appendices_w2.doc 108KB Jun 05 2011 09:30:46 PM
Teks penuh
Gambar





Garis besar
Dokumen terkait
Ground and excited state CASPT 2 geometry optimisations of small organic molecules.
Conformational energies are calculated in the rigid geometry model with ICFF (red dashed lines) and MMFF94s (blue dotted lines) “local” energy functions at different values of
MATRIX WITH THE DISCRETE VALUES FOR EACH ROTATION ANGLE AND ITS CORRESPONDING ENERGY VALUE FOR THE FIRST ROTATION FOR BASIC STRUCTURE. Labels in bold were not used in
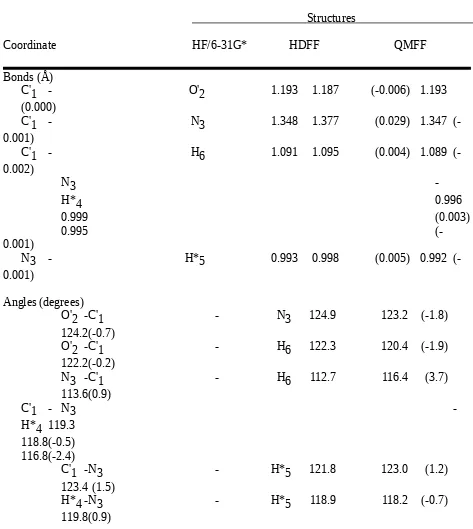
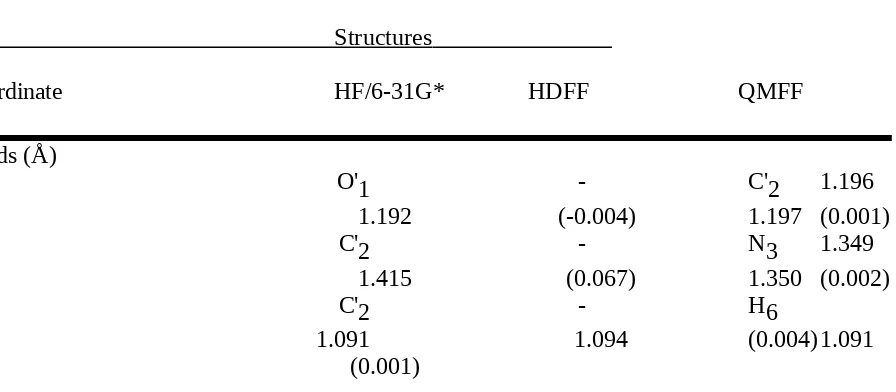
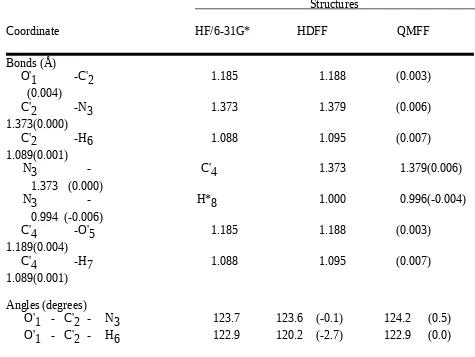
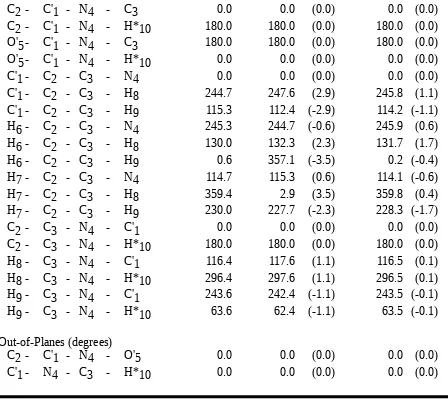
The bond lengths were allowed to refine in an interval that was 0.05 Å larger than the maximum values of all occurences of the respective bond, the bond stretching force constants
bromide, compared to the scaled structures, gave rms differences of 0.004 Å and 1.16 degrees, 0.003 Å and 1.68 degrees, and 0.005 Å and 1.42 degrees, respectively, for the bond
The electrostatic potential points used are those of the merged sets of points used in the CHELP, CHELPG, and Merz-Kollman methods, except for the two copper complexes where
Table with details of the experimental structures used for the force field refinement For the definition of structural parameters see Chart
QCISD(T) energies at the MP2 optimized geometry and the B3LYP energies at the B3LYP optimized geometries, dipole moments calculated at the HF/6-31G* level of theory are in Debyes... a
