VALIDASI METODE KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK PADA PENETAPAN KADAR KURKUMIN DALAM SEDIAAN KAPSUL LUNAK OBAT HERBAL TERSTANDAR MEREK
RHEUMAKUR®
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Farmasi
oleh: Benny Nugroho NIM: 078114027
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA
YOGYAKARTA
ii
VALIDASI METODE KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK PADA PENETAPAN KADAR KURKUMIN DALAM SEDIAAN KAPSUL LUNAK OBAT HERBAL TERSTANDAR MEREK
RHEUMAKUR®
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Farmasi
oleh: Benny Nugroho NIM: 078114027
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA
YOGYAKARTA
v
HALAMAN PERSEMBAHAN
Perjalanan Jauh Ribuan Mil
Dimulai Dari Langkah Pertama
Sebuah hasil perjuangan dan semangat...
Kupersembahkan untuk
Keluargaku
viii
PRAKATA
Puji dan syukur penulis panjatkan kepada Tuhan Yang Maha Esa atas segala berkat dan karunia-Nya sehingga penulis dapat menyelesaikan skripsi yang berjudul Validasi Metode Kromatografi Cair Kinerja Tinggi Fase Terbalik Pada Penetapan Kadar Kurkumin Dalam Sediaan Kapsul Lunak Obat Herbal Terstandar Merek Rheumakur® yang disusun sebagai salah satu syarat untuk mencapai gelar Sarjana Farmasi di Fakultas Farmasi Universitas Sanata Dharma Yogyakarta.
Dalam serangkain proses skripsi yang telah dijalani, penulis mendapatkan bantuan dari banyak pihak. Pada kesempatan ini penulis ingin menyampaikan ucapan terima kasih kepada:
1. Mama tercinta yang telah memberikan doa dan semangat semoga cepat selesai skripsi dan kuliahnya.
2. Ipang Djunarko, S.Si., Apt., M.Sc. selaku Dekan Fakultas Farmasi Universitas Sanata Dharma Yogyakarta dan sekaligus sebagai dosen pembimbing akademik atas perhatian dan semangat yang diberikan kepada penulis selama menjalani serangkaian proses skripsi.
3. Christine Patramurti, M.Si., Apt. selaku dosen pembimbing atas segala perhatian, dukungan, sentilan, dan semangat yang terus memotivasi penulis dari proses kerja awal hingga proses akhir penyusunan skripsi ini.
ix
5. Yohanes Dwiatmaka, M.Si. selaku dosen penguji atas saran dan kritik yang membangun.
6. Rini Dwi Astuti, M.Sc., Apt. selaku Kepala Laboratorium Farmasi Universitas Sanata Dharma yang telah memberikan izin kepada penulis untuk melakukan penelitian di laboratorium.
7. Prof. Dr. Sudibyo, M.S., Apt. yang telah memberikan senyawa baku kurkumin untuk penelitian yang dilakukan oleh penulis.
8. Mas Bimo, Pak Timbol, Mas Kunto, Mas Parlan atas segala canda dan bantuannya selama peneliti bekerja di laboratorium Kimia Analisis Instrumental.
9. Segenap dosen pengajar, staf kesekretariatan, staf keamanan, dan laboran Fakultas Farmasi Universitas Sanata Dharma atas dukungan dan bantuannya dalam menyelesaikan skripsi ini.
10.Dian Prahara Florentino Wara dan Katarina Kusmiyanti sebagai rekan kerja satu tim penulis, atas segala kerja sama sebelum penelitian, selama penelitian, dan dalam proses penyusunan naskah skripsi atas kebersamaannya di saat senang dan susah.
11.Lala, Toro, Katitik, Seno, Tere, Eliz, Yunita, Venny, dan Lilis sebagai bagian dari tim besar penulis, atas segala kebersamaan sebelum proses penelitian, selama proses penelitian, dan masukan serta ilmu pengetahuan yang sangat berarti dalam proses penyusunan naskah skripsi.
x
13.Teman-teman seperjuangan penulis selama proses penelitian: Wicak, Siwi, Sere atas segala canda, dan semangat kepada penulis.
14.Lia Natalia Setiomulyo yang telah memberikan segenap rasa percaya, dorongan, dan semangat kepada penulis.
15.Teman-teman FST 2007 atas kebersamaan bersama yang selalu mengenang di hati penulis.
16.Semua orang yang telah membantu penulis dan tidak dapat disebutkan satu per satu, terima kasih atas segala bentuk perhatian dan bantuan yang diberikan.
Penulis menyadari bahwa skripsi ini belumlah sempurna, untuk itu penulis sangat mengharapkan kritik dan saran yang membangun dari para pembaca yang diharapkan dapat menyempurnakan skripsi ini. Akhir kata, semoga Tuhan Yang Maha Esa memberikan berkat-Nya kepada semua pihak yang telah membantu penulis dalam menyelesaikan skripsi ini. Semoga skripsi ini dapat memberikan manfaat bagi perkembangan ilmu pengetahuan. Ad Maiorem Dei Gloriam.
Yogyakarta, Maret 2011
xi
DAFTAR ISI
Halaman
HALAMAN JUDUL... ii
HALAMAN PERSETUJUAN PEMBIMBING... iii
HALAMAN PENGESAHAN... iv
HALAMAN PERSEMBAHAN... v
PERNYATAAN KEASLIAN KARYA... vi
PERNYATAAN PERSETUJUAN PUBLIKASI ILMIAH... vii
PRAKATA... viii
A. Obat Herbal Terstandar (OHT)...
xii
B. Kapsul Lunak... C. Kurkumin... D. Spektrofotometri Visibel...
1. Definisi spektrofotometri visibel dan konsep dasar radiasi elektromagnetik... 2. Tipe transisi elektron... 3. Analisis kualitatif dan kuantitatif spektrofotometri
visibel... E. Kromatografi Cair Kinerja Tinggi (KCKT)... 1. Definisi dan tinjauan umum KCKT... 2. Variabel-variabel dalam KCKT... 3. Kromatografi partisi... 4. Waktu retensi... 5. Pemisahan puncak dalam kromatografi... 6. Analisis kualitatif dan analisis kuantitatif... F. Validasi Metode Analisis Instrumental... 1. Tinjauan umum validasi metode analisis instrumental... 2. Parameter validasi metode analisis instrumental... G. Landasan Teori...
A. Jenis dan Rancangan Penelitian... B. Variabel Penelitian...
xiii 2. Pembuatan pelarut metanol pH 4... 3. Pembuatan larutan baku kurkumin... 4. Penetapan panjang gelombang (λ) maksimum kurkumin... 5. Preparasi sampel...
A. Pembuatan Fase Gerak KCKT... B. Pembuatan Pelarut dan Stabilitas Kurkumin... C. Pembuatan Larutan Baku Kurkumin... D. Penentuan Panjang Gelombang Maksimum Kurkumin... E. Analisis Kualitatif Berdasarkan Waktu Retensi (tR)
xiv
F. Pembuatan Kurva Baku Kurkumin... G. Validasi Metode Analisis... BAB V. KESIMPULAN DAN SARAN...
xv
DAFTAR TABEL
Halaman
Tabel I. Nilai indeks polaritas pelarut... 17
Tabel II. Kategori metode pengujian validitas data... 24
Tabel III. Parameter validasi yang dipersyaratkan untuk validasi metode analisis... 25 Tabel IV. Kriteria penerimaan akurasi pada konsentrasi analit yang berbeda... 27 Tabel V. Kriteria penerimaan presisi berdasar kadar analit... 29
Tabel VI. Penentuan kurva baku kurkumin... 49
Tabel VII. Penentuan kurva baku kurkumin hasil modifikasi... 50
Tabel VIII. Hasil penetapan recovery (%) baku kurkumin... 55
Tabel IX. Hasil perhitungan nilai recovery (%) dengan standard addition method... 56
Tabel X. Hasil pengukuran coefficient of variation larutan baku kurkumin... 57
xvi
DAFTAR GAMBAR
Halaman
Gambar 1. Logo Obat Herbal Terstandar... 7
Gambar 2. Struktur dari kurkumin... 9
Gambar 3. Diagram tingkat energi elektronik... 12
Gambar 4. Ilustrasi instrumen KCKT... 15
Gambar 5. Reaksi silanasi... 21
Gambar 6. Kromatogram pemisahan dua senyawa secara KCKT... 23
Gambar 7. Kromatogram respon analit dalam campuran multikomponen... 25
Gambar 8. Linearitas dengan koefisien korelasi (r) > 0,999... 26
Gambar 9. Reaksi degradasi kolom C18 pada pH asam ≤ 2... 41
Gambar 10. Gugus metilen aktif pada kurkumin... 42
Gambar 11. Gugus kromofor dan auksokrom pada struktur kurkumin... 44
Gambar 12. Spektra panjang gelombang maksimum kurkumin... 44
Gambar 13. Gugus polar dan non polar pada struktur kurkumin... 46
Gambar 14. Interaksi hidrogen antara kurkumin dengan fase gerak metanol : asam asetat glasial 2% (95:5)... 47
Gambar 15. Interaksi kurkumin dengan fase diam oktadesilsilan... 47
Gambar 16. Kromatogram baku kurkumin (a) dan kurkumin dalam sampel (b)... 48
xvii
merek Rheumakur® pada konsentrasi tinggi
(6,5ppm)... 53 Gambar 19. Respon sampel sebelum diadisi (kiri) dan sampel setelah
diadisi (kanan)... 56 Gambar 20. Respon baku kurkumin pada konsentrasi tinggi
xviii
DAFTAR LAMPIRAN
Halaman Lampiran 1. Pernyataan jaminan keaslian senyawa kurkumin standar
hasil sintesis... 65
Lampiran 2. Data penimbangan bahan... 66
Lampiran 3. Skema pembuatan larutan baku kurkumin dan contoh perhitungan kadar larutan baku yang digunakan... 67
Lampiran 4. Optimasi stabilitas larutan baku kurkumin berdasar pH... 68
Lampiran 5. Spektra panjang gelombang maksimum kurkumin... 68
Lampiran 6. Kromatogram baku kurkumin untuk kurva baku... 69
Lampiran 7. Data penentuan kurva baku kurkumin... 72
Lampiran 8. Persamaan dan gambar kurva baku kurkumin... 73
Lampiran 9. Kromatogram baku kurkumin untuk validasi metode.... 74
Lampiran 10. Perolehan nilai AUC untuk validasi metode dan contoh perhitungan konsentrasi terukur baku kurkumin ... 81
Lampiran 11. Contoh perhitungan persen perolehan kembali (recovery) dan coefficient of varriation (CV) baku kurkumin... 82
Lampiran 12. Kromatogram sampel dan sampel adisi... 84
xix
VALIDASI METODE KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK PADA PENETAPAN KADAR KURKUMIN DALAM SEDIAAN KAPSUL LUNAK OBAT HERBAL TERSTANDAR MEREK
RHEUMAKUR®
INTISARI
Kurkumin merupakan senyawa yang memiliki aktifitas farmakologis sebagai antiinflamasi dan salah satunya terkandung dalam Obat Herbal Terstandar (OHT) merek Rheumakur®. Aktifitas farmakologi kurkumin tergantung pada ketepatan dan keseragaman dosis. Untuk menjamin ketepatan dosis OHT diperlukan metode penjaminan kualitas kandungan zat aktif.
Penelitian yang dilakukan bersifat non eksperimental deskriptif. Kromatografi Cair Kinerja Tinggi (KCKT) fase terbalik dapat digunakan sebagai metode penjaminan kualitas kandungan kurkumin dalam OHT. Validasi metode KCKT fase terbalik menggunakan sistem yang optimal dengan fase diam oktadesilsilan (C18), fase gerak metanol p.a. : asam asetat glasial 2% (95:5 v/v),
kecepatan alir 1,0 ml/menit dengan detektor visibel pada panjang gelombang 432 nm.
Parameter validitas metode yang digunakan adalah selektivitas, linearitas, akurasi, presisi, dan rentang. Hasil penelitian menunjukkan bahwa metode KCKT fase terbalik memiliki selektifitas yang baik dengan adanya pemisahan sempurna dari peak sampel dan linearitas yang baik dengan nilai koefisien korelasi 0,9996 pada konsentrasi 1,5-6,5 ppm. Nilai recovery dan CV untuk baku kurkumin pada konsentrasi 1,5 ppm; 3,5 ppm; dan 6,5 ppm berturut-turut adalah 94,3922-97,9863% dan 0,7088%; 86,3053-91,5140% dan 0,6414%; dan 98,7074-102,9929% dan 0,3687%, sedangkan untuk recovery standard addition method adalah 98,4635-102,2448% dan 1,6705%. Berdasarkan hasil tersebut, metode KCKT fase terbalik pada penetapan kadar kurkumin dalam sediaan kapsul lunak OHT merek Rheumakur® memenuhi parameter validitas yang baik.
xx
VALIDATION OF HIGH PERFORMANCE LIQUID CHROMATOGRAPHY REVERSE PHASE IN CURCUMIN QUANTIFICATION IN SOFT CAPSULE OF SCIENTIFIC BASED
HERBAL MEDICINE RHEUMAKUR®
ABSTRACT
Curcumin is a compound that has pharmacological activity as anti inflammantory and are found mainly in Scientific Based Herbal Medicine (SBHM). Curcumin pharmacology's activity depends on the accuracy and uniformity of the dose. To guarantee that SBHM's dose is accuracy, it required assurance method which is qualified active substance.
The research that has conducted is non experimental descriptive. High Performance Liquid Chromatography (HPLC) reverse phase can be used as a method of quality assurance content of curcumin in SBHM. Validation of HPLC reverse phase with optimal system conditions using the optimal stationary phase octadecylsylane (C18), mobile phase methanol p.a.: glacial acetic acid 2% (95:5
v/v), flow rate 1,0 ml/min with visible detector at wavelength 432 nm.
Parameter validity of the method used is the selectivity, linearity, SBHM Rheumakur® meets good validity parameters.
1
BAB I
PENGANTAR
A. Latar Belakang
Kecenderungan gaya hidup back to nature menyebabkan sebagian besar masyarakat mulai beralih dari penggunaan obat moderen ke penggunaan obat tradisional untuk menunjang kesehatan tubuh. Pertimbangan berdasarkan pada aspek ekonomi dan keamanan bagi kesehatan bisa menjadi dua alasan mendasar di mana masyarakat mulai beralih ke penggunaan obat tradisional. Persentase penggunaan obat tradisional dari tahun ke tahun terus mengalami peningkatan, baik di negara berkembang maupun negara maju. World Health Organization (WHO) menyebutkan bahwa hingga 80% dari penduduk negara-negara di Asia dan Afrika telah menggunakan pengobatan tradisional sebagai pengobatan utama (WHO, 2008).
kanker. Salah satu contoh OHT mengandung kurkumin yang beredar di pasaran yaitu kapsul lunak Rheumakur® yang berkhasiat sebagai anti rheumatik.
Aktifitas farmakologi kurkumin tergantung pada ketepatan dan keseragaman dosis. Untuk menjamin ketepatan dosis suatu sediaan obat herbal tradisional diperlukan penjaminan kualitas kandungan zat aktif dalam sediaan obat herbal tradisional jenis OHT. Jaminan kualitas terhadap produk obat herbal tradisional sangat dibutuhkan seiring dengan meningkatnya permintaan terhadap produk obat herbal tradisional jenis OHT yang mengandung kurkumin dalam bentuk sediaan padat, khususnya Rheumakur®. Tujuan dari penjaminan kualitas adalah terciptanya produk yang berkhasiat dan aman pada obat herbal tradisional khususnya OHT yang mengandung kurkumin, di mana ditunjukkan oleh ketepatan dan keseragaman dosis setiap proses produksinya.
Beberapa penelitian mengenai analisis kurkumin secara KCKT yang telah dilakukan antara lain menggunakan fase diam C18 dan fase gerak metanol :
asam asetat glasial 2% (90:10 v/v) dengan detektor visibel (Widjaja, 2011), fase diam amino bonded dan fase gerak etanol absolut dengan detektor visibel (Sumule, 2007). Hal utama yang membedakan penelitian ini dengan penelitian sebelumnya terletak pada komposisi fase gerak dan kecepatan alir yang digunakan.
Berdasarkan penelusuran pustaka yang dilakukan oleh peneliti, validasi metode KCKT untuk penetapan kadar pada sediaan kapsul lunak OHT merek Rheumakur® yang mengandung kurkumin belum pernah dilakukan. Pemilihan metode KCKT fase terbalik didasarkan pada selektivitas metode tersebut dimana memiliki kemampuan untuk memisahkan suatu senyawa dari campuran sampel yang multikomponen dalam kadar yang kecil.
Pada penelitian ini dilakukan proses validasi terhadap sistem KCKT fase terbalik hasil optimasi dalam rangkaian penelitian penetapan kadar kurkumin dalam sediaan kapsul lunak OHT merek Rheumakur®, yaitu: optimasi, validasi, dan penetapan kadar. Berdasarkan hasil optimasi diperoleh kondisi optimal sistem KCKT fase terbalik menggunakan fase diam C18 dan fase gerak metanol : asam
asetat glasial 2% (95:5 v/v) dengan kecepatan alir 1,0 ml/menit yang selanjutnya digunakan sebagai sistem acuan pada proses validasi ini.
untuk menetapkan kadar kurkumin dalam sediaan kapsul lunak OHT merek Rheumakur®.
1. Permasalahan
Berdasarkan latar belakang tersebut, maka permasalahan yang muncul adalah apakah metode kromatografi cair kinerja tinggi fase terbalik menggunakan sistem yang sudah dioptimasi pada penetapan kadar kurkumin dalam sediaan kapsul lunak Obat Herbal Terstandar merek Rheumakur® memenuhi persyaratan validitas seperti: selektivitas, linearitas, akurasi, presisi, dan rentang?
2. Keaslian penelitian
3. Manfaat penelitian
a. Manfaat metodologis. Hasil penelitian ini memberikan satu alternatif metode baru yang memiliki validitas yang baik dalam menetapkan kadar kurkumin dalam sediaan kapsul lunak Obat Herbal Terstandar merek Rheumakur® menggunakan metode kromatografi cair kinerja tinggi fase terbalik menggunakan sistem optimal dengan validitas yang baik.
b. Manfaat praktis. Penelitian ini diharapkan dapat digunakan untuk menetapkan kadar kurkumin dalam sediaan Obat Herbal Terstandar bentuk kapsul lunak merek Rheumakur® yang beredar di pasaran.
B. Tujuan Penelitian
6
BAB II
PENELAAHAN PUSTAKA
A. Obat Herbal Terstandar (OHT)
Menurut Undang-Undang No.23 tahun 1992 tentang kesehatan bab I pasal I ayat (10) obat tradisional adalah bahan atau ramuan bahan yang berupa bahan tumbuhan, bahan hewan, bahan mineral, sediaan cairan (galenik) atau campuran dari bahan tersebut yang secara turun temurun telah digunakan untuk pengobatan berdasarkan pengalaman.
Berdasarkan Keputusan Kepala Badan Pengawas Obat dan Makanan (BPOM) Republik Indonesia nomor: HK.00.05.4.2411, penggolongan obat bahan alam Indonesia berdasarkan cara pembuatan serta jenis klaim penggunaan dan tingkat pembuktian khasiat, dikelompokkan menjadi jamu, Obat Herbal Terstandar (OHT), dan fitofarmaka.
Obat Herbal Terstandar adalah sediaan obat bahan alam yang telah dibuktikan keamanan dan khasiatnya secara ilmiah dengan uji praklinik dan bahan bakunya telah distandarisasi. Standarisasi merupakan serangkaian parameter, prosedur, dan cara pengukuran yang hasilnya berupa paradigma mutu sesuai standar dan jaminan stabilitas produk (Badan Pengawasan Obat dan Makanan RI, 2005).
1. Aman sesuai dengan persyaratan yang ditetapkan 2. Klaim khasiat dibuktikan secara ilmiah atau pra klinik
3. Telah dilakukan standarisasi terhadap bahan baku yang digunakan dalam produk jadi
4. Memenuhi persyaratan mutu yang berlaku
(Direktorat Jenderal Pengawasan Obat dan Makanan RI, 2005).
Gambar 1. Logo Obat Herbal Terstandar (Badan Pengawas Obat dan Makanan RI, 2004)
B.Kapsul Lunak
Kapsul adalah sediaan padat yang terdiri dari obat dalam cangkang keras atau lunak yang dapat larut. Cangkang umumnya terbuat dari gelatin,tetapi dapat juga terbuat dari pati atau bahan lain yang sesuai. Cangkang gelatin lunak sedikit lebih tebal dibanding kapsul cangkang keras dan dapat diplastisasi dengan penambahan senyawa poliol. Umumnya kapsul cangkang lunak berisi cairan, khususnya bahan aktif di mana dilarutkan atau disuspensikan dalam bahan pembawa cair.
bobot rata-rata tiap isi kapsul. Perbedaan dalam persen bobot isi tiap kapsul terhadap bobot rata-rata tiap isi kapsul tidak lebih dari 7,5 % (Direktorat Jenderal Pengawasan Obat dan Makanan RI, 1995).
C.Kurkumin
Kurkumin merupakan salah satu jenis kandungan dalam kurkuminoid selain demetoksikurkumin, dan bis-demetoksikurkumin. Kurkumin secara kuantitas merupakan komponen terbesar dalam kurkuminoid yang memberikan warna kuning (Dandekar and Patravale, 2009).
Dari ketiga senyawa kurkuminoid, kurkumin merupakan komponen terbesar. Kandungan kurkumin dalam kunyit mencapai 50-60% sedangkan demetoksikurkumin dan bis-demetoksikurkumin hanya terdapat dalam jumlah kecil sehingga seringkali kadar total kurkuminoid dihitung sebagai persen (%) kurkumin. Berdasarkan alasan tersebut, beberapa penelitian lebih ditekankan pada kurkumin (Parinussa dan Timotius, 2002).
independent), menghambat perkembangbiakan sel kanker kolon in vitro, leukimia
dan sel kanker kulit. Sebagai antivirus, kurkumin bekerja dengan cara menghambat enzim integrase HIV-1, protease HIV-1 sehingga menghambat transaktivasi HIV-1. Kurkumin juga memiliki aktivitas sebagai imunostimulan dengan kemampuannya meningkatkan sintesis antibodi IgG dan meningkatkan sitotoksisitas Natural Killer Cells. Kurkumin juga dikenal sebagai anti inflamasi, bekerja dengan cara menghambat aktivitas enzim lipooksigenase dan siklooksigenase (Bermawie, Rahardjo, Wahyuno, dan Ma’mun, 2006).
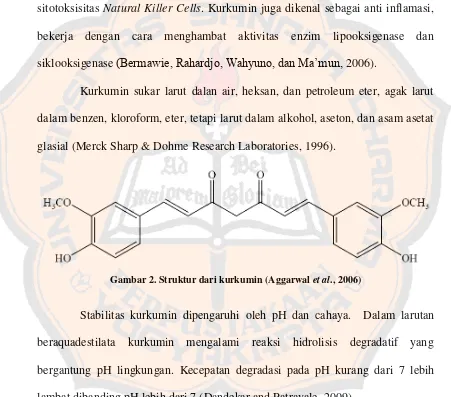
Kurkumin sukar larut dalan air, heksan, dan petroleum eter, agak larut dalam benzen, kloroform, eter, tetapi larut dalam alkohol, aseton, dan asam asetat glasial (Merck Sharp & Dohme Research Laboratories, 1996).
Gambar 2. Struktur dari kurkumin (Aggarwal et al., 2006)
Stabilitas kurkumin dipengaruhi oleh pH dan cahaya. Dalam larutan beraquadestilata kurkumin mengalami reaksi hidrolisis degradatif yang bergantung pH lingkungan. Kecepatan degradasi pada pH kurang dari 7 lebih lambat dibanding pH lebih dari 7 (Dandekar and Patravale, 2009).
berwarna coklat kemerahan yang pekat sampai warna kuning muda (Sharma et al. , 2005). Kurkumin bersifat tidak larut dalam air, tetapi larut dalam alkohol dan asam asetat glasial. Kurkumin akan terdegradasi pada pH di atas 7,2 dan oleh sinar ultra violet. Adanya cahaya dapat menyebabkan terjadinya degradasi fotokimia karena kurkumin memiliki gugus metilen aktif (-CH2-) di antara dua
gugus keton pada senyawa tersebut (Tonnesen dan Karlsen, 1985).
Secara spektrofotometri, absorbansi maksimum kurkumin (λmax) di metanol yaitu 430 nm. Kurkumin berwarna kuning pada pH 2,5 sampai 7 dan berwarna merah pada pH > 7. Fluoresensi dari kurkumin terjadi pada asetonitril
(λmax = 524 nm), etanol (λmax = 549 nm), atau micellar solution (λmax = 557 nm),
tetapi di toluen (λmax = 460, 488 nm) (Aggarwal et al., 2006).
Analisis kurkumin secara kuantitatif pada tahun 1983 dilakukan dengan Kromatografi Cair Kinerja Tinggi (KCKT) menggunakan detektor fluorometer. Adanya gugus polar dan gugus kromofor pada kurkumin secara kuantitatif dapat dianalisis menggunakan KCKT fase terbalik dengan kolom oktadesilsilan dan detektor ultraviolet-visibel (Musfiroh, Indriyati, Susilawati, dan Percekawati, 2009).
D.Spektrofotometri Visibel
1. Definisi spektrofotometri visibel dan konsep dasar radiasi
elektromagnetik
gelombang 380-780 nm (Direktorat Jenderal Pengawasan Obat dan Makanan RI, 1995).
Radiasi elektromagnetik pada daerah visibel dapat dianggap sebagai energi yang merambat dalam bentuk gelombang. Panjang gelombang merupakan jarak linier dari suatu titik pada satu gelombang ke titik yang bersebelahan pada gelombang yang berdekatan.
Prinsip kerja spektrofotometri visibel berdasarkan interaksi antara radiasi elektromagnetik dengan atom, ion, atau molekul. Serapan atom menyebabkan peralihan elektronik atau transisi elektronik, yaitu peningkatan energi elektron dari tingkat dasar (ground state) ke tingkat energi yang lebih tinggi (excited state). Transisi ini terjadi bila energi yang dihasilkan oleh radiasi sama dengan energi yang diperlukan untuk melakukan transisi (Rohman dan Gandjar, 2007).
2. Tipe transisi elektron
Ada empat tipe transisi elektron yang dapat terjadi yaitu: σ σ*, n
σ*, n π*, dan π π*. Transisi elektron (σ σ*) pada suatu elektron di dalam
Transisi elektron (n σ*) terjadi pada senyawa organik jenuh yang mengandung atom-atom yang memiliki elektron bukan ikatan (elektron n). Energi yang diperlukan untuk transisi jenis ini lebih kecil dibanding transisi σ σ* sehingga sinar yang diserap pun mempunyai panjang gelombang lebih panjang, yaitu sekitar 150-250 nm (Rohman dan Gandjar, 2007).
Gambar 3. Diagram tingkat energi elektronik (Skoog et al, 1998)
Transisi elektron n π* dan π π* merupakan transisi yang paling cocok untuk analisis sebab sesuai dengan panjang gelombang antara 200-700 nm. Secara teknis dapat diaplikasikan pada spektrofotometer. Untuk memungkinkan terjadinya jenis transisi ini, molekul organik harus mempunyai gugus fungsional yang tidak jenuh sehingga ikatan rangkap dalam gugus tersebut memberikan orbital π yang diperlukan (Rohman dan Gandjar, 2007). Kedua transisi ini membutuhkan adanya kromofor dan auksokrom dalam struktur molekulnya.
Kromofor merupakan gugus fungsional tak jenuh yang menyediakan orbital π
intensitas serapan maksimum, cirinya adalah heteroatom yang langsung terikat pada kromofor (Sastrohamidjojo, 2002).
3. Analisis kualitatif dan kuantitatif spektrofotometri visibel
Pada analisis kualitatif dengan metode spektrofotometri visibel yang dapat ditentukan ada dua yaitu: pemeriksaan kemurnian spektrum visibel dan penentuan panjang gelombang serapan maksimum (Mulja dan Suharman, 1995).
Dalam analisis kuantitatif, suatu berkas radiasi dikenakan pada cuplikan (larutan sampel) dan intensitas sinar radiasi yang diteruskan diukur besarnya. Radiasi yang diserap oleh cuplikan ditentukan dengan membandingkan intensitas sinar yang diteruskan dengan intensitas sinar yang diserap jika tidak ada jenis penyerap lainnya. Intensitas atau kekuatan radiasi cahaya sebanding dengan jumlah foton yang melalui satu satuan luas penampang perdetik.
Serapan dapat terjadi jika radiasi yang mengenai cuplikan memiliki energi yang sama dengan energi yang dibutuhkan untuk menyebabkan terjadinya perubahan tenaga. Kekuatan radiasi juga mengalami penurunan dengan adanya penghamburan dan pemantulan cahaya, akan tetapi penurunan karena hal ini sangat kecil dibanding dengan proses penyerapan.
T=It
Io
=10−ε.C.b
Menurut Watson (1999), nilai daya serap (a) dapat dinyatakan sebagai sehingga persamaan hukum Lambert-Beer dapat ditulis menjadi:
A=1
T=ε.C.b Keterangan:
T= persen transmitan
It= intensitas radiasi yang diteruskan Io= intensitas radiasi yang datang
ε= daya serap molar (L mol-1 cm-1)
C= konsentrasi larutan (mol L-1) b= tebal kuvet (cm)
A=serapan/absorbansi
(Rohman dan Gandjar, 2007) Dalam hukum Lambert-Beer tersebut terdapat beberapa pembatasan, yaitu: sinar yang digunakan dianggap monokromatis, penyerapan terjadi dalam suatu volume yang mempunyai penampang luas yang sama, senyawa yang menyerap dalam larutan tidak tergantung terhadap senyawa lain dalam larutan tersebut, tidak terjadi peristiwa fluoresensi atau fosforesensi, dan indeks bias tidak tergantung pada konsentrasi larutan (Rohman dan Gandjar, 2007).
E.Kromatografi Cair Kinerja Tinggi (KCKT)
1. Definisi dan tinjauan umum KCKT
Kromatografi Cair Kinerja Tinggi (KCKT) merupakan salah satu metode kromatografi cair yang fase geraknya dialirkan secara cepat dengan bantuan tekanan dan hasilnya dideteksi dengan detektor. Pada alat ini diperlukan sistem pompa tekanan tinggi yang mengalirkan fase gerak dari bejana fase gerak ke kolom dengan tekanan tinggi sampai 300 atmosfer sehingga pada awalnya
(1)
kromatografi ini disebut High Pressure Liquid Chromatography (Direktorat Jenderal Pengawasan Obat dan Makanan RI, 1995). Pada akhir tahun 1970, perkembangan instrumen ini dapat menghasilkan pemisahan yang baik atau menghasilkan penampilan peak yang baik sehingga sistem ini lebih dikenal dengan KCKT (Kromidas, 2000).
KCKT merupakan teknik analisis yang paling banyak digunakan, karena sensitivitas dari metode ini menghasilkan determinasi kuantitatif yang akurat (Skoog, Holler, dan Nieman, 1998). Maksud dan tujuan analisis dengan KCKT hanya ada dua hal yaitu memperoleh pemisahan yang baik dalam proses yang relatif singkat (Mulja dan Suharman, 1995). Keterbatasan metode KCKT adalah jika analit yang akan dianalisis sangat kompleks maka resolusi yang baik sulit diperoleh (Rohman dan Gandjar, 2007). Peralatan KCKT dapat dilihat pada gambar:
2. Variabel-variabel dalam KCKT
1. Fase gerak. KCKT dapat dikembangkan dengan pelarut tunggal maupun campuran pelarut. Pemisahan dengan fase gerak tunggal disebut elusi isokratik, sedangkan pemisahan dengan dua fase gerak dengan berbagai perubahan komposisi disebut elusi gradien. Fase gerak untuk pemisahan secara KCKT harus murni untuk mencegah adanya peak pengganggu yang dapat tumpang tindih dengan peak analit, tidak bereaksi dengan kemasan, dapat melarutkan cuplikan, viskositasnya rendah (tidak lebih dari 50cP), memungkinkan memperoleh kembali cuplikan dengan mudah bila diperlukan, tidak mudah terbakar, toksisitasnya rendah, dan memiliki harga yang wajar (Skoog, Holler, dan Nieman, 1985).
Fase gerak KCKT harus bebas dari gas yang terlarut karena dapat mempengaruhi respon detektor sehingga memunculkan sinyal palsu dan akan mempengaruhi kolom (Gritter, Bobbit, Schwarting, 1991). Maka peralatan degassing diperlukan untuk menghilangkan gas yang terlarut di dalam fase gerak
(Dean, 1995).
dapat bereampur dengan air. Pemodifikasi organik yang banyak digunakan adalah metanol, asetonitril, dan tetrahidrofuran (Munson, 1984).
Kepolaran pelarut dinyatakan dalam bentuk P’ (indeks polaritas).
Besarnya polaritas campuran pelarut dapat dihitung dengan persamaan berikut:
P’ = ФaP’a+ ФbP’b
Keterangan:
Фa=fraksi volume pelarut a
Фb = fraksi volume pelarut b
P’a = kepolaran pelarut a murni
P’b = kepolaran pelarut b murni
P’ = kepolaran campuran pelarut
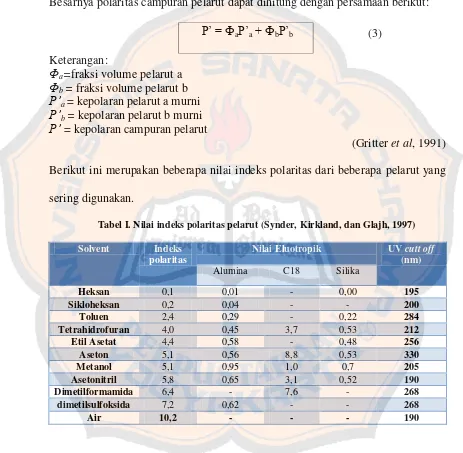
(Gritter et al, 1991) Berikut ini merupakan beberapa nilai indeks polaritas dari beberapa pelarut yang sering digunakan.
Tabel I. Nilai indeks polaritas pelarut (Synder, Kirkland, dan Glajh, 1997)
Solvent Indeks polaritas
Nilai Eluotropik UV cutt off (nm) suatu pelarut maka semakin mudah untuk mengelusi analit. Sedangkan semakin besar indeks polaritas pelarut maka semakin polar pelarut tersebut.
2. Fase diam. Kolom pada KCKTmerupakan bagian yang sangat penting karena pemisahan komponen-komponen sampel akan terjadi di dalam kolom. Keberhasilan pemisahan komponen-komponen sampel akan sangat bergantung pada keadaan kolom (Mulja dan Suharman, 1995).
Kolom pada KCKT dapat berupa gelas atau baja tidak berkarat. Kolom gelas dapat menahan tekanan sampai 50 atmosfer. Panjang kolom bervariasi antara 15-150cm, pengisi kolom biasanya adalah silika gel, alumina, dan elit (Khopkar, 1990).
Diameter kolom dibuat 3-5mm dengan tujuan supaya kepekaannya lebih teliti, menghemat fase gerak, memperluas kemampuan detektor, dan mengurangi jumlah sampel yang dianalisis. Untuk mendapatkan fase yang non polar, silika gel direaksikan dengan klorosilan (Mulja dan Suharman, 1995). Oktadesil silika (ODS) merupakan fase diam yang paling banyak dipakai karena mampu memisahkan senyawa-senyawa dengan kepolaran yang rendah, sedang, maupun tinggi (Rohman dan Gandjar, 2007).
Kolom kromatografi yang baik diharapkan mempunyai kemampuan tinggi dalam hal pemisahan, cepat dalam operasi, dan mempunyai kapasitas yang tinggi.
3. Detektor. Detektor diperlukan untuk mengindera adanya komponen cuplikan di dalam eluen kolom dan mengukur jumlahnya. Beberapa persyaratan yang harus dimiliki detektor adalah sensitivitas yang tinggi, kestabilan, reprodusibilitas yang baik, memberikan respon yang linear terhadap konsentrasi solut, dapat bekerja pada suhu kamar, mudah didapat, dan mudah pemakaiannya oleh operator (Ahuja, and Dong, 2005).
sedikit senyawa yang mempunyai sifat tersebut (Munson, 1984; Willard dkk, 1988).
3. Kromatografi partisi
Pada kromatografi partisi, fase diam dapat polar atau non polar. Bila fase diam polar dan fase gerak non polar disebut kromatografi partisi fase normal, sedangkan bila fase diam non polar dan fase gerak polar dinamakan kromatografi partisi fase terbalik.
Kecepatan migrasi analit dalam fase diam ditentukan oleh perbandingan distribusinya (D) yang bergantung pada afinitas relatif pada fase diam dan fase gerak. Dalam kromatografi, D didefinisikan sebagai perbandingan konsentrasi analit dalam fase diam (Cs) dan dalam fase gerak (Cm) (Rohman dan Gandjar, 2007).
D = Cs Cm
Kolom yang biasa digunakan dalam kromatografi partisi fase terbalik adalah kolom dengan kemasan fase terikat yang memiliki sifat stabil karena fase diamnya terikat secara kimia pada penyangga, sehingga tidak mudah terelusi oleh fase gerak. Penyangga pada fase terikat biasanya terbuat dari silika yang sudah diseragamkan, berpori, dan umumnya mempunyai diameter 3,5 atau 10 µm (Skogg et al., 1998).
Pada KCKT partisi fase terbalik biasanya mengandung bagian organik yang terikat secara kimia dengan gugus silanol pada permukaan silika. Bagian
organik tersebut umumnya hidrokarbon rantai panjang, sehingga fase gerak umumnya polar. Gugus silanol permukaan dapat direaksikan dengan berbagai cara menempelkan berbagai jenis gugus organik. Kemasan fase terikat dengan tipe ikatan siloksan (Si-O-Si-O) dibuat dengan mereaksikan organoklorosilan dengan gugus silanol pada permukaan silika gel. Reaksi silanasi sebagai berikut:
Reaksi tersebut digunakan untuk membuat isian kolom oktadesilsilan (ODS) gugus silanol dan oktadesilklorosilan sebagai berikut:
Gambar 5. Reaksi silanasi (Harris, 1999)
Gugus yang ditempelkan pada silanol pada umumnya dalah hidrokarbon rantai panjang. Panjang pendeknya rantai karbon mempengaruhi tertambatnya senyawa pada fase diam.
Fase gerak yang sering digunakan adalah campuran metanol atau asetonotril dengan air atau dengan larutan buffer. Untuk analit yang bersifat asam atau basa lemah, peranan pH sangat penting karena jika pH fase gerak tidak diatur maka analit akan mengalami ionisasi sehingga ikatan dengan fase diam akan menjadi lemah jika dibandingkan dengan bentuk tidak terionisasi, spesies yang terionisasi akan terelusi lebih cepat (Rohman dan Ganjar, 2007).
4. Waktu retensi
Waktu yang dibutuhkan oleh senyawa untuk bergerak melalui kolom menuju detektor disebut sebagai waktu retensi. Waktu retensi diukur berdasarkan
waktu di mana sampel diinjeksikan sampai sampel menunjukkan ketinggian puncak yang maksimum dari senyawa itu. Senyawa-senyawa yang berbeda memiliki waktu retensi yang berbeda. Untuk beberapa senyawa, waktu retensi akan sangat bervariasi dan bergantung pada tekanan yang digunakan (karena itu akan berpengaruh pada laju alir dari pelarut), kondisi dari fase diam (tidak hanya terbuat dari material apa, tetapi juga pada ukuran partikel), komposisi yang tepat dari pelarut, dan temperatur pada kolom. Itu berarti bahwa kondisi harus dikontrol secara hati-hati jika menggunakan waktu retensi sebagai saran untuk mengidentifikasi senyawa-senyawa (Ahuja and Dong, 2005).
5. Pemisahan puncak dalam kromatografi
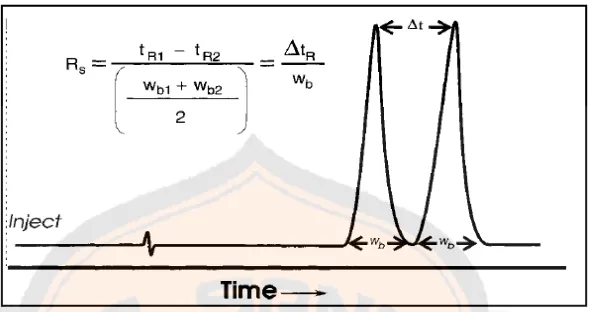
Sasaran akhir dari analisis menggunakan instrumen KCKT adalah pemisahan satu atau lebih analit dari komponen lain dalam sampel sehingga dapat diperoleh informasi kuantitatif dari masing-masing analit. Resolusi adalah derajat pemisahan dari dua puncak analit yang berdekatan.
Faktor resolusi adalah ukuran pemisahan dari 2 puncak. Daya pisah (R) dapat diukur dengan persamaan:
Rs=1 (tR2-tR)
2(W1-W2)
Keterangan:
Rs = resolusi
tR1 dan tR2 = waktu retensi senyawa diukur pada titik maksimum puncak
W1 dan W2= lebar alas puncak
Gambar 6. Kromatogram pemisahan dua senyawa secara KCKT (Ahuja and Dong, 2005)
Nilai Rs sebesar 1,5 menunjukkan bahwa baseline resolution tercapai dengan pemisahan dari dua puncak dengan ukuran yang sama sehingga diperoleh perhitungan yang dapat dipercaya. Dalam penelitian, nilai Rs sebesar 1 menunjukkan pemisahan yang sudah memadai (Ahuja and Dong, 2005).
6. Analisis kualitatif dan analisis kuantitatif
Analisis kualitatif pada KCKT pada prinsipnya mengacu pada waktu retensi peak kromatogram yang dianalisis. Pengamatan waktu retensi kromatogram analit dilakukan dengan jalan membandingkan dengan waktu retensi standar acuan (reference standard) (Mulja dan Suharman, 1995).
terpengaruh oleh pelebaran peak. Oleh karena itu, pengukuran berdasarkan luas peak lebih disukai dari pada tinggi peak (Noegrohati, 1994).
F. Validasi Metode Analisis Instrumental
1. Tinjauan umum validasi metode analisis instrumental
Validasi metode analisis adalah suatu prosedur yang digunakan untuk membuktikan apakah suatu metode analisis memenuhi persyaratan yang ditentukan atau tidak (United States Pharmacopeial Convention, 2005).
Validasi diperlukan untuk setiap metode baru atau metode yang diubah untuk memastikan bahwa metode tersebut mampu memberikan reprodusibilitas dan realibilitas yang baik, meskipun digunakan oleh operator yang berbeda menggunakan peralatan yang sama di laboratorium yang sama atau berbeda (Dong, 2005). USP 28 mencantumkan beberapa kategori uji umum yang harus memenuhi validitas data, yaitu:
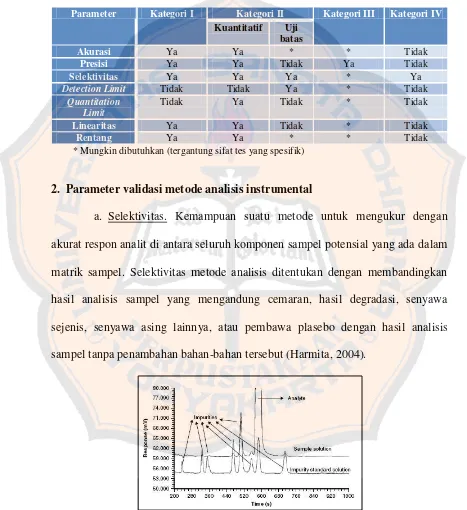
Tabel II. Kategori metode pengujian validitas data (United States Pharmacopeial Convention, 2007)
Kategori Keterangan
I Metode analitik yang digunakan untuk mengukur secara kuantitatif sejumlah besar komponen dari serbuk obat atau senyawa aktif (termasuk preservatif)
II Metode analitik yang digunakan untuk penentuan kemurnian dalam bentuk serbuk obat atau penentuan senyawa degradasi
III Metode analitik yang digunakan untuk penentuan sifat-sifat khusus seperti kecepatan disolusi dan pelepasan obat
IV Metode analitik yang digunakan untuk mengidentifikasi sediaan farmasi
kategori. Pengelompokan menjadi 4 kategori tergantung pada sifat tes yang dilakukan untuk tujuan validasi metode analisis.
Tabel III. Parameter validasi yang dipersyaratkan untuk validasi metode analisis (United States Pharmacopeial Convention, 2007)
Parameter Kategori I Kategori II Kategori III Kategori IV Kuantitatif Uji
* Mungkin dibutuhkan (tergantung sifat tes yang spesifik)
2. Parameter validasi metode analisis instrumental
a. Selektivitas. Kemampuan suatu metode untuk mengukur dengan akurat respon analit di antara seluruh komponen sampel potensial yang ada dalam matrik sampel. Selektivitas metode analisis ditentukan dengan membandingkan hasil analisis sampel yang mengandung cemaran, hasil degradasi, senyawa sejenis, senyawa asing lainnya, atau pembawa plasebo dengan hasil analisis sampel tanpa penambahan bahan-bahan tersebut (Harmita, 2004).
Selektivitas metode analisis yang terbaik dapat dibuktikan melalui pemisahan puncak-puncak yang berdekatan dengan perhitungan nilai resolusi (Rs). Nilai Rs sebesar 1,5 menunjukkan bahwa baseline resolution tercapai dengan pemisahan dari dua puncak dengan ukuran yang sama sehingga diperoleh perhitungan yang dapat dipercaya (Chan, Lam, Lee, and Zhang, 2004).
b. Linearitas. Kemampuan metode analisis memberikan respon yang secara langsung, proporsional terhadap konsentrasi analit dalam sampel dengan rentang yang ada. Untuk memperoleh linearitas antara respon analit dengan konsentrasi, data penelitian yang diperoleh harus dimasukkan dalam persamaan matematika (United States Pharmacopeial Convention, 2005). Persyaratan data linearitas yang dapat diterima adalah jika memenuhi nilai koefisien korelasi (r) > 0,999 (Mulja dan Hanwar, 2003).
Gambar 8. Linearitas dengam koefisien korelasi (r) > 0,999 (Chan et al., 2004)
Linearitas ditunjukkan langsung oleh pengenceran dari larutan stok standar dari konsentrasi yang berbeda-beda dalam rentang tertentu. Linearitas terbaik dievaluasi secara perhitungan matematika dari respon sinyal terhadap konsentrasi analit (Chan et al., 2004).
analit yang ditambahkan (United States Pharmacopeial Convention, 2005). Tabel di bawah ini merupakan kriteria penerimaan akurasi berdasarkan kadar analit (Yuwono dan Indrayanto, 2005).
Tabel IV. Kriteria penerimaan akurasi pada konsentrasi analit yang berbeda (United States Pharmacopeial Convention, 2007)
Konsentrasi analit (%) Unit Akurasi (%recovery)
100 100% 98-102 merekomendasikan menggunakan minimal sembilan kali pengujian pada tiga konsentrasi rentang tertentu dengan masing-masing konsentrasi tiga kali pengulangan (Chan et al., 2004).
Akurasi dapat ditentukan dengan dua cara, yaitu: metode simulasi (spiked placebo recovery) dan metode penambahan baku (standard addition method).
sampel kemudian dianalisis kembali. Selisih kedua hasil dibandingkan dengan kadar yang sebenarnya (Harmita, 2004).
Metode penambahan analit (standard addition method) ke dalam sampel dapat digunakan untuk menentukan konsentrasi suatu analit dengan konsentrasi yang kecil dalam sampel multikomponen seperti pada cairan biologis. Perubahan respon instrumen terhadap sampel menunjukkan adanya perubahan konsentrasi analit (International Union of Pure and Applied Chemistry, 2002).
d. Presisi. Derajat kesesuaian antara hasil uji individual yang diperoleh dari pengambilan sampel yang berulang dari suatu sampel yang homogen dengan menggunakan suatu metode analisis. Presisi dinyatakan dengan coefficient of
variation (CV) atau relative standard deviation (RSD) (United States
Pharmacopeial Convention, 2005).
Presisi suatu metode dapat dikategorikan menjadi tiga macam, yaitu: reproducibility, intermediate precision, dan repeatability. Reproducibility adalah
keseksamaan metode bila analisis dikerjakan di laboratorium yang berbeda. Intermediate precision adalah keseksamaan metode jika analisis dikerjakan di
Tabel V. Kriteria penerimaan presisi berdasar kadar analit (United States
e. Rentang. Suatu metode analisis diartikan sebagai interval antara kadar terendah sampai tertinggi analit yang dapat diukur secara kuantitatif menggunakan metode analisis tertentu dan menghasilkan ketelitian dan ketepatan, dan linearitas yang mencukupi (United States Pharmacopeial Convention, 2005). Rentang bukan merupakan parameter validasi yang dapat diukur secara independen, tetapi harus disimpulkan dari penelaahan terhadap data yang dikumpulkan dari linearitas, akurasi, dan presisi (Ahuja and Dong, 2005).
G. Landasan Teori
aktif yang tidak berubah-ubah. Salah satu bentuk sediaan OHT yang beredar di masyarakat adalah kapsul lunak dengan senyawa aktif kurkumin.
Kurkumin merupakan senyawa berwarna kuning yang memiliki kelarutan yang baik di dalam metanol dan asam asetat glasial. Kurkumin dapat dianalisis dengan menggunakan metode KCKT fase terbalik dengan sistem yang telah dioptimasi dengan kolom oktadesilsilan dan detektor visibel. Jenis dan komposisi fase gerak yang digunakan adalah metanol p.a. dan asam asetat glasial 2% (95:5 v/v) di mana kurkumin dapat terlarut di dalamnya. Penggunaan metode KCKT memiliki kelebihan dalam hal selektivitas yang baik. Hal ini didukung dengan adanya proses validasi sebagai tahap kedua dalam rangkaian penetapan kadar kurkumin dalam sediaan kapsul lunak OHT. Validasi perlu dilakukan untuk memberikan hasil dengan reprodusibilitas dan realibilitas yang baik. Validasi suatu metode analisis ditentukan oleh parameter validasi, yaitu: selektivitas, linearitas, akurasi, presisi, dan rentang.
H. Hipotesis
31
BAB III
METODE PENELITIAN
A. Jenis dan Rancangan Penelitian
Penelitian ini berdasarkan judul mengikuti jenis dan rancangan penelitian non eksperimental deskriptif karena tidak ada manipulasi terhadap subyek uji dan hanya mendeskripsikan keadaan yang ada.
B. Variabel Penelitian
1. Variabel bebas dalam penelitian ini adalah kondisi optimal dari sistem kromatografi cair kinerja tinggi fase terbalik yaitu fase gerak dan kecepatan alir.
2. Variabel tergantung dalam penelitian ini adalah parameter validasi seperti selektivitas, linearitas, akurasi, presisi, dan rentang.
3. Variabel pengacau terkendali
a. Pelarut yang digunakan yaitu metanol memiliki pH 4 dengan kemurnian yang tinggi (pro analysis).
b. Perlakuan terhadap larutan baku kurkumin terhindar dari cahaya.
C. Definisi Operasional
metanol p.a. dan asam asetat glasial 2% (95:5 v/v) dengan kecepatan alir 1,0 ml/menit.
2. Kadar kurkumin dinyatakan dalam satuan part per million (ppm).
3. Validasi metode analisis merupakan serangkaian prosedur untuk menentukan apakah metode analisis kategori I yang digunakan memenuhi parameter selektivitas, linearitas, akurasi, presisi, linearitas dan rentang.
D. Bahan-bahan Penelitian
Bahan-bahan yang digunakan dalam penelitian adalah kurkumin baku hasil sintesis Prof. Dr. Sudibyo Martono, M.S., Apt. yang telah dikonfirmasi strukturnya dengan metode spektroskopi 1H-NMR dan Mass Spectra dengan titik lebur 181,2-182,4°C (Sudibyo, 2000), metanol pro analysis 1.06009.2500 EMSURE® ACS, ISO, Reag., Ph Eur (E. Merck), asam asetat glasial pro analysis 1.00063.2500 EMPARTA® ACS (E. Merck), aquabidestilata, sampel kapsul lunak Obat Herbal Terstandar merek Rheumakur®.
E. Alat-alat Penelitian
25EE181KSJ (B115Y620), seperangkat komputer (merk Dell B6RDZ1S Connexant System RD01-D850 A03-0382 JP France S.A.S, UPS Prolink Model PR0650P , printer HP Deskjet D2566 HP-024-000 625 730), ultrasonikator (merk Retsch tipe T460 no V935922013 Ey), syringe, neraca analitik, penyaring
Milipore, mikropipet Socorex, vakum, organic solvent membrane filter
(Whatman) ukuran pori 0,45 µm; diameter 47 mm, indikator pH (E-Merck), seperangkat alat gelas (Pyrex).
F. Tata Cara Penelitian
1. Pembuatan fase gerak
Jenis dan komposisi fase gerak yang digunakan adalah metanol p.a dan asam asetat glasial 2% dengan perbandingan 95:5 v/v. Komponen fase gerak masing-masing disaring menggunakan organic solvent membrane filter untuk metanol, dan anorganic solvent membrane filter untuk asam asetat glasial 2% dengan bantuan pompa vakum kemudian diawaudarakan menggunakan ultrasonikator selama 15 menit. Selanjutnya pencampuran kedua komponen fase gerak dilakukan secara gradien di dalam instumen KCKT dengan perbandingan metanol p.a. : asam asetat glasial 2% sebesar 95:5 v/v.
2. Pembuatan pelarut metanol pH 4
3. Pembuatan larutan baku kurkumin
a. Pembuatan larutan stok kurkumin. Lebih kurang 10,0 mg baku kurkumin ditimbang seksama dan dimasukkan ke dalam labu takar 10,0 ml kemudian dilarutkan dengan metanol pH 4 hingga batas tanda sehingga diperoleh konsentrasi 1000 ppm.
b. Pembuatan larutan intermediet kurkumin. Diambil 1,0 ml larutan stok kurkumin dan dimasukkan ke dalam labu ukur 10,0 ml kemudian diencerkan dengan metanol pH 4 sampai batas tanda sehingga diperoleh konsentrasi 100 ppm. c. Pembuatan seri larutan baku kurkumin. Larutan intermediet kurkumin dipipet 150; 250; 350; 450; 550; dan 650 µl dan masing-masing dimasukkan dalam labu takar 10,0 ml dan diencerkan dengan metanol pH 4 sampai batas tanda sehingga didapatkan konsentrasi 1,5; 2,5; 3,5; 4,5; 5,5; dan 6,5 ppm. Larutan kemudian disaring dengan Milipore dan diawaudarakan dengan ultrasonikator selama 15 menit.
4. Penetapan panjang gelombang (λ) maksimum kurkumin
gelombang maksimumnya dan selanjutnya digunakan sebagai panjang gelombang deteksi pada sistem KCKT.
5. Preparasi sampel
Sebanyak 50,0 mg sampel kapsul lunak OHT merek Rhemakur® ditimbang dan dilarutkan dengan 10,0 mL metanol pH 4. Sampel selanjutnya diekstraksi dengan pengadukan secara mekanik dan konstan selama 15 menit menggunakan magnetic stirrer. Diambil sebanyak 1,0 mL dari larutan tersebut dan dimasukkan dalam labu 10,0 mL diencerkan dengan metanol pH 4 sampai tanda. Preparasi dilakukan di tempat yang terlindung dari cahaya.
6. Validasi metode analisis
a. Penentuan selektivitas. Sebanyak masing-masing 10 µl larutan baku kurkumin dan larutan ekstrak sampel Rheumakur® yang telah diawaudarakan dengan ultrasonikator selama 15 menit diinjeksikan ke dalam sistem KCKT fase terbalik dengan jenis dan komposisi fase gerak metanol p.a. : asam asetat glasial 2% (95:5 v/v) pada kecepatan alir 1,0 ml/menit. Repetisi dilakukan sebanyak 5 kali. Perhitungan resolusi dilakukan apabila terdapat 2 puncak kromatogram yang saling berdekatan dengan memasukkan selisih waktu retensi dan lebar peak ke dalam rumus perhitungan resolusi.
ultrasonikator selama 15 menit. Kemudian diinjeksikan pada sistem KCKT fase terbalik menggunakan dengan jenis dan komposisi fase gerak metanol p.a. : asam asetat glasial 2% (95:5 v/v) pada kecepatan alir 1,0 ml/menit. Replikasi dilakukan sebanyak 5 kali. Kromatogram akan menunjukkan luas area kurkumin untuk masing-masing konsentrasi. Luas area diplotkan dengan masing-masing konsentrasi untuk membuat persamaan regresi linier dengan persamaan y = bx + a. Dari kurva hubungan antara luas area dan konsentrasi akan diperoleh nilai koefisien korelasi (r) sebagai parameter untuk menentukan linieritasnya.
c. Penentuan akurasi dan presisi larutan baku kurkumin. Sebanyak 10,0 µl larutan baku kurkumin dengan kadar 0,5; 3,5; dan 6,5 ppm yang telah disaring dengan Milipore dan diawaudarakan selama 15 menit diinjeksikan pada sistem KCKT fase terbalik dengan jenis dan komposisi fase gerak metanol p.a. : asam asetat glasial 2% (95:5 v/v) pada kecepatan alir 1,0 ml/menit. Selanjutnya dihitung kadar terukur dari masing-masing seri larutan baku dengan memasukkan nilai respon yang diperoleh ke dalam persamaan kurva baku. Replikasi dilakukan sebanyak 5 kali.
Millipore dan diawaudarakan menggunakan ultrasonikator selama 15 menit.
Sebanyak 10 µl dari tiap larutan diinjeksikan pada sistem KCKT fase terbalik dengan jenis dan komposisi fase gerak metanol p.a. : asam asetat glasial 2% (95:5 v/v) pada kecepatan alir 1,0 ml/menit. Repetisi dilakukan sebanyak 5 kali. Kadar baku kurkumin yang ditambahkan dalam sampel dihitung dengan memasukkan selisih nilai respon sampel adisi dan respon sampel ke dalam persamaan kurva baku.
e. Penentuan rentang. Rentang diperoleh dari hasil perhitungan data linearitas, akurasi, dan presisi.
G. Analisis Hasil
1. Selektivitas
Selektivitas ditentukan dengan daya resolusi dari puncak kromatogram yang dihasilkan oleh baku kurkumin dan kemampuan metode KCKT fase terbalik dengan sistem yang telah dioptimasi untuk memisahkan kurkumin dengan senyawa lain dalam sampel kapsul lunak OHT merek Rheumakur®. Selektivitas ditentukan dengan perhitungan resolusi menggunakan rumus:
)
tR1= waktu retensi puncak pertama
tR2= waktu retensi puncak kedua
w1= lebar dasar peak pertama
w2= lebar dasar peak kedua
Nilai Rs= 1,0 menunjukkan puncak kromatogram baku kurkumin tidak terpisah sampai ke baseline, nilai Rs= 1,5 menunjukkan puncak telah terpisah sampai baseline, dan nilai Rs > 1,5 menunjukkan pemisahan puncak-puncak telah sempurna antara satu sama lain.
2. Linearitas
Linearitas dilihat dari harga koefisien korelasi (r) hasil pengukuran seri baku kurkumin. Rentang ditentukan dari kadar larutan baku kurkumin konsentrasi terkecil hingga terbesar. Persyaratan data linearitas yang dapat diterima adalah jika memenuhi nilai koefisien korelasi (r) > 0,999.
3. Akurasi
Akurasi metode analisis dinyatakan dengan nilai % recovery yang dihitung dari konsentrasi terukur dibandingkan dengan konsentrasi teoritis (kadar sebenarnya) dikalikan 100%.
Recovery= konsentrasi terukur
konsentrasi teoritis ×100%
4. Presisi
Presisi metode analisis dinilai berdasarkan Coefficient of Variation (CV) yang dihitung dengan cara sebagai berikut:
CV= SD
𝑋 ×100%
Keterangan:
SD = Standard Deviation 𝑋 = kadar rata-rata
𝐶𝑉 = Coefficient of Variation
5. Rentang
Rentang merupakan interval antara kadar terendah sampai tertinggi analit yang dapat diukur secara kuantitatif menggunakan metode analisis tertentu dan menghasilkan linearitas, akurasi, dan presisi yang baik.
40
BAB IV
HASIL DAN PEMBAHASAN
A. Pembuatan Fase Gerak KCKT
Sistem KCKT yang digunakan dalam validasi metode penetapan kadar kurkumin dalam sediaan kapsul lunak OHT merek Rheumakur® secara KCKT fase terbalik karena menggunakan fase gerak yang bersifat lebih polar dibandingkan fase diamnya. Jenis dan komposisi fase gerak yang digunakan dalam penelitian ini adalah campuran metanol p.a. dan asam asetat glasial 2% dengan perbandingan 95:5 (v/v) dengan indeks polaritas sebesar 5,351. Metanol p.a. dan asam asetat glasial 2% dipilih sebagai fase gerak karena keduanya dapat
melarutkan kurkumin dengan baik.
Sebelum digunakan, komponen fase gerak disaring menggunakan penyaring Whatman dengan bantuan pompa vakum untuk menyaring partikel-partikel yang dapat menyumbat kolom. Selanjutnya diawaudarakan menggunakan ultrasonikator untuk menghilangkan gelembung udara yang dapat mengganggu tekanan pompa pada instrumen KCKT. Gelembung udara dapat mengakibatkan tekanan pada pompa tidak stabil sehingga mempengaruhi proses pembacaan sinyal dalam instrumen KCKT.
sehingga tidak akan merusak kolom oktadesilsilan (C18) pada instrumen KCKT.
Penggunaan campuran metanol p.a. dan asam asetat glasial 2% (95:5 v/v) sebagai fase gerak memiliki nilai pH 4 sehingga tidak akan merusak kolom kromatografi.
Pada pH terlalu asam (pH ≤ 2), C18 bereaksi dengan asam sehingga oktadesilsilan
kembali ke bentuk silanol (Gambar 9).
Si O Si (CH2)17CH3 Si OH Cl Si (CH)
17CH3
H2O/H
H+
Gambar 9. Reaksi degradasi kolom C18 pada pH asam ≤ 2
B. Pembuatan Pelarut dan Stabilitas Kurkumin
Menurut Tonnesen dan Karlsen (1995), stabilitas kurkumin dipengaruhi oleh pH dan cahaya. Kurkumin stabil dalam pH asam yang secara kenampakan fisik menunjukkan warna kuning sesuai dengan warna asli kurkumin. Pada kondisi lingkungan basa, kurkumin akan mengalami degradasi membentuk asam ferulat dan feruloil metan. Pengujian stabilitas kurkumin dilakukan pada kondisi lingkungan suasana asam pada berbagai pH. Metanol p.a. memiliki pH 5 dan asam asetat glasial 2% memiliki pH 2. Pelarut baku kurkumin yang digunakan adalah metanol p.a dengan penambahan sejumlah tertentu asam asetat glasial 2% untuk mengatur suasana asam. Hasil optimasi pengujian stabilitas kurkumin berdasarkan pH menunjukkan bahwa kurkumin stabil pada suasana asam dengan pH 4. Pada suasana asam (pH 4) kurkumin akan stabil pada bentuk molekulnya.
lama-kelamaan senyawa kurkumin dapat terputus ikatannya pada gugus metilen aktif menjadi trans-6-(4’-hidroksi-3-metoksifenil)-2,3-diokso-5-heksanal, asam ferulat, feruloilmetan, dan vanilin yang tidak berwarna.
Gambar 10. Gugus metilen aktif pada kurkumin
C.Pembuatan Larutan Baku Kurkumin
Larutan baku kurkumin dibuat dengan melarutkan baku kurkumin dalam pelarut metanol p.a pada pH 4. Larutan baku yang dibuat pada penelitian ini terdiri dari 3 macam, yaitu: larutan stok, larutan intermediet, dan larutan seri baku. Larutan stok dibuat dengan konsentrasi 1000 ppm, sedangkan larutan intermediet dibuat dengan konsentrasi 100 ppm. Larutan seri baku dibuat dalam enam konsentrasi berbeda, yaitu: 1,5; 2,5; 3,5; 4,5; 5,5; dan 6,5 ppm.
D.Penentuan Panjang Gelombang Serapan Maksimum Kurkumin
Penentuan panjang gelombang serapan maksimum kurkumin bertujuan untuk mengetahui pada panjang gelombang berapa kurkumin memberikan absorbansi maksimum pada detektor visibel. Selain itu, untuk mengetahui intermediate precision dari metode yang digunakan yaitu ketepatan pada kondisi
Penentuan panjang gelombang serapan maksimum kurkumin dilakukan dengan menggunakan konsentrasi larutan baku kurkumin dengan konsentrasi 0,4; 1,0; dan 1,6 ppm. Pelarut yang digunakan dalam penentuan panjang gelombang maksimum kurkumin sama dengan pelarut yang digunakan untuk analisis pada sistem KCKT. Panjang gelombang serapan maksimum dilakukan dengan mengukur absorbansi kurkumin pada daerah visibel. Menurut Aggarwal et al., (2006), panjang gelombang serapan maksimum (λmaks.) kurkumin adalah 430 nm. Suatu senyawa untuk dapat ditetapkan kadarnya pada panjang gelombang visibel harus memiliki gugus kromofor dan auksokrom, di mana kedua gugus ini bertanggung jawab dalam penyerapan radiasi. Kurkumin memiliki gugus kromofor dan aukokrom pada strukturnya sehingga dapat memberikan serapan pada daerah visibel (Gambar 11). Kromofor merupakan gugus fungsional tak jenuh yang mengandung elektron π dan jika terkena radiasi elektromagnetik,
mudah tereksitasi ke tingkat energi yang lebih tinggi yaitu orbital π*. Auksokrom
Gambar 11. Gugus kromofor dan auksokrom pada struktur kurkumin
Keterangan:
: gugus kromofor : gugus auksokrom
Spektra yang dihasilkan pada penetapan panjang gelombang maksimum kurkumin menggunakan 3 konsentrasi berbeda ditunjukkan pada gambar di bawah ini:
1 2
3
Gambar 12. Spektra panjang gelombang maksimum kurkumin
Keterangan:
1= konsentrasi rendah (0,4 ppm) 2= konsentrasi tengah (1,0 ppm) 3= konsentrasi tinggi (1,6 ppm)
konsentrasi maka absorban juga semakin bertambah. Spektra ketiga larutan baku kurkumin dengan konsentrasi 0,4; 1,0; dan 1,6 ppm diperoleh panjang gelombang maksimum berturut-turut sebesar 432, 433, dan 432 nm. Spektra panjang gelombang maksimum kurkumin (gambar 12) menunjukkan pada konsentrasi larutan baku kurkumin yang berbeda-beda tetap menunjukkan pola spektra yang sama meskipun terjadi peningkatan absorbansi di mana ditunjukkan dengan panjang gelombang maksimum dari tiap konsentrasi. Panjang gelombang maksimum kurkumin terletak pada panjang gelombang 432 nm sesuai spektra yang ditunjukkan pada larutan baku konsentrasi rendah (0,4 ppm) dan konsentrasi tinggi (1,6 ppm). Menurut Farmakope Indonesia III, penyimpangan panjang gelombang maksimum yang diperbolehkan sebesar ± 2 nm dari panjang gelombang maksimum teoritis sehingga panjang gelombang maksimum yang diperoleh dari pengukuran masih dapat digunakan untuk validasi metode penetapan kadar kurkumin dalam sediaan kapsul lunak OHT merek Rheumakur® secara KCKT fase terbalik.
E.Analisis Kualitatif Berdasarkan Waktu Retensi (tR) Kurkumin
Analisis kualitatif pada KCKT pada prinsipnya mengacu pada waktu retensi (tR) peak kromatogram yang dianalisis. Tujuan dari pengamatan waktu
retensi (tR) dari kurkumin adalah untuk mengetahui waktu yang dibutuhkan
kurkumin saat melewati fase diam dengan bantuan fase gerak.
partisi dari kurkumin terhadap fase diam dan fase geraknya. Kurkumin memiliki sisi polar dan non polar pada strukturnya. Pada penelitian ini sistem kromatografi yang digunakan adalah kromatografi partisi fase terbalik di mana fase gerak yang digunakan lebih polar dibanding fase diam. Oleh karena itu, senyawa yang cenderung bersifat non polar menyebabkan senyawa akan lebih lama melewati kolom sehingga waktu retensinya akan lebih besar.
Dilihat dari strukturnya, kurkumin memiliki gugus polar dan non polar yang berinteraksi dengan fase diam dan fase gerak.
HO
O O
OH
OH3C CH3O
Gambar 13. Gugus polar dan non polar pada struktur kurkumin
Keterangan gambar : : gugus polar
: gugus non polar
O
Gambar 14. Interaksi hidrogen antara kurkumin dengan fase gerak metanol:asam asetat glasial 2% (95:5)
Gambar 15 . Interaksi Van Der Waals kurkumin dengan fase diam oktadesilsilan
Dari gambar 14 dan 15 menunjukkan bahwa interaksi kurkumin dengan fase gerak lebih kuat dibandingkan dengan fase diam. Hal ini dipengaruhi oleh adanya interaksi hidrogen antara kurkumin dengan fase gerak.
Rheumakur® memiliki tR 2,680 menit. Oleh karena itu dapat dipastikan bahwa
dalam sampel kapsul OHT merek Rheumakur® terdapat kandungan kurkumin.
(a)
(b)
Gambar 16. Kromatogram baku kurkumin (a) dan kurkumin dalam sampel (b)
F.Pembuatan Kurva Baku Kurkumin
Pembuatan kurva baku kurkumin bertujuan untuk mendapatkan persamaan regresi linear sehingga dapat digunakan dalam analisis kuantitatif. Kurva baku menyatakan hubungan linier antara konsentrasi dengan Area Under Curve (AUC) di mana dengan meningkatnya konsentrasi maka akan
Penelitian ini menggunakan 6 seri konsentrasi larutan baku kurkumin, yaitu: 1,5; 2,5; 3,5; 4,5; 5,5; dan 6,5 ppm yang masing-masing direplikasi 3 kali. Pembuatan kurva baku dilakukan replikasi sebanyak 3 kali dengan tujuan untuk mendapatkan kurva baku dengan nilai koefisien korelasi paling baik. Ada beberapa pertimbangan yang diperhatikan dalam pemilihan data persamaan kurva baku beberapa replikasi yaitu berdasarkan pada nilai r terhitung, nilai A (intersept), nilai B (slope), dan SE (standard error). Dalam penelitian ini, parameter utama yang digunakan adalah berdasarkan nilai r terhitung yang didapatkan yaitu 0,9996 di mana r yang didapatkan lebih besar dari nilai r linearitas analisis yaitu > 0,999 untuk minimal 5 seri konsentrasi (ICH, 1995). Nilai r semakin mendekati 1 menunjukkan semakin baik linearitas persamaan yang didapat, sehingga semakin baik hubungan antara peningkatan konsentrasi dengan peningkatan respon yaitu AUC. Berikut tabel data perolehan AUC seri baku kurkumin dari 3 kali replikasi.
Tabel VI. Penentuan kurva baku kurkumin
Replikasi 1 Replikasi 2 Replikasi 3
C (ppm) AUC C (ppm) AUC C (ppm) AUC
Dari hasil penentuan kurva baku kurkumin, ketiga persamaan regresi linear yang
diperoleh memiliki nilai α yang lebih dari 45° sehingga bila dibuat kurva baku
hubungan konsentrasi seri baku kurkumin dengan AUC akan dihasilkan kurva
dengan kemiringan kurang baik dengan α sebesar 89,99°. Oleh karena itu,
dilakukan modifikasi yaitu konsentrasi baku kurkumin dikalikan 60.000.000 sehingga diperoleh hasil seperti yang tersaji pada tabel VII.
Tabel VII. Penentuan kurva baku kurkumin hasil modifikasi
Replikasi 1 Replikasi 2 Replikasi 3
C (mg/ml)x6.107 AUC C (mg/ml)x6.107 AUC C (mg/ml)x6.107 AUC 90000 80587 88200 88101 91800 99579 150000 140888 147000 151483 153000 154468 210000 192509 205800 234079 214200 227391 270000 246752 264600 290285 275400 285619 330000 316510 323400 383911 336600 350697 390000 383164 382200 463126 397800 413947 A = -12578,6
Berikut grafik kurva baku yang dihasilkan:
Gambar 17. Kurva baku kurkumin
Kurva baku kurkumin dengan nilai α sebesar 46,01° dapat menunjukkan
korelasi yang baik antara AUC dengan konsentrasi di mana semakin tinggi konsentrasi maka nilai AUC juga semakin meningkat sehingga persamaan kurva baku yang dihasilkan dapat digunakan untuk perhitungan kadar kurkumin. Nilai r yang mendekati satu menggambarkan adanya korelasi yang baik antara variabel bebas (konsentrasi) dan variabel tergantung (AUC).
G.Validasi Metode Analisis
Validasi metode analisis merupakan suatu proses untuk membuktikan apakah suatu metode memiliki validitas yang baik sesuai dengan parameter-parameter yang diujikan. Tujuan dilakukan validasi metode adalah untuk membuktikan dan menjamin bahwa metode analisis yang digunakan memiliki validitas yang baik sehingga hasilnya dapat dipercaya.
y=1,0358x + 1711,5
0 100000 200000 300000 400000 500000
Menurut USP XXVIII validasi metode dilakukan minimum 9 kali penentuan mencakup range tertentu, misal 3 macam konsentrasi dan setiap konsentrasi direplikasi 3 kali (United States Pharmacopeial Convention, 2000). Dalam penelitian ini digunakan 3 macam konsentrasi yang direplikasi 5 kali yaitu konsentrasi rendah 1,5 ppm; konsentrasi tengah 3,5 ppm; dan konsentrasi tinggi 6,5 ppm. Pemilihan konsentrasi rendah, tengah, tinggi dari kurva baku adalah untuk mewakili keseluruhan konsentrasi yang dibuat yaitu antara konsentrasi 1,5 sampai 6,5 ppm.
Parameter-parameter yang dibutuhkan dalam validasi metode ditentukan oleh kategori metode analisis yang digunakan. Pada penelitian ini termasuk dalam kategori I menurut USP 28 karena penelitian yang dilakukan merupakan metode analisis kuantitatif yang digunakan untuk mengukut secara kuantitatif sejumlah besar komponen dari serbuk obat atau senyawa aktif (termasuk preservatif) dalam sediaan obat jadi. Dengan demikian, parameter-parameter validasi yang dibutuhkan dalam penelitian ini adalah selektivitas, linearitas, akurasi, presisi, dan rentang.
1. Selektivitas
selektivitas yang baik jika nilai Rs yang dihasilkan > 1,5. Pada penelitian yang dilakukan tidak dilakukan perhitungan nilai Rs karena pada kromatogram yang diperoleh (Gambar 18) menunjukkan peak yang dominan dari kurkumin dalam sampel. Proses preparasi sampel kapsul lunak OHT merek Rheumakur® dengan cara ekstraksi menggunakan pelarut metanol p.a. pH 4. Selanjutnya dilakukan pengadukan mekanik selama 15 menit untuk melarutkan kurkumin yang terdapat dalam sampel. Komponen lain yang tidak larut dalam metanol akan tersaring dalam penyaring sehingga tidak ikut diinjeksikan ke dalam instrumen KCKT. Komponen lain yang larut metanol memberikan respon dalam kromatogram, namun peak yang muncul memiliki signal kecil sehingga tidak mengganggu peak kurkumin. Oleh karena itu, perhitungan Rs tidak dilakukan. Hasil penentuan selektivitas dari kromatogram kurkumin dalam sampel yang diperoleh menunjukkan bahwa metode ini dapat dikatakan memiliki selektivitas yang baik.