INTISARI
Obat yang beredar di pasaran dapat dibagi menjadi obat generik dan obat merk dagang. Kedua jenis obat tersebut harus terjamin keamanan dan khasiatnya. Dalam penelitian ini, dilakukan perbandingan antara obat merk dagang dan obat generik dengan pendekatan farmakokinetika. Penelitian ini bertujuan untuk membandingkan bioavailabilitas obat merk dagang dan obat generik pada kelinci putih jantan.
Penelitian ini merupakan penelitian eksperimental murni dengan rancangan eksperimental silang. Sampel yang digunakan adalah tablet parasetamol (generik), tablet Pyrexin®, dan tablet Progesic® yang diberikan kepada kelinci putih jantan dengan desain cross over. Metode yang digunakan untuk menetapkan kadar parasetamol adalah metode Chafetz et al. (1971) yang telah dimodifikasi.
Hasil yang diperoleh diolah menjadi parameter bioavailabilitas menggunakan program STRIPE (Johnston and Woolard, 1983, yang telah dimodifikasi oleh Jung), kemudian dianalisis statistik dengan metode ANOVA taraf kepercayaan 90%. Hasil penelitian ini adalah nilai AUC(0-inf) (μg.menit/ml) tablet parasetamol generik : 21029,077 + 3336,122; tablet Pyrexin® : 16666,110 + 1456,821; dan tablet Progesic® : 33823,687 + 5640,811. Nilai Cmax (μg/ml) tablet parasetamol generik : 179,743 + 21,631; tablet Pyrexin® : 116,717 + 10,018; dan tablet Progesic® : 236,037 + 15,762. Nilai tmax (menit) tablet parasetamol generik : 24,733 + 1,943; tablet Pyrexin® : 46,433 + 3,353; dan tablet Progesic® : 33,600 + 3,637. Jadi dapat disimpulkan bahwa bioavailabilitas tablet parasetamol (generik), tablet Pyrexin®, dan tablet Progesic® tidak sama.
Kata kunci : bioavailabilitas, parasetamol, farmakokinetika
ABSTRACT
The medicine could be classified as generic and brand-name medicine. The safety and efficacy of those tablets should be guaranteed. In this research, brand-name and generic medicine were compared by pharmacokinetics approach. The purpose of this research was comparing bioavailability of brand-name tablets to generic tablet on male-white rabbits.
This research was pure cross experimental research. The samples used in this research were generic paracetamol tablet, Pyrexin® tablet and Progesic tablet®. Those tablets were given to male-white rabbits. This research used cross over design and Chafetz et al. (1971) method to determine concentration of drug in the blood.
The result was converted to bioavailability values by STRIPE (Johnston and Woolard, 1983, modified by Jung) program, then the bioavailability values were analyzed by ANOVA method with 90% confidence intervals. The result showed that AUC(0-inf) (μg.minute/ml) of generic paracetamol tablet : 21029,077 + 3336,122; Pyrexin® tablet : 16666,110 + 1456,821; and Progesic®tablet : 33823,687 + 5640,811. Cmax (μg/ml) of generic paracetamol tablet : 179,743 + 21,631; Pyrexin® tablet : 116,717 + 10,018; and Progesic® tablet : 236,037 + 15,762. tmax (minute) of generic paracetamol tablet : 24,733 + 1,943; Pyrexin® tablet : 46,433 + 3,353; and Progesic® tablet : 33,600 + 3,637. So, it can be concluded that the bioavailability of generic paracetamol tablet, Pyrexin® tablet, and Progesic® tablet was different.
Key words : bioavailability, paracetamol, pharmacokinetics
PERBANDINGAN BIOAVAILABILITAS
TABLET PYREXIN® DAN TABLET PROGESIC® DENGAN
TABLET PARASETAMOL (GENERIK) PADA KELINCI PUTIH JANTAN
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Ilmu Farmasi
Oleh :
Clara Jeviana Sri Widyarini
NIM : 038114007
FAKULTAS FARMASI UNIVERSITAS SANATA DHARMA
PERBANDINGAN BIOAVAILABILITAS
TABLET PYREXIN® DAN TABLET PROGESIC® DENGAN
TABLET PARASETAMOL (GENERIK) PADA KELINCI PUTIH JANTAN
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Ilmu Farmasi
Oleh :
Clara Jeviana Sri Widyarini
NIM : 038114007
FAKULTAS FARMASI UNIVERSITAS SANATA DHARMA
YOGYAKARTA 2007
Jika kita tidak dibimbing oleh Roh Allah, kita bekerja hanya demi kesia-siaan belaka, tanpa makna, terasa hambar apapun yang kita kerjakan…
(St. Yohanes Maria Vianney)
Karya ini kupersembahkan untuk :
My Jesus Christ...
Terima kasih Tuhan atas penyertaan-Mu, Kau selalu menguatkanku saat ku lemah,
Kau selalu mencukupkan kebutuhanku saat ku kekurangan, Kau selalu mengangkatku saat kujatuh....
Papa & Mama tercinta
Kakakku tersayang
Almamaterku…. Fakultas Farmasi Universitas Sanata Dharma
Yogyakarta
Untuk segala sesuatu ada masanya,
Untuk apapun di bawah langit ada waktunya ...
.... IIaammeemmbbuuaattsseeggaallaasseessuuaattuuiinnddaahhppaaddaawwaakkttuunnyyaa..
(Pengkhotbah 3)
PRAKATA
Puji syukur dan terima kasih kepada Tuhan Yang Maha Kasih atas berkat
yang selalu diberikan sehingga penulis dapat menyelesaikan skripsi ini. Skripsi ini
dimaksudkan sebagai salah satu syarat memperoleh gelar sarjana farmasi dari
Fakultas Farmasi, Universitas Sanata Dharma Yogyakarta.
Selama proses pembuatan dan penyelesaian skripsi ini, penulis telah banyak
mendapatkan bantuan baik materi maupun dorongan dari berbagai pihak. Oleh sebab
itu, pada kesempatan ini penulis mengucapkan terima kasih kepada :
1. Papa dan Mama yang senantiasa mendoakan dan memberikan dorongan
kepada penulis.
2. Ibu Rita Suhadi, M. Si., Apt., selaku Dekan Fakultas Farmasi Universitas
Sanata Dharma Yogyakarta.
3. Bapak Drs. Mulyono, Apt., selaku dosen pembimbing I dan penguji yang
telah banyak membantu, mengarahkan dan memberi motivasi kepada
penulis sehingga penulis dapat menyelesaikan skripsi ini.
4. Ibu Christine Patramurti, M. Si., Apt., selaku penguji yang memberikan
saran dan masukan kepada penulis.
5. Ibu C. M. Ratna Rini Nastiti, S. Si., Apt., selaku penguji yang memberikan
saran dan masukan kepada penulis.
6. Bapak Yosef Wijoyo, M.Si., Apt., yang telah memberikan saran-saran yang
positif dan membangun kepada penulis.
7. Para laboran : Mas Heru, Mas Parjiman, Mas Kayat (Laboratorium
Farmakokinetika-Biofarmasetika), Pak Musrimin (Laboratorium Formulasi
dan Teknologi Sediaan Padat), Mas Wagiran (Laboratorium Farmakognosi
Fitokimia), Pak Mukmin (Laboratorium Kimia Analisis Instrumental) yang
telah banyak mendampingi dan membantu kelancaran selama penulis
melakukan penelitian.
8. Mas Robert di Jakarta atas bantuannya memberikan bahan penelitian.
9. Kakakku Ardiatmoko yang selalu memberi motivasi agar penulis tidak patah
semangat dan atas pinjaman laptopnya.
10.Untuk Vincilia “Yeyen” Indriyani atas segala kerja sama, pengetahuan, dan
pemikiran selama menempuh pendidikan dan menyelesaikan skripsi ini serta
kebersamaan perjuangan menyelesaikan PKM.
11.Kepada teman-teman Farmasi 2003 USD yang telah berjuang bersama,
terutama kepada Arnie, Marga, Vita, Mita, Nanda, Raya, Eta, Ria, Galuh,
Tina, Adi, dan Andhika “Ble-q”.
12.Untuk Surya, Angga, Galih, Fanny dan Essy atas kebersamaan kita selama
melaksanakan penelitian di laboratorium.
13.Untuk Alfons, Dewi, Erlisa, Teddy, dan teman-teman KKN (Abit, Mas
Bayu, Titin, Jane, Ratna, Iis, Vicky, “Nyak” Alfonsa, dan Nani).
14.Dan untuk kawan-kawanku, Acay, Dhamet, Indra, Eci, Rinto, Punto, Poke,
Beny, Bowo, Angga “Too-cool” serta semua teman dari SMU Pangudi
Luhur Van Lith Muntilan gen. X, terima kasih untuk dukungan dan
dorongannya.
Kepada seluruh pihak yang telah membantu dan tidak dapat disebutkan satu
per satu, penulis dengan tulus mengucapkan terima kasih. Penulis sangat menyadari
bahwa skripsi ini belum sempurna, maka kritikan dan saran atas skripsi ini
merupakan sesuatu yang berharga bagi penulis dan bagi perkembangan pengetahuan
di bidang farmasi. Terima kasih.
Penulis
INTISARI
Obat yang beredar di pasaran dapat dibagi menjadi obat generik dan obat merk dagang. Kedua jenis obat tersebut harus terjamin keamanan dan khasiatnya. Dalam penelitian ini, dilakukan perbandingan antara obat merk dagang dan obat generik dengan pendekatan farmakokinetika. Penelitian ini bertujuan untuk membandingkan bioavailabilitas obat merk dagang dan obat generik pada kelinci putih jantan.
Penelitian ini merupakan penelitian eksperimental murni dengan rancangan eksperimental silang. Sampel yang digunakan adalah tablet parasetamol (generik), tablet Pyrexin®, dan tablet Progesic® yang diberikan kepada kelinci putih jantan
dengan desain cross over. Metode yang digunakan untuk menetapkan kadar
parasetamol adalah metode Chafetz et al. (1971) yang telah dimodifikasi.
Hasil yang diperoleh diolah menjadi parameter bioavailabilitas
menggunakan program STRIPE (Johnston and Woolard, 1983, yang telah
dimodifikasi oleh Jung), kemudian dianalisis statistik dengan metode ANOVA taraf kepercayaan 90%. Hasil penelitian ini adalah nilai AUC(0-inf) (μg.menit/ml) tablet
parasetamol generik : 21029,077 + 3336,122; tablet Pyrexin® : 16666,110 +
1456,821; dan tablet Progesic® : 33823,687 + 5640,811. Nilai Cmax (μg/ml) tablet
parasetamol generik : 179,743 + 21,631; tablet Pyrexin® : 116,717 + 10,018; dan
tablet Progesic® : 236,037 + 15,762. Nilai tmax (menit) tablet parasetamol generik :
24,733 + 1,943; tablet Pyrexin® : 46,433 + 3,353; dan tablet Progesic® : 33,600 +
3,637. Jadi dapat disimpulkan bahwa bioavailabilitas tablet parasetamol (generik), tablet Pyrexin®, dan tablet Progesic® tidak sama.
Kata kunci : bioavailabilitas, parasetamol, farmakokinetika
ABSTRACT
The medicine could be classified as generic and brand-name medicine. The safety and efficacy of those tablets should be guaranteed. In this research, brand-name and generic medicine were compared by pharmacokinetics approach. The purpose of this research was comparing bioavailability of brand-name tablets to generic tablet on male-white rabbits.
This research was pure cross experimental research. The samples used in this research were generic paracetamol tablet, Pyrexin® tablet and Progesic tablet®. Those tablets were given to male-white rabbits. This research used cross over design and Chafetz et al. (1971) method to determine concentration of drug in the blood.
The result was converted to bioavailability values by STRIPE (Johnston
and Woolard, 1983, modified by Jung) program, then the bioavailability values were analyzed by ANOVA method with 90% confidence intervals. The result showed that AUC(0-inf) (μg.minute/ml) of generic paracetamol tablet : 21029,077 + 3336,122;
Pyrexin® tablet : 16666,110 + 1456,821; and Progesic®tablet : 33823,687 + 5640,811. Cmax (μg/ml) of generic paracetamol tablet : 179,743 + 21,631; Pyrexin® tablet :
116,717 + 10,018; and Progesic® tablet : 236,037 + 15,762. tmax (minute) of generic
paracetamol tablet : 24,733 + 1,943; Pyrexin® tablet : 46,433 + 3,353; and Progesic® tablet : 33,600 + 3,637. So, it can be concluded that the bioavailability of generic
paracetamol tablet, Pyrexin® tablet, and Progesic® tablet was different.
Key words : bioavailability, paracetamol, pharmacokinetics
DAFTAR ISI
HALAMAN JUDUL ... ii
HALAMAN PERSETUJUAN PEMBIMBING ... iii
HALAMAN PENGESAHAN ... iv
HALAMAN PERSEMBAHAN ... v
PRAKATA ... vi
PERNYATAAN KEASLIAN KARYA ... ix
INTISARI ... x
ABSTRACT ... xi
DAFTAR ISI ... xii
DAFTAR TABEL ... xvi
DAFTAR GAMBAR ... xviii
DAFTAR LAMPIRAN ... xx
BAB I PENGANTAR A. Latar Belakang ... 1
1. Permasalahan ... 2
2. Keaslian Penelitian... 2
3. Manfaat ... 3
B. Tujuan ... 3
1. Tujuan Umum ... 3
2. Tujuan Khusus ... 3
BAB II PENELAAHAN PUSTAKA A. Bioavailabilitas dan Bioekivalensi ... 4
1. Definisi ... 4
2. Faktor-Faktor yang Mempengaruhi Bioavailabilitas ... 8
3. Bioavailabilitas dan Disolusi In Vitro ... 18
4. Obat ... 19
B. Parasetamol ... 19
C. Farmakokinetika ... 23
1. Definisi ... 23
2. Strategi Penelitian Farmakokinetika ... 25
D. Nasib Obat di Dalam Tubuh ... 27
1. Absorpsi ... 27
2. Distribusi ... 29
3. Biotransformasi ... 29
4. Ekskresi ... 30
E. Dasar-Dasar Perhitungan Farmakokinetika ... 31
1. Model Kompartemen ... 31
2. Parameter Farmakokinetika ... 32
F. Darah ... 36
1. Plasma Darah ... 36
2. Denaturasi Protein Plasma ... 37
G. Kolorimetri ... 38
1. Definisi ... 38
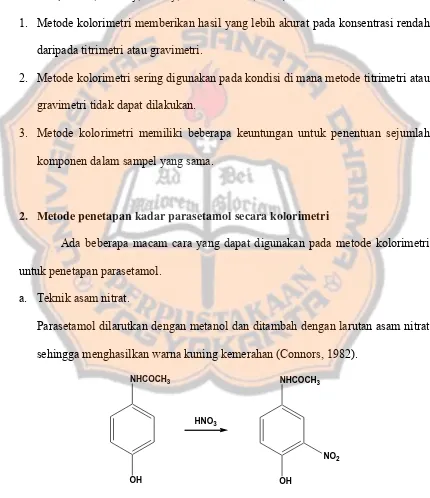
2. Metode Penetapan Kadar Parasetamol secara Kolorimetri ... 39
H. Desain Cross Over ... 42
I. Keterangan Empiris ... 42
BAB III METODOLOGI PENELITIAN A. Jenis dan Rancangan Penelitian ... 43
B. Variabel dan Definisi Operasional ... 43
1. Variabel Penelitian ... 43
2. Definisi Operasional ... 45
C. Bahan Penelitian ... 45
D. Alat Penelitian ... 46
E. Tata Cara Penelitian ... 46
1. Uji Pendahuluan Tablet ... 46
2. Pembuatan Larutan ... 49
3. Pembuatan Larutan Parasetamol ... 50
4. Cara Perolehan Plasma Darah ... 50
5. Optimasi Metode ... 51
6. Orientasi Dosis dan Waktu Pengambilan Sampel Darah ... 53
7. Perlakuan Hewan Uji ... 54
F. Analisis Hasil ... 56
1. Kesahihan Metode ... 56
2. Perhitungan Parameter Bioavailabilitas ... 57
3. Cara Penafsiran dan Penyimpulan Hasil Penelitian ... 57
BAB IV HASIL DAN PEMBAHASAN A. Uji Pendahuluan Tablet ... 58
1. Uji Keseragaman Bobot ... 59
2. Uji Kekerasan ... 60
3. Uji Kerapuhan ... 61
4. Uji Waktu Hancur ... 62
5. Uji Disolusi ... 63
B. Cara Perolehan Plasma Darah ... 67
C. Optimasi Metode ... 68
1. Penentuan Operating Time (OT) ... 72
2. Penentuan Panjang Gelombang Maksimum ... 74
3. Pembuatan Kurva Baku ... 75
4. Penentuan Nilai Perolehan Kembali, Kesalahan Sistematik, dan Kesalahan Acak ... 76
D. Orientasi Dosis dan Waktu Pengambilan Sampel Darah ... 78
E. Perbandingan Bioavailabilitas ... 79
1. Kadar Parasetamol dalam Plasma ... 79
2. AUC(0-inf) ... 83
3. Cmax ... 84
4. tmax ... 86
5. Kriteria Bioekivalen ... 87
BAB V KESIMPULAN DAN SARAN
A. Kesimpulan ... 93
B. Saran ... 94
DAFTAR PUSTAKA ... 95
LAMPIRAN ... 100
BIOGRAFI PENULIS ... 133
DAFTAR TABEL
Tabel I Konsep Desain Cross Over ... 54
Tabel II Parameter-Parameter Farmakokinetika ... 57
Tabel III Hasil Uji Keseragaman Bobot Tablet ... 59
Tabel IV Hasil Uji Kekerasan Tablet ... 61
Tabel V Hasil Uji Kerapuhan Tablet ... 61
Tabel VI Hasil Uji Waktu Hancur Tablet ... 62
Tabel VII Data Persamaan Kurva Baku Disolusi ... 64
Tabel VIII Data Disolusi Tablet ... 65
Tabel IX Kemiripan Profil Disolusi ... 66
Tabel X Data Persamaan Kurva Baku ... 76
Tabel XI Nilai Perolehan Kembali, Kesalahan Sistematik, dan Kesalahan Acak ... 77
Tabel XII Kadar Parasetamol dalam Plasma Setelah Pemberian Produk Obat ... 80
Tabel XIII ln Kadar Parasetamol dalam Plasma Setelah Pemberian Produk Obat ... 80
Tabel XIV Nilai Parameter Bioavailabilitas ... 82
Tabel XV Uji Post-Hoc Nilai AUC(0-inf) ... 83
Tabel XVI Uji Post-Hoc Nilai C(max) ... 85
Tabel XVII Uji Post-Hoc Nilai t(max) ... 86
Tabel XVIII Perbandingan Parameter Bioavailabilitas ... 88
Tabel XIX Hasil Penimbangan Tablet ... 100
Tablet XX Seri Kadar Larutan Intermediet Parasetamol dalam Pembuatan Kurva Baku Uji Disolusi ... 101
Tabel XXI Hasil Perhitungan Disolusi Tablet Parasetamol Generik .... 102
Tabel XXII Hasil Perhitungan Disolusi Tablet Parasetamol Pyrexin® .. 102
Tabel XXIII Hasil Perhitungan Disolusi Tablet Parasetamol Progesic® . 102
Tabel XXIV Perhitungan Persentase Kumulatif Obat Terlarut ... 105
Tabel XXV Konversi Perhitungan Dosis antar Jenis Hewan ... 107
Tabel XXVI Seri Kadar Larutan Intermediet Parasetamol dalam
Pembuatan Kurva Baku ... 110
Tabel XXVII Seri Kadar Larutan Intermediet Parasetamol dalam Plasma 110
Tabel XXVIII Konsentrasi Larutan Parasetamol untuk Penentuan Nilai Perolehan Kembali, Kesalahan Sistematik, dan Kesalahan Acak ... 112
Tabel XXIX Hasil Pengolahan STRIPE untuk Tablet Generik 1 ... 114
Tabel XXX Hasil Pengolahan STRIPE untuk Tablet Generik 2 ... 115
Tabel XXXI Hasil Pengolahan STRIPE untuk Tablet Generik 3 ... 116
Tabel XXXII Hasil Pengolahan STRIPE untuk Tablet Pyrexin® 1 ... 117
Tabel XXXIII Hasil Pengolahan STRIPE untuk Tablet Pyrexin® 2 ... 118
Tabel XXXIV Hasil Pengolahan STRIPE untuk Tablet Pyrexin® 3 ... 119
Tabel XXXV Hasil Pengolahan STRIPE untuk Tablet Progesic® 1 ... 120
Tabel XXXVI Hasil Pengolahan STRIPE untuk Tablet Progesic® 2 ... 121
Tabel XXXVII Hasil Pengolahan STRIPE untuk Tablet Progesic® 3 ... 122
Tabel XXXVIII Harga Rata-Rata Parameter Farmakokinetika ... 125
Tabel XXXIX Perhitungan Rata-Rata Parameter Bioavailabilitas untuk Penentuan Bioekivalensi ... 126
DAFTAR GAMBAR
Gambar 1 Proses Laju Bioavailabilitas Obat ... 7
Gambar 2 Struktur Parasetamol ... 20
Gambar 3. Metabolisme Parasetamol ... 22
Gambar 4 Proses Obat dalam Tubuh untuk Menimbulkan Efek ... 24
Gambar 5 Proses Farmakokinetika Obat di dalam Tubuh ... 27
Gambar 6 Reaksi Parasetamol dengan Asam Nitrat ... 39
Gambar 7 Reaksi Hidrolisis Parasetamol menjadi p-aminofenol ... 40
Gambar 8 Reaksi Pembentukan Warna pada Metode Chafetz et al. (1971) 41
Gambar 9 Kurva Hubungan antara Kadar Parasetamol dengan Serapan pada Uji Disolusi ... 65
Gambar 10 Profil Disolusi ... 66
Gambar 11 Reaksi antara Asam Klorida dengan Natrium Nitrit Membentuk Ion Nitrosonium ... 69
Gambar 12 Reaksi antara Parasetamol dengan Ion Nitrosonium Membentuk 2-nitro-4-asetamidofenol Beserta Gugus Kromofor dan Auksokromnya ... 69
Gambar 13 Mekanisme Reaksi antara Parasetamol dengan Ion Nitrosonium 70
Gambar 14 Reaksi antara Asam Nitrit dengan Asam Sulfamat ... 71
Gambar 15 Reaksi Penetralan Asam dan Pembentukan Ion Fenolat dalam Suasana Basa ... 71
Gambar 16 Mekanisme Reaksi antara 2-nitro-4-asetamidofenol dengan Natrium Hidroksida ... 72
Gambar 17 Pengukuran Operating Time (OT) Larutan Parasetamol dalam Plasma Kadar 100 μg/ml ... 73
Gambar 18 Pengukuran Operating Time (OT) Larutan Parasetamol dalam Plasma Kadar 400 μg/ml ... 73
Gambar 19 Pengukuran Panjang Gelombang Maksimum Larutan Parasetamol dalam Plasma Kadar 100 μg/ml ... 74
Gambar 20 Pengukuran Panjang Gelombang Maksimum Larutan
Parasetamol dalam Plasma Kadar 400 μg/ml ... 75
Gambar 21 Kurva Hubungan antara Kadar Parasetamol dengan Serapan .. 76
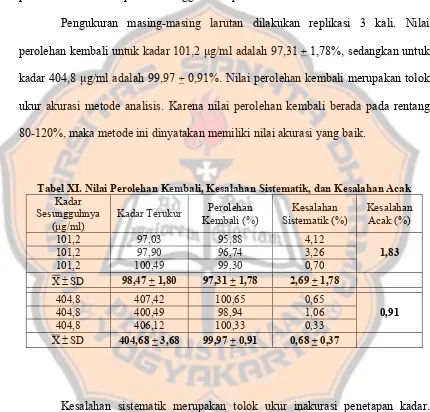
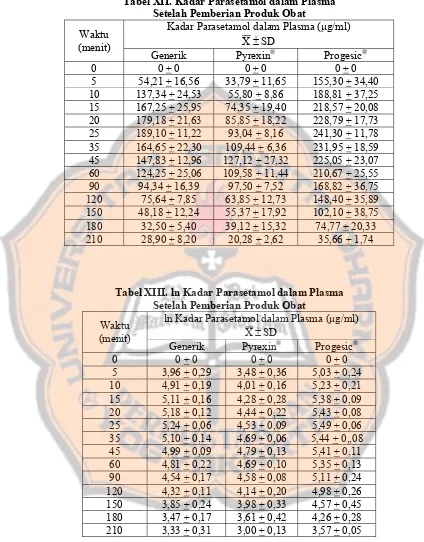
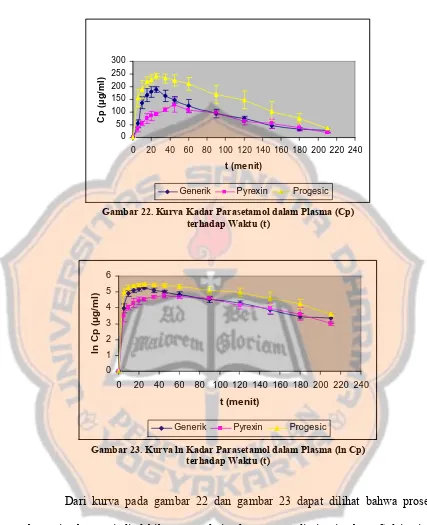
Gambar 22 Kurva Kadar Parasetamol dalam Plasma (Cp) terhadap Waktu
(t) ... 81
Gambar 23 Kurva ln Kadar Parasetamol dalam Plasma (ln Cp) terhadap
Waktu (t) ... 81
Gambar 24 Profil Disolusi Tablet Paraseamol (Generik) (A), Tablet
Pyrexin® (B), dan Tablet Progesic® (C) ... 104
Gambar 25 Kurva Kadar Parasetamol dalam Plasma (Cp) vs. Waktu (t)
pada Tablet Parasetamol Generik (A), Tablet Pyrexin® (B), dan
Tablet Progesic® (C) ... 123
Gambar 26 Kurva ln Kadar Parasetamol dalam Plasma (ln Cp) vs. Waktu
(t) pada Tablet Parasetamol Generik (A), Tablet Pyrexin® (B),
dan Tablet Progesic® (C) ... 124
DAFTAR LAMPIRAN
Lampiran 1 Hasil Penimbangan Tablet untuk Uji Keseragaman Bobot ... 100
Lampiran 2 Data Kurva Baku Disolusi Tablet ... 101
Lampiran 3 Hasil Uji Disolusi Tablet ... 102
Lampiran 4 Contoh Cara Perhitungan Data Disolusi Tablet ... 103
Lampiran 5 Grafik Uji Disolusi Tablet ... 104
Lampiran 6 Contoh Cara Perhitungan Faktor Kemiripan Profil Disolusi .... 105
Lampiran 7 Contoh Perhitungan Pembuatan Larutan Obat ... 106
Lampiran 8 Tabel Konversi Perhitungan Dosis Antar Jenis Hewan dan
Perhitungan Dosis Awal untuk Orientasi Dosis ... 107
Lampiran 9 Operating Time Larutan Parasetamol dalam Plasma dengan
Kadar 100 μg/ml (A) dan 400 μg/ml (B) ... 108
Lampiran 10 Panjang Gelombang Maksimum Larutan Parasetamol dalam
Plasma dengan Kadar 100 μg/ml (A) dan 400 μg/ml (B) ... 109
Lampiran 11 Data Kurva Baku Parasetamol ... 110
Lampiran 12 Kurva Baku ... 111
Lampiran 13 Pembuatan Larutan untuk Penentuan Nilai Perolehan
Kembali, Kesalahan Sistematik, dan Kesalahan Acak ... 112
Lampiran 14 Sertifikat Analisis Parasetamol ... 113
Lampiran 15 Hasil Pengolahan STRIPE untuk Tablet Generik ... 114
Lampiran 16 Hasil Pengolahan STRIPE untuk Tablet Pyrexin® ... 117
Lampiran 17 Hasil Pengolahan STRIPE untuk Tablet Progesic® ... 120
Lampiran 18 Kurva Kadar Parasetamol dalam Plasma (Cp) vs. Waktu (t) .... 123
Lampiran 19 Kurva ln Kadar Parasetamol dalam Plasma (ln Cp) vs. Waktu (t) 124
Lampiran 20 Harga Rata-Rata Parameter Farmakokinetika ... 125
Lampiran 21 Perhitungan Rata-Rata Parameter Bioavailabilitas untuk
Penentuan Bioekivalensi ... 126
Lampiran 22 Analisis Statistik (SPSS 14.0) ... 127
BAB I PENGANTAR
A. Latar Belakang
Obat yang beredar di pasaran dapat dibedakan menjadi dua macam, yaitu
obat generik dan obat bermerk dagang. Obat generik merupakan obat jadi yang
dipasarkan dengan nama umum (nama generik) bahan aktifnya sedangkan obat
bermerk dagang merupakan obat jadi yang dipasarkan dengan nama dagang yang
dipakai oleh masing-masing produsen (Anonim, 2000).
Setiap produsen pasti melakukan promosi untuk masing-masing produknya
sehingga harga obat bermerk dagang umumnya lebih mahal daripada obat generik
(Anonim, 2000). Fenomena yang sering terjadi adalah dokter jarang meresepkan obat
generik yang harganya lebih murah, sedangkan pasien cenderung untuk memilih obat
bermerk dagang dengan anggapan bahwa harga yang lebih mahal akan memberikan
efek terapeutik yang lebih baik.
Semua obat, baik obat generik maupun obat bermerk dagang, harus terjamin
keamanan dan khasiatnya. Hal tersebut dapat diuji secara farmakokinetika dan
farmakodinamika. Pendekatan farmakokinetika membicarakan tentang nasib obat
tersebut di dalam tubuh, meliputi proses absorpsi, distribusi, biotransformasi, dan
ekskresi sedangkan pendekatan farmakodinamika membicarakan tentang efek yang
ditimbulkan obat tersebut di dalam tubuh. Selama ini, kebanyakan pasien dan tenaga
kesehatan memandang obat hanya dari sisi farmakodinamika tanpa mengetahui sisi
diketahui sebab proses farmakokinetika berpengaruh terhadap keseluruhan aksi obat,
termasuk efek terapeutik yang dihasilkan.
Dalam penelitian ini, dilakukan perbandingan antara obat bermerk dagang
terhadap obat generik secara farmakokinetika, yaitu dengan membandingkan
parameter-parameter bioavailabilitas obat bermerk dagang terhadap obat generik
pada kelinci putih jantan. Sampel yang digunakan adalah beberapa tablet yang
mengandung parasetamol sebagai zat aktif tunggal, yaitu tablet parasetamol
(generik), tablet Pyrexin®, dan tablet Progesic®. Penulis memilih parasetamol sebab
parasetamol banyak digunakan dalam obat bebas dan obat bebas terbatas sebagai
analgesik-antipiretik yang dapat diperoleh dengan mudah oleh pasien.
1. Permasalahan
Masalah yang diangkat dari latar belakang tersebut adalah apakah tablet
parasetamol (generik), tablet Pyrexin®, dan tablet Progesic® memiliki bioavailabilitas
yang sama ?
2. Keaslian penelitian
Sejauh yang penulis ketahui, masalah tersebut belum pernah diteliti dalam
penelitian di lingkungan Universitas Sanata Dharma Yogyakarta dan Universitas
3. Manfaat
Manfaat teoritis. Penelitian ini diharapkan dapat memberikan informasi
tentang bioavailabilitas tablet parasetamol (generik), tablet Pyrexin®, dan tablet
Progesic® pada kelinci putih jantan.
B. Tujuan Tujuan dari penelitian ini adalah :
1. Tujuan umum
Untuk mengetahui bioavailabilitas tablet parasetamol (generik), tablet
Pyrexin®, dan tablet Progesic® pada kelinci putih jantan.
2. Tujuan khusus
Untuk mengetahui ada tidaknya perbedaan bioavailabilitas antara tablet
parasetamol (generik), tablet Pyrexin®, dan tablet Progesic® pada kelinci putih
BAB II
PENELAAHAN PUSTAKA
Berkaitan dengan penelitian Perbandingan Bioavailabilitas Tablet Pyrexin®
dan Tablet Progesic® dengan Tablet Parasetamol (Generik) pada Kelinci Putih
Jantan, maka dalam bab ini ditelaah tentang Bioavailabilitas dan Bioekivalensi,
Parasetamol, Farmakokinetika, Nasib Obat di Dalam Tubuh, Dasar-Dasar
Perhitungan Farmakokinetika, Darah, Kolorimetri, dan Desain Cross Over.
A. Bioavailabilitas dan Bioekivalensi 1. Definisi
Bioavailabilitas (ketersediaan hayati) merupakan persentase dan kecepatan
zat aktif dalam suatu produk obat yang mencapai/tersedia dalam sirkulasi sistemik
dalam bentuk utuh/aktif setelah pemberian produk obat tersebut. Bioavailabilitas
dapat diukur dari kadarnya dalam darah terhadap waktu atau dari ekskresinya dalam
urin (Anonim, 2004b).
Terdapat dua macam bioavailabilitas, yaitu bioavailabilitas absolut dan
bioavailabilitas relatif. Bioavailabilitas absolut merupakan perbandingan
bioavailabilitas obat yang diberikan secara ekstravaskular terhadap bioavailabilitas
obat yang diberikan secara intravaskular, sedangkan bioavailabilitas relatif
merupakan perbandingan bioavailabilitas produk obat terhadap pembanding (selain
Istilah ekivalensi atau kesetaraan digunakan dalam perbandingan suatu
produk obat dengan produk obat lainnya. Ada beberapa istilah ekivalensi menurut
Malinowski (2000).
a. Ekivalensi kimia.
Jika dua atau lebih bentuk sediaan mengandung obat seperti yang tertera pada
etiket.
b. Ekivalensi klinik.
Jika obat yang sama dalam dua atau lebih bentuk sediaan memberikan efek in
vivo yang identik, yang dapat dilihat dari respon farmakologi atau kontrol
terhadap gejala atau penyakit.
c. Ekivalensi terapeutik.
Ekivalensi terapeutik berarti bahwa dua merk obat diharapkan menghasilkan efek
klinik yang sama.
d. Bioekivalensi.
Jika obat dalam dua atau lebih bentuk sediaan yang sejenis mencapai sirkulasi
sistemik dengan jumlah dan kecepatan yang relatif sama.
e. Ekivalensi farmasetik.
Jika dua produk obat mengandung zat aktif yang sama dalam bentuk sediaan dan
kekuatan yang sama.
Bioekivalensi merupakan perbandingan bioavailabilitas dari dua atau lebih
produk obat. Dua produk atau formulasi yang mengandung zat aktif sama dikatakan
Menurut Pedoman Uji Bioekivalensi Badan POM RI, dua produk obat disebut
bioekivalen jika keduanya mempunyai ekivalensi farmasetik atau merupakan
alternatif farmasetik dan pada pemberian dengan dosis molar yang sama akan
menghasilkan bioavailabilitas yang sebanding sehingga efeknya akan sama, baik
dalam hal efikasi maupun keamanan. Dua produk obat mempunyai ekivalensi
farmasetik jika keduanya mengandung zat aktif yang sama dalam jumlah dan bentuk
sediaan yang sama. Dua produk obat merupakan alternatif farmasetik jika keduanya
mengandung zat aktif yang sama tetapi berbeda dalam bentuk kimia (garam, ester,
dsb.) atau bentuk sediaan atau kekuatan.
Studi bioavailabilitas digunakan untuk menunjukkan efek sifat fisika kimia
komponen obat dan bentuk sediaan terhadap farmakokinetika obat. Studi
bioekivalensi digunakan untuk membandingkan bioavailabilitas obat dengan zat aktif
yang sama dari berbagai produk obat. Apabila produk obat tersebut bioekivalen
maka efikasi dan profil keamanan produk-produk obat tersebut dapat dianggap sama
dan dapat digantikan satu dengan yang lain (Shargel, Wu-Pong, and Yu, 2005).
Respon farmakologis pada umumnya terkait dengan konsentrasi obat pada
reseptor sehingga ketersediaan obat dari bentuk sediaan merupakan faktor yang
penting dalam menentukan efikasi obat. Konsentrasi obat pada tempat aksi biasanya
tidak dapat diukur secara langsung sehingga kebanyakan studi bioavailabilitas
melibatkan pengukuran konsentrasi obat di dalam darah atau urin. Hal ini
berdasarkan pada suatu anggapan bahwa obat pada tempat aksi berada dalam
Obat dalam bentuk sediaan padat yang ditujukan untuk penggunaan sistemik
umumnya mengalami absorpsi melalui suatu rangkaian proses, yaitu disintegrasi
produk obat yang diikuti pelepasan obat, pelarutan obat dalam media aqueous, dan
absorpsi melewati membran sel menuju sirkulasi sistemik (Shargel et al., 2005).
Di dalam proses tersebut, kecepatan obat mencapai sistem sirkulasi
ditentukan oleh tahap yang paling lambat. Tahap yang paling lambat di dalam
rangkaian proses kinetik disebut tahap penentu kecepatan (rate limiting step).
Bentuk sediaan padat
disintegrasi deagregasi
Granul Partikel kecil
Gambar 1. Proses laju bioavailabilitas obat (Malinowski, 2000)
Untuk obat-obat yang mempunyai kelarutan kecil dalam air, laju pelarutan biasanya
merupakan tahap yang paling lambat sehingga menjadi penentu kecepatan terhadap
bioavailabilitas obat (Shargel et al., 2005).
Studi bioavailabilitas dilakukan terhadap bahan obat aktif yang telah
disetujui maupun obat dengan efek terapeutik yang belum disetujui oleh Food and
Drug Administration (FDA) untuk dipasarkan. Dalam menyetujui suatu produk obat
untuk dipasarkan, FDA harus memastikan bahwa produk obat tersebut aman dan
efektif sesuai label indikasi penggunaan. Selain itu, produk obat juga harus
memenuhi seluruh standar yang digunakan dalam identitas, kekuatan, kualitas dan
kemurnian (Shargel et al., 2005).
Disolusi obat Disolusi obat
Larutan obat
Absorpsi
Obat dalam darah
Untuk meyakinkan bahwa standar-standar tersebut telah dipenuhi, FDA
menghendaki studi bioavailabilitas/farmakokinetika dan bila perlu persyaratan
bioekivalensi untuk semua produk (Shargel et al., 2005). Akibat perkembangan studi
bioavailabilitas dan bioekivalensi, maka diperlukan suatu kepastian bahwa produk
generik bioekivalen terhadap produk dagang sehingga produk generik tidak perlu
diragukan lagi jika diresepkan oleh dokter (Chereson, 1999).
2. Faktor-faktor yang mempengaruhi bioavailabilitas
Bioavailabilitas sangat dipengaruhi oleh proses absorpsi. Obat-obat yang
diberikan secara oral harus diabsorpsi terlebih dahulu sebelum memberikan efek
terapeutik. Faktor-faktor yang mempengaruhi proses absorpsi adalah sebagai berikut.
a. Rute dan cara pemberian.
Obat yang diberikan secara oral, subkutan, intramuskular, intradermal,
hipodermal atau intraperitoneal memerlukan proses absorpsi. Beberapa obat
yang diberikan secara oral akan termetabolisme pada saluran pencernaan dalam
jumlah yang besar sehingga hanya sedikit obat yang dapat mencapai sirkulasi
sistemik. Kebanyakan obat yang diberikan secara oral juga mengalami first- pass
effect sehingga tidak semua obat yang diberikan akan diabsorpsi (Wagner, 1975).
b. Dosis dan aturan dosis.
Dosis yang diberikan harus diperhatikan agar konsentrasi obat dalam darah dapat
c. Efek bentuk sediaan.
Bentuk sediaan obat dapat mempengaruhi laju dan jumlah obat yang mencapai
sirkulasi sistemik.
1) Sifat fisika kimia obat.
a) Faktor yang mempengaruhi kelarutan.
Laju pelarutan obat dijelaskan dengan persamaan Noyes-Whitney
(Proudfoot, 1990) :
C)
= laju disolusi partikel obat
D = koefisien difusi A = luas permukaan efektif h = tebal lapisan difusi
Cs = kelarutan jenuh obat pada lapisan difusi C = konsentrasi obat pada cairan gastrointestinal
Faktor-faktor yang mempengaruhi kelarutan adalah sebagai berikut.
(1) Bentuk kristal, amorf, polimorfi, solvate.
Polimorfi.
Banyak obat memiliki lebih dari satu bentuk kristal. Hal ini disebut
dengan istilah polimorfi, sedangkan masing-masing bentuk kristal
disebut dengan istilah polimorf. Bentuk polimorf metastabil memiliki
kelarutan dalam air paling besar (Proudfoot, 1990).
Amorf
bentuk amorf biasanya lebih larut dan laju disolusinya lebih cepat
Solvate
solvate adalah bentuk kristal yang terbentuk ketika obat berikatan
dengan molekul pelarut (solvent). Jika pelarutnya air, maka bentuk
solvate dinamakan hidrat. Biasanya semakin besar solvation pada
kristal, maka kelarutan dan laju disolusinya akan menurun (Proudfoot,
1990).
(2) Asam bebas, basa bebas, atau bentuk garam.
Bentuk asam bebas, basa bebas dan bentuk garam dapat
mempengaruhi kelarutan obat. Sebagai contoh : garam logam alkali
dari asam organik lemah (misal : natrium atau kalium warfarin) akan
terdisolusi lebih cepat daripada bentuk asam lemahnya. Serupa
dengan itu, garam asam mineral dari basa lemah (misal : amina atau
sulfat) akan terdisolusi dengan lebih cepat daripada basa lemahnya
(Wagner, 1975).
(3) Nilai pKa.
Pengaruh nilai pKa dalam kelarutan obat dapat dijelaskan dalam
persamaan Krebs & Speakman :
untuk asam monobasa : SpH = S0 (1+10(pH-pKa)) (2)
untuk basa monoasam : SpH = S0 (1+10(pKa-pH)) (3)
Keterangan :
SpH = kelarutan pada pH tertentu
S0 = kelarutan intrinsik (kelarutan bentuk tak terion)
(4) Kompleksasi, solid solution, eutectics.
Laju dan jumlah obat yang diabsorpsi tergantung pada konsentrasi
efektif obat. Kompleksasi dapat mempengaruhi konsentrasi efektif
obat pada cairan gastrointestinal. Contoh kompleksasi yang terjadi
adalah antara mucin dengan obat-obat tertentu (misal streptomisin)
yang membentuk kompleks yang tidak dapat diabsorpsi (Proudfoot,
1990).
(5) Surfaktan.
Surfaktan memiliki efek yang bervariasi pada laju disolusi dan
absorpsi. Biasanya surfaktan menurunkan tegangan permukaan
sehingga laju disolusi akan meningkat. Namun jika konsentrasi
surfaktan sudah di atas critical micelle concentrations, maka
surfaktan akan membentuk micelle dengan obat sehingga laju absorpsi
obat akan menurun sebab obat yang dapat diabsorpsi hanya obat
dalam bentuk bebas (Wagner, 1975).
b) Faktor yang mempengaruhi transport obat.
(1) Nilai pKa dan pH.
Banyak obat mengandung substituen lipofilik dan hidrofilik.
Obat-obat yang lebih larut dalam lemak akan lebih mudah melewati
membran sel daripada obat yang kurang larut lemak. Bagi obat yang
bersifat sebagai elektrolit lemah, besarnya ionisasi mempengaruhi laju
Ionisasi suatu elekrolit lemah tergantung pada nilai pKa dan pH yang
dijelaskan dalam persamaan Handerson-Hasselbach :
untuk asam lemah :
(2) Ada tidaknya muatan.
Muatan pada obat dapat mempengaruhi transport obat menembus
membran. Berdasarkan penelitian Benet dkk., ternyata bentuk ion dari
obat juga dapat menembus membran (Wagner, 1975).
(3) Koefisien partisi.
Semakin besar koefisien partisi obat antara membran dan lumen,
maka laju absorpsi akan semakin besar pula (Wagner, 1975).
(4) Molal volume, monomeric atau micellar, dan difusivitas.
Laju difusi micelle lebih lambat daripada laju difusi monomeric
(Wagner, 1975).
(5) Stagnant water layer (aqueous diffusion layer).
Perpindahan obat melewati aqueous diffusion layer antara luminal dan
permukaan membran dapat menjadi rate limiting step dalam proses
2) Faktor farmasetika dan pembuatan bentuk sediaan padat.
a) Ukuran partikel dan luas permukaan spesifik.
Laju disolusi obat berbanding langsung dengan luas permukaan spesifik
(Wagner, 1975). Penurunan ukuran partikel akan menyebabkan
peningkatan luas permukaan spesifik (York, 1990). Laju disolusi, laju
absorpsi, keseragaman kandungan dalam bentuk sediaan dan stabilitas
bentuk sediaan tergantung pada ukuran partikel dan ukuran distribusinya.
b) Static electrification.
Beberapa proses seperti pencampuran dan penyalutan dapat menghasilkan
static electrification. Hal ini dapat menyebabkan terjadinya agregasi
partikel dan terjadinya unmixing (tidak tercampurnya) obat. Agregasi
menyebabkan penurunan luas permukaan sehingga laju disolusi menjadi
lebih lambat (Wagner, 1975).
c) Tipe bentuk sediaan.
Pada umumnya, urutan laju absorpsi obat dalam bentuk sediaan dari yang
tercepat hingga terlambat adalah larutan, suspensi, tablet, tablet salut
gula, dan tablet salut enterik. Namun urutan tersebut dapat berubah jika
obat terdegradasi oleh asam di lambung (Wagner, 1975).
d) Tipe dan jumlah bahan tambahan.
Secara umum, penggunaan bahan tambahan yang tidak larut air akan
menyebabkan laju disolusi dan absorpsi obat menjadi lebih lambat
dibandingkan dengan penggunaan bahan tambahan yang larut air. Hal ini
larut air sehingga obat menjadi lebih hidrofob. Penambahan garam netral
akan meningkatan disolusi obat (Wagner, 1975).
e) Ukuran granul dan distribusi ukurannya.
Granulasi merupakan salah satu proses dalam pembuatan tablet. Proses
disintegrasi tablet diasumsikan melalui 2 tahap, yaitu tablet menjadi
granul dan granul menjadi partikel kecil. Oleh karena itu, ukuran granul
dan distribusi ukurannya menjadi penting untuk diperhatikan (Wagner,
1975).
f) Tipe dan jumlah bahan penghancur.
Bahan penghancur akan mengembang oleh adanya air dan mendesak
tablet untuk hancur. Semakin banyak jumlah bahan penghancur yang
digunakan, maka tablet semakin mudah hancur (Wagner, 1975).
g) Waktu pencampuran.
Dalam proses pencampuran terdapat waktu optimum, di mana setelah
waktu optimum terlewati, obat menjadi tidak tercampur lagi (Wagner,
1975).
h) Tekanan dan kecepatan kompresi.
Tekanan kompresi merupakan faktor penentu waktu hancur dan laju
disolusi obat dari tablet (Wagner, 1975).
i) Penyalutan (salut film, salut gula, salut enterik).
Tablet salut film terdisolusi lebih cepat daripada tablet salut gula. Tablet
enterik tidak larut pada lambung, namun larut pada usus halus (Wagner,
1975).
j) Efek matriks.
Dalam tablet lepas lambat, obat dicampur dengan wax atau polimer
sintetik yang inert dan tidak dapat diabsorpsi di saluran pencernaan, yang
disebut dengan matriks. Saat tablet tersebut diberikan secara oral, cairan
akan masuk ke dalam matriks dan dengan perlahan akan melarutkan obat
dari matriks (Wagner, 1975).
k) Tipe dan jumlah surfaktan.
Surfaktan dapat menurunkan tegangan antarmuka antara obat dengan
media disolusi sehingga dapat meningkatkan laju disolusi (Wagner,
1975).
l) Kondisi lingkungan selama pembuatan.
Jika obat mudah terhidrolisis, maka stabilitas bentuk sediaan dapat
dipengaruhi oleh kondisi lingkungan selama pembuatan (Wagner, 1975).
m) Kondisi saat penyimpanan dan lama penyimpanan.
Stabilitas obat dalam bentuk sediaan tertentu dapat diuji dengan uji
stabilitas bentuk sediaan dengan peningkatan temperatur (Wagner, 1975).
d. Faktor fisiologis.
1) Waktu transit obat.
Semakin lama obat berada di usus halus, maka semakin banyak obat yang
diabsorpsi dengan asumsi bahwa obat stabil pada cairan intestinal (Proudfoot,
2) Laju pengosongan lambung.
Kebanyakan obat diabsorpsi secara optimal pada usus halus. Penurunan laju
pengosongan lambung akan menurunkan laju absorpsi obat dan menunda
waktu onset obat. Laju pengosongan lambung juga penting untuk obat yang
mudah terdegradasi di lambung. Semakin lama obat berada di lambung, maka
semakin banyak obat yang terdegradasi sehingga bioavailabilitasnya akan
menurun. Adanya makanan akan menurunkan laju pengosongan lambung
sehingga absorpsi obat akan tertunda (Proudfoot, 1990).
3) Luas permukaan area efektif pada tempat absorpsi.
Usus halus memiliki luas permukaan area efektif terbesar karena adanya vili
dan mikrovili. Oleh karena itu, mayoritas obat akan diabsorpsi secara
maksimum pada usus halus, meskipun pH cairan intestinal bukan merupakan
kondisi optimum untuk absorpsi obat-obat asam lemah/basa lemah.
Sebaliknya, luas permukaan lambung dan usus besar relatif kecil karena tidak
memiliki vili dan mikrovili (Proudfoot, 1990).
4) Laju aliran darah.
Aliran darah pada saluran pencernaan merupakan faktor yang penting untuk
membawa obat ke sirkulasi sistemik kemudian ke tempat kerja. Di dalam
usus terdapat pembuluh-pembuluh darah mesentrika. Obat dilepaskan ke hati
melalui vena porta hepatika dan kemudian menuju ke sirkulasi sistemik. Jika
laju aliran darah mesentrika menurun, maka bioavailabilitas obat juga akan
5) Nilai pH cairan pada saluran pencernaan.
Nilai pH cairan bervariasi di sepanjang saluran pencernaan. pH lambung
1-3,5; pH usus halus 5-8 (pH duodenum 5-6, pH ileum 8); pH usus besar 8.
Derajat ionisasi obat dipengaruhi oleh nilai pH. Bentuk tak terion akan
diabsorpsi lebih cepat daripada bentuk terion. Perubahan nilai pH pada
saluran pencernaan (karena adanya makanan atau faktor lain) dapat
menyebabkan perubahan jumlah bentuk tak terion sehingga dapat
mempengaruhi absorpsinya (Proudfoot, 1990).
6) Aktivitas enzimatik.
Obat yang diberikan secara oral dan ditujukan untuk sirkulasi sistemik
biasanya mengalami first pass effect, di mana obat akan termetabolisme
sebelum mencapai sirkulasi sistemik. First pass effect menyebabkan
penurunan bioavailabilitas (Proudfoot, 1990).
7) Mukus dan glycocalyx.
Molekul obat harus melalui unstirred aqueous layer, lapisan mukus, dan
glycocalyx untuk mencapai mikrovili. Glycocalyx adalah bagian yang
menyatu dengan mikrovili, berfungsi sebagai penyalut bagi mikrovili dan
tersusun atas mukopolisakarida (Proudfoot, 1990).
8) Ada tidaknya makanan pada saluran pencernaan.
Makanan dapat mempengaruhi absorpsi obat dengan beberapa mekanisme, di
antaranya mengubah laju pengosongan lambung, memacu sekresi asam dan
absorpsi, membentuk kompleks dengan obat, meningkatkan viskositas pada
saluran pencernaan (Proudfoot, 1990).
9) Lain-lain : konsentrasi elektrolit, tegangan permukan dan tegangan
antarmuka, emulsifying agents dan complexing agents (misal : garam
empedu), posisi anatomi tubuh dan aktivitas relatif, suhu tubuh, integritas
membran gastrointestinal, tekanan hidrostatik dan intralumenal, kapasitas
buffer, tonisitas (Wagner, 1975).
3. Bioavailabilitas dan disolusi in vitro
Disolusi adalah proses di mana bahan obat padat larut dalam pelarut. Uji
disolusi dapat menentukan bioavailabilitas suatu obat jika terdapat korelasi yang baik
antara uji in vitro dan in vivo. Korelasi in vitro dan in vivo yang dimaksud adalah
hubungan antara karakteristik biologi obat (efek farmakodinamika atau konsentrasi
obat dalam plasma) dan karakteristik fisika kimia produk obat (Shargel et al., 2005).
Korelasi in vitro dan in vivo ini penting untuk diketahui agar dalam
menentukan bioavailabilitas suatu obat cukup dengan uji in vitro saja, tidak perlu
dengan uji in vivo. Selama ini, uji bioavailabilitas secara in vivo memerlukan waktu
yang lama, biaya yang relatif tinggi, serta terdapat beberapa masalah dalam
pemberian obat kepada subjek uji sehat/pasien (Chereson, 1999).
Parameter uji in vitro yang paling dekat hubungannya dengan
bioavailabilitas adalah laju disolusi. Obat yang masuk ke dalam tubuh dapat
bentuk sediaannya (laju disolusi) akan menentukan kecepatan dan atau jumlah obat
yang terabsorpsi (Chereson, 1999).
4. Obat
Menurut S. P. Menkes R. I. No. 193/Keb/VII/71, obat adalah suatu bahan
atau paduan bahan-bahan yang digunakan dalam menetapkan diagnosa, mencegah,
mengurangi, menghilangkan, menyembuhkan penyakit atau gejala penyakit, luka,
atau kelainan badaniah dan rohaniah pada manusia atau hewan, memperelok badan
atau bagian badan manusia (Lestari, Rahayu, Rya, Suhardjono, Maisunah, Soewarni,
dkk., 2002).
Obat generik adalah obat dengan nama resmi yang ditetapkan dalam
Farmakope Indonesia untuk zat berkhasiat yang dikandungnya. Nama generik adalah
nama obat berdasarkan International Nonproprietary Name (I.N.N.) yang ditetapkan
WHO. Nama generik berlaku di negara manapun dan boleh diproduksi oleh setiap
industri, sedangkan obat paten yaitu obat jadi dengan nama dagang yang terdaftar
atau nama pembuat atau yang dikuasakannya dan dijual dalam bungkus asli dari
pabrik yang memproduksinya. Nama dagang adalah nama khas yang dilindungi
hukum yaitu merk terdaftar atau Proprietary Name (Lestari dkk., 2002).
B. Parasetamol
Parasetamol memiliki beberapa sinonim, di antaranya asetaminofen,
p-acetamidophenol, dan N-acetyl-p-aminophenol (Connors, Amidon, and Stella, 1986).
C8H9NO2 dihitung terhadap zat anhidrat. Parasetamol berupa serbuk hablur, putih,
tidak berbau, berasa sedikit pahit. Tablet parasetamol mengandung parasetamol
(C8H9NO2) tidak kurang dari 90,0% dan tidak lebih dari 110,0% dari jumlah yang
tertera pada etiket (Anonim, 1995).
HO NHCOCH3
Parasetamol
BM = 151,16
Gambar 2. Struktur parasetamol (Anonim, 1995)
Kelarutan parasetamol adalah mudah larut dalam etanol (95%) P dan dalam
propilenglikol P; larut dalam air mendidih, dalam natrium hidroksida 1N, dan dalam
aseton P; agak sukar larut dalam air dan dalam gliserol P (Anonim, 1979; Anonim
1995). Parasetamol tidak larut dalam benzen dan eter (Connors et al., 1986).
Titik lebur parasetamol adalah 169°C-172°C. Dalam larutan jenuh, pH
parasetamol adalah sekitar 5,3-6,5. Parasetamol memiliki nilai pKa 9,51.
Parasetamol sangat stabil dalam larutan air dan stabil dalam larutan dengan nilai pH
5-7. Parasetamol dapat membentuk kompleks dengan polyethyleneglycol (PEG) 4000
dan polyvynylpyrrolidone (PVP). Kompleks ini akan meningkatkan kelarutan
parasetamol dalam air dan kecepatan disolusi parasetamol. Parasetamol akan
menghasilkan efek terbaik dalam campuran parasetamol dan PEG dengan
perbandingan parasetamol : PEG = 1 : 2 b/b (Connors et al., 1986; Hanson, 2000).
Parasetamol diabsorpsi secara cepat dan lengkap melalui saluran
pencernaan. Absorpsi parasetamol menurun jika asupan parasetamol diikuti dengan
Anonim, 2005a). McGilveray dan Mattok (1972) menemukan bahwa adanya
makanan akan menurunkan absorpsi parasetamol. Pemberian makanan bersama 1
gram parasetamol ternyata menurunkan kecepatan absorpsi menjadi lima kali lebih
lambat daripada pemberian parasetamol pada manusia puasa. Makanan yang
mengandung karbohidrat dan pektin dapat menurunkan kecepatan absorpsi
parasetamol. Sebaliknya, keadaan puasa ternyata meningkatkan kecepatan absorpsi
parasetamol walaupun tidak mempengaruhi jumlah total yang diabsorpsi.
Waktu onset parasetamol kurang dari 1 jam dengan durasi 4-6 jam (Lacy et
al., 2003). Parasetamol memiliki tmax 0,5-2 jam. Parasetamol terdistribusi hampir ke
seluruh cairan tubuh (Anonim, 2004a). Di dalam plasma, sebanyak 20-50%
parasetamol akan terikat oleh protein plasma (Lacy et al., 2003). Volume distribusi
parasetamol menurut Melmon & Morelli (1992) adalah 0,94 l/kg. Besarnya
konsentrasi efektif minimum (KEM) parasetamol adalah 10-20 μg/ml, sedangkan
konsentrasi toksik minimum (KTM) adalah 300 μg/ml (Benet, 1992).
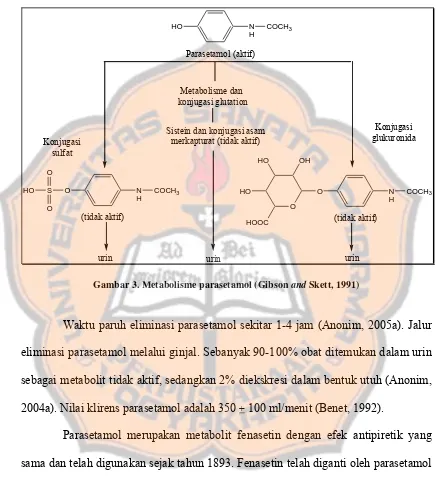
Sebanyak 90-95% parasetamol dimetabolisme oleh hati, dalam reaksi
konjugasi glutation, konjugasi glukuronida, dan konjugasi sulfat. Metabolit hasil
konjugasi tersebut merupakan metabolit yang tidak aktif secara farmakologis
(Gibson and Skett, 1991). Proses metabolisme parasetamol dapat dilihat pada
gambar 3.
Sebagian lainnya dimetabolisme oleh enzim sitokrom P450 menjadi
metabolit toksik yang berbahaya bagi sel hati (Anonim, 2004a). Glutation di dalam
tubuh dapat berikatan dengan metabolit ini dan membuatnya menjadi tidak toksik.
dosis parasetamol terlalu tinggi tetap bersifat toksik (Bowman and Rand, 1990;
(tidak aktif) (tidak aktif)
Metabolisme dan konjugasi glutation
Sistein dan konjugasi asam merkapturat (tidak aktif)
urin urin urin
Gambar 3. Metabolisme parasetamol (Gibson and Skett, 1991)
Waktu paruh eliminasi parasetamol sekitar 1-4 jam (Anonim, 2005a). Jalur
eliminasi parasetamol melalui ginjal. Sebanyak 90-100% obat ditemukan dalam urin
sebagai metabolit tidak aktif, sedangkan 2% diekskresi dalam bentuk utuh (Anonim,
2004a). Nilai klirens parasetamol adalah 350 + 100 ml/menit (Benet, 1992).
Parasetamol merupakan metabolit fenasetin dengan efek antipiretik yang
sama dan telah digunakan sejak tahun 1893. Fenasetin telah diganti oleh parasetamol
dalam banyak sediaan. Namun sampai sekarang tidak dijamin sempurna bahwa
pemberian parasetamol dalam waktu lama lebih kurang toksik terhadap ginjal
Efek analgesik parasetamol serupa dengan salisilat yaitu menghilangkan
atau mengurangi nyeri ringan sampai sedang. Parasetamol menurunkan suhu tubuh
dengan mekanisme yang diduga juga berdasarkan efek sentral seperti salisilat
(Wilmana, 2003). Parasetamol memiliki efek analgesik antipiretik yang sama dengan
aspirin. Parasetamol merupakan obat pilihan bagi pasien yang memerlukan efek
analgesik sedang atau antipiretik dan bagi pasien yang kontraindikasi dengan aspirin,
yaitu pasien yang hipersensitif terhadap aspirin, pasien yang mempunyai riwayat
ulcer, pasien dengan penyakit gout, anak yang terinfeksi virus, dan pasien yang
sedang mengkonsumsi antikoagulan (Anonim, 2001a).
C. Farmakokinetika 1. Definisi
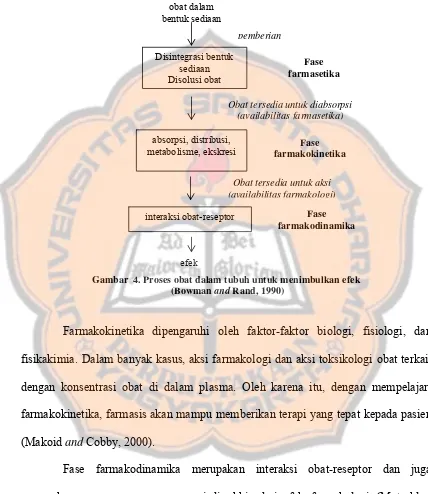
Proses yang berawal dari pemberian obat hingga efek yang ditimbulkan oleh
obat dapat dibagi menjadi tiga fase, yaitu fase farmasetika, farmakokinetika, dan
farmakodinamika. Tahap-tahap tersebut dapat dilihat pada gambar 4.
Fase farmasetika meliputi hancurnya bentuk sediaan obat dan larutnya
bahan obat. Oleh karena itu, fase ini terutama ditentukan oleh sifat-sifat galenik obat
(Mutschler, 1999).
Fase farmakokinetika meliputi proses invasi (absorpsi, distribusi) dan proses
eliminasi (biotransformasi, ekskresi) (Mutschler, 1999). Farmakokinetika merupakan
ilmu yang menggambarkan rentang waktu perpindahan obat masuk ke dalam tubuh,
Shargel et al. (2005), farmakokinetika mempelajari kinetika absorpsi, distribusi dan
eliminasi obat.
obat dalam
Obat tersedia untuk diabsorpsi (availabilitas farmasetika)
efek
Obat tersedia untuk aksi (availabilitas farmakologi)
Gambar 4. Proses obat dalam tubuh untuk menimbulkan efek (Bowman and Rand, 1990)
Farmakokinetika dipengaruhi oleh faktor-faktor biologi, fisiologi, dan
fisikakimia. Dalam banyak kasus, aksi farmakologi dan aksi toksikologi obat terkait
dengan konsentrasi obat di dalam plasma. Oleh karena itu, dengan mempelajari
farmakokinetika, farmasis akan mampu memberikan terapi yang tepat kepada pasien
(Makoid and Cobby, 2000).
Fase farmakodinamika merupakan interaksi obat-reseptor dan juga
merupakan proses-proses yang menjadi akhir dari efek farmakologi (Mutschler,
2. Strategi penelitian farmakokinetika
Definisi dari strategi penelitian farmakokinetika (SPF) adalah rencana yang
disusun sebelum meneliti tahap farmakokinetika obat untuk memperoleh informasi
tentang nasib obat dalam tubuh secara kuantitatif. Objek penelitian farmakokinetika
adalah tahap farmakokinetika obat dengan parameter farmakokinetika sebagai tolok
ukurnya. Parameter farmakokinetika adalah besaran yang diturunkan secara
matematik dari hasil pengukuran kadar obat atau metabolitnya di dalam darah atau
urin (Suryawati dan Donatus, 1998).
SPF meliputi tahap-tahap sebagai berikut.
a. Pemilihan rancangan uji coba.
b. Pemilihan subjek uji dan jumlahnya.
c. Pemilihan cuplikan hayati.
d. Pemilihan metode analisis penetapan kadar.
Metode analisis ini memiliki syarat-syarat sebagai berikut.
1) Selektivitas
Selektivitas adalah kemampuan metode analisis untuk membedakan suatu
obat dengan metabolitnya, obat lain dan kandungan endogen cuplikan
hayati.
2) Sensitivitas
Sensitivitas berkaitan dengan kadar terendah yang dapat diukur dengan
metode analisis yang digunakan. Hal ini diperlukan karena dalam
data kadar obat dari waktu ke waktu atau dari kadar tertinggi sampai kadar
terendah dalam cuplikan hayati yang digunakan.
3) Ketelitian dan ketepatan
Ketelitian dan ketepatan ini akan menentukan kesahihan hasil penetapan
kadar. Ketepatan (akurasi) ditunjukkan oleh kemampuan metode
memberikan hasil pengukuran sedekat mungkin dengan nilai yang
sesungguhnya. Ketelitian (presisi) menunjukkan kedekatan hasil
pengukuran berulang pada cuplikan hayati yang sama.
e. Pemilihan takaran dosis dan bentuk sediaan obat.
Takaran dosis yang diberikan harus menjamin dapat diukurnya kadar obat atau
metabolitnya pada rentang waktu tertentu sehingga diperoleh data yang cukup
memadai untuk analisis farmakokinetika.
f. Pemilihan lama dan banyaknya waktu pengambilan cuplikan hayati.
Apabila menggunakan cuplikan darah, sebaiknya pengambilan dilakukan
sebanyak 3-5 kali t½ eliminasi obat yang diuji. Hal ini disebabkan karena pada
kondisi tersebut, 99,2%-99,9% obat telah diekskresi. Frekuensi pengambilan
cuplikan obat sebaiknya dilakukan setidaknya 3 kali pada tahap absorpsi, 3 kali
di sekitar puncak, 3 kali pada tahap distribusi, dan 3 kali pada tahap eliminasi.
g. Analisis dan evaluasi hasil.
Langkah-langkah ini meliputi analisis sederetan kadar obat utuh atau
metabolitnya dalam darah atau urin, analisis statistika dan evaluasi.
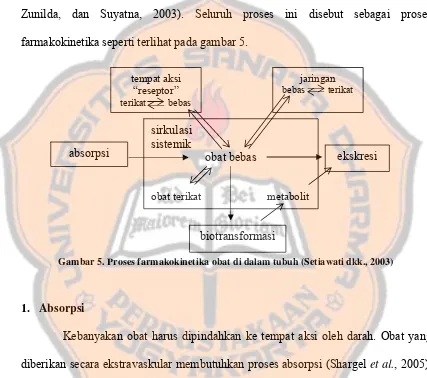
D. Nasib Obat di Dalam Tubuh
Obat yang masuk ke dalam tubuh umumnya mengalami absorpsi, distribusi,
dan pengikatan untuk sampai di tempat kerja dan menimbulkan efek. Kemudian,
dengan atau tanpa biotransformasi, obat diekskresi dari dalam tubuh (Setiawati,
Zunilda, dan Suyatna, 2003). Seluruh proses ini disebut sebagai proses
farmakokinetika seperti terlihat pada gambar 5.
tempat aksi “reseptor” terikat bebas
jaringan bebas terikat
sirkulasi sistemik
obat bebas
obat terikat metabolit
biotransformasi
ekskresi absorpsi
Gambar 5. Proses farmakokinetika obat di dalam tubuh (Setiawati dkk., 2003)
1. Absorpsi
Kebanyakan obat harus dipindahkan ke tempat aksi oleh darah. Obat yang
diberikan secara ekstravaskular membutuhkan proses absorpsi (Shargel et al., 2005).
Absorpsi merupakan proses penyerapan obat dari tempat pemberian, menyangkut
kelengkapan dan kecepatan proses tersebut. Kelengkapan dinyatakan dalam persen
dari jumlah obat yang diberikan (Setiawati dkk., 2003).
Absorpsi menggambarkan laju obat meninggalkan tempat pemberian dan
jumlah obat yang tersedia. Oleh karena itu, menurut para ahli klinis parameter
digunakan untuk menggambarkan jumlah obat yang mencapai tempat aksi atau
cairan tubuh. Sebagai contoh, obat yang diberikan per oral harus diabsorpsi terlebih
dahulu dari lambung dan usus halus. Absorpsi ini dipengaruhi oleh sifat bentuk
sediaan dan sifat fisika kimia obat. Obat juga akan mengalami metabolisme di hati
sebelum akhrinya mencapai sirkulasi sistemik. Akibatnya, sejumlah obat yang
diberikan dan diabsorpsi akan menjadi tidak aktif atau berubah bentuk. Jika kapasitas
metabolisme di hati besar, maka bioavailabilitas akan berkurang (disebut sebagai
first-pass effect) (Wilkinson, 2001).
Mekanisme absorpsi dapat terjadi secara difusi pasif, difusi terfasilitasi,
transpor aktif atau pinositosis, fagositosis dan persorpsi. Absorpsi obat melalui
saluran cerna pada umumnya terjadi secara difusi pasif. Absorpsi mudah terjadi bila
obat dalam bentuk non-ion dan mudah larut dalam lemak.
Mekanisme difusi pasif dijelaskan dengan Hukum Fick (Proudfoot, 1990) :
)
Konsentrasi obat di dalam darah jauh lebih kecil daripada konsentrasi obat dalam
saluran cerna (CGI >> CB). Kondisi ini disebut dengan kondisi “sink” yang
memastikan bahwa perbedaan konsentrasi tetap terjaga selama proses absorpsi
2. Distribusi
Organ target bagi obat biasanya bukan darah sehingga obat harus dapat
menembus jaringan untuk dapat memberi efek yang diharapkan (Clark and Smith,
1993). Setelah diabsorpsi, obat akan didistribusikan ke seluruh tubuh melalui
sirkulasi darah. Selain tergantung aliran darah, distribusi obat juga ditentukan oleh
sifat fisika kimianya (Setiawati dkk., 2003).
Distribusi obat dibedakan atas 2 fase berdasarkan penyebarannya dalam
tubuh. Distribusi fase pertama terjadi segera setelah penyerapan, yaitu ke organ yang
perfusinya sangat baik, misalnya jantung, hati, ginjal, dan otak. Selanjutnya,
distribusi fase kedua jauh lebih luas yaitu mencakup jaringan yang perfusinya tidak
secepat organ di atas misalnya otot, visera, kulit dan jaringan lemak (Setiawati dkk.,
2003).
Obat yang mudah larut dalam lemak akan melintasi membran sel dan
terdistribusi ke dalam sel, sedangkan obat yang tidak larut lemak akan sulit
menembus membran sehingga distribusinya terbatas terutama di cairan ekstrasel.
Selain itu, distribusi juga dibatasi oleh ikatan obat pada protein plasma dan hanya
obat bebas yang dapat berdifusi dan mencapai keseimbangan. Derajat ikatan obat
pada protein plasma ditentukan oleh afinitas obat terhadap protein, kadar obat, dan
kadar proteinnya (Setiawati dkk., 2003).
3. Biotransformasi
Biotransformasi atau metabolisme obat adalah proses perubahan struktur
molekul obat diubah menjadi lebih polar sehingga lebih mudah larut dalam air dan
kurang larut dalam lemak sehingga lebih mudah diekskresi melalui ginjal. Selain itu,
pada umumnya obat menjadi inaktif sehingga biotransformasi sangat berperan dalam
mengakhiri kerja obat (Setiawati dkk., 2003).
Reaksi biokimia yang terjadi dapat dibedakan menjadi reaksi fase I dan fase
II. Proses yang termasuk reaksi fase I adalah oksidasi, reduksi, dan hidrolisis. Reaksi
fase I ini mengubah obat menjadi metabolit yang lebih polar. Reaksi fase II yang
disebut juga reaksi sintetik merupakan konjugasi obat atau metabolit hasil reaksi fase
I dengan substrat endogen misalnya asam glukuronat, sulfat, asetat, atau asam amino.
Hasil konjugasi ini bersifat lebih polar dan lebih mudah terionisasi sehingga lebih
mudah diekskresi (Setiawati dkk., 2003).
Sebagian besar biotransformasi obat dikatalis oleh enzim mikrosom hati,
demikian pula biotransformasi asam lemak, hormon steroid, dan bilirubin. Untuk itu
obat harus larut lemak agar dapat melintasi membran, masuk ke dalam retikulum
endoplasma dan berikatan dengan enzim mikrosom (Setiawati dkk., 2003).
4. Ekskresi
Ekskresi suatu obat dan metabolitnya menyebabkan penurunan konsentrasi
bahan berkhasiat dalam tubuh. Ekskresi dapat terjadi tergantung pada sifat fisika
kimia (bobot molekul, harga pKa, kelarutan, tekanan uap) senyawa yang diekskresi
(Mutschler, 1999). Obat dikeluarkan dari tubuh melalui berbagai organ ekskresi
metabolit polar diekskresi lebih cepat daripada obat larut lemak, kecuali pada
ekskresi melalui paru (Setiawati dkk., 2003).
Organ ekskresi yang terpenting adalah ginjal. Ekskresi meliputi 3 proses
berikut : filtrasi di glomerulus, sekresi aktif di tubuli proksimal, dan reabsorpsi pasif
di tubuli proksimal dan distal (Setiawati dkk., 2003). Selain melalui ginjal, ekskresi
obat juga dapat terjadi melalui empedu dan usus (feses), kulit (keringat), air liur, air
mata, air susu, paru-paru (udara ekspirasi) dan rambut (Mutschler, 1999; Setiawati
dkk., 2003). Ekskresi obat melalui kulit dan turunannya tidak begitu penting. Pada
ibu menyusui, eliminasi obat dan metabolitnya dalam air susu dapat menyebabkan
intoksikasi yang membahayakan bagi bayi (Mutschler, 1999).
E. Dasar-Dasar Perhitungan Farmakokinetika 1. Model kompartemen
Tubuh terdiri dari banyak kompartemen. Masing-masing sel tubuh dan
bagian-bagian dari sel merupakan kompartemen yang kecil. Dalam farmakokinetika,
yang disebut dengan kompartemen adalah organ-organ dan jaringan di mana
kecepatan absorpsi dan klirens obat adalah sama (Clark and Smith, 1993). Model
kompartemen adalah suatu hubungan matematika yang menggambarkan perubahan
konsentrasi terhadap waktu dalam sistem tubuh (Mutschler, 1999).
a. Model satu kompartemen. Pada model satu kompartemen, obat akan
segera terdistribusi ke dalam ruang distribusi secara merata setelah pemakaian. Jika
proses eliminasi mungkin terjadi, maka model satu kompartemen disebut terbuka
b. Model dua kompartemen. Pada model dua atau lebih kompartemen,
distribusi obat ke dalam ruang distribusi terjadi dengan kecepatan yang
berbeda-beda. Dengan demikian dapat dibedakan menjadi kompartemen pusat, yang secara
kinetika bersifat seperti darah (organ transpor) dan kompartemen perifer. Bila
pertukaran zat antara suatu kompartemen perifer dan kompartemen pusat sangat
lambat, maka disebut kompartemen dalam (Mutschler, 1999).
2. Parameter farmakokinetika
Parameter farmakokinetika diperoleh dari perubahan konsentrasi obat dan
metabolitnya dalam cairan darah (darah, plasma, dan serum) dan dalam urin terhadap
waktu. Kedua cairan tersebut mudah dilewati dan konsentrasi dalam darah, yaitu alat
transpornya, mencerminkan proses kinetika dalam organisme (Mutschler, 1999).
Untuk memperoleh kurva konsentrasi terhadap waktu sebagai hasil dari
berbagai bagian proses farmakokinetika yang berbeda-beda perlu dilakukan
penentuan konsentrasi obat berulang-ulang (Mutschler, 1999). Dalam membuat
kurva konsentrasi terhadap waktu untuk suatu obat, suatu bentuk sediaan tertentu
akan diberikan kepada sekelompok pasien dan sampel darah pasien itu akan diambil
pada periode waktu yang telah ditentukan. Jumlah obat dalam sampel darah ini
kemudian akan dianalisis dan dibuat grafik konsentrasi darah terhadap waktu (Ansel
and Prince, 2006). Kurva konsentrasi darah terhadap waktu dapat digunakan untuk
menentukan atau membuat parameter-parameter berikut (Ansel and Prince, 2006;
a. Area Under the Curve (AUC). Nilai AUC biasanya dihitung dari profil
kurva konsentrasi plasma terhadap waktu. Nilai AUC menggambarkan jumlah obat
di dalam tubuh dan dapat dihitung dengan aturan trapezoid.
Luas area trapezoid = (tn+1 – tn) . (Cn+ Cn+1) / 2 (7)
Keterangan :
tn+1 = waktu saat n+1 (menit) tn = waktu saat n (menit)
Cn = konsentrasi pada waktu tn (μg/ml) Cn+1 = konsentrasi pada waktu tn+1 (μg/ml)
Jumlah semua area trapezoid merupakan nilai AUC(0-t). Untuk menghitung total
AUC (AUC(0-∞)), maka dilakukan ekstrapolasi bagian akhir area setelah titik akhir
pengukuran (AUC(t-∞)). Prosedur ini sahih jika bagian ekstrapolasi area lebih kecil
dari 10% AUC(0-t) dan sebaiknya data tidak dipakai jika bagian ekstrapolasi lebih
besar dari 20% AUC(0-t).
b. Volume distribusi (Vd). Volume distribusi adalah volume hipotetis cairan
tubuh yang akan diperlukan untuk melarutkan jumlah total obat pada konsentrasi
yang sama seperti yang ditemukan dalam darah.
Cp D
Vd= (8)
Keterangan :
Vd = volume distribusi (ml) D = dosis (mg)
Cp = kadar obat dalam plasma (μg/ml)
Volume distribusi dapat dianggap sebagai volume plasma, cairan ekstraseluler, atau
cairan tubuh total. Jumlah total obat dalam tubuh dapat dihitung dari konsentrasi obat
yang terdistribusi ke jaringan juga besar atau dapat juga obat terkonsentrasi pada
jaringan tertentu.
c. Klirens (Cl). Klirens merupakan volume darah atau plasma yang dapat
dibersihkan dari obat per satuan waktu. Klirens total diperoleh dari hasil kali tetapan
laju eliminasi (kel) dan volume distribusi (Vd)
Cl = Vd . kel (9)
atau dari hasil bagi dosis (D) dengan AUC.
AUC
Jika obat hanya dieliminasi oleh satu organ, maka klirens total sama dengan klirens
organ tersebut. Namun biasanya nilai klirens total melibatkan beberapa jalur yang
terdiri dari beberapa organ klirens juga. Jalur terpenting adalah hepatik (ClH) dan
ginjal (ClR) sehingga rumus klirens total menjadi :
Cl = ClH + ClR + Clx (11)
Keterangan :
Cl = klirens total (ml/menit) ClH = klirens hepatik (ml/menit) ClR = klirens ginjal (ml/menit) ClX = klirens organ lain (ml/menit)
d. Waktu paruh eliminasi (t½ eliminasi). Nilai t½ eliminasi merupakan waktu
kadar obat dalam darah atau plasma menjadi setengah dari kadar awal.