehingga akan menurunkan jumlah
obat yang terabsorpsi yang berarti menurunkan bioavailabilitas (Proudfoot,
1990)
g. Keberadaan makanan di saluran pencernaan
Mekanisme makanan dalam mempengaruhi bioavailabilitas obat yaitu dengan
mengubah kecepatan pengosongan lambung, menyebabkan terjadinya
stimulasi sekresi gastrointestinal, kompetisi antara komponen makanan dan
obat, kompleksasi obat dengan komponen dalam makanan, meningkatkan
viskositas dari isi gastrointestinal, dan dapat mengubah aliran darah ke hati
(Proudfoot, 1990; Wagner, 1975).
h. Faktor-faktor lain : kecepatan aliran darah, agen pengemulsi dan
pengkompleks, tegangan permukaan dan tegangan interfasial, gross
tekanan hidrostatik dan intralumenal, kapasitas buffer, dan tonisitas (Wagner,
1975).
2. Distribusi
Setelah diabsorpsi, obat akan didistribusi ke seluruh tubuh melalui sirkulasi
darah (Setiawati, Zulnida, Suyatna, 2002). Distribusi merupakan proses perpindahan
obat dari sirkulasi sistemik menuju ke jaringan dan organ tubuh serta ke cairan tubuh
lainnya seperti cairan interstitial dan cairan intercellular (Wilkinson, 2001).
Distribusi obat dibedakan atas 2 fase berdasarkan penyebarannya di dalam
tubuh. Distribusi fase pertama berjalan dengan cepat yaitu ke organ-organ yang
perfusinya cepat seperti hati, ginjal, dan otak. Distribusi fase kedua memerlukan
waktu lebih lama sebelum mencapai keseimbangan konsentrasi obat di jaringan
dengan yang di dalam darah, yaitu ke organ-organ yang perfusinya tidak secepat
organ di atas seperti otot, visera, kulit, dan jaringan lemak (Setiawati dkk., 2002;
Wilkinson, 2001).
3. Biotransformasi atau metabolisme
Biotransformasi atau metabolisme obat ialah proses perubahan struktur
kimia obat yang terjadi dalam tubuh dan dikatalisis oleh enzim. Pada proses ini
molekul obat diubah menjadi lebih polar artinya lebih mudah larut dalam air dan
kurang larut dalam lemak sehingga lebih mudah diekskresi melalui ginjal. Selain itu,
mengakhiri kerja obat (Setiawati dkk., 2002). Biotransformasi terjadi pada hati,
saluran cerna, ginjal, dan paru-paru (Wilkinson, 2001).
Reaksi biotransformasi obat dapat dibedakan menjadi 2 fase. Reaksi fase I
adalah reaksi fungsional yang mengubah obat menjadi metabolit yang lebih polar,
meliputi reaksi oksidasi, reduksi, dan hidrolisis (Setiawati dkk., 2002). Reaksi fase II
merupakan reaksi biosintetik (konjugasi). Reaksi ini merupakan reaksi konjugasi obat
atau metabolit hasil reaksi fase pertama dengan menggunakan substrat endogen
seperti asam glukuronat, sulfat, glutation, asam amino atau asetat. Konjugat yang
dihasilkan akan bersifat polar, inaktif dan dengan cepat dapat diekskresi melalui urin
dan feses (Setiawati dkk., 2002; Wilkinson, 2001).
4. Ekskresi
Ekskresi merupakan peristiwa pengeluaran obat dan atau metabolitnya dari
dalam tubuh. Ginjal merupakan organ terpenting untuk mengekskresi obat dan
metabolitnya. Obat dikeluarkan dari tubuh dalam bentuk metabolit hasil
biotransformasi atau dalam bentuk asalnya. Obat atau metabolit polar diekskresi lebih
cepat daripada obat larut lemak (Setiawati dkk., 2002).
Ekskresi di sini merupakan resultante dari 3 proses, yakni filtrasi di
glomerulus, sekresi aktif di tubuli proksimal dan reabsorpsi pasif di tubuli proksimal
dan distal. Selain melalui ginjal, ekskresi dapat terjadi pada paru-paru, hati, kelenjar
ludah, dan kelenjar susu, keringat, air mata, dan rambut (Setiawati., 2002; Wilkinson,
C. Bioekivalensi 1. Definisi
Ekivalensi dapat didefinisikan antara lain :
a. Ekivalensi kimia menunjukkan dua atau lebih sediaan obat mengandung
jumlah yang sama yang tertera pada label (kurang lebih pada rentang
tertentu) (Malinowski, 2000).
b. Ekivalensi klinik terjadi ketika obat yang sama dari dua atau lebih sediaan
obat menunjukkan efek in vivo yang identik yang terukur dari respon
farmakologik atau dari kontrol gejala atau penyakit (Malinowski, 2000).
c. Ekivalensi terapeutik menyatakan bahwa dua merek produk obat diharapkan
akan menghasilkan hasil klinik yang sama (Malinowski, 2000). Dua produk
obat mempunyai ekivalensi terapeutik jika keduanya mempunyai ekivalensi
farmaseutik atau merupakan alternatif farmaseutik dan pada pemberian
dengan dosis molar yang sama akan menghasilkan efikasi klinik dan
keamanan yang sebanding. Dengan demikian, ekivalensi / inekivalensi
terapeutik seharusnya ditunjukkan dengan uji klinik (Anonim, 2004 b).
Ekivalensi farmasetik
d. ditujukan pada dua produk dengan kesamaan bentuk
sediaan, zat aktif dan jumlah zat aktif (Malinowski, 2000; Anonim 2004 b).
e. Alternatif farmasetik jika keduanya mengandung zat aktif yang sama tetapi
berbeda dalam bentuk kimia (garam, ester, dsb.) atau bentuk sediaan atau
kekuatan (Anonim, 2004 b; Chereson, 1999).
sediaan yang sama mencapai sirkulasi sistemik dengan kecepatan dan
jumlah yang sama atau bisa disebut memiliki bioavailabilitas yang sama.
Bioekivalensi ditunjukkan jika keduanya mempunyai ekivalensi farmasetik
atau merupakan alternatif farmasetik dan pada pemberian dengan dosis
molar yang sama akan menghasilkan bioavailabilitas yang sebanding
sehingga efeknya akan sama, dalam hal efikasi maupun keamanan
(Malinowski, 2000; Anonim, 2004 b).
2. Studi Bioavailabilitas dan Bioekivalensi
Studi bioavailabilitas dilakukan baik terhadap bahan obat aktif yang telah
disetujui maupun terhadap obat dengan efek terapeutik yang belum disetujui oleh
Food and Drug Administration (FDA) untuk dipasarkan. FDA dalam menyetujui suatu produk obat untuk dipasarkan harus yakin bahwa produk obat tersebut aman
dan efektif sesuai label indikasi penggunaan serta harus memenuhi seluruh standar
yang digunakan dalam identitas, kekuatan, kualitas dan kemurnian (Shargel et al.,
2005).
Untuk meyakinkan bahwa standar-standar tersebut telah dipenuhi, FDA
menghendaki studi bioavailabilitas / farmakokinetika dan bila perlu persyaratan
bioekivalensi untuk semua produk (Shargel et al., 2005). Dalam tahun-tahun terakhir
ini, studi bioavailabilitas dan bioekivalensi dilakukan untuk menurunkan biaya
kesehatan dengan cara meningkatkan pemakaian obat generik. Oleh sebab itu,
dagang (Chereson, 1999).
Bioavailabilitas dari produk obat sering menentukan efikasi terapeutik dari
obat tersebut karena hal ini mempengaruhi onset, intensitas, dan durasi dari respon
terapeutik obat tersebut (Chereson, 1999). Pada studi bioekivalensi, dibutuhkan suatu
formulasi obat sebagai standar pembanding yang hendaknya mengandung obat aktif
terapeutik dalam formulasi yang paling banyak berada dalam sistemik (yakni larutan
atau suspensi), dalam jumlah sama, dan hendaknya diberikan dengan rute sama
seperti formulasi yang dibandingkan (Shargel et al., 2005).
Bioekivalensi dapat dilakukan menggunakan uji in vitro jika uji in vitro
memiliki korelasi yang baik dengan data bioavailabilitas secara in vivo. Selain itu, uji
bioekivalensi dapat dilakukan melalui studi farmakodinamika melalui uji
perbandingan klinis (Malinowski, 2000).
3. Korelasi in vitro dan in vivo
Korelasi in vitro dan in vivo yang dimaksud adalah hubungan antara karakteristik biologi obat (efek farmakodinamika atau konsentrasi obat dalam plasma)
dan karakteristik fisika kimia produk obat (Shargel et al., 2005). Korelasi in vitro dan
in vivo ini penting untuk diketahui agar dalam menentukan bioavailablitas suatu obat cukup dengan uji in vitro saja, tidak perlu dengan uji in vivo. Selama ini, uji
bioavailabilitas secara in vivo memerlukan waktu yang lama, biaya yang relatif tinggi,
serta terdapat beberapa masalah dalam pemberian obat kepada subjek uji sehat/pasien
Parameter uji in vitro yang paling dekat hubungannya dengan
bioavailabilitas adalah kecepatan disolusi. Obat yang masuk ke dalam tubuh dapat
diabsorpsi jika sudah dalam bentuk larutan sehingga kecepatan obat untuk larut dari
bentuk sediaannya (laju disolusi) akan menentukan kecepatan dan atau jumlah obat
yang terabsorpsi (Chereson, 1999).
D. Dasar-dasar Farmakokinetika 1. Definisi
Farmakokinetika adalah ilmu yang mempelajari kinetika absorpsi, distribusi
dan eliminasi (terdiri dari metabolisme dan ekskresi) dari obat. Studi farmakokinetika
meliputi pendekatan eksperimental dan teoritis. Aspek eksperimental dari
farmakokinetika meliputi perkembangan teknik pengambilan sampel biologis, metode
analisis obat dan metabolitnya, dan prosedur pengolahan data. Aspek teoritis dari
farmakokinetika meliputi perkembangan model farmakokinetika yang digunakan
untuk memprediksikan proses disposisi yang terjadi setelah pemberian obat. Aplikasi
dari metode statistik termasuk dalam studi farmakokinetika yaitu untuk penetapan
parameter farmakokinetika dan interpretasi data (Shargel, Wu-Pong, B. C. Yu, 2005).
2. Model Farmakokinetika
Model farmakokinetika adalah struktur hipotesis yang digunakan untuk
menggambarkan kecepatan dari proses absorpsi, distribusi, dan eliminasi obat
Model farmakokinetika digunakan untuk menginterpretasikan data-data
farmakokinetika (Shargel et al., 2005).
Kompartemen dianggap sebagai sebuah jaringan atau kumpulan jaringan
yang memiliki kesamaan aliran darah dan afinitas terhadap obat. Di setiap
kompartemen, obat dianggap terdistribusi secara seragam. Model ini merupakan suatu
sistem terbuka apabila obat dapat dieliminasi dari tubuh (Shargel et al., 2005).
Kompartemen farmakokinetik ini tidak berhubungan dengan lokasi secara anatomi
tubuh namun hanya parameter operasional yang diturunkan secara matematis
(Mutschler, Derendorf, Schäfer-Korting, Elrod and Estes, 1995).
Model satu kompartemen menunjukkan bahwa setelah pemberian, obat
terdistribusi secara langsung. Model dua atau lebih kompartemen, terjadi distribusi
obat ke dalam ruang distribusi yang dapat dilewatinya dengan kecepatan
berbeda-beda (Mutschler et al., 1995). Model dua kompartemen, obat dapat berpindah antara
kompartemen sentral ke dan dari kompartemen perifer (jaringan). Kompartemen
sentral menggambarkan plasma dan organ yang memiliki perfusi tinggi dan secara
cepat seimbang dengan obat. Jumlah total obat di dalam tubuh dapat dihitung dari
jumlah obat di dalam kompartemen sentral ditambah dengan obat di dalam
kompartemen jaringan (Shargel et al., 2005).
3. Parameter Farmakokinetika
Parameter farmakokinetika adalah konstanta yang menunjukkan profil obat
farmakokinetika diperoleh dari profil kinetika dari obat yang dapat diperoleh melalui
kurva konsentrasi obat terhadap waktu. Konsentrasi obat dapat diukur sebagai fungsi
terhadap waktu di beberapa cairan tubuh seperti darah, plasma, serum, saliva, dan
urin. Konsentrasi obat dalam darah mencerminkan perubahan kinetika di sirkulasi
sistemik. Untuk mendapatkan kurva konsentrasi obat terhadap waktu maka perlu
dilakukan pengukuran konsentrasi obat berulangkali pada beberapa titik waktu
(Mutschler et al., 1995). Parameter-parameter farmakokinetika antara lain:
a. AUC (Area under the curve)
AUC merupakan ukuran dari jumlah obat di dalam tubuh dan dapat
dihitung dengan menggunakan rumus trapezoid, yaitu :
[
]
t n-1 n(
n n-1t t t
2 C C AUC n
1
-n −
+
=
)
(7)AUC = area di bawah kurva
tn = waktu pengamatan dari konsentrasi obat Cn
tn-1 = waktu pengamatan sebelumnya yang berhubungan dengan konsentrasi
obat Cn-1 (Mutschler et al., 1995).
Rumus trapezoid ini menganggap titik-titik data berada pada suatu
fungsi linier. Jika titik-titik data tersebar secara luas, maka lengkung dari garis
akan menyebabkan kesalahan yang besar dalam memperkirakan area. Pada
suatu waktu area di bawah kurva kadar plasma-waktu diekstrapolasikan
Dalam hal ini area tersisa,
[
]
k C AUC t pn
tn =
∞ (8)
Cpn = konsentrasi dalam plasma terakhir pada tn
k = slop yang diperoleh dari bagian akhir kurva (Shargel et al., 2005).
Untuk menghitung AUC total (AUC∞) maka dilakukan ekstrapolasi
bagian akhir area setelah titik terakhir yang diukur (AUCtn - ∞). Prosedur yang
digunakan disebut sahih bila bagian ekstrapolasi tersebut kira-kira di bawah
10% dari AUC total dan tidak boleh digunakan bila melebihi 20% dari AUC
total (Mutschler et al., 1995).
b. Volume distribusi (Vd)
Parameter ini menunjukkan volume penyebaran obat dalam tubuh
dengan kadar plasma atau serum. Vd tidak perlu menunjukkan volume
penyebaran obat yang sesungguhnya ataupun volume secara anatomik, tetapi
hanya volume imajinasi dimana tubuh dianggap sebagai 1 kompartemen yang
terdiri dari plasma atau serum, dan Vd menghubungkan jumlah obat dalam
tubuh dengan kadarnya dalam plasma atau serum (Setiawati, 2002).
DB = Vd . C (9) p
D = jumlah obat dalam tubuh B
Tubuh dapat dianggap sebagai suatu sistem dengan volume yang
konstan. Oleh karena itu, volume distribusi untuk suatu obat umumnya
konstan (Shargel et al., 2005). Volume distribusi besar menunjukkan jumlah
obat yang terdistribusi ke dalam jaringan besar atau terkonsentrasi di jaringan
tertentu (Mutschler, et al., 1995).
c. Bersihan total (Klirens / Cl)
Klirens adalah volume plasma yang dibersihkan dari obat per satuan
waktu oleh seluruh tubuh (ml/menit). Parameter ini menunjukkan kemampuan
tubuh untuk mengeliminasi obat. Untuk obat dengan kinetika orde satu, Cl
merupakan bilangan konstan pada kadar obat yang biasa ditemukan dalam
klinik.
oral oral IV
IV
AUC F.D AUC
D
Cl= = (10)
el d k V
Cl= ⋅ (11)
D = Dosis
F = Fraksi obat yang
terabsorpsi
AUC = Area under the curve Vd = Volume distribusi
Umumnya bersihan total merupakan hasil beberapa bersihan bagian
bersama-sama, yang terpenting adalah bersihan ginjal (ClR) dan bersihan hati
(ClH) (Mutschler, 1991).
d. Waktu paruh eliminasi (t ) dan kecepatan eliminasi ½
Waktu paruh eliminasi adalah waktu yang diperlukan untuk turunnya
kadar obat dalam plasma atau serum pada fase eliminasi (setelah fase absorpsi
dan distribusi) menjadi separuhnya. Untuk obat-obat dengan kinetika orde
reaksi satu, t½ ini merupakan bilangan konstan, tidak tergantung dari besarnya
dosis, interval pemberian, kadar plasma maupun cara pemberian (Setiawati,
2002).
el el k
0,693 k
2 ln t
2
1 = = (12)
kel adalah konstanta kecepatan eliminasi.
Waktu paruh eliminasi adalah parameter farmakokinetik yang berbeda
dengan waktu paruh dari efek atau waktu yang diperlukan untuk menjadikan
efek farmakologi menjadi separuh dengan efek semula (Mutschler et al.,
1995).
Kecepatan eliminasi merupakan kecepatan pengeluaran per satuan
2 1 t
2 ln
kel = (13)
e. Bioavailabilitas
Parameter ini menunjukkan fraksi dari dosis obat yang mencapai
peredaran darah sistemik dalam bentuk aktif (Setiawati, 2002). Faktor yang
menentukan bioavailabilitas adalah kecepatan dan jumlah obat yang dilepas
dari bentuk sediaannya, kecepatan dan jumlah obat yang mengalami absorpsi,
dan besarnya efek lintas pertama (Mutschler et al., 1995).
Besarnya bioavailabilitas absolut dapat dihitung dengan cara sebagai
berikut :
( )
% 100 AUCAUC F
i.v x ⋅
= (14)
F = bioavailabilitas absolut
AUC = AUC pemberian nonsistemik x
= AUC pemberian intravaskuler AUCiv
Dalam kasus apabila dosis dan formulasi untuk rute pemberian i.v
tidak ada, maka dapat ditentukan bioavailabilitas relatif yang diperoleh
dengan cara :
( )
% 100 AUCAUC F
standar x
rel= ⋅ (15)
= AUC pemberian nonsistemik AUCx
AUCstandar = AUC produk standar
Parameter untuk menggambarkan kecepatan absorpsi adalah
konsentrasi obat dalam plasma maksimum (Cmaks) dan selang waktu antara
pemberian obat hingga mencapai konsentrasi maksimum dalam plasma
(tmaks) (Mutschler et al., 1995). 4. Strategi Penelitian Farmakokinetika
Strategi penelitian farmakokinetika (SPF) adalah rencana yang disusun
sebelum meneliti tahap farmakokinetika obat, guna memperoleh informasi tentang
nasib obat dalam tubuh secara kuantitatif. Objek penelitian farmakokinetika adalah
tahap farmakokinetika obat dengan parameter farmakokinetika sebagai tolok ukurnya.
Parameter farmakokinetika adalah besaran yang diturunkan secara matematik dari
hasil pengukuran kadar obat atau metabolitnya di dalam darah atau urin (Suryawati
dan Donatus, 1998).
Tahap-tahap SPF meliputi :
1. Pemilihan rancangan uji coba.
2. Pemilihan subjek uji dan jumlahnya.
3. Pemilihan cuplikan hayati.
4. Pemilihan metode analisis penetapan kadar.
Syarat-syarat metode analisis yaitu :
selektivitas adalah kemampuan metode analisis untuk membedakan suatu
obat dengan metabolitnya, obat lain dan kandungan endogen cuplikan
hayati.
b. sensitivitas
sensitivitas berkaitan dengan kadar terendah yang dapat diukur dengan
metode analisis yang digunakan. Hal ini diperlukan karena untuk
menghitung parameter farmakokinetika suatu obat diperlukan kadar obat
tertinggi sampai terendah pada rentang waktu tertentu.
c. ketelitian dan ketepatan
ketelitian dan ketepatan ini akan menentukan kesahihan hasil penetapan
kadar. Ketepatan (accuracy) ditunjukkan oleh kemampuan metode
memberikan hasil pengukuran sedekat mungkin dengan nilai yang
sesungguhnya. Ketelitian (precision) menunjukkan kedekatan hasil
pengukuran berulang pada cuplikan hayati yang sama.
5. Pemilihan takaran dosis dan bentuk sediaan obat.
Takaran dosis yang diberikan harus menjamin dapat diukurnya kadar obat atau
metabolitnya pada rentang waktu tertentu sehingga diperoleh data yang cukup
memadai untuk analisis farmakokinetika.
6. Pemilihan lama dan banyaknya waktu pengambilan cuplikan hayati.
Bila menggunakan cuplikan darah, sebaiknya pengambilan dilakukan sebanyak 3
– 5 kali t ½ eliminasi obat yang diuji. Hal ini disebabkan karena pada kondisi
obat sebaiknya dilakukan setidaknya 3 kali pada tahap absorpsi, 3 kali di sekitar
puncak, 3 kali pada tahap distribusi, dan 3 kali pada tahap eliminasi.
7. Analisis dan evaluasi hasil.
Langkah-langkah ini meliputi analisis sederetan kadar obat utuh atau
metabolitnya dalam darah atau urin, analisis statistika dan evaluasi (Suryawati
dan Donatus, 1998).
E. Desain Cross Over
Desain cross over merupakan desain blok secara acak dimana tiap blok menerima lebih dari satu formulasi obat pada waktu yang berbeda. Keuntungan dari
desain cross over pada studi bioavailabilitas-bioekivalensi adalah tiap subjek bertindak sebagai kontrol sendiri, desain ini menghilangkan variasi biologik
antarsubjeknya, dan dengan randomisasi yang tepat maka hal ini akan memberikan
kalkulasi yang paling baik mengenai perbedaan tiap formulasi (Chow and Jen-Pei,
2000)
Perlakuan pertama dan perlakuan kedua dipisahkan oleh periode washout
yang cukup untuk eliminasi produk obat yang pertama diberikan (biasanya lebih dari
lima kali waktu paruh terminal dari obat, atau lebih lama jika mempunyai metabolit
aktif dengan waktu paruh yang lebih panjang). Jika obat mempunyai kecepatan
eliminasi yang sangat bervariasi antarsubjek, periode washout yang lebih lama
diperlukan untuk memperhitungkan kecepatan eliminasi yang lebih rendah pada
(> 24 jam), dapat dipertimbangkan penggunaan desain dua kelompok paralel
(Anonim, 2004 b).
F. Parasetamol
Parasetamol mengandung tidak kurang dari 98,0 % dan tidak lebih dari
101,0 % C H NO8 9 2, dihitung terhadap zat anhidrat. Parasetamol berupa serbuk hablur
putih, tidak berbau, rasa sedikit pahit. Larut dalam air mendidih dan dalam NaOH 1
N, mudah larut dalam etanol. Tablet parasetamol mengandung parasetamol tidak
kurang dari 90,0% dan tidak lebih dari 110,0% dari jumlah yang tertera pada etiket
(Anonim, 1995). Larut dalam 70 bagian air, dalam 7 bagian etanol (95 %) P, dalam
13 bagian aseton P, dalam 40 bagian gliserol P, dan dalam 9 bagian propilenglikol P,
larut dalam larutan alkali hidroksida. Khasiat dan penggunaan : analgetikum,
antipiretikum (Anonim, 1979).
NHCOCH3 HO
N-(4-hydroxyphenyl)acetamide
Gambar 4. Struktur Parasetamol (Anonim, 1995)
Titik lebur parasetamol 169 °C – 172 °C, tidak larut dalam benzen dan eter,
namun larut dalam larutan basa hidroksida. Parasetamol memiliki pH 5,3 sampai 6,5
pada larutan jenuh. Parasetamol sangat stabil dalam larutan berair dengan pH 5-7.
2000).
Melalui uji klinis, telah terbukti bahwa makanan dapat menurunkan tingkat
absorpsi parasetamol. Pada keadaan puasa secara nyata dapat meningkatkan
kecepatan absorpsi parasetamol walaupun tidak mempengaruhi jumlah total yang
diabsorpsi (McGilveray and Mattok, 1972). Menurut Lacy, Armstrong, Goldman, dan
Lance (2003), parasetamol cepat diabsorpsi dan hampir sempurna, namun apabila
parasetamol dikonsumsi diikuti dengan makanan berkarbohidrat tinggi akan terjadi
penundaan absorpsi yang berarti menunjukkan penurunan kecepatan absorpsi.
Menurut Proudfoot (1990), makanan akan menurunkan laju pengosongan lambung
sehingga akan menunda onset parasetamol.
Onset dari parasetamol relatif cepat, yaitu kurang dari 1 jam, sedangkan
durasinya sekitar 4 – 6 jam. Parasetamol memiliki tmax 0,5 – 2 jam. Obat ini tersebar
ke seluruh cairan tubuh. Availabilitas oral parasetamol adalah 88 ± 15% (Benet,
1992). Dalam plasma, 20 – 50% parasetamol akan terikat oleh protein plasma
(Anonim, 2004 a ; Lacy et al., 2003). Volume distribusi dari parasetamol adalah 0,94
L/kg (Melmon and Morelli, 1992) atau pada manusia 70 kg, volume distribusinya
sekitar 67 ± 8 L (Benet, 1992).
Parasetamol memiliki t½ sebesar 1 sampai 4 jam (Anonim, 2005 c). Dalam
urin, terdapat 90 – 100% metabolit tidak aktif, namun kadang ditemukan 3%
parasetamol dalam bentuk utuh (Anonim, 2004 a ; Mutschler et al., 1995). Efek
analgesik antipiretik dari parasetamol akan timbul apabila konsentrasinya dalam
(Kadar Efek Minimum) parasetamol adalah bila kadar parasetamol dalam darah
adalah sebesar 10µg/ml hingga 20 µg/ml, sedangkan nilai KTM (Kadar Toksik
Minimum) parasetamol adalah bila kadar parasetamol dalam plasma lebih besar dari
300 µg/mL (Benet, 1992).
Parasetamol akan dimetabolisme oleh enzim mikrosom hati. Sebagian
parasetamol (80%) dikonjugasi dengan asam glukuronat dan sebagian kecil lainnya
dengan asam sulfat. Selain itu, parasetamol juga dapat mengalami hidroksilasi.
Metabolit hasil hidroksilasi ini, dapat menimbulkan methemoglobinemia dan
hemolisis eritrosit (Wilmana, 2002). Klirens parasetamol adalah 250 ml/menit sampai
450 ml/menit. Klirens parasetamol akan turun apabila terjadi disfungsi hati. Klirens
akan meningkat bila terjadi hipertiroidsm (Melmon and Morelli, 1992).
Parasetamol mengalami metabolisme fase kedua yang menghasilkan
inaktivasi farmakologis dari obat induk. Seperti yang terlihat pada gambar 5,
parasetamol mengalami konjugasi glutation, glukuronida, dan konjugasi sulfat, dan
sebagai hasilnya konjugasi fase kedua tidak aktif secara farmakologis (Gibson and
Skett, 1991).
Parasetamol dalam dosis berlebihan dapat menyebabkan kematian karena
akan menghasilkan nekrosis pada hati, tapi dosis terapi tidak akan menyebabkan
hepatotoksik. Dosis kecil dari parasetamol akan dieliminasi melalui proses konjugasi
yang kemudian diikuti dengan ekskresi, tapi pada dosis yang berlebihan enzim yang
berperan mengalami saturasi maka obat akan mengalami proses metabolisme yang
Hidroksilamin akan bereaksi nonenzimatik dengan glutation dan kemudian
akan didetoksifikasi. Namun, karena jumlah glutation di hati terbatas maka apabila
parasetamol dikonsumsi berlebihan maka hidroksilamin yang mungkin terbentuk
akan bereaksi dengan makromolekul dan merusak struktur dan fungsinya sehingga
akan menyebabkan kerusakan sel. Kerusakan sel ini dapat dicegah dengan pemberian
sulfidril nukleofilik yang akan bereaksi dengan hidroksilamin elektrofilik dan untuk
mencegah hilangnya glutation secara berlebihan dapat digunakan systeamin dan
dimerkaprol (Benet, 1992).
HO HN COCH3
O HN COCH3
S O
O
HO O HN COCH3
O OH HO
HO
HOOC
Parasetamol (aktif)
Konjugasi sulfat
Konjugasi glukuronida
(tidak aktif) (tidak aktif)
Metabolisme dan konjugasi glutation
Sistein dan konjugasi asam merkapturat (tidak aktif)
ekskresi urin ekskresi urin ekskresi urin
Gambar 5. Metabolisme parasetamol (Gibson and Skett, 1991)
Sinonim parasetamol di antaranya asetaminofen, p-acetamidophenol,
N-acetyl-p-aminophenol (Connors et al., 1986). Parasetamol merupakan derivat para
amino fenol, suatu metabolit fenasetin dengan efek antipiretik yang sama (Wilmana,
menghilangkan atau mengurangi nyeri ringan sampai sedang. Parasetamol
menurunkan suhu tubuh dengan mekanisme yang diduga juga berdasarkan efek
sentral seperti salisilat (Wilmana, 2003).
G. Darah
Darah terdiri atas unsur-unsur padat, yaitu eritrosit, leukosit serta trombosit
yang tersuspensi di dalam suatu media cair yakni plasma. Pada darah normal, jumlah
plasma mencapai 55% dari volume darah. Konsentrasi total protein dalam plasma
manusia kurang lebih 77,5 g/dL, dan membentuk bagian utama unsur-unsur padat
plasma.
Begitu darah membeku (mengalami koagulasi), fase cair yang tertinggal
dinamakan serum. Serum sudah tidak lagi mengandung faktor-faktor pembekuan
(termasuk fibrinogen) (Murray, Granner, Mayes, and Rodwell, 1990). Plasma
dihasilkan dari melakukan sentrifugasi pada darah dan ditambahkan ke dalamnya
bahan antikoagulan Chamberlain, 1995).
Plasma ma