PENGEMBANGAN METODE SPECTRAL ALIGNMENT BERBASIS
FUZZY UNTUK PENGOREKSIAN SEQUENCE DNA
DARI NEXT GENERATION SEQUENCER
KANA SAPUTRA S
SEKOLAH PASCASARJANA INSTITUT PERTANIAN BOGOR
PERNYATAAN MENGENAI TESIS DAN
SUMBER INFORMASI SERTA PELIMPAHAN HAK CIPTA
Dengan ini saya menyatakan bahwa disertas berjudul Pengembangan Metode
Spectral Alignment Berbasis Fuzzy untuk Pengoreksian Sequence DNA dari Next Generation Sequencer adalah benar karya saya dengan arahan dari komisi pembimbing dan belum diajukan dalam bentuk apapun kepada perguruan tinggi mana pun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir tesis ini.
Dengan ini saya melimpahkan hak cipta dari karya tulis saya kepada Institut Pertanian Bogor.
Bogor, Agustus 2015
Kana Saputra S
RINGKASAN
KANA SAPUTRA S. Metode Spectral Alignment Berbasis Fuzzy untuk Pengoreksian Sequence DNA dari Next Generation Sequencer. Dibimbing oleh WISNU ANANTA KUSUMA dan AGUS BUONO.
Teknologi sequencing terus berkembang dari traditional Sanger Shotgun sequencing menjadi Next Generation Sequencing (NGS). Teknologi ini menghasilkan short read dalam jumlah yang banyak dan membutuhkan waktu yang relatif singkat dalam sekali menjalankan program. Teknologi yang digunakan saat ini masih menghasilkan kesalahan atau error dalam proses pembacaan urutan sekuen DNA. Kesalahan pembacaan urutan sekuen DNA dapat mengakibatkan akurasi data yang rendah dan menambah waktu pada saat proses DNA sequence assembly. Permasalahan ini dapat diatasi dengan melakukan pendeteksian dan pengoreksian DNA sequencing error.
Metode untuk mendeteksi dan mengoreksi DNA sequencing error
menggunakan metode spectral alignment. Metode tersebut hanya menggunakan aspek frekuensi kemunculan tuple (multiplicity). Penelitian lain untuk mendeteksi dan mengoreksi DNA sequencing error menggunakan model statistika dengan melihat aspek kualitas basa. Dalam penelitian ini akan menggabungkan aspek
multiplicity dan kualitas basa. Kemudian muncul permasalahan bagaimana menentukan batas multiplicity dan kualitas tuple yang tidak mengandung error.
Fuzzy inference system (FIS) dipilih karena dapat mengatasi permasalahan ini. FIS digunakan untuk mengklasifikasikan sebuah tuple masuk ke dalam solid atau weak tuples.
Metode spectral alignment berbasis fuzzy diimplementasikan sebagai tahap
preprocessing sebelum proses DNA sequence assembly. Keberhasilan proses pendeteksian dan pengoreksian DNA sequencing error dilihat dari aspek perhitungan jumlah nodes. Perhitungan jumlah nodes dihasilkan oleh Velvet assembler. Untuk memastikan akurasi data set yang telah dikoreksi maka akan dilakukan proses evaluasi. Evaluasi dilakukan dengan melihat kualitas contigs dan perhitungan similarity antara data set yang telah dikoreksi dengan data reference. Data reference diperoleh dari National Center for Biotechnology Information (NCBI). Tool yang digunakan untuk menghitung similarity menggunakan Basic Local Alignment Search (BLAST).
Penelitian ini berhasil mendapatkan model fuzzy inference system (FIS) yang sesuai untuk mengklasifikasikan tuple ke dalam solid atau weak tuple yang menjadi inputan untuk metode spectral alignment sebagai tahapan preprocessing. Data set yang dikoreksi menggunakan metode spectral alignment berbasis fuzzy
menghasilkan jumlah nodes yang lebih sedikit dibandingkan dengan data set yang belum dikoreksi (uncorrected read) dan data set yang hanya dikoreksi menggunakan metode spectral alignment dengan mempertahankan akurasi data dan kualitas contigs. Ini menunjukkan bahwa pendeteksian dan pengoreksian DNA
sequencing error menggunakan menggunakan metode spectral alignment berbasis
fuzzy dapat menyederhanakan graf dibandingkan dengan hanya menggunakan metode spectral alignment.
SUMMARY
KANA SAPUTRA S. Fuzzy-based Spectral Alignment Method for Correcting DNA Sequence from Next Generation Sequencing. Supervised by WISNU ANANTA KUSUMA and AGUS BUONO.
Sequencing technology continues to evolve from traditional Sanger Shotgun sequencing into the Next Generation Sequencing (NGS). This technology produces short read in large numbers and requires a relatively short time in a running program. The technology still generates an error in the DNA sequencing process. DNA sequencing error could result low data accuracy and increase time of process of DNA sequence assembly. This problem can be handled by performing DNA sequencing error detection and correction.
The method used for detecting and correcting DNA sequencing errors in this research was spectral alignment method. This method uses only frequency of tuple occurrence aspect (multiplicity). Another reserach for detecting and correcting DNA sequencing errors was done statistical models to look at the bases quality aspect. In this research we will combine aspects of multiplicity and bases quality. Then the problem is how to define the limits of multiplicity and tuple quality that contains no errors. Fuzzy Inference System (FIS) has been able to overcome these problems. FIS is used to classify a tuple into the solid or weak tuples.
Fuzzy based spectral alignment method is implemented as a preprocessing step before the process of DNA sequence assembly. The success of detection and correction DNA sequencing error is seen from the aspect of the calculation of total nodes. Calculation of the total nodes produced by Velvet assembler. To ensure accuracy of the data set that has been corrected, an evaluation process will be conducted. Evaluation is done by looking at the quality of contigs and calculation of similarity between data sets that have been corrected with reference data. Reference data was obtained from the National Center for Biotechnology Information (NCBI). The tool used to calculate the similarity using the Basic Local Alignment Search (BLAST).
This research successfully obtained a model of fuzzy inference system (FIS) appropriate to classify a tuple into a solid or weak tuples as input for spectral alignment method as a preprocessing step. Data sets were corrected using fuzzy based spectral alignment method generated fewer number of nodes compared with data sets that have not been corrected (uncorrected read) and the data set that is only corrected using the spectral alignment method to maintain data accuracy and quality of the contigs. This shows that the detection and correction DNA sequencing error using fuzzy based spectral alignment method can simplify graph as compared to just using spectral alignment method.
© Hak Cipta Milik IPB, Tahun 2015
Hak Cipta Dilindungi Undang-Undang
Dilarang mengutip sebagian atau seluruh karya tulis ini tanpa mencantumkan atau menyebutkan sumbernya. Pengutipan hanya untuk kepentingan pendidikan, penelitian, penulisan karya ilmiah, penyusunan laporan, penulisan kritik, atau tinjauan suatu masalah; dan pengutipan tersebut tidak merugikan kepentingan IPB
Tesis
sebagai salah satu syarat untuk memperoleh gelar Magister Ilmu Komputer
pada
Program Studi Ilmu Komputer
PENGEMBANGAN METODE SPECTRAL ALIGNMENT BERBASIS
FUZZY UNTUK PENGOREKSIAN SEQUENCE DNA
DARI NEXT GENERATION SEQUENCER
SEKOLAH PASCASARJANA INSTITUT PERTANIAN BOGOR
BOGOR 2015
PRAKATA
Puji dan syukur penulis panjatkan kepada Allah subhanahu wa ta’ala atas segala karunia-Nya sehingga karya ilmiah ini berhasil diselesaikan. Tema yang dipilih dalam penelitian yang dilaksanakan sejak bulan September 2014 ini ialah pengoreksian DNA sequencing error, dengan judul Pengembangan Metode
Spectral Alignment Berbasis Fuzzy untuk Pengoreksian Sequence DNA dari Next Generation Sequencer.
Terima kasih penulis ucapkan kepada Bapak Dr Eng Wisnu Ananta Kusuma ST MT dan Bapak Dr Ir Agus Buono MSi MKom selaku pembimbing, serta Bapak Irman Hermadi SKom MS PhD yang telah banyak memberi saran. Di samping itu, penghargaan penulis sampaikan kepada M. Syafiuddin Usman, Auriza Akbar, dan Abrar Istiadi dari Laboratorium Apllied Computing yang telah membantu selama penelitian ini. Ungkapan terima kasih juga disampaikan kepada ayahanda Aiptu Saparuddin Saragih, ibunda Salmina, adinda Putri Sasalia S, serta seluruh keluarga dan ilkom angkatan 15, atas segala doa dan kasih sayangnya.
Semoga karya ilmiah ini bermanfaat.
Bogor, Agustus 2015
DAFTAR ISI
DNA Sequencing Error 4
FASTQ 5
Metode Spectral Alignment 5
4 HASIL DAN PEMBAHASAN 15
Data set 15
Analisis Metode Spectral Alignment 15
Analisis Metode Spectral Alignment Berbasis Fuzzy 16
Pemodelan fuzzy 16
Pengklasifikasian tuples 19
Evaluasi 20
Perhitungan jumlah nodes 20
Evaluasi kualitas contigs 21
Evaluasi similarity 22
5 SIMPULAN DAN SARAN 23
Simpulan 23
Saran 23
Ucapan Terima Kasih 23
DAFTAR PUSTAKA 24
LAMPIRAN 26
DAFTAR TABEL
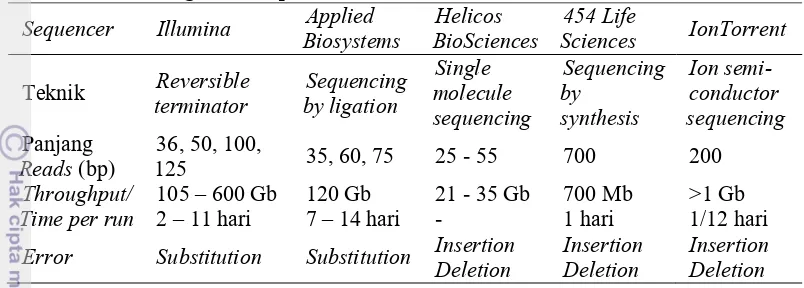
1 Perbandingan Next Generation Sequencing (NGS) pada tahun 2012 5
2 Definisi variabel input dan output 11
3 Karakteristik data set 15
4 Jumlah solid dan weak tuples (Metode spectral alignment) 15
5 Himpunan fuzzy 16
6 Kombinasi rules 18
7 Jumlah solid tuples dan weak tuples (Metode spectral alignment berbasis
fuzzy) 19
8 Penentuan range untuk solid tuples 20
9 Evaluasi perhitungan jumlah nodes 21
10 Hasil evaluasi kualitas contigs untuk k = 17 21 11 Hasil evaluas kualitasi contigs untuk k = 21 22
DAFTAR GAMBAR
1 Contoh isi dari file FASTQ 5
2 Tahapan penelitian metode spectral alignment 6
3 Representasi kurva membership function trapesium 7 4 Tahapan penelitian metode spectral alignment berbasis fuzzy 9
5 Interface dari DNA Data Bank of Japan (DDBJ) 10
6 Contoh konversi kode ASCII menjadi skor Phred 10
7 Representasi dari variabel input dan output 11
DAFTAR LAMPIRAN
1 Model fuzzy inference system (FIS) 1 yang telah diimplementasikan 27 2 Model fuzzy inference system (FIS) 2 yang telah diimplementasikan 28 3 Model fuzzy inference system (FIS) 3 yang telah diimplementasikan 29 4 Model fuzzy inference system (FIS) 4 yang telah diimplementasikan 30 5 Source Code (Menghitung multiplicity dan kualitas tuple) 31 6 Source Code (Menghitung kedekatan antara weak tuple dengan solid
tuple menggunakan jarak Levenshtein) 34
7 Source Code (Menggantikan weak tuple yang terdapat di data set
menjadi solid tuple - string matching) 36
8 Source Code (Menghitung number of contigs, N50 size, dan maximum
1
PENDAHULUAN
Latar Belakang
Dalam ilmu biologi dan kesehatan, deoxyribonucleic acid (DNA) merupakan makromolekul yang sangat penting. DNA berfungsi untuk menyimpan semua informasi tentang genetika dari makhuk hidup (Bryce & Pacini 1998). Proses
sequencing diperlukan untuk mengetahui urutan DNA. Dengan kata lain,
sequencing merupakan suatu proses pembacaan urutan DNA. Hasil yang diperoleh berupa sekuen DNA yang dapat digunakan untuk menemukan gen, menemukan daerah yang memiliki kode untuk suatu protein yang spesifik, dan dapat membandingkan homologous DNA sequences dari organisme yang berbeda (Rogers 2011). Proses sequencing saat ini juga diterapkan untuk berbagai sampel tumor dalam upaya untuk mengidentifikasi mutasi terkait dengan kanker (Chong et al. 2012).
Saat ini teknologi untuk melakukan sequencing terus berkembang dari
traditional Sanger Shotgunsequencing menjadi next generation sequencing (NGS). NGS yang digunakan masih menghasilkan kesalahan atau error dalam pembacaan urutan sekuen DNA. NGS yang memiliki throughput tinggi dan waktu sequencing
yang cepat telah dikembangkan, misalnya Solexa/Illumina, Applied Biosystems SoLiD, dan Roche/454 Life Sciences yang dapat menghasilkan jutaan short reads
setiap program dijalankan. Jutaan short reads yang dihasilkan oleh NGS masih memungkinkan terjadinya kesalahan pembacaan (error). Terdapat beberapa jenis
error yang dihasilkan oleh sequencer, yaitu substitution, insertion, dan deletion
(Chevreux 2005). Hasil sequencing yang dibaca menggunakan Illumina merupakan salah satu dari teknologi NGS paling terkenal dan umum digunakan menghasilkan
reads dengan panjang berkisar 35-125 bp. Pembacaan urutan DNA mengandung kesalahan (error) berkisar antara 0,5-2,5%, sebagian besar berupa substitution error (Kelley et al. 2010).
Kesalahan pembacaan (error sequencing) yang dihasilkan oleh Illumina adalah substitution (Liu et al. 2012). Error ini dapat mengakibatkan terbentuknya graf yang memiliki cabang sehingga menambah jumlah node yang dihasilkan. Hal ini didukung oleh pendapat Miller et al. (2010) yang menyebutkan bahwa
sequencing error menyebabkan graf yang dihasilkan pada proses DNA sequence assembly menjadi lebih kompleks. Oleh karena itu, pengoreksian error sangat penting dilakukan untuk meningkatkan akurasi DNA yang dihasilkan oleh NGS (Yang et al. 2012) dan mengurangi kompleksitas graf. Mengenai hal tersebut Pevzner et al. (2001) telah mengembangkan metode untuk mendeteksi dan mengoreksi DNA sequencing errors, yaitu metode spectral alignment. Selanjutnya metode spectral alignment dikembangkan oleh Shi et al. (2009) menggunakan CUDA dan Caesar et al. (2013) berdasarkan frekuensi kemunculan tuple
(multiplicity). Selain itu, Wijaya et al. (2009) telah mengembangkan tool
2
Dari beberapa penelitian di atas, belum ada yang menggabungkan aspek
multiplicity dan kualitas tuple. Permasalahan yang mucul dalam kasus ini adalah bagaimana menentukan range multiplicity dan kualitas tuple yang baik. Metode yang dapat digunakan untuk mengatasi masalah penentuan range tersebut adalah logika fuzzy. Logika fuzzy mampu menangani ketidakjelasan dan ketidakpastian dari berbagai variabel inputan (Thamrin 2012). Logika fuzzy yang digunakan adalah
fuzzy inference system (FIS) untuk memproses kedua aspek tersebut. Penelitian menggunakan FIS sebelumnya telah diterapkan oleh Qidway et al. (2007) untuk kasus memprediksi Failed Back Surgery Syndrome (FBSS) dengan tingkat akurasi 88%. Penelitian lain mengenai FIS dilakukan oleh Othman et al. (2002) untuk mengintegrasikan kapasitas produksi dan keseimbangan muatan selama aktifitas penjadwalan dan Abdullah et al. (2012) untuk mengklasifikasikan likelihoods dari pembelian asuransi kesehatan. Oleh karena itu, penelitian ini akan mencoba menerapkan beberapa model FIS untuk mengurangi kompleksitas graf. Model FIS yang terbentuk diharapkan merupakan model FIS yang sesuai.
Penelitian ini bertujuan menerapkan dan memperoleh model FIS yang sesuai untuk mendeteksi dan mengoreksi DNA sequencing error menggunakan metode spectral alignment sebagai tahapan preprocessing sebelum proses DNA
assembly dilakukan. Model FIS berfungsi untuk mengklasifikasikan sebuah tuple
menjadi solid tuple atau weak tuple. Untuk mengevaluasi metode ini, reads yang telah dikoreksi akan dirakit menggunakan Velvet assembler untuk melihat penurunan jumlah nodes, dan tingkat akurasi berdasarkan similarity yang menunjukkan keberhasilan pengoreksian DNA sequencing error.
Perumusan Masalah
Berdasarkan uraian pada latar belakang, maka rumusan masalah dalam penelitian ini adalah bagaimana meningkatkan akurasi data hasil pengoreksian menggunakan metode spectral alignment berbasis fuzzy dan bagaimana mengklasifikasikan tuple ke dalam solid tuples atau weak tuples menggunakan model FIS berdasarkan multiplicity dan kualitas tuple yang menjadi input untuk metode spectral alignment yang dilakukan oleh Caesar et al. (2013).
Tujuan Penelitian Tujuan dari penelitian ini adalah:
1. Mendapatkan model FIS yang sesuai untuk mengklasifikasikan tuple ke dalam
solid tuples atau weak tuples.
2. Memperbaiki metode spectral alignment yang diterapkan oleh Caesar et al.
(2013) dengan menerapkan FIS yang dapat menyederhanakan graf dengan mempertahankan akurasi dan kualitas contigs.
3 Manfaat Penelitian
Manfaat yang diperoleh dari penelitian ini adalah memperoleh data DNA dari suatu organisme yang lebih akurat dan mengurangi kompleksitas graf yang terbentuk setelah proses assembly.
Ruang Lingkup Penelitian Ruang lingkup dari penelitian ini adalah:
1. Mengoreksi kesalahan substitusi (error substitution). 2. Fokus pada hasil akurasi, tanpa memperhatikan efisiensi.
4
2
TINJAUAN PUSTAKA
Deoxyribonucleic acid (DNA)
Deoxyribonucleic acid (DNA) pertama kali ditemukan pada pertengahan abad kedelapan belas, ketika dokter Swiss dan biokimia Friedrich Miescher mengisolasi inti dari sel-sel darah putih dalam nanah pada perban kotor (Lewis 2010). DNA atau asam deoksiribosa nukleat (ADN) merupakan tempat penyimpanan informasi genetik. DNA merupakan makromolekul polinukleotida yang tersusun atas polimer nukleotida yang berulang-ulang, tersusun rangkap, membentuk DNA heliks ganda dan berpilin ke kanan. Ada tiga komponen utama penyusun DNA, yaitu basa nitrogen, fosfat, dan pentosa (Nelson & Cox 2008). Informasi aktual yang terkandung dalam DNA dikodekan oleh empat basa berbeda:
Adenine (A), Cytosine (C), Guanine (G) dan Thymine (T) (Higgs & Attwood 2005). DNA ini biasanya ditemukan sebagai untai ganda (double helix) dan memiliki fungsi sebagai tempat penyimpanan informasi genetik dari suatu makhluk hidup.
DNA Sequencing
DNA Sequencing atau pengurutan DNA adalah proses atau teknik penentuan urutan basa pada suatu molekul DNA (Alphey 1997). Urutan tersebut dikenal sebagai sekuens DNA, yang merupakan informasi paling mendasar suatu gen atau genom karena mengandung instruksi yang dibutuhkan dalam pembentukan tubuh makhluk hidup. DNA sequencing dapat dimanfaatkan untuk menentukan identitas maupun fungsi gen atau fragmen DNA lainnya dengan membandingkan sekuens-nya dengan sekuens DNA lain yang sudah diketahui. Alasan mendasar mengetahui urutan molekul DNA adalah untuk membuat prediksi tentang fungsinya dan memfasilitasi manipulasi molekul.
Ada dua metode yang dapat digunakan untuk mengurutkan molekul DNA. Metode Maxam-Gilbert dan metode Sanger. Kedua metode tersebut menghasilkan fragmen-fragmen DNA dengan panjang bervariasi. Teknik yang digunakan adalah gel-gel poliakrilamid pendenaturasi (denaturing polyacrylamide gels). Gel agarosa dapat memisahkan molekul-molekul DNA dengan perbedaan panjang 30-50 basa, sedangkan gel poliakrilamid dapat memisahkan molekul-molekul DNA dengan perbedaan panjang satu basa. Gel-gel pendenaturasi menyebabkan molekul DNA menjadi beruntai tunggal dan tetap dalam keadaan seperti itu sepanjang proses elektroforesis. Gel pendenaturasi mengandung urea dan dijalankan dengan suhu yang ditinggikan. Kedua hal tersebut mendorong terjadinya pemisahan kedua untai molekul DNA.
DNA Sequencing Error
5
Sequencing (NGS) yang umum digunakan terlihat pada Tabel 1 (Liu et al. 2012; Yang et al. 2012).
Tabel 1 Perbandingan NGS pada tahun 2012
Sequencer Illumina Biosystems Applied BioSciences Helicos Sciences 454 Life IonTorrent
Teknik terminator Reversible by ligation Sequencing molecule Single sequencing
Error Substitution Substitution Insertion Deletion Insertion Deletion Insertion Deletion
Tabel 1 menunjukkan bahwa untuk setiap sequencer masih mengandung
error dengan jenis error yang berbeda. Penelitian ini hanya fokus terhadap data hasil sequencing menggunakan Illumina. Jenis error yang dihasilkan Illumina adalah substitution error.
FASTQ
Dalam bidang DNA sequencing format FASTQ telah umum digunakan untuk menyimpan data DNA. Format FASTQ adalah format berbasis teks untuk menyimpan urutan biologis (urutan basa). Pada FASTQ terdapat tambahan informasi, yaitu kode skor kualitas basa. Skor kualitas basa dikodekan dengan karakter American Standard Code for Information Interchange (ASCII) tunggal agar lebih singkat. FASTQ pada awalnya dikembangkan di Wellcome Trust Sanger Institute oleh oleh Jim Mullikin (Cock et al. 2009). Contoh isi dari file FASTQ dapat dilihat pada Gambar 1.
Gambar 1 Contoh isi dari file FASTQ
Gambar 1 menunjukkan contoh data yang tersimpan dalam format FASTQ. Baris pertama menunjukkan kode read, baris kedua menunjukkan read, baris ketiga menunjukkan kode kualitas basa, dan baris keempat menunjukkan kualitas basa.
Metode Spectral Alignment
6
menggunakan CUDA dan Caesar et al. (2013) berdasarkan frekuensi kemunculan
tuple (multiplicity). Penelitian ini akan memperbaiki metode spectral alignment
yang dilakukan oleh Caesar et al. (2013).
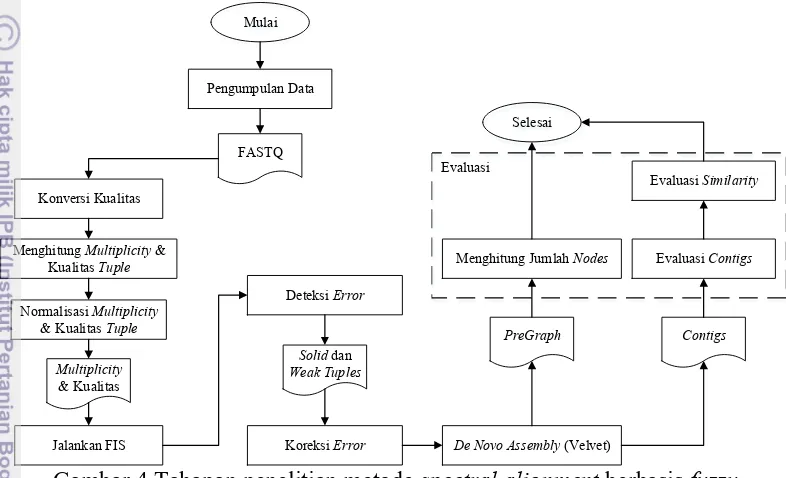
Tahapan penelitian yang dilakukan pada metode spectral alignment seperti yang terlihat pada Gambar 2.
Mulai
Gambar 2 Tahapan penelitian metode spectral alignment
Metode spectral alignment ini masih memiliki kelemahan, sehingga dibutuhkan perbaikan. Beberapa kelemahan yang ditemukan adalah:
1. Kesulitan dalam menentukan batas multiplicity untuk mengklasifikasikan sebuah tuple masuk ke dalam solid atau weak tuples.
2. Tahapan evaluasi hanya menghitung jumlah nodes tanpa memperhatikan kualitas contigs dan akurasi data yang terbentuk setelah proses DNA
assembly.
3. Aspek yang digunakan sebagai justifikasi hanya dari sisi multiplicity tanpa memperhatikan kualitas basa.
Dari beberapa kelemahan yang ada, maka salah satu dari tujuan penelitian ini adalah memperbaiki metode spectral alignment tersebut dengan menambahkan aspek kualitas basa, menerapkan metode fuzzy inference system (FIS) untuk mengklasifikasikan sebuah tuple masuk ke dalam solid atau weak tuples, dan menambah tahapan evaluasi untuk melihat dan mempertahankan kualitas contigs
7 Logika Fuzzy
Logika Fuzzy (Fuzzy Logic) merupakan modifikasi dari teori himpunan dimana setiap anggotanya memiliki derajat keanggotaan yang bernilai kontinu antara 0 sampai 1 yang pertama kali dikenalkan oleh Lotfi A Zadeh pada tahun 1965 (Kusumadewi 2002). Penggunaan logika fuzzy dipilih karena memiliki kelebihan sebagai berikut :
1. Konsep logika fuzzy mudah dimengerti, karena konsep matematis yang mendasari penalaran fuzzy sangat sederhana dan mudah dimengerti.
2. Logika fuzzy sangat fleksibel.
3. Logika fuzzy memiliki toleransi terhadap data yang tidak tepat. 4. Logika fuzzy memodelkan fungsi nonlinier yang sangat kompleks.
5. Dengan logika fuzzy dapat dibangun dan diaplikasikan pengalaman para pakar secara langsung tanpa melalui proses pelatihan.
6. Logika fuzzy didasarkan pada bahasa alami. Himpunan fuzzy
Himpunan fuzzy adalah teknik yang secara matematis mampu mengekspresikan keambiguan dalam bahasa (Marimin 2002). Contohnya jika seseorang dikatakan muda, kita tidak dapat mendefinisikan dengan tepat berapa tahunkah seseorang dikatakan muda. Dengan himpunan fuzzy ini, kasus keambiguan di atas dapat ditangani.
Himpunan fuzzy didasarkan pada gagasan untuk memperluas jangkauan fungsi karakteristik sedemikian hingga fungsi tersebut akan mencakup bilangan real pada interval [0,1]. Dengan kata lain, nilai keanggotaannya menunjukkan bahwa semesta pembicaraan tidak hanya berada pada 0 atau 1, tetapi nilai tersebut juga terletak diantaranya.
Fungsi keanggotaan (Membership function)
Fungsi keanggotaan (membership function) adalah suatu kurva yang menunjukkan pemetaan titik input data ke dalam nilai keanggotaannya (derajat keanggotaan) yang memiliki interval antara 0 sampai 1. Kurva membership function
yang digunakan dalam penelitian ini adalah trapesium. Representasi kurva trapesium ini pada dasarnya mirip dengan kurva segitiga, yaitu memiliki segmen garis lurus, tidak halus pada titik-titik sudut yang ditentukan oleh parameter. Kurva trapesium memiliki 4 parameter yaitu a, b, c, dan d. Representasi kurva trapesium dapat dilihat pada Gambar 3.
1
a b c d
8
Membership function untuk kurva trapesium ada pada persamaan (1).
( ; , , , ) =
Fuzzy inference system (FIS)
Fuzzy inference system adalah sistem komputasi berdasarkan pada konsep teori fuzzy, dan aturan if-then fuzzy. Sistem ini telah sukses pada beberapa bidang seperti klasifikasi data, analisa keputusan, sistem pakar, dan pattern recognition. Dalam metode FIS, input dapat berupa nilai fuzzy atau nilai crisp tetapi output selalu berupa himpunan fuzzy. Dalam beberapa kasus kita perlu output sebagai crisp, terutama ketika FIS digunakan sebagai kontrol. Namun, dalam penelitian ini dimana FIS digunakan untuk mengkasifikasikan sebuah tuple ke dalam weak atau
solid tuple dibutuhkan nilai crisp. Dalam hal ini diperlukan metode defuzzifikasi untuk mengekstrak suatu nilai crisp yang merepresentasikan kondisi terbaik himpunan fuzzy.
Metode mamdani
Metode Mamdani sering dikenal sebagai metode Max-Min. Metode ini diperkenalkan oleh Ebrahim Mamdani pada tahun 1975. Untuk mendapatkan output menurut Kusumadewi (2002), diperlukan 4 tahapan, yaitu:
1. Pembentukan himpunan fuzzy 2. Aplikasi fungsi implikasi
3. Komposisi rules (Metode Max, Metode Additive, Metode Probabilistik OR) 4. Penegasan (defuzzifikasi)
Proses defuzzifikasi
Input dari proses defuzzifikasi adalah suatu himpunan fuzzy yang diperoleh dari komposisi rules fuzzy, sedangkan output yang dihasilkan merupakan suatu bilangan pada domain himpunan fuzzy tersebut. Sehingga jika diberikan suatu himpunan fuzzy dalam range tertentu, maka harus dapat diambil suatu nilai crisp
tertentu sebagai output. Ada beberapa metode defuzzifikasi yang bisa digunakan pada komposisi aturan Mamdani, antara lain: metode centroid, metode smallest of maximum, dan lainnya. Penelitian ini menggunakan metode centroid dengan rumus seperti yang terlihat pada persamaan (2).
9
3
METODE
Tahapan Penelitian
Metode pada penelitian ini terdiri atas beberapa tahapan proses. Tahapan penelitian digambarkan dalam bagan yangditampilkan pada Gambar 4.
Menghitung Multiplicity & Kualitas Tuple
Gambar 4 Tahapan penelitian metode spectral alignment berbasis fuzzy
Gambar 4 menunjukkan langkah-langkah yang akan dilakukan dalam penelitian ini. Penelitian ini dimulai dari pengumpulan data dalam format FASTQ sampai dengan tahapan evaluasi untuk melihat keberhasilan dari proses pendeteksian dan pengoreksian DNA sequencing error.
Pengumpulan Data
Tahapan awal akan menelusuri data dalam format FASTQ dari DNA Data Bank of Japan (DDBJ). Data yang akan digunakan merupakan data hasil
10
Gambar 5 Interface dari DNA Data Bank of Japan (DDBJ) (Sumber : http://www.ddbj.nig.ac.jp/)
Konversi Kualitas Basa
Kualitas basa dalam file FASTQ berupa kode American Standard Code for Information Interchange (ASCII). Oleh karena itu, kode tersebut akan dilakukan konversi dari kode ASCII ke bilangan (skor Phred). Akan tetapi sebelum mengkonversi kode ASCII menjadi skor Phred, perlu diperhatikan platform yang digunakan dan jenis Phred. Setelah diperoleh informasi mengenai jenis platform
dan Phred, maka kode ASCII dapat langsung dikonversi menjadi skor Phred. Contoh konversi kode ASCII menjadi skor Phred dapat dilihat pada Gambar 6.
Gambar 6 Contoh konversi kode ASCII menjadi skor Phred
Menentukan Multiplicity dan Kualitas Tuple
Multiplicity adalah banyaknya kemunculan tuple dengan panjang tertentu. Panjang tuple yang digunakan dalam penelitian ini adalah 5 sesuai dengan penelitian sebelumnya yang dilakukan oleh Caesar et al. (2013). Penentuan kualitas
11 Normalisasi Multiplicity dan Kualitas Tuple
Proses normalisasi untuk hasil perhitungan multiplicity dan kualitas tuple
dibutuhkan karena range data keduanya terlalu jauh. Normalisasi menggunakan metode Max-Min dengan rumus seperti pada persamaan (3).
=
(3)Keterangan:
- = data yang telah dinormalisasi - x = data awal
- l = data minimum - u = data maksimum
Implementasi Fuzzy Inference System (FIS)
Hasil perhitungan multiplicity dan kualitas tuple akan menjadi inputan untuk
fuzzy inference system (FIS). Dari proses tersebut diharapkan memperoleh model yang sesuai agar dapat mengklasifikasikan tuple ke dalam solid tuples atau weak tuples.
Pendefinisian variabel input dan output
Tahapan ini akan menentukan variabel input dan output yang akan digunakan untuk mengklasifikasikan tuple ke dalam solid tuples atau weak tuples. Pendefinisian variabel input dan output dapat dilihat pada Tabel 2.
Tabel 2 Definisi variabel input dan output Fungsi Nama Variabel Keterangan
Input MultiplicityKualitas Frekuensi kemunculan Kualitas tuple tuple Output Keputusan Penentuan solid atau weak tuple
Setelah menentukan variabel input dan output, maka selanjutnya akan ditentukan membership function dan model FIS yang dapat mengklasifikasikan
tuple ke dalam solid tuples atau weak tuples. Representasi dari diagram FIS dapat dilihat pada Gambar 7.
12
Gambar 7 menunjukkan ikon varibel input yang terdiri atas 2 inputan, yaitu
multiplicity dan kualitas tuple dan 1 output, yaitu variabel keputusan. Setelah pendefinisian variabel maka selanjutnya akan menentukan himpunan fuzzy, menentukan kombinasi rules, dan proses defuzzifikasi.
Pendefinisian himpunan fuzzy
Tahapan ini akan menentukan himpunan fuzzy yang digunakan pada variabel input dan output. Dalam penelitian ini himpunan fuzzy untuk variabel input adalah “Rendah”, “Sedang”, dan “Tinggi”, sedangkan untuk variabel output berupa keputusan suatu tuple diklasifikasikan ke dalam solid tuple atau weak tuple.
Penentuan kombinasi rules
Penentuan kombinasi rules dibuat berdasarkan seluruh kemungkinan yang mungkin terjadi. Ini dilakukan karena tidak adanya informasi mengenai variabel yang digunakan.
Proses defuzzifikasi
Proses terakhir adalah defuzzifikasi, yaitu proses untuk mendapatkan nilai
crisp dari suatu himpunan fuzzy. Nilai tersebut akan dijadikan sebagai penentuan dalam mengklasifikasikan suatu tuple ke dalam solid tuple atau weak tuple.
Deteksi Error
Tahap ini merupakan pendeteksian DNA sequencing error berdasarkan
multiplicity dan kualitas tuple. Kedua aspek tersebut akan menjadi inputan untuk memperoleh model FIS yang sesuai, sehingga dapat mengklasifikasikan suatu tuple
ke dalam solid tuple atau weak tuple. Weak tuple tersebut merupakan himpunan
tuple yang mengandung error sehingga harus dikoreksi.
Koreksi Error
Proses pengoreksian DNA sequencing error dilakukan berdasarkan skor yang menyatakan jarak kedekatan antara tuple yang berada dalam himpunan weak tuples dengan tuple yang berada dalam himpunan solid tuples. Jarak yang akan digunakan adalah jarak Levenshtein (Levenshtein VI 1966). Jarak Levenshtein digunakan untuk menentukan skor yang menyatakan kedekatan antara dua string
13 Evaluasi
Proses evaluasi dilakukan untuk melihat keberhasilan proses pendeteksian dan pengoreksian DNA sequencing error. Proses evaluasi dilakukan dengan cara menghitung jumlah nodes. Untuk mendukung kebenaran dari hasil perhitungan jumlah nodes maka ada dua aspek tambahan yang digunakan, yaitu evaluasi kualitas contigs dan evaluasi similarity. Proses evaluasi ini akan menggunakan
Velvetassembler. Velvetassembler adalah sebuah tool untuk merakit data set hasil koreksi menggunakan metode spectral alignment dan metode spectral alignment
berbasis fuzzy.
Perhitungan jumlah nodes
Ada dua file berisi graf De Bruijn yang saling berhubungan hasil dari Velvet assembler (Zerbino & Birney 2008). Dua file tersebut adalah “PreGraph” dan “LastGraph”. Kedua file tersebut mengandung jumlah nodes yang merepresentasikan graf De Bruijn. Dalam penelitian ini hanya file “PreGraph” yang menjadi perhatian, karena file tersebut merupakan file yang berisi graf De Bruijn yang belum dilakukan error removal oleh Velvet assembler. Jadi, file tersebut digunakan untuk melihat indikasi keberhasilan dari metode yang digunakan. Evaluasi kualitas contigs
File hasil assembly menggunakan Velvet assembler juga menghasilkan file
contigs. Contigs terbentuk dari hasil reads yang saling overlap. Untuk memperoleh
contigs terbaik, ada tiga aspek yang diperhatikan. Ketiga aspek tersebut adalah N50
size, number of contigs, dan maximum contig length (Kusuma et al. 2011). Setelah perhitungan ketiga aspek tersebut, dibutuhkan evaluasi akurasi data yang telah dikoreksi. Akurasi data set yang telah dikoreksi akan dihitung kedekatan (similarity) dengan data reference. Program dijalankan menggunakan command prompt dengan cara mengetik perintah:
assembly_quality_stats --no_hist contigs.fa Keterangan :
- --no_hist : perintah untuk tidak mencetak grafik - Contigs.fa : nama file inputan
Evaluasi similarity
Perhitungan similarity dilakukan untuk melihat akurasi dari data set yang telah dikoreksi. Akurasi diperoleh dengan cara menghitung kedekatan (similarity) antara data set (contigs) dengan data reference. Similarity dihitung untuk ketiga data set tersebut dengan masing-masing data reference. Data reference diperoleh dari National Center for Biotechnology Information (NCBI). Data reference
merupakan data yang menjadi rujukan dengan tingkat kebenaran yang tinggi.
14
blastn -subject data_ref.fasta -query data_set.fasta -out hasil_blastn
Keterangan :
- subject : perintah untuk memanggil data reference dari NCBI - data_ref.fasta : data reference dari NCBI
- query : perintah untuk memanggil data set yang digunakan - data_set.fasta : data set yang digunakan
- out : perintah untuk mencetak data hasil blastn
- hasil_blastn : file hasil menjalankan program blastn
Lingkungan Implementasi
Sistem perangkat lunak yang digunakan adalah: a. Sistem operasi: Windows 8 Pro
b. MATHLAB_R2013A : Menentukan model FIS menggunakan Toolbox. c. Notepad ++ : Editor
d. Pyhton 3.4.0 : Menentukan multiplicity dan kualitas, string matching, menghitung jarak Levenshtein, dan evaluasi contigs.
e. Velvet : Melakukan DNA assembly. f. BLASTN : Menghitung similarity.
Spesifikasi perangkat keras yang digunakan adalah:
a. Processor Intel (R) Core (TM) i3-3217U CPU @ 1.80GHz 1.8GHz b. Memori 4,00 GB RAM
15
4
HASIL DAN PEMBAHASAN
Data set
Data set diperoleh dari DNA Data Bank of Japan (DDBJ) dengan panjang
read adalah 100 bp dan jumlah read bervariasi. Data set direpresentasikan dalam format FASTQ. Karakteristik data set yang digunakan dapat dilihat pada Tabel 3. Tabel 3 Karakteristik data set
Organisme Jumlah Reads Mean (bp)
Caenorhabditis elegans 27887 100
Drosophila melanogaster 76364 100
Streptococcus anginosus 1741 100
Tabel 3 menunjukkan karakteristik data set yang merupakan hasil
sequencing menggunakan Illumina sequencer. Reads yang dihasilkan oleh Illumina memiliki sequencing error dan simbol selain Adenine (A), Cytosine (C), Guanine (G), dan Thymine (T), yaitu N. N adalah sebuah simbol yang menunjukkan ketidakmampuan sequencer dalam menentukan basa. Penelitian ini fokus pada
substitution error.
Analisis Metode Spectral Alignment
Berdasarkan penelitian yang dilakukan oleh Caesar et al. (2013), kriteria
tuple yang termasuk ke dalam weak tuples adalah tuple yang memiliki multiplicity
kurang dari atau sama dengan 10 dan/atau terdapat karakter N, sebaliknya untuk
solid tuples. Adapun hasil perhitungan jumlah solid dan weak tuples untuk metode
spectral alignment dapat dilihat pada Tabel 4. Tabel 4 Jumlah solid dan weak tuples
Organisme Jumlah tuple Solid Tuples Weak
Caenorhabditis elegans 3124 1024 2100
Drosophila melanogaster 3124 1024 2100
Streptococcus anginosus 2851 1022 1829
Tabel 4 menunjukkan bahwa terdapat data set yang memiliki jumlah tuple
yang berbeda. Ini terjadi karena jumlah tuple diperoleh berdasarkan variasi basa yang terdapat pada data set. Jumlah solid tuples dan weak tuples untuk
Caenorhabditis elegans dan Drosophila melanogaster adalah sama karena hampir semua kemungkinan kombinasi tuple terbentuk, sedangkan Streptococcus anginosus tidak semua kemungkinan kombinasi tuple terbentuk. Untuk ketiga data set menunjukkan jumlah weak tuples lebih banyak dibandingkan solid tuples, ini menunjukkan error tuple lebih banyak dibandingkan tuple yang tidak mengandung
16
Analisis Metode Spectral Alignment Berbasis Fuzzy
Pemodelan fuzzy
Penelitian ini menggunakan model fuzzy inference system (FIS). Model FIS ini menggunakan Mamdani inference (Musi 2009) yang memungkinkan sistem untuk mengambil satu set nilai input crisp dan menerapkan satu set fuzzy rules
untuk memperoleh suatu nilai output. Berikut adalah langkah-langkah untuk mengklasifikasikan tuple ke dalam solid atau weak tuple.
Langkah 1: Pendefinisian variabel input and output
Multiplicity and kualitas tuple merupakan variabel input untuk Mamdani inference
dan output dari sistem merupakan keputusan suatu tuple diklasifikasikan ke dalam
weak tuple atau solid tuple.
Langkah 2: Pendefinisian himpunan fuzzy
Untuk menentukan parameter, maka terlebih dahulu ditentukan himpunan fuzzy
untuk setiap variabel. Himpunan fuzzy untuk variabel input didefinisikan menjadi tiga istilah linguistic, yaitu “Rendah”, “Sedang”, dan “Tinggi”, sedangkan variabel output menjadi dua istilah linguistic, yaitu “weak tuple” dan “solid tuple”. Himpunan fuzzy untuk setiap variabel dapat dilihat pada Tabel 5.
Tabel 5 Himpunan fuzzy
Variabel Nama Himpunan Fuzzy Parameter
Multiplicity (x) Rendah Sedang [0 0 0.1 0.2] [0.1 0.3 0.5 0.8]
Tinggi [0.4 0.6 1 1]
Kualitas (y) Rendah Sedang [0 0 0.1 0.2] [0.1 0.3 0.5 0.8]
Tinggi [0.4 0.6 1 1]
Keputusan (z) Weak Tuples Solid Tuples [0 0 0.1 0.5] [0.1 0.3 1 1]
Tabel 5 menunjukkan bahwa parameter untuk setiap himpunan fuzzy. Variabel input memiliki parameter yang sama. Parameter tersebut mempengaruhi nilai output
fuzzy. Parameter tersebut diperoleh berdasarkan hasil try and error.
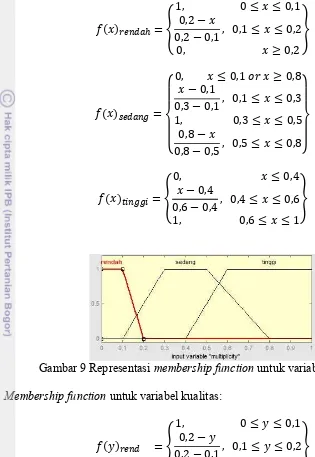
Setiap variabel direpresentasikan dengan menggunakan kurva trapesium. Representasi variabel tersebut dapat dilihat pada Gambar 8 sampai dengan Gambar 10.
17
Membership function untuk variabel multiplicity:
( ) =
Gambar 9 Representasi membership function untuk variabel kualitas
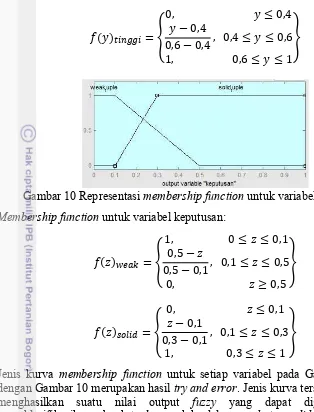
Membership function untuk variabel kualitas:
18
Gambar 10 Representasi membership function untuk variabel keputusan
Membership function untuk variabel keputusan:
( ) = dengan Gambar 10 merupakan hasil try and error. Jenis kurva tersebut yang dapat menghasilkan suatu nilai output fuzzy yang dapat digunakan untuk mengklasifikasikan sebuah tuple masuk ke dalam weak atau solid tuple.
Langkah 3: Pendefinisian fuzzy rules
Langkah berikutnya adalah pendefinisian If-Then rules untuk menjelaskan perilaku sistem. Rules didesain untuk mendeskripsikan pentingnya variabel keputusan. Pada langkah ini, multiplicity dan kualitas dari suatu tuple menjadi masukan ke dalam sistem. Berbasis pada expert knowledge, permasalahan ini dinyatakan ke dalam istilah logical rules. Kombinasi rules yang digunakan dapat dilihat pada Tabel 6. Tabel 6 Kombinasi rules
Kode Rules
[R1] IF kualitas tuple rendah AND multiplicity rendah THEN Weak tuple
[R2] IF kualitas tuple rendah AND multiplicity sedang THEN Weak tuple
[R3] IF kualitas tuple rendah AND multiplicity tinggi THEN Solid tuple
[R4] IF kualitas tuple sedang AND multiplicity rendah THEN Weak tuple
[R5] IF kualitas tuple sedang AND multiplicity sedang THEN Weak tuple
[R6] IF kualitas tuple sedang AND multiplicity tinggi THEN Solid tuple
[R7] IF kualitas tuple tinggi AND multiplicity rendah THEN Solid tuple
[R8] IF kualitas tuple tinggi AND multiplicity sedang THEN Solid tuple
19 Jumlah rules yang proporsional untuk FIS adalah jumlah membership function
dipangkatkan dengan jumlah variabel input (Tang & Shozo 1999). Penelitian ini menggunakan 9 rules yang diperoleh dari 3 membership functions dipangkatkan dengan 2 variabel input.
Langkah 4: Proses defuzzifikasi
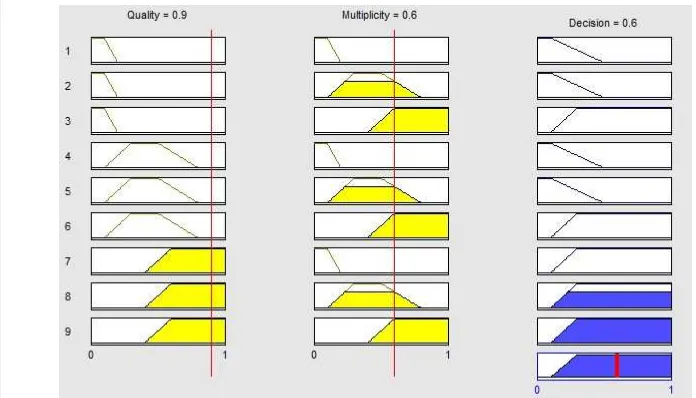
Langkah defuzzifikasi dibutuhkan untuk mengkonversi semua inputan ke dalam 3 istilah linguistic yang dapat digunakan untuk mengklasifikasikan tuples. Proses defuzzifikasi mentransformasikan himpunan fuzzy ke dalam nilai crisp. Sebagai contoh, jika sebuah tuple memiliki multiplicity adalah 0,6 dan kualitasnya adalah 0,9 maka hasil defuzzifikasi menunjukkan nilai output 0,6. Salah satu hasil proses defuzzifikasi dapat dilihat pada Gambar 11.
Gambar 11 Representasi salah satu hasil proses defuzzifikasi Pengklasifikasian tuples
Sebuah tuple yang memiliki nilai output fuzzy lebih dari 0,4 dan tidak mengandung karakter N diklasifikasikan sebagai solid tuple, selain dari itu diklasifikasikan sebagai weak tuple. Jumlah solid dan weak tuples dapat dilihat pada Tabel 7.
Tabel 7 Jumlah solid tuples dan weak tuples
Organisme Jumlah tuple Solid Weak Tuples
Caenorhabditis elegans 3124 1022 2102
Drosophila melanogaster 3124 1022 2102
Streptococcus anginosus 2851 1024 1827
Tabel 7 menunjukkan pengelompokkan tuple ke dalam solid dan weak tuples. Weak tuples yang terbentuk lebih dominan dibandingkan solid tuples. Ini menunjukkan masih banyak terdapat error tuples. Terdapat perbedaan antara jumlah solid tuples dan weak tuples yang dibentuk oleh metode spectral alignment
20
dibandingkan metode spectral alignment. Ini menunjukkan model FIS lebih banyak mendeteksi error tuples dibandingkan metode spectral alignment.
Dari pengelompokan tuple kedalam solid tuples dan weak tuples, maka dapat ditentukan range untuk kualitas tuple dan multiplicity. Penentuan range untuk
solid tuples dapat dilihat pada Tabel 8.
Tabel 8 Penentuan range untuk solid tuples
Organisme Multiplicity Model FIS Kualitas
Caenorhabditis elegans 5.467 – 13.243 12,1 – 30,25
Drosophila melanogaster 5.877 – 14.679 16 – 36,98
Streptococcus anginosus 218 - 542 15 – 35
Tabel 8 menunjukkan range dari kualitas tuple dan multiplicity. Ketiga organisme memiliki range kualitas tuple dan multiplicity yang berbeda karena perbedaan jumlah reads dan variasi kombinasi basa. Dari ketiga organisme tersebut diperoleh range dari solid tuples untuk kualitas tuple adalah 12,1 – 36,98 dan
multiplicity adalah 218 – 14.679, selain itu masuk ke dalam weak tuples. Penentuan
range belum dapat dispesifikkan, karena range sangat bergantung kombinasi basa yang terdapat pada data set.
Evaluasi
Proses evaluasi menggunakan Velvetassembler. Evaluasi dilakukan untuk melihat keberhasilan dari proses pendeteksian dan pengoreksian DNA sequencing error. Proses evaluasi dilakukan dengan cara menghitung jumlah nodes. Untuk mendukung kebenaran dari hasil perhitungan jumlah nodes maka ada dua aspek tambahan yang digunakan, yaitu evaluasi kualitas contigs dan evaluasi similarity. Perhitungan jumlah nodes
Perhitungan jumlah nodes dilakukan untuk melihat kompleksitas graf yang terbentuk. Perhitungan jumlah nodes dilakukan untuk ketiga organisme menggunakan Velvet. Data set yang menjadi inputan adalah data set yang belum dikoreksi (uncorrected read), data set yang dikoreksi menggunakan metode
spectral alignment, dan data set yang dikoreksi menggunakan metode spectral alignment berbasis fuzzy. Data set yang menjadi inputan dalam format FASTA.
Untuk setiap proses DNA sequence assembly menggunakan Velvet, parameter panjang hash harus ditentukan. Panjang hash adalah panjang dari k-mers
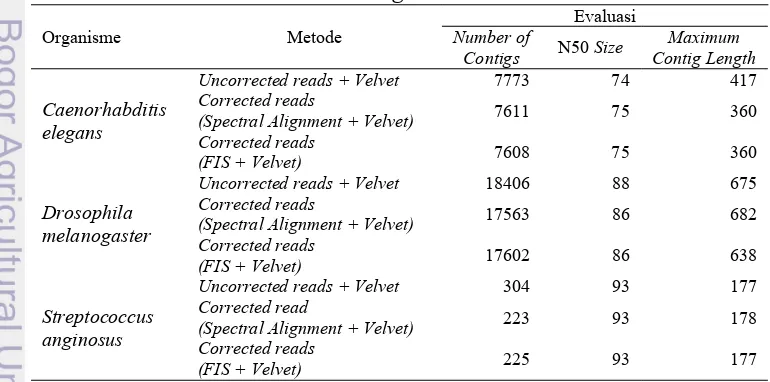
21 Tabel 9 Evaluasi perhitungan jumlah nodes
Organisme Metode 17 19 21 Nilai 23 k 25 27 29
(Spectral Alignment) 66999 39341 28904 23707 19706 17435 15704
Corrected read
Tabel 9 menunjukkan bahwa pengurangan jumlah nodes hanya terpenuhi untuk nilai k tertentu. Pengurangan jumlah nodes untuk metode spectral alignment
berbasis fuzzy hanya terjadi untuk nilai k = {17, 21}. Ini menunjukkan bahwa kompleksitas graf yang dibentuk oleh data set yang telah dikoreksi menggunakan metode spectral alignment berbasis fuzzy untuk nilai k = {17, 21} lebih rendah dibandingkan dengan data set yang belum dikoreksi (uncorrected read) dan data set yang dikoreksi hanya menggunakan metode spectral alignment. Untuk memastikan hasil tersebut, dibutuhkan proses evaluasi untuk melihat akurasi data dan kualitas contigs yang sama atau tidak berbeda jauh dari metode spectral alignment.
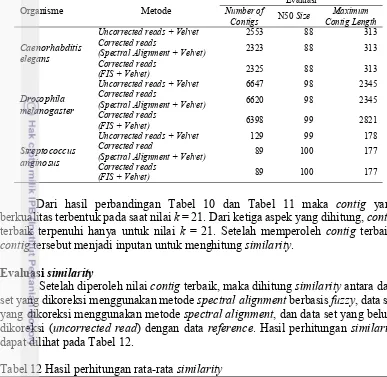
Evaluasi kualitas contigs
Tahapan evaluasi ini dilakukan untuk mendapatkan contig yang berkualitas. Terdapat tiga aspek yang diperhatikan, yaitu N50 size, number of contigs, dan
maximum contig length. Number of contigs terkecil adalah number of contigs
terbaik, N50 size terbesar adalah N50 size terbaik, dan maximum contig length
terbesar adalah maximum contig length terbaik. Perbandingan ketiga aspek tersebut dilakukan pada contig untuk nilai k = {17, 21}. Hasil perhitungan untuk ketiga aspek tersebut dapat dilihat pada Tabel 10 dan Tabel 11.
Tabel 10 Hasil evaluasi kualitas contigs untuk k = 17
Organisme Metode Number of Evaluasi
Contigs N50 Size Contig Length Maximum
Caenorhabditis
(Spectral Alignment + Velvet) 17563 86 682
22
Tabel 11 Hasil evaluasi kualitas contigs untuk k = 21
Organisme Metode Number of Evaluasi
Contigs N50 Size Contig Length Maximum
Caenorhabditis
(Spectral Alignment + Velvet) 6620 98 2345
Corrected reads berkualitasterbentuk pada saat nilai k = 21. Dari ketiga aspek yang dihitung, contig
terbaik terpenuhi hanya untuk nilai k = 21. Setelah memperoleh contig terbaik,
contig tersebut menjadi inputan untuk menghitung similarity. Evaluasi similarity
Setelah diperoleh nilai contig terbaik, maka dihitung similarity antara data set yang dikoreksi menggunakan metode spectral alignment berbasis fuzzy, data set yang dikoreksi menggunakan metode spectral alignment, dan data set yang belum dikoreksi (uncorrected read) dengan data reference. Hasil perhitungan similarity
dapat dilihat pada Tabel 12.
Tabel 12 Hasil perhitungan rata-rata similarity
Organisme Uncorrected Rata-rata similarity
reads (Spectral Alignment) Corrected reads Corrected reads (FIS) Caenorhabditis elegans 96,58 % 96,33 % 96,42 %
Drosophila melanogaster 97,51 % 97,51 % 97,54 %
Streptococcus anginosus 98,79 % 99,41 % 99,41 %
23
5
SIMPULAN DAN SARAN
Simpulan
Penelitian ini berhasil memperoleh model fuzzy inference system (FIS) yang sesuai untuk mengklasifikasikan tuple ke dalam solid tuple atau weak tuple yang menjadi inputan untuk metode spectral alignment sebagai tahapan preprocessing. Data set yang dikoreksi menggunakan metode spectral alignment berbasis fuzzy
menghasilkan jumlah nodes yang lebih sedikit dibandingkan dengan data set yang belum dikoreksi (uncorrected read) dan data set yang hanya dikoreksi menggunakan metode spectral alignment untuk nilai k = 21 dengan mempertahankan akurasi data dan kualitas contigs. Ini menunjukkan bahwa pendeteksian dan pengoreksian DNA sequencing error menggunakan menggunakan metode spectral alignment berbasis fuzzy dapat menyederhanakan graf dibandingkan dengan hanya menggunakan metode spectral alignment.
Saran
Saran untuk penelitian lanjutan adalah memilih data yang memiliki coverage 40x – 50x, mengoptimalkan parameter-parameter model FIS dengan menggunakan metode optimasi, dan menggunakan parallel untuk mempercepat waktu komputasi.
Ucapan Terima Kasih
24
DAFTAR PUSTAKA
Abdullah L, Rahman MNA. 2012. Employee Likelihood of Purchasing Health Insurance using Fuzzy Inference System. International Journal of Computer Science Issues. 9(2): 112-116.
Alphey L. 1997. DNA Sequencing From Experimental Methods To Bioinformatics. BIOS Scientific Publishers Limited. ISBN:0-387-91509-5.
Bryce CFA, Pacini D. 1998. The Structure and Function of Nucleic Acids. Holbrooks Printers Ltd: Portsmouth, U.K. ISBN: 0-90449-834-4.
Caesar N, Kusuma WA, Wijaya SH. 2013. DNA Sequencing Error Correction using Spectral Alignment. ICACSIS. ISBN: 978-979-1421-19-5.
Chevreux B. 2005. MIRA: An Automated Genome and EST Assembler. German Cancer Research Center Heidelberg, Department of Molecular Biophysics. page.18.
Chong ML, Ku CS, Wu M, Soong R. 2012. Characterising Somatic Mutations in Cancer Genome by Means of Next-generation Sequencing. In: eLS. John Wiley & Sons, Ltd: Chichester. DOI: 10.1002/9780470015902.a0023379.
Cock PJA, Field CJ, Goto N, Heuer ML, Rice PM. 2009. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Research, 2010, Vol. 38, No. 6 1767–1771. DOI:10.1093/nar/gkp1137.
Higgs PG, Attwood TK. 2005. Bioinformatics and Molecular Evolution. Blackwell Publishing. ISBN 1–4051–0683–2.
Illumina, Inc. 2011. Quality Scores for Next-Generation Sequencing (Assessing sequencing accuracy using Phred quality scoring). URL:
http://res.illumina.com/documents/products/technotes/technote_q-scores.pdf. [15 September 2014].
Kelley DR, Michael CS, Steven LS. 2010. Quake: Quality-Aware Detection and Correction of Sequencing Errors. Genome Biology. DOI:10.1186/gb-2010-11-11-r116.
Kusuma WA, Ishida T, Akiyama Y. 2011. A combined approach for de novo dna sequence assembly of very short reads. IPSJ Transactions on Bioinformatics. Kusumadewi S. 2002. Analisis dan Desain Sistem Fuzzy Menggunakan Toolbox
Matlab. Graha Ilmu. Yogyakarta
Levenshtein VI. 1966. Binary Codes Capable of Correcting Deletions, Insertions and Reversals. Soviet Physics Doklady. 10(8):707.
Lewis R. 2010. Human Genetics: Concepts & Applications ninth edition. McGraw-Hill Companies. ISBN−10:0−39−023244−0.
Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M. 2012. Comparison of Next-Generation Sequencing Systems. Journal of Biomedicine and Biotechnology. Volume 2012, Article ID 251364, 11 pages. DOI:10.1155/2012/251364.
Marimin. 2002. Teori dan Aplikasi Sistem Pakar Dalam Teknik Manajerial. IPB Press. Bogor. Indonesia.
25 Musi IID. 2009. Pengembangan Fuzzy Inferensi Sistem Untuk Seleksi Metode
Peningkatan Perolehan Minyak Tingkat Lanjut. Tesis. Ilmu Komputer, Institut Pertanian Bogor.
Nelson DL, Cox MM. 2008. Lehninger: Principal of Biochemistry (Fifth Edition). W.H Freeman and Company: New York.
Othman Z, Subari K, Morad N. 2002. Application of Fuzzy Inference Systems and Genetic Algorithm in Integrated Process Planning and Scheduling. International Journal of The Computer, The Internet, and Management. 10(2): 81 - 96. Pevzner PA, Tang H, Waterman MS. 2001. An eulerian path approach to dna
fragment assembly. Proceedings of the National Academy of Sciences. 98(17): 9748–9753.
Qidway U, Shamim MS, Raquib F, Enam A. 2007. Failed Back Surgery Syndrome (FBSS) Prediction Using Fuzzy Inference System (FIS). IEEE International Conference on Signal Processing and Communications. 1-4244-1236-6/07/$25.00.
Rogers K. 2011. New Thinking about Genetics. New York: Britannica Educational Publishing, hlm. 132.
Shi H, Bertil S, Weiguo L, Wolfgang MW. 2009. Accelerating Error Correction in High-Throughput Short-Read DNA Sequencing Data with CUDA. IEEE. Tang K, Shozo T. 1999. Optimization of fuzzy inference system rules by using the
genetic algorithm and its application to the bond rating. Journal of the Operations Research Society of Japan. 42(3): 302-315.
Thamrin F. 2012. Studi inferensi fuzzy tsukamoto untuk penentuan faktor pembebanan trafo PLN. Tesis. Universitas Diponegoro Semarang.
Wijaya E, Frith MC. 2009. Recount: Expectation Maximization Based Error Correction Tool For Next Generation Sequencing Data. Computational Biology Research Center.
Yang X, Chockalingam SP, Aluru S. 2012. A Survey of Error-Correction Methods For Next-Generation Sequencing. Briefings In Bioinformatics. DOI:10.1093/bib/bbs01.
26
27 Lampiran 1 Model fuzzy inference system (FIS) 1 yang pernah diimplementasikan Variabel kualitas
Variabel multiplicity
28
Lampiran 2 Model fuzzy inference system (FIS) 2 yang pernah diimplementasikan Variabel kualitas
Variabel multiplicity
29 Lampiran 3 Model fuzzy inference system (FIS) 3 yang pernah diimplementasikan Variabel kualitas
Variabel multiplicity
30
Lampiran 4 Model fuzzy inference system (FIS) 4 yang pernah diimplementasikan Variabel kualitas
Variabel multiplicity
31 Lampiran 5 Source Code (Menghitung multiplicity dan kualitas tuple)
# Membuka file fastq
33 # mencetak informasi hasil proses
fhhasil = open ('hasil_awal(SRR022835).txt', 'w'); fhhasil.write ("===================================== \n");
fhhasil.write (" Tuples Frek Kualitas \n"); fhhasil.write
("====================================== \n"); for i in range(0,len(tupl)):
34
Lampiran 6 Source Code (Menghitung kedekatan antara weak tuple dengan solid tuple menggunakan jarak Levenshtein)
#membaca file solid tuples dan weak tuples
fh = open('solid.txt') # membaca file solid tuples fg = open('weak.txt') # membaca file weak tuples solid = fh.readlines(); # membaca data perbaris pada file solid
weak = fg.readlines(); # membaca data perbaris pada file weak
for i in range (0,len(solid)):
solid[i] = solid[i].replace('\n','') # menghapus \n pada data
for i in range (0,len(weak)):
weak[i] = weak[i].replace('\n','') # menghapus \n pada data
fh.close() # tutup fg.close() # tutup
# fungsi untuk menghitung jarak levenshtein dengan skor 1 untuk setiap perubahan
def lev_dist(weak,solid):
if len(weak) > len(solid): weak,solid = solid,weak
distances = range(len(weak) + 1)
for index2,char2 in enumerate(solid): newDistances = [index2+1]
# mencetak informasi hasil proses perhitungan skor untuk stiap tuples
35
for i in range (0,len(weak)):
skor = lev_dist(weak[i],solid[0]) minSkor = skor;
minSolid = solid[0]
for j in range (0,len(solid)):
skor = lev_dist(weak[i],solid[j]) if minSkor > skor:
minSkor = skor
minSolid = solid[j] minSolidHsl = minSolidHsl + [minSolid];
fhhasil.write (" "+str(minSolidHsl[i])+" \n");
36
Lampiran 7 Source Code (Menggantikan weak tuple yang terdapat di data set menjadi solid tuple – string matching)
# membaca file sol_akhir dan weak fs = open('sol_akhir.txt')
fw = open('weak.txt')
#list solid dan weak tuple s = [];
# mereplace tuple yg termasuk kedalam weak fh = open('file_organisme.fastq')
fhhasil = open ('hasil_akhir.fasta', 'w');
37 jmlberubah = 0;
for i in range(0,jloop): t = seq[i:i+n];
idx = 0;
ketemu = False;
while (idx<=len(w)-1) and (not(ketemu)): if t == w[idx]:
ketemu = True else:
idx = idx + 1; if ketemu:
seq = seq.replace(t,s[idx]); jmlberubah = jmlberubah + 1; print(jmlberubah)
if i < jloop-1:
hsl = hsl + seq[i]; else:
if ketemu :
hsl = hsl + s[idx] else:
hsl = hsl + t; if jmlberubah == 0 :
ok = True;
loopbsr = loopbsr + 1; seq = hsl;
fhhasil.write(seq+'\n')
38
Lampiran 8 Source Code (Menghitung number of contigs, N50 size, dan maximum contig length)
'''
Author: Travis Poulsen Date: 09 Feb. 2013
Generate some basic statistics/information about an assembly.
This script will generate a file named 'assembly_stats.txt' that will
include number of contigs, min/max/mean contig lengths, and the N50 for
the assembly. It also generates a histogram of contig lengths as a png.
Needs: numpy, scipy, matplotlib, and Biopython Expected input: A fasta file of assembled contigs. Options:--no_hist: generate the stats only, no histogram.
-v, --verbose: print the stats to the screen as well as to the file.
'''
import os, sys, numpy, argparse from Bio import SeqIO
# Main function: generates content if called as a stand alone.
def main():
parser =
argparse.ArgumentParser(description='Generate basic statistics about a collection of contigs.')
parser.add_argument('input', help='fasta file containing the contigs for analysis.')
parser.add_argument('-v', '--verbose', action='store_true', help = 'Print statistics to screen as well as save to an output file.')
parser.add_argument('--no_hist',
39 # Plot the histogram of contig lengths with the specified axis labels and
40
# Get the N50 of the contigs. This is the sequence length at which point
# half of the bases in the entire assembly are contained in contigs of a
# smaller size. def getN50(sizes): bases = []
for read in sizes:
for i in range(read):
bases.append(read) return numpy.median(bases)
41
RIWAYAT HIDUP
Penulis dilahirkan di Sigli, Aceh Pidie pada tanggal 16 Agustus 1990. Penulis merupakan anak pertama dari dua bersaudara pasangan Aiptu Saparuddin Saragih dan Salmina. Penulis mengenyam pendidikan dasar di SD Negeri Neulop Aceh Pidie dan SD Bhayangkari Banda Aceh (1996-2002). Kemudian, penulis melanjutkan pendidikan menengahnya di SMP Negeri 7 Banda Aceh dan SMP Negeri 15 Medan (2002-2004). Pada tahun 2008, penulis menamatkan pendidikan di SMA Negeri 21 Medan. Penulis melanjutkan studi di Universitas Syiah Kuala (Unsyiah) Banda Aceh melalui jalur Seleksi Penerimaan Mahasiswa Baru (SPMB) di Program Studi Pendidikan Matematika, Fakultas Keguruan dan Ilmu Pendidikan. Selanjutnya enulis mendapatkan kesempatan untuk melanjutkan studi di Institut Pertanian Bogor (IPB) di Program Studi Ilmu Komputer dan mendapatkan beasiswa BPP-DN dari DIKTI.