UIN SYARIF HIDAYATULLAH JAKARTA
Validasi Metode Analisis N-(hidroksietil)-
p
-metoksi
sinamamida dalam Plasma secara
In Vitro
menggunakan
Kromatografi Cair Kinerja Tinggi (KCKT)
SKRIPSI
Farmasi
ELSA RAHMI
1112102000034
FAKULTAS KEDOKTERAN DAN ILMU KESEHATAN
PROGRAM STUDI FARMASI
i
UIN SYARIF HIDAYATULLAH JAKARTA
Validasi Metode Analisis N-(hidroksietil)-
p
-metoksi
sinamamida dalam Plasma secara
In Vitro
menggunakan
Kromatografi Cair Kinerja Tinggi (KCKT)
SKRIPSI
Diajukan sebagai salah satu syarat memperoleh gelar Sarjana Farmasi
ELSA RAHMI
1112102000034
FAKULTAS KEDOKTERAN DAN ILMU KESEHATAN
PROGRAM STUDI FARMASI
v ABSTRAK
Nama : Elsa Rahmi NIM : 1112102000034 Program Studi : Farmasi
Judul Skripsi : Validasi Metode Analisis N-(hidroksietil)-p-metoksi sinamamida dalam Plasma secara In Vitro menggunakan Kromatografi Cair Kinerja Tinggi (KCKT).
N-(hidroksietil)-p-metoksi sinamamida merupakan senyawa hasil modifikasi etil
p-metoksisinamat melalui reaksi amidasi dengan menggunakan etanolamin. Substitusi gugus ester pada etil p-metoksisinamat menjadi bentuk amida terbukti meningkatkan aktifitas antiinflamasi. Kadar N-(hidroksietil)-p-metoksi sinamamida perlu diukur dan dipantau, sehingga keamanan, dosis dan efikasi dari penggunaan N-(hidroksietil)-p-metoksi sinamamida sebagai agen antiinflamasi dapat dipastikan. Tujuan dari penelitian ini adalah untuk memperoleh kondisi optimum dan metode bioanalisis yang tervalidasi untuk analisis N-(hidroksietil)-p-metoksi sinamamida dalam plasma secara in vitro menggunakan KCKT dengan
Diode Array Detector (DAD). N-(hidroksietil)-p-metoksi sinamamida diekstraksi dari plasma dengan metode pengendapan protein menggunakan metanol. Metanol ditambahkan ke dalam plasma dengan perbandingan metanol-plasma 4:1 kemudian divortex selama 20 detik dan disentrifugasi selama 10 menit dengan kecepatan 3000 rpm. Sistem kromatografi terdiri dari kolom Dionex C18 (150 x 4,6 mm, 3 µm) dengan fase gerak metanol-akuabides perbandingan 40:60 dengan laju alir 1,0 mL/menit. Senyawa dideteksi pada panjang gelombang 290 nm. Pada rentang konsentrasi 5,04 − 40,32 µL dihasilkan kurva kalibrasi yang linier dengan nilai koefisien korelasi (r) sebesar 0,9962. Nilai akurasi (% diff) dari metode ini berada diantara -1,813% sampai 11,954% dengan nilai presisi (KV) antara 0,439% sampai 3,895% dan uji perolehan kembali antara 98,187% sampai 111,954%.
ABSTRACT
Name : Elsa Rahmi Study Program : Farmasi
Title : Validation of Analytical Method of N-(hydroxyethyl)-p -methoxycinnamamide in Plasma In Vitro by High Performance Liquid Chromatography (HPLC).
N-(hydroxyethyl)-p-methoxycinnamamide is a modified compound of ethyl p
methoxycinnamate (EPMC) through amidation reaction using ethanolamine. Substitution of the ester of EPMC to amide can increase the anti-inflammatory activity. The level of N-(hydroxyethyl)-p-methoxycinnamamide in the plasma needs to be quantified and monitored so that the safety, dosage, and efficacy of the use of N-(hydroxyethyl)-p-methoxycinnamamide as an anti-inflammatory agent can be ascertained. The aim of this study was to obtain the optimum conditions and validates methods for analysis of N-(hydroxyethyl)-p -methoxycinnamamide in plasma in vitro using HPLC with Diode Array Detector
(DAD). N-(hydroxyethyl)-p-methoxycinnamamide was extracted from plasma by protein deproteination using methanol. Chromatographic system consisted of a Dionex C18 column (150 x 4,6 mm, 3 µm) with an isocratic mobile phase of the methanol-aquabidest ratio of 40:60 with a flow rate of 1,0 mL/min. Samples were detected at a wavelength of 290 nm. Linearity was established for range concentration of 5,04 − 40,32 µL with a correlation coefficient (r) of 0,9962. Accuracy (% diff) of this method is between -1,813% to 11,954% with precision (CV) between 0,439% to 3,895% and test recovery between 98,187% to 111,954%.
vii
KATA PENGANTAR
Puji syukur saya ucapkan kehadirat Allah SWT, yang telah memberikan rahmat, karunia serta nikmat iman dan islam yang tak terhingga, Shalawat serta salam kepada Nabi Muhammad SAW. Syukur atas limpahan nikmat dan kasih sayang-Nya, sehingga penulis dapat menyelesaikan penyusunan skripsi yang
berjudul “Validasi Metode Analisis N-(hidroksietil)-p-metoksi sinamamida dalam
Plasma secara In Vitro menggunakan Kromatografi Cair Kinerja Tinggi (KCKT).” bertujuan guna memperoleh gelar Sarjana Farmasi pada Fakultas Kedokteran dan Ilmu Kesehatan Universitas Islam Negeri Syarif Hidayatullah Jakarta.
Pada penyelesaian penelitian dan penyusunan skripsi ini, penulis mendapat bantuan dari berbagai pihak. Oleh karena itu, penulis ingin mengucapkan rasa terima kasih kepada semua pihak yang telah membantu dan mengarahkan, yaitu kepada :
1. Kedua orang tua, ibunda tersayang Elmi dan ayahanda Guslimar, yang selalu memberikan kasih sayang, doa yang tak pernah putus, semangat serta dukungan baik moril dan materil.
2. Bapak Supandi, M.Si.,Apt selaku pembimbing I dan Ibu Ismiarni Komala, M.Sc., Ph.D., Apt selaku pembimbing II, yang telah meluangkan waktu, tenaga dan pikiran serta dengan sabar membimbing dan memberikan saran sehingga penulis dapat menyelesaikan skripsi ini.
3. Dr. H. Arif Sumantri, SKM, M.Kes, selaku Dekan Fakultas Kedokteran dan Ilmu Kesehatan Universitas Islam Negeri Syarif Hidayatullah Jakarta.
4. Ibu Dr.Nurmeilis, M.si.,Apt selaku Ketua Program Studi Farmasi Fakultas Kedokteran dan Ilmu Kesehatan Universitas Islam Negeri Syarif Hidayatullah Jakarta.
5. Ibu/Bapak Dosen Program Studi Farmasi Fakultas Kedokteran dan Ilmu Kesehatan Universitas Islam Negeri Syarif Hidayatullah Jakarta atas ilmu dan pengetahuan selama penulis menempuh pendidikan.
DAFTAR ISI
Halaman
HALAMAN JUDUL ... i
HALAMAN PERNYATAAN ORISINALITAS ... ii
HALAMAN PERSETUJUAN PEMBIMBING ... iii
HALAMAN PENGESAHAN SKRIPSI ... iv
ABSTRAK ... v
ABSTRACT ... vi
KATA PENGANTAR ... vii
HALAMAN PERNYATAAN PERSETUJUAN PUBLIKASI ... ix
DAFTAR ISI ... x
2.1 N-(hidroksietil)-p-metoksi sinamamida ... 4
2.2 Plasma ... 5
2.4.2.8 Komputer, Integrator, atau Rekorder ... 15
2.5 Uji Kesesuaian Sistem ... 15
2.6.2 Pengembangan Metode Bioanalisis ... 18
2.6.2.1Selektivitas ... 19
xi
3.3.1 Pembuatan Larutan Induk N-(hidroksietil)-p-metoksi sinamamida konsentrasi 1000 µg/mL ... 26
3.3.2 Pembuatan Fase Gerak ... 26
3.3.3 Penentapan Panjang Gelombang Analisis ... 26
3.3.4 Optimasi Kondisi Analisis ... 27
3.3.4.1 Pemilihan Komposisi Fase Gerak ... 27
3.3.5 Uji Kesesuaian Sistem ... 27
3.3.6 Penetapan Metode Ekstraksi ... 27
3.3.7 Validasi Metode Bioanalisis N-(hidroksietil)-p -metoksi sinamamida dalam Plasma secara in vitro ... 28
3.3.7.1 Pengukuran Batas Kuantitasi Terendah (LLOQ) ... 28
4.1 Penetapan Panjang Gelombang Analisis ... 31
4.2 Optimasi Kondisi analisis ... 31
4.2.1 Pemilihan Komposisi Fase Gerak ... 31
4.3 Uji Kesesuaian Sistem ... 34
4.4 Penetapan Metode Ekstraksi ... 36
4.5 Validasi Metode Analisis N-(hidroksietil)-p-metoksi sinamamida dalam Plasma secara In Vitro ... 38
4.5.1 Pengukuran Batas Kuantifikasi Terendah(LLOQ) ... 38
4.5.2 Pembuatan Kurva Kalibrasi dan Uji Linieritas dalam Plasma secara In Vitro... 38
4.5.3 Uji Selektivitas ... 39
BAB V KESIMPULAN DAN SARAN ... 42
5.1 Kesimpulan ... 42
5.2 Saran ... 42
xiii
DAFTAR TABEL
Tabel 3.1 Komposisi Fase Gerak ... 26 Tabel 4.1 Data hubungan antara waktu retensi, jumlah lempeng teoritis
dan asimetrisitas terhadap perubahan komposisi fase gerak ... 34 Tabel 4.2 Hasil uji rata-rata kesesuaian sistem ... 35 Tabel 4.3 Hasil optimasi metode ekstraksi N-(hidroksietil)-p-metoksi
sinamamida dalam plasma ... 37 Tabel 4.4 Hasil uji akurasi, presisi dan perolehan kembali hari ke-1 ... 41 Tabel 5.1 Data hasil uji kesesuaian sistem ... 50 Tabel 5.2 Data pengukuran kurva kalibrasi N-(hidroksietil)-p-metoksi
sinamamida dalam plasma untuk penentuan LLOQ ... 51 Tabel 5.3 Data kurva kalibrasi N-(hidroksietil)-p-metoksi sinamamida
dalam plasma ... 52 Tabel 5.4 Data uji selektivitas N-(hidroksietil)-p-metoksi sinamamida
dalam plasma ... 54 Tabel 5.5 Data uji akurasi, perolehan kembali (%recovery), dan presisi
N-(hidroksietil)-p-metoksi sinamamida dalam plasma, Hari ke-1 ... 55 Tabel 5.6 Data uji akurasi, perolehan kembali (%recovery), dan presisi
DAFTAR GAMBAR
Halaman
Gambar 2.1 Struktur kimia N-(hidroksietil)-p-metoksi sinamamida ... 4
Gambar 2.2 Diagram Alat dan Komponen KCKT ... 10
Gambar 4.1 Kromatogram N-(hidroksietil)-p-metoksi sinamamida menggunakan fase gerak metanol 100% ... 32
Gambar 4.2 Kromatogram N-(hidroksietil)-p-metoksi sinamamida menggunakan fase gerak metanol : akubides (70:30) ... 32
Gambar 4.3 Kromatogram N-(hidroksietil)-p-metoksi sinamamida menggunakan fase gerak metanol : akuabides (60:40) ... 32
Gambar 4.4 Kromatogram N-(hidroksietil)-p-metoksi sinamamida menggunakan fase gerak metanol : akuabides (40:60) ... 33
Gambar 4.5 Kromatogram plasma blangko dengan perbandingan metanol - plasma (1:1) ... 37
Gambar 4.6 Kromatogram N-(hidroksietil)-p-metoksi sinamamida dalam plasma dengan perbandingan metanol - plasma (1:1) ... 37
Gambar 4.7 Kromatogram plasma blangko dengan perbandingan metanol - plasma (4:1) ... 37
Gambar 4.8 Kromatogram N-(hidroksietil)-p-metoksi sinamamida dalam plasma dengan perbandingan metanol - plasma (4:1) ... 37
Gambar 4.9 Kurva Kalibrasi N-(hidroksietil)-p-metoksi sinamamida dalam plasma ... 39
Gambar 5.1 Spektrum serapan N-(hidroksietil)-p-metoksi sinamamida pada spektofotometer UV-Vis ... 48
Gambar 5.2 Kromatogram N-(hidroksietil)-p-metoksi sinamamida ... 49
Gambar 5.3 Kromatogram Plasma Blangko ... 49
Gambar 5.4 Kromatogram N-(hidroksietil)-p-metoksi sinamamida dalam plasma ... 49
Gambar 5.5 Kurva Kalibrasi N-(hidroksietil)-p-metoksi sinamamida dalam plasma ... 51
Gambar 5.6 Kurva Kalibrasi N-(hidroksietil)-p-metoksi sinamamida dalam plasma ... 52
Gambar 5.7 Alat Kromatografi Cair Kinerja Tinggi Dionex Ultimate® 3000 ... 57
Gambar 5.8 Kolom KCKT ... 58
Gambar 5.9 Plasma ... 58
Gambar 5.10 Pot penyimpanan sampel ... 58
Gambar 5.11 Vial penyuntikan KCKT ... 58
Gambar 5.12 Plasma mengandung senyawa untuk disentrifugasi ... 58
Gambar 5.13 Penyaring fase gerak ... 58
Gambar 5.14 Sonikator ... 58
xv
DAFTAR LAMPIRAN
Lampiran 1 Alur penelitian ... 46
Lampiran 2 Perhitungan pembuatan larutan ... 47
Lampiran 3 Spektrum serapan N-(hidroksietil)-p-metoksi sinamamida pada spektrofotometer UV-Vis ... 48
Lampiran 4 Kromatogram Hasil Analisis ... 49
Lampiran 5 Uji kesesuaian sistem ... 50
Lampiran 6 Pengukuran Batas Kuantifikasi Terendah (LLOQ) ... 51
Lampiran 7 Hasil pengukuran kurva kalibrasi N-(hidroksietil)-p-metoksi sinamamida dalam plasma ... 52
Lampiran 8 Uji selektivitas N-(hidroksietil)-p-metoksi sinamamida dalam plasma ... 54
Lampiran 9 Uji akurasi dan presisi N-(hidroksietil)-p-metoksi sinamamida dalam plasma ... 55
Lampiran 10 Rumus-rumus ... 56
Lampiran 11 Alat Kromatografi Cair Kinerja Tinggi (KCKT) ... 57
BAB I
PENDAHULUAN
1.1 Latar Belakang
Etil p-metoksisinamat (EPMS) memiliki potensi yang besar sebagai dasar sintesis untuk turunan sinamat karena memiliki gugus fungsi ester yang sangat reaktif sehingga mudah ditransformasikan dengan gugus fungsi lainnya seperti gugus amina (Barus, 2009). N-(hidroksietil)-p-metoksi sinamamida merupakan hasil modifikasi etil p-metoksisinamat melalui reaksi amidasi dengan etanolamin. Penelitian sebelumnya yang dilakukan oleh Muhammad Reza (2015), telah melaporkan mengenai hubungan struktur aktivitas hasil modifikasi EPMS terhadap aktivitas anti-inflamasi melalui reaksi amidasi dengan etanolamin menunjukkan bahwa penambahahan gugus amida terbukti meningkatkan aktivitas anti-inflamasi. Peningkatan aktifitas tersebut mejadikan N-(hidroksietil)-p-metoksi sinamamida ini layak untuk dikembangkan lebih lanjut serta bisa menjadi agen terapi yang menjanjikan (kandidat obat) karena aktifitas yang dimilikinya.
Setelah diperoleh kandidat obat, maka selanjutnya kandidat obat tersebut akan melalui serangkaian uji yang memakan waktu yang panjang dan biaya yang tidak sedikit sebelum diresmikan sebagai obat oleh Badan pemberi izin. Uji yang harus ditempuh oleh kandidat obat adalah uji praklinik dan uji klinik. Uji praklinik merupakan persyaratan uji untuk kandidat obat, dari uji ini diperoleh informasi tentang efikasi (efek farmakologi), profil farmakokinetik dan toksisitas kandidat obat. Setelah dinyatakan mempunyai manfaat dan aman pada hewan percobaan, barulah dapat diujikan pada manusia (uji klinik) (Sukandar, 2006).
2
UIN Syarif Hidayatullah Jakarta
dan/atau biofarmasetik dan uji farmakologi klinik (Food and Drug Administration, 2001; Harahap. Y., 2010).
Pengukuran analit dalam matriks biologis harus divalidasi. Validasi metode bioanalisis mencakup semua prosedur yang menunjukkan bahwa metode yang digunakan untuk pengukuran kuantitatif analit yang berasal dalam matriks biologis, seperti darah, plasma, serum, atau urin, dapat dipercaya dan dapat dilakukan ulang (reproducible) untuk penggunaan yang diinginkan. Beberapa parameter validasi metode meliputi limit deteksi (LOD) dan limit kuantifikasi (LOQ), selektivitas, linearitas, akurasi, dan presisi, stabilitas (Food and Drug Administration, 2001).
Pada penelitian ini, akan dilakukan validasi metode analisis N-(hidroksietil)-p-metoksi sinamamida dalam plasma secara in vitro
menggunakan Kromatografi Cair Kinerja Tinggi (KCKT) dengan detektor DAD (Diode Array Detector). Analisis kadar senyawa dalam plasma merupakan hal yang kompleks karena plasma mengandung sejumlah unsur endogen, sehingga perlu untuk memisahkan analit yang akan dianalisis dari unsur endogen tersebut (Swarbrick dan Boylan, 1988). Metode ekstraksi yang digunakan pada penelitian ini adalah metode pengendapan protein menggunakan metanol. Metode KCKT fase balik dengan menggunakan fase diam oktadesil silika (ODS atau C18) merupakan metode yang populer digunakan saat ini untuk menetapkan kadar obat baik dalam bentuk sediaan atau dalam sampel hayati. Hal ini dikarenakan metode ini bersifat selektif, memiliki sensitifitas dan spesifisitas yang tinggi, serta sederhana dalam perlakuannya (Harmita, 2006; Gandjar dan Rohman, 2007).
1.2 Perumusan masalah
a. Bagaimanakah optimasi kondisi analisis N-(hidroksietil)-p-metoksi sinamamida menggunakan KCKT?
b. Bagaimanakah metode ekstraksi untuk analisis N-(hidroksietil)-
c. Apakah metode analisis N-(hidroksietil)-p-metoksi sinamamida dalam plasma secara in vitro menggunakan KCKT memiliki nilai validitas yang sesuai dengan persyaratan?
1.3 Tujuan Penelitian
a. Memperoleh kondisi optimum metode analisis N-(hidroksietil)-
p-metoksi sinamamida menggunakan KCKT.
b. Memperoleh metode ekstraksi untuk analisis N-(hidroksietil)-
p-metoksi sinamamida dalam plasma secara in vitro menggunakan KCKT.
c. Memperoleh metode yang valid untuk analisis N-(hidroksietil)-
p-metoksi sinamamida dalam plasma secara in vitro menggunakan KCKT.
1.4 Manfaat Penelitian
Penelitian ini diharapkan mampu mendapatkan metode analisis yang valid untuk N-(hidroksietil)-p-metoksi sinamamida dalam plasma secara in vitro menggunakan KCKT.
.
4 UIN Syarif Hidayatullah Jakarta
BAB II
TINJAUAN PUSTAKA
2.1 N-(hidroksietil)-p-metoksi sinamamida
Struktur kimia :
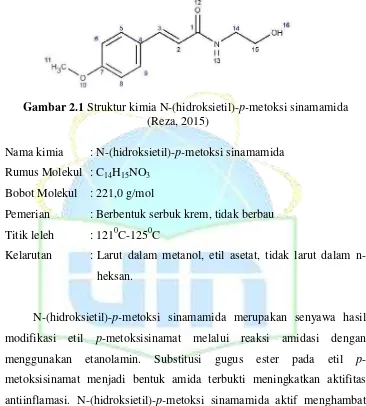
Gambar 2.1 Struktur kimia N-(hidroksietil)-p-metoksi sinamamida (Reza, 2015)
Nama kimia : N-(hidroksietil)-p-metoksi sinamamida Rumus Molekul : C14H15NO3
Bobot Molekul : 221,0 g/mol
Pemerian : Berbentuk serbuk krem, tidak berbau Titik leleh : 1210C-1250C
Kelarutan : Larut dalam metanol, etil asetat, tidak larut dalam n-heksan.
2.2 Plasma
Darah terdiri atas plasma dan sel-sel darah. Sebagian besar sel darah merupakan sel darah merah atau eritrosit, sedangkan jumlah sel darah putih atau leukosit relatif sangat sedikit, yaitu 0,2% dari jumlah eritrosit. Disamping eritrosit dan leukosit, ada partikel lain yang disebut trombosit. Trombosit sangat berguna pada proses penggumpalan darah (Pudjiadi, 1994).
Apabila darah yang sebelumnya telah diberi antikoagulan dilakukan sentrifugasi, maka sel-sel darah merah akan mengendap sedangkan plasma akan berada dalam bentuk cairan bening atau supernatan di atasnya (Pudjiadi, 1994).
Volume rata-rata plasma pada pria adalah 55%, pada wanita 58% dari volume darah (Sherwood, 1996). Plasma manusia mengandung 90-92% air. Peranan air dalam darah sangat besar, sebab disamping sebagai pelarut zat - zat, air diperlukan untuk menjaga tekanan darah, kondisi osmotik, dan pengatur suhu tubuh dengan meratakan panas tubuh (Pudjiadi, 1994).
6
UIN Syarif Hidayatullah Jakarta
2.3 Analisis Obat dalam Plasma
Farmakokinetik obat berhubungan dengan paparan obat terhadap tubuh dan perubahan konsentrasi obat terhadap waktu. Darah merupakan salah satu cairan biologis yang bisa diperoleh dan konsentrasi obat dapat dianalisis berulang kali dalam beberapa waktu setelah pemberian obat. Namun, konsentrasi obat dalam darah utuh jarang digunakan untuk melihat profil farmakokinetik obat karena darah merupakan sistem fisik yang sangat kompleks. Darah mengandung sel darah merah, sel darah putih, dan platelet yang tersuspensi dalam air plasma. Darah dengan penghilangan unsur sel tersebut dengan cara sentrifugasi setelah sebelumnya ditambahkan antikoagulan sehingga mendapatkan plasma, atau dengan penggumpalan darah sehingga diperoleh serum, lebih disukai. Alasan lain fokus terhadap konsentrasi plasma dalam farmakokinetik adalah karena plasma merupakan suatu cairan kompleks yang terdapat dalam sistem sirkulasi yang berfungsi sebagai medium transportasi zat-zat yang di angkut dalam darah (Ganong, 2001; Rosenbaum, 2011). Selain itu, intensitas efek farmakologi atau efek toksik suatu obat seringkali dikaitkan dengan konsentrasi obat pada reseptor, yang biasanya terdapat dalam sel-sel jaringan. Oleh karena sebagian besar sel-sel-sel-sel jaringan diperfusi oleh cairan jaringan atau plasma, maka pemeriksaan kadar obat dalam plasma merupakan suatu metode yang sesuai untuk pemantauan pengobatan (Shargel, 2005).
Ada beberapa teknik penyiapan sampel yang biasa digunakan untuk analisis dalam matriks plasma, antara lain :
a. Pengendapan protein
Pada metode ini, digunakan asam atau pelarut organik yang bercampur dengan air untuk menghilangkan protein dengan cara denaturasi atau presipitasi. Asam seperti asam trikloroasetat (TCA) dan asam perklorat merupakan pengendap protein yang sangat efisien untuk mengendapkan protein pada konsentrasi 5-20%. Pelarut organik seperti metanol, asetonitril, aseton, dan etanol meskipun kurang efisien dalam mengendapkan protein, namun pelarut-pelarut tersebut telah secara luas digunakan untuk bioanalisis karena kompatibel dengan fase gerak KCKT serta dapat mengekstraksi senyawa berdasarkan prinsip kepolaran. Pelarut organik dapat menurunkan solubilitas protein sehingga protein akan mengendap (Evans, 2004).
b. Ekstraksi cair-cair
8
UIN Syarif Hidayatullah Jakarta
c. Ekstraksi fase padat
Prinsip mekanisme pemisahan dan isolasi yang digunakan dalam ekstraksi fase padat adalah fase terbalik, fase normal, dan ion exchange, sama seperti yang digunakan dalam KCKT. Prinsip umum ekstraksi fase padat yaitu adsorbsi obat dari larutan ke dalam adsorben atau fase diam. Adsorben yang digunakan pada ekstraksi ini terdiri dari partikel silika ukuran 40-60 µm yang berikatan membentuk fase hidrokarbon. C18 merupakan adsorben yang memiliki kapasitas paling baik dalam mengadsorbsi analit. Ekstraksi fase padat adalah suatu teknik yang dapat mengatasi beberapa masalah yang ditemui pada ekstraksi cair-cair. Ektraksi fase padat secara umum melalui 5 tahap proses, diantaranya pengkondisian (conditioning), penyeimbangan fase diam (equlibration), memasukkan sampel (loading), pencucian dan elusi sampel (washing dan elution). (Evans, 2004 ; Harahap, Y., 2010).
2.4 Kromatografi Cair Kinerja Tinggi
2.4.1 Teori Dasar KCKT
kromatografi. Pemisahan solut dipengaruhi oleh distribusi solut dalam fase gerak dan fase diam. Penggunaan kromatografi cair membutuhkan penggabungan berbagai macam kondisi operasional secara tepat seperti jenis kolom, fase gerak, panjang dan diameter kolom, kecepatan alir fase gerak, suhu kolom, dan ukuran sampel.
Kegunaan umum KCKT adalah untuk pemisahan sejumlah senyawa organik, anorganik, maupun senyawa biologis, analisis ketidakmurnian (impurities); analisis senyawa-senyawa tidak mudah menguap (non volatile); penentuan molekul-molekul netral, ionik, maupun zwitter ion; isolasi dan pemurnian senyawa; pemisahan senyawa-senyawa yang memiliki struktur hampir sama; pemisahan senyawa-senyawa dalam jumlah sekelumit (trace elements), dalam jumlah banyak, dan dalam skala proses industri. KCKT merupakan metode yang tidak destruktif dan dapat digunakan baik untuk analisis kualitatif maupun kuantitatif (Gandjar dan Rohman, 2007).
Dalam analisis farmasi, metode KCKT merupakan metode yang sangat populer untuk menetapkan kadar senyawa obat baik dalam bentuk sediaan maupun dalam sampel hayati. Hal ini disebabkan KCKT merupakan metode yang memberikan sensitifitas dan spesifisitas yang tinggi.
Kelebihan metode KCKT jika dibandingkan dengan Kromatografi Cair tradisional, yaitu (Johnson dan Stevenson, 1991):
a. Cepat;
b. Daya pisah baik; c. Peka; detektor unik;
10
UIN Syarif Hidayatullah Jakarta
2.4.2 Instrumentasi
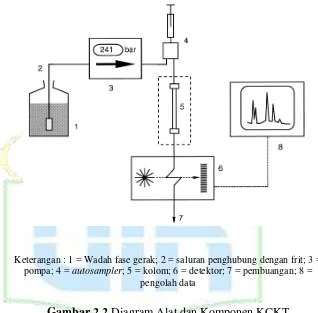
Instrumentasi KCKT pada dasarnya terdiri atas delapan komponen pokok, yaitu wadah fase gerak, sistem penghantaran fase gerak dan alat untuk memasukkan sampel, kolom, detektor, wadah penampung buangan fase gerak, dan komputer atau integrator atau perekam (Gandjar dan Rohman, 2007).
Keterangan : 1 = Wadah fase gerak; 2 = saluran penghubung dengan frit; 3 =
pompa; 4 = autosampler; 5 = kolom; 6 = detektor; 7 = pembuangan; 8 =
pengolah data
Gambar 2.2 Diagram Alat dan Komponen KCKT
(Sumber : Practical High-Performance Liquid Chromatography 4th edition,
2004)
2.4.2.1Wadah Fase Gerak pada KCKT
Wadah fase gerak harus bersih dan lembam (inert). Wadah ini biasanya dapat menampung fase gerak antara 1-2 liter pelarut. Fase gerak sebelum digunakan harus dilakukan
2.4.2.2Fase Gerak pada KCKT
Fase gerak atau eluen biasanya terdiri atas campuran pelarut yang dapat bercampur, berperan dalam daya elusi dan resolusi. Daya elusi dan resolusi ini ditentukan oleh polaritas keseluruhan pelarut, polaritas fase diam, dan sifat komponen-komponen sampel. Untuk fase normal (fase diam lebih polar daripada fase gerak), kemampuan elusi meningkat dengan meningkatnya polaritas pelarut (Gandjar dan Rohman, 2007).
2.4.2.3Pompa pada KCKT
Pompa yang digunakan untuk KCKT harus inert
terhadap fase gerak. Pompa berfungsi untuk mengalirkan eluen ke dalam kolom. Pompa yang digunakan sebaiknya mampu memberikan tekanan sampai 5000 psi dan mampu mengalirkan fase gerak dengan kecepatan alir 3 mL/menit. Untuk tujuan preparatif, pompa yang digunakan harus mampu mengalirkan fase gerak dengan kecepatan 20 mL/menit.
Tujuan penggunaan pompa atau sistem penghantaran fase gerak adalah untuk menjamin proses penghantaran fase gerak berlangsung secara tepat, reprodusibel, konstan, dan bebas dari gangguan. Terdapat 2 jenis pompa dalam KCKT, yaitu pompa dengan tekanan konstan dan pompa dengan aliran fase gerak yang konstan (Gandjar dan Rohman, 2007).
2.4.2.4Penyuntikan Sampel pada KCKT
12
UIN Syarif Hidayatullah Jakarta
Pada saat penyuntikan, katup diputar sehingga fase gerak mengalir melewati keluk sampel dan memasukkan sampel ke kolom (Gandjar dan Rohman, 2007).
2.4.2.5Kolom pada KCKT
Kolom merupakan jantung kromatografi. Keberhasilan atau kegagalan analisis bergantung pada pilihan kolom dan kondisi kerja yang tepat. Kolom dapat dibagi menjadi dua kelompok, yaitu kolom analitik dan kolom preparatif. Kolom umumnya terbuat dari tembaga tahan karat dan biasanya dioperasikan pada temperatur kamar, tetapi bisa juga digunakan temperatur lebih tinggi, terutama untuk kromatografi pertukaran ion dan kromatografi eksklusi (Johnson dan Stevenson, 1991).
Terdapat beberapa hal yang perlu diperhatikan dalam memilih kolom, yaitu (Harmita, 2006) :
a. Panjang kolom b. Diameter kolom c. Pengisi kolom d. Fase gerak e. Tekanan kolom
2.4.2.6Fase Diam pada KCKT
Kebanyakan fase diam pada KCKT berupa silika yang dimodifikasi secara kimiawi, silika yang tidak dimodifikasi, atau polimer-polimer stiren dan divinil benzen. Permukaan silika adalah polar dan sedikit asam karena adanya residu gugus silanol (Si-OH).
Hasil reaksi yang diperoleh disebut dengan silika fase terikat yang stabil terhadap hidrolisis karena terbentuk ikatan-ikatan siloksan (Si-O-O-Si). Silika yang dimodifikasi ini mempunyai karakteristik kromatografi dan selektifitas yang berbeda jika dibandingkan dengan silika yang tidak dimodifikasi.
Oktadesil silika (ODS atau C18) merupakan fase diam yang paling banyak digunakan karena mampu memisahkan senyawa-senyawa dengan kepolaran yang rendah, sedang, maupun tinggi (Gandjar dan Rohman, 2007).
2.4.2.7Detektor
Detektor pada KCKT dikelompokkan menjadi 2 golongan, yaitu detektor universal (yang mampu mendeteksi zat secara umum, tidak bersifat spesifik, dan tidak bersifat selektif) seperti detektor indeks bias dan detektor spektrometri massa; dan golongan detektor yang spesifik, seperti detektor UV-Vis, detektor fluoresensi, dan elektrokimia.
Idealnya, suatu detektor harus mempunyai karakteristik sebagai berikut:
a. Mempunyai respon terhadap solut yang cepat dan reprodusibel,
b. Mempunyai sensitifitas yang tinggi, yakni mampu mendeteksi solut pada kadar yang sangat kecil,
c. Stabil dalam pengoperasiannya,
14
UIN Syarif Hidayatullah Jakarta
e. Signal yang dihasilkan berbanding lurus dengan konsentrasi solut pada kisaran yang luas (kisaran dinamis linier), dan
f. Tidak peka terhadap perubahan suhu dan kecepatan alir fase gerak.
Beberapa detektor yang sering digunakan pada KCKT : a. Detektor Spektrofotometri UV-Vis
Detektor ini didasarkan pada adanya penyerapan radiasi ultraviolet (UV) dan sinar tampak (Vis) pada kisaran panjang gelombang 190-800 nm oleh spesies solut yang mempunyai struktur-struktur atau gugus-gugus kromoforik. Detektor spektrofotometri UV-Vis dapat berupa detektor dengan panjang gelombang tetap (merupakan detektor yang paling sederhana) serta detektor dengan panjang gelombang bervariasi.
b. Detektor photodiode-array (PDA)
Detektor PDA merupakan detektor UV-Vis dengan berbagai keistimewaaan. Detektor ini mampu memberikan kumpulan kromatogram secara simultan pada panjang gelombang yang berbeda pada sekali proses (single run). Selama proses berjalan, suatu kromatogram pada panjang gelombang yang diinginkan (biasanya antara 190-400) dapat ditampilkan.
c. Detektor Flouresensi
flouresensi ini sangat spesifik. Disamping itu, detektor ini juga sangat sensitif dibandingkan dengan detektor UV.
d. Detektor Indeks Bias
Detektor ini akan merespon setiap perbedaan indeks bias antara analit (zat terlarut) dengan pelarutnya (fase geraknya). Penggunaan detektor ini terutama untuk senyawa-senyawa yang tidak mempunyai gugus kromofor.
e. Detektor Elektrokimia
Detektor ini bekerja berdasarkan oksidasi dan reduksi senyawa organik (termasuk obat) secara elektrokimia pada elektroda yang cocok.
(Gandjar dan Rohman, 2007).
2.4.2.8Komputer, Integrator, atau Rekorder
Alat pengumpul data seperti komputer, integrator, atau rekorder dihubungkan dengan detektor. Alat ini akan mengukur sinyal elektronik yang dihasilkan oleh detektor lalu memplotkannya sebagai suatu kromatogram yang selanjutnya dapat dievaluasi oleh analis (Gandjar dan Rohman, 2007).
2.5 Uji Kesesuaian Sistem
Persyaratan-16
UIN Syarif Hidayatullah Jakarta
persyaratan kesesuian sistem biasanya dilakukan setelah dilakukan pengembangan metode dan validasi metode.
United States Pharmacopeia (USP) menentukan parameter yang dapat digunakan untuk menentukan kesesuaian sistem sebelum analisis. Parameter-parameter yang digunakan meliputi: jumlah lempeng teoritis (N), asimetrisitas, faktor kapasitas (k’ atau α) dan nilai standar deviasi relatif (RSD) tinggi puncak dan luas puncak dari serangkaian injeksi. Pada umumnya, paling tidak ada 2 kriteria yang biasanya dipersyaratkan untuk menunjukkan kesesuaian sistem suatu metode. Parameter yang berguna untuk uji kesesuaian sistem adalah keberulangan penyuntikan larutan baku yang dinyatakan dalam standar deviasi relatif (RSD) yang dinyatakan dalam persen bila tidak dinyatakan lain dalam monografi baku yang digunakan dengan nilai RSD kurang dari 2% (Farmakope Indonesia edisi IV).
2.6 Validasi Metode Analisis
Validasi metode menurut United States Pharmacopeia (USP) dilakukan untuk menjamin bahwa metode analisis akurat, spesifik, reprodusibel, dan tahan pada kisaran analit yang akan dianalisis.
Suatu metode analisis harus divalidasi untuk melakukan verifikasi bahwa parameter-parameter kinerjanya cukup mampu untuk mengatasi masalah analisis. Oleh karena itu suatu metode harus divalidasi ketika: a. Metode baru dikembangkan untuk mengatasi masalah analisis
tertentu,
b. Metode yang sudah baku direvisi untuk menyesuaikan perkembangan atau karena munculnya suatu masalah yang mengarahkan bahwa metode baku tersebut harus direvisi,
c. Penjaminan mutu yang mengindikasikan bahwa metode baku telah berubah seiring dengan berjalannya waktu,
e. Untuk mendemonstrasikan kesetaraan antar 2 metode, seperti antara metode baru dan metode baku.
(Gandjar dan Rohman, 2007).
Validasi metode bioanalisis ini digunakan pada studi farmakologi klinis, pengujian ketersediaan hayati (bioavailabilitas) dan bioekuivalensi, serta uji farmakokinetika. Metode analisis yang selektif dan sensitif sangat penting untuk evaluasi obat dan metabolitnya (analit) secara kuantitatif untuk keberhasilan studi farmakologi pre-klinis dan klinis. Validasi metode bioanalisis mencakup semua prosedur yang menunjukkan bahwa metode yang digunakan untuk analisis analit secara kuantitatif di dalam matriks biologis, seperti darah, plasma, serum, atau urin dapat dipercaya dan reprodusibel sesuai tujuan penggunaannya.
2.6.1 Tipe dan Tingkatan Validasi
Dalam bioanalisis, validasi metode dibagi menjadi 3 kategori, yaitu (Food and Drug Administration, 2001):
2.6.1.1Validasi Lengkap
Validasi lengkap penting dilakukan saat melakukan pengembangan dan implementasi metode bioanalisis untuk pertama kali. Validasi lengkap juga penting untuk obat-obat baru dan dilakukan jika ada metabolit yang ditambahkan pada suatu penetapan kadar.
2.6.1.2 Validasi Parsial
18
UIN Syarif Hidayatullah Jakarta
a. Metode bioanalisis yang di pindahkan antar laboratorium atau analis,
b. Perubahan dalam metode analisis (misal perubahan dalam sistem deteksi),
c. Perubahan dalam antikoagulan yang digunakan dalam cairan biologis,
d. Perubahan matriks (misal dari plasma menjadi urin), e. Perubahan dalam prosedur proses sampel,
f. Perubahan spesies pada matriks yang sama (misal dari
rat plasma menjadi mouse plasma), g. Perubahan dalam rentang konsentrasi,
h. Perubahan instrumen dan/ atau software yang digunakan, i. Volume sampel yang terbatas (misal pada studi pediatri), j. Matriks yang jarang.
2.6.1.3 Validasi Silang
Validasi silang merupakan perbandingan parameter validasi ketika 2 atau lebih metode bioanalisis digunakan untuk mendapatkan data pada studi yang sama ataupun studi yang berbeda. Salah satu contoh dari validasi silang adalah keadaan dimana metode bioanalisis yang telah tervalidasi dianggap sebagai referensi dan metode bioanalisis hasil revisi dijadikan sebagai pembanding. Perbandingan harus dilakukan dua arah.
2.6.2 Pengembangan Metode Bioanalisis
Pengembangan dan penetapan metode bioanalisis meliputi : 2.6.2.1Selektivitas
Selektivitas adalah kemampuan metode analisis untuk membedakan dan mengukur secara kuantitatif analit dengan adanya komponen lain di dalam sampel. Untuk selektivitas, analisis sampel blangko dari matriks biologi yang sesuai (seperti plasma, urin, atau matriks lainnya) harus dilakukan sedikitnya dari enam sumber yang berbeda. Tiap sampel blangko harus diuji terhadap interferensi, dan selektivitas harus dipastikan pada kadar batas kuantifikasi terendah (LLOQ). Jika suatu metode mengukur lebih dari satu analit, maka tiap analit harus diuji untuk memastikan tidak ada interferensi (Food and Drug Administration, 2001).
2.6.2.2Kurva Kalibrasi
Kurva kalibrasi merupakan hubungan antara respon instrumen dan konsentrasi analit yang diketahui. Kurva kalibrasi disiapkan dalam matriks biologi yang sama dengan sampel, dengan cara menambahkan sejumlah analit dengan konsentrasi yang diketahui ke dalam matriks. Rentang konsentrasi standar dipilih berdasarkan literatur atau penelitian. Pembuatan kurva kalibrasi harus mencakup sampel blangko (matriks tanpa baku dalam), sampe zero
(matriks dengan baku dalam), dan 6 sampai 8 non-zero
sampel pada rentang konsentrasi standar, termasuk LLOQ.
a. Lower Limit of Quantification (LLOQ)
20
UIN Syarif Hidayatullah Jakarta
reprodusibel dengan nilai presisi ≤20% dan akurasi 80-120%.
b. Kurva Kalibrasi/ Kurva Standar/ Konsentrasi-Respon Syarat kurva kalibrasi agar dapat diterima, yaitu nilai nilai deviasi sebesar ≤20% dari konsentrasi LLOQ dan nilai nilai deviasi sebesar ≤15% dari konsentrasi standar selain LLOQ. Sedikitnya empat dari enam standar non-zero berada diatas kriteria diatas, termasuk LLOQ dan konsentrasi tertinggi dari kurva kalibrasi.
(Food and Drug Administration, 2001)
2.6.2.3Batas Deteksi (limit of detection, LOD)
Batas deteksi didefinisikan sebagai konsentrasi analit terendah dalam sampel yang masih dapat dideteksi, meskipun tidak selalu dapat dikuantifikasi. LOD merupakan batas uji yang secara spesifik menyatakan apakah analit diatas atau dibawah nilai tertentu. Definisi batas deteksi yang paling umum digunakan dalam kimia analisis adalah kadar analit yang memberikan respon sebesar respon blangko (yb) ditambah dengan 3 simpangan baku blangko
(3Sb).
LOD dapat dihitung berdasarkan pada standar deviasi (SD) respon dan kemiringan (slope, S) kurva baku pada level yang mendekati LOD sesuai dengan rumus, LOD = 3,3 (SD/S) (Gandjar dan Rohman, 2007).
2.6.2.4Batas Kuantifikasi (limit of quantification, LOQ)
operasional metode yang digunakan. Seperti halnya LOD, LOQ juga diekspresikan sebagai konsentrasi (dengan akurasi dan presisi juga dilaporkan). Untuk menentukan LOQ, dapat menggunakan perhitungan yang didasarkan pada standar deviasi respon (SD) dan slope (S) kurva baku sesuai rumus : LOQ = 10 (SD/S) (Gandjar dan Rohman, 2007).
2.6.2.5Akurasi
Akurasi adalah suatu metode analisis yang menggambarkan kedekatan nilai rata-rata hasil uji yang diperoleh dengan nilai konsentrasi analit sebenarnya. Akurasi ditentukan oleh analisis berulang dari sampel yang telah diketahui kadar analit yang terkandung didalamnya (Food and Drug Administration, 2001). ICH merekomendasikan pengukuran minimal menggunakan 3 kali pengukuran (Ravichandranravichandran, V., Shalini S., Sundram K. M., Harish Rajak, 2010). Uji akurasi minimal menggunakan tiga konsentrasi pada rentang yang telah direkomendasikan, yaitu pada konsentrasi rendah (tiga kali LLOQ), sedang, dan tinggi dari kurva standar. Perbedaan nilai rata-rata harus + 15% terhadap nilai sebenarnya, kecuali pada LLOQ tidak boleh lebih dari 20% (Food and Drug Administration, 2001).
2.6.2.6Presisi
22
UIN Syarif Hidayatullah Jakarta
menggunakan tiga konsentrasi pada rentang yang telah direkomendasikan, yaitu pada konsentrasi rendah (tiga kali LLOQ), sedang, dan tinggi dari kurva standar. Pengukuran presisi dikelompokkan menjadi within-run (selama waktu analisis), intra-batch precision atau repeatabilitas (dalam satu kali analisis) dan between-run, inter-batch precision
atau repeatabilitas (bila metode dilakukan oleh analis, alat, reagen, dan laboratorium yang berbeda). Perbedaan nilai rata-rata harus tidak lebih dari 15% terhadap nilai sebenarnya, kecuali pada LLOQ tidak boleh lebih dari 20% (Food and Drug Administration, 2001).
2.6.2.7Perolehan Kembali
Perolehan kembali suatu analit adalah respon detektor yang diperoleh dari jumlah analit yang ditambahkan dan diekstraksi dari matriks biologi dibandingkan dengan respon detektor analit yang diketahui konsentrasinya. Perolehan kembali analit tidak harus 100% namun tingkat perolehan kembali analit dan baku dalam harus konsisten, presisi, dan reprodusibel dengan rentang syarat 80-120%. Uji perolehan kembali harus dilakukan dengan membandingkan hasil analisis sampel pada tiga konsentrasi (rendah, sedang, dan tinggi) yang diekstraksi dari matriks biologi dengan baku yang tidak diekstraksi yang mewakili perolehan kembali 100% (Food and Drug Administration,
2001).
2.6.2.8Stabilitas
matriks dan sistem penyimpanan tersebut dan tidak dapat diekstrapolasikan ke matriks dan sistem penyimpanan lain.
Semua penentuan stabilitas harus menggunakan sampel yang disiapkan dari larutan stok analit yang dibuat baru dalam matriks biologis yang bebas analit dan bebas dari interferensi. Larutan stok analit harus disiapkan dalam pelarut yang sesuai pada konsentrasi yang diketahui.
Penentuan uji stabilitas dapat menggunakan beberapa cara, antara lain :
a. Stabilitas Beku-Cair
Stabilitas analit dapat ditentukan setelah tiga siklus beku-cair. Sedikitnya tiga larutan senyawa dari setiap konsentrasi rendah, sedang, dan tinggi di simpan pada kondisi penyimpanan yang diinginkan selama 24 jam kemudian dikeluarkan dan dibiarkan hingga mencair pada temperatur ruang. Setelah semua mencair, sampel dibekukan kembali selama 12-24 jam pada kondisi yang sama. Siklus beku-cair ini harus diulang sebanyak 2 kali lagi, kemudian dianalisis setelah tiga siklus. Jika analit tidak stabil pada temperatur penyimpanan, maka uji dapat dilakukan dengan menyimpan sampel pada suhu -700C selama tiga siklus beku-cair.
b. Stabilitas Jangka Pendek
24
UIN Syarif Hidayatullah Jakarta
c. Stabilitas Jangka Panjang
Waktu penyimpanan pada evaluasi stabilitas jangka panjang harus melebihi waktu pertama kali sampel di kumpulkan sampai waktu terakhir sampel dianalisis. Stabilitas jangka panjang ditentukan dengan menyimpan sedikitnya tiga larutan senyawa dari setiap konsentrasi rendah, sedang, dan tinggi dicairkan pada kondisi yang sama seperti uji sampel. Konsentrasi dari semua sampel dibandingkan dengan rata-rata nilai perolehan kembali yang sesuai dengan konsentrasi standar dari hari pertama uji stabilitas jangka panjang.
d. Stabilitas Larutan Stok
Stabilitas larutan stok obat dan baku dalam harus dievaluasi pada temperatur ruang selama minimal 6 jam. Jika larutan stok dibekukan selama periode tertentu, perlu dicatat stabilitasnya. Setelah itu, dilakukan uji stabilitas dengan membandingkan respon instrumen terhadap larutan yang baru dibuat.
e. Stabilitas Post-Preparatif
Stabilitas dari sampel yang telah diproses, termasuk waktu sampel berada dalam autosampler. Stabilitas obat dan baku dalam harus ditentukan selama waktu analisis untuk setiap batch dalam validasi sampel dengan menentukan konsentrasi berdasarkan kalibrasi standar.
BAB III
METODE PENELITIAN
3.1 Tempat dan Waktu Penelitian
Penelitian dilaksanakan di Laboratorium Farmakognosi dan Fitokimia, Laboratorium Penelitian II, Laboratorium Kimia Obat, Laboratorium Penelitian I, Laboratorium Analisa Obat dan Pangan Halal Fakultas Kedokteran dan Ilmu Kesehatan Universitas Islam Negeri Syarif Hidayatullah Jakarta. Penlitian dimulai dari bulan Maret sampai Juni 2016.
3.2 Alat dan Bahan
3.2.1 Alat
Kromatografi Cair Kinerja Tinggi (Dionex UltiMate® 3000) dilengkapi dengan; pompa, autosampler, kolom Acclaim® Polar Advantage II (C18; 3 µm; 4,6 x 150 mm), detektor DAD (Diode Array Detector), dan program komputer PC (Chromeleon®). Spektrofotometer Ultaviolet-Visibel (Hitachi U-2910), sentrifus (Eppendorf 5417R) dengan tabung sentrifugasi, syringe filter
(Sartorius, RC 0,45 µm), timbangan analitik kepekaan 220 g – 1 mg (AND-GH202), vorteks, mikropipet (Rainin 20-200 µL dan 100-1000 µL), dry vacuum pump/compressor (Welch®), tabung vacutainer, lemari pendingin, sonikator (Elmasonic®), dan alat-alat gelas.
3.2.2 Bahan
26
UIN Syarif Hidayatullah Jakarta
3.3 Prosedur Kerja
3.3.1 Pembuatan Larutan Induk N-(hidroksietil)-p-metoksi sinamamida konsentrasi 1008 µg/mL
Ditimbang sebanyak 50,4 mg N-(hidroksietil)-p-metoksi sinamamida. Dilarutkan ke dalam metanol hingga 50,0 mL sehingga diperoleh konsentrasi 1008 µg/mL. Pengenceran dilakukan untuk memperoleh larutan dengan konsentrasi tertentu.
3.3.2 Pembuatan Fase Gerak
Beberapa komposisi fase gerak dibuat dengan mengkombinasikan metanol dan akuabides dengan komposisi yang dapat dilihat pada tabel 3.1.
Tabel 3.1 Komposisi Fase Gerak
No. Metanol Akuabides
1 100 -
2 70 30
3 60 40
4 40 60
Fase gerak yang telah dibuat disaring menggunakan vakum dan filter 0,45 um. Gas yang terdapat di dalam larutan dihilangkan menggunakan sistem penghilang gas (degasser).
3.3.3 Penentapan Panjang Gelombang Analisis
3.3.4 Optimasi Kondisi Analisis
3.3.4.1 Pemilihan Komposisi Fase Gerak
Larutan induk N-(hidroksietil)-p-metoksi sinamamida diencerkan dengan metanol hingga diperoleh konsentrasi 10,08 µg/mL. Sebanyak 20,0 µL supernatan disuntikkan ke dalam KCKT menggunakan komposisi fase gerak dalam variasi perbandingan diatas dengan laju alir 1,0 mL/menit, dan dideteksi pada panjang gelombang terpilih. Kemudian dicatat waktu retensi (tR), luas puncak, dihitung jumlah lempeng teoritis (N), HETP (height equivalent theoritical plate), dan asimetrisitas. Hasil analisis yang diperoleh dari beberapa perbandingan fase gerak dibandingkan.
3.3.5 Uji Kesesuaian Sistem
Larutan yang mengandung N-(hidroksietil)-p-metoksi sinamamida pada konsentrasi 10,08 µg/mL disiapkan. Sebanyak 20,0 µL supernatan disuntikkan ke dalam KCKT pada kondisi analisis terpilih. Kemudian dihitung jumlah lempeng teoritis (N), HETP, asimetrisitas dan nilai RSD (standar deviasi relatif) pada lima kali penyuntikan.
3.3.6 Penetapan Metode Ekstraksi (Polson, 2002)
28
UIN Syarif Hidayatullah Jakarta
Dibuat larutan N-(hidroksietil)-p-metoksi sinamamida 10,08 µg/mL dalam plasma. Lalu diambil 0,5 mL campuran tersebut dan dilakukan ekstraksi sesuai dengan perbandingan metanol-plasma (1:1) dan (4:1), lalu sebanyak 20,0 µL supernatan diinjeksikan ke dalam KCKT. Kemudian dilakukan pengamatan kromatogram plasma mengandung N-(hidroksietil)-p-metoksi sinamamida dengan membandingkan luas area, resolusi, jumlah lempeng teoritis, dan asimetrisitas puncak N-(hidroksietil)-p-metoksi sinamamida dari masing-masing perbandingan tersebut.
3.3.7 Validasi Metode Analisis N-(hidroksietil)-p-metoksi sinamamida dalam Plasma secara In Vitro
3.3.7.1 Pengukuran Batas Kuantifikasi Terendah(LLOQ)
Larutan N-(hidroksietil)-p-metoksi sinamamida dalam plasma dengan konsentrasi 10,08 µg/mL; 15,12 µg/mL; 20,16 µg/mL; 30,24 µg/mL; dan 40,32 µg/mL disiapkan. Kemudian masing-masing larutan diekstraksi sesuai dengan prosedur. Sebanyak 20 µL supernatan disuntikkan ke dalam KCKT pada kondisi analisis terpilih. Dibuat kurva kalibrasi, ditentukan persamaan garis regresi linier dan koefisien korelasinya, kemudian dihitung nilai LOQ. Setelah diperoleh nilai LOQ, dibuat larutan N-(hidroksietil)-p-metoksi sinamamida dalam plasma dengan konsentrasi ½ atau ¼ dari nilai LOQ tersebut.
3.3.7.2 Pembuatan Kurva Kalibrasi dan Uji Linieritas dalam Plasma secara In Vitro
pada kondisi analisis terpilih. Setelah itu dianalisis regresi luas puncak terhadap konsentrasi N-(hidroksietil)-p-metoksi sinamamida dalam plasma dari masing-masing konsentrasi dan dibuat kurva kalibrasi dengan persamaan garis regresi linier (y = a + bx). Dihitung pula koefisien korelasi (r) dari kurva tersebut, kemudian dihitung lengukuran batas deteksi (LOD) dan limit batas kuantifikasi (LOQ).
Nilai LOD dan LOQ dihitung dengan menggunakan data kalibrasi. LOQ diperoleh dengan rumus :
LOQ =
Sedangkan LOD diperoleh dengan rumus :
LOD =
dimana adalah simpangan baku residual, b adalah
slope dari persamaan regresi.
3.3.7.3 Uji Selektivitas
Sebanyak 20,0 µL plasma hasil deproteinasi yang mengandung N-(hidroksietil)-p-metoksi sinamamida pada konsentrasi LLOQ (5,04 µg/mL) disuntikkan ke dalam KCKT dengan kondisi analisis terpilih. Diulang sebanyak enam kali menggunakan enam plasma dari sumber yang berbeda. Kemudian dihitung koefisien variasinya (KV)
dengan nilai ≤20% dan akurasi (%diff) dengan nilai ± 20%.
30
UIN Syarif Hidayatullah Jakarta
dalam KCKT pada kondisi analisis terpilih. Prosedur tersebut diulangi sebanyak tiga kali untuk masing-masing konsentrasi. Kemudian dihitung persentase akurasi (%diff), perolehan kembali (% recovery) dan nilai koefisien variasinya (KV) pada masing-masing konsentrasi larutan tersebut. Nilai rata-rata % diff yang disyaratkan adalah + 15%, dan nilai koefisien variasi (KV) yang disyaratkan tidak lebih dari 15%. Adapun % recovery dihitung dengan membandingkan nilai terukur dari konsentrasi N-(hidroksietil)-p-metoksi sinamamida dalam plasma dengan nilai yang sebenarnya dikalikan 100%. Nilai % recovery
BAB IV
HASIL DAN PEMBAHASAN
4.1 Penetapan Panjang Gelombang Analisis
Pada penelitian ini, penetapan panjang gelombang analisis dilakukan dengan menggunakan alat spektrofotometer UV-Vis. Sebanyak 5,04 µg/mL larutan N-(hidroksietil)-p-metoksi sinamamida diukur pada panjang gelombang 200 nm hingga 400 nm. Diperoleh spektrum serapan maksimumnya. Spektrum serapan yang dihasilkan memperlihatkan bahwa N-(hidroksietil)-p-metoksi sinamamida berada pada panjang gelombang sinar UV, yaitu 290 nm. Hal ini disebabkan karena N-(hidroksietil)-p-metoksi sinamamida mempunyai gugus kromofor yang terdeteksi pada daerah UV. Spektrum N-(hidroksietil)-p-metoksi sinamamida pada spektrofotometer UV-Vis dapat dilihat pada lampiran 3 gambar 5.1. Pemilihan panjang gelombang analisis ini berguna untuk meningkatkan selektivitas dan sensitivitas analisis sampel yang digunakan. Panjang gelombang ini kemudian yang digunakan pada instrumen Kromatografi Cair Kinerja Tinggi (KCKT) untuk mendeteksi sampel pada analisis N-(hidroksietil)-p-metoksi sinamamida dalam plasma secara in vitro.
4.2 Optimasi Kondisi Analisis
4.2.1 Pemilihan Komposisi Fase Gerak
32
UIN Syarif Hidayatullah Jakarta
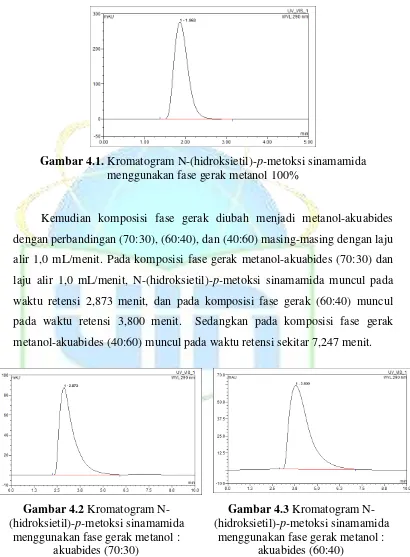
N-(hidroksietil)-p-metoksi sinamamida diujikan pada beberapa komposisi fase gerak. Komposisi fase gerak yang pertama kali diujikan adalah metanol 100% dengan laju alir 1,0 mL/menit. Pada komposisi fase gerak ini, diperoleh kromatogram tunggal dengan waktu retensi sekitar 1,863 menit.
Gambar 4.1. Kromatogram N-(hidroksietil)-p-metoksi sinamamida menggunakan fase gerak metanol 100%
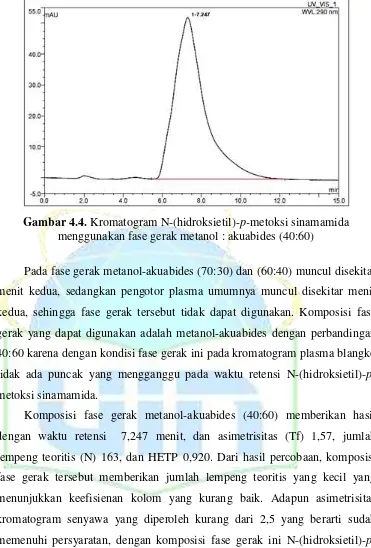
Kemudian komposisi fase gerak diubah menjadi metanol-akuabides dengan perbandingan (70:30), (60:40), dan (40:60) masing-masing dengan laju alir 1,0 mL/menit. Pada komposisi fase gerak metanol-akuabides (70:30) dan laju alir 1,0 mL/menit, N-(hidroksietil)-p-metoksi sinamamida muncul pada waktu retensi 2,873 menit, dan pada komposisi fase gerak (60:40) muncul pada waktu retensi 3,800 menit. Sedangkan pada komposisi fase gerak metanol-akuabides (40:60) muncul pada waktu retensi sekitar 7,247 menit.
Gambar 4.2 Kromatogram N-(hidroksietil)-p-metoksi sinamamida
menggunakan fase gerak metanol : akuabides (70:30)
Gambar 4.3 Kromatogram N-(hidroksietil)-p-metoksi sinamamida
Gambar 4.4. Kromatogram N-(hidroksietil)-p-metoksi sinamamida menggunakan fase gerak metanol : akuabides (40:60)
Pada fase gerak metanol-akuabides (70:30) dan (60:40) muncul disekitar menit kedua, sedangkan pengotor plasma umumnya muncul disekitar menit kedua, sehingga fase gerak tersebut tidak dapat digunakan. Komposisi fase gerak yang dapat digunakan adalah metanol-akuabides dengan perbandingan 40:60 karena dengan kondisi fase gerak ini pada kromatogram plasma blangko tidak ada puncak yang mengganggu pada waktu retensi N-(hidroksietil)-
p-metoksi sinamamida.
Komposisi fase gerak metanol-akuabides (40:60) memberikan hasil dengan waktu retensi 7,247 menit, dan asimetrisitas (Tf) 1,57, jumlah lempeng teoritis (N) 163, dan HETP 0,920. Dari hasil percobaan, komposisi fase gerak tersebut memberikan jumlah lempeng teoritis yang kecil yang menunjukkan keefisienan kolom yang kurang baik. Adapun asimetrisitas kromatogram senyawa yang diperoleh kurang dari 2,5 yang berarti sudah memenuhi persyaratan, dengan komposisi fase gerak ini N-(hidroksietil)-
34
UIN Syarif Hidayatullah Jakarta
Tabel. 4.1 Data hubungan antara waktu retensi, jumlah lempeng teoritis dan asimetrisitas terhadap perubahan komposisi fase gerak
Fase Gerak (v/v) Metanol-akuabides (70:30) 2,873 77 1,948 2,38 Metanol-akuabides (60:40) 3,800 63 2,381 2,22 Metanol-akuabides (40:60) 7,247 163 0,920 1,57
Dari hasil optimasi ini, maka diperoleh suatu kondisi analisis N-(hidroksietil)-p-metoksi sinamamida di dalam plasma dengan ketentuan sebagai berikut:
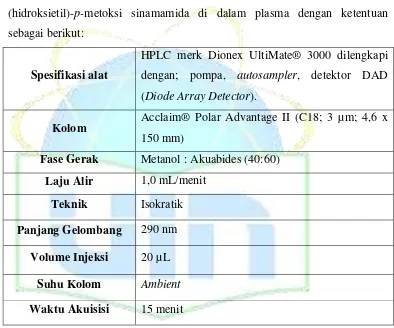
Spesifikasi alat
HPLC merk Dionex UltiMate® 3000 dilengkapi dengan; pompa, autosampler, detektor DAD (Diode Array Detector).
Kolom Acclaim® Polar Advantage II (C18; 3 µm; 4,6 x 150 mm)
Fase Gerak Metanol : Akuabides (40:60) Laju Alir 1,0 mL/menit
Teknik Isokratik Panjang Gelombang 290 nm
Volume Injeksi 20 µL Suhu Kolom Ambient
Waktu Akuisisi 15 menit
4.3 Uji Kesesuaian Sistem
Parameter yang berguna untuk uji kesesuaian sistem adalah keberulangan penyuntikan larutan baku yang dinyatakan dalam standar deviasi relatif (RSD) yang dinyatakan dalam persen bila tidak dinyatakan lain dalam monografi baku yang digunakan dengan nilai RSD kurang dari 2% (Farmakope Indonesia edisi IV).
Menurut USP, ada beberapa parameter yang dijadikan rujukan untuk menunjukkan bahwa metode telah sesuai dengan sistem yang tersedia. Parameter-parameter tersebut meliputi: jumlah lempeng teoritis (N), asimetrisitas, faktor kapasitas dan nilai standar deviasi relatif (RSD) tinggi puncak dan luas area dari serangkaian injeksi. Suatu metode dinyatakan memenuhi syarat uji kesesuaian sistem bila minimal ada 2 parameter yang memenuhi persyaratan dari beberapa parameter yang diujikan.
Dari uji kesesuaian sistem, diperoleh rata-rata waktu retensi N-(hidroksietil)-
p-metoksi sinamamida muncul pada menit 7,258 dengan rata-rata nilai area N-(hidroksietil)-p-metoksi sinamamida pada 5 kali penyuntikan adalah 65,062 mAu dengan % RSD luas area sebesar 0,042%. Data uji kesesuaian sistem dapat dilihat pada tabel 4.2 dan data selengkapnya tercantum dalam lampiran 5 tabel 5.1.
Tabel 4.2 Hasil uji rata-rata kesesuaian sistem analisis N-(hidroksietil)-p-metoksi sinamamida menggunakan fase gerak metanol : akuabides (40:60 (v/v))
Nilai jumlah lempeng teoritis dan asimetrisitas menunjukkan kinerja kolom dalam memisahkan komponen dengan menggunakan metode tersebut. Semakin besar nilai lempeng teoritis berarti semakin efisien suatu kolom dalam memisahkan komponen menggunakan metode tersebut. Asimetrisitas menunjukkan bentuk puncak N-(hidroksietil)-p-metoksi sinamamida yang simetris atau tidak memiliki pengekoran. Dari uji kesesuaian sistem ini, fase gerak yang ditetapkan telah memberikan hasil parameter yang telah memenuhi persyaratan uji kesesuaian sistem, kecuali untuk parameter lempeng teoritis yang kurang dari kondisi ideal.
Parameter Syarat Hasil yang diperoleh Kesimpulan RSD waktu retensi <2% 0,493 % ✓
36
UIN Syarif Hidayatullah Jakarta
4.4 Penetapan Metode Ekstraksi
Sebelum dianalisis menggunakan KCKT, N-(hidroksietil)-p-metoksi sinamamida dalam plasma perlu diekstraksi terlebih dahulu, terutama protein yang ada dalam plasma. Ekstraksi N-(hidroksietil)-p-metoksi sinamamida dalam plasma dilakukan dengan menggunakan pelarut organik, yaitu metanol. Penyiapan sampel dengan menggunakan metanol sebagai pengendap protein ini bertujuan untuk memisahkan analit dari gangguan yang ada dalam plasma seperti protein dan senyawa endogen lainnya. Penambahan larutan organik seperti metanol pada larutan protein dalam air akan menurunkan konstanta dielektrik air yang meningkatkan tarikan antara molekul-molekul bermuatan dan memfasilitasi interaksi elektrostatik protein. Selain itu pelarut organik juga akan menggantikan beberapa molekul air disekitar daerah hidrofob dari permukaan protein yang berasosiasi dengan protein sehingga menurunkan konsentrasi air dalam larutan dengan demikian kelarutan protein akan menurun dan memungkinkan terjadinya pengendapan.
Pada penelitian ini, ekstraksi dilakukan dengan menambahkan sejumlah metanol ke dalam plasma. Komposisi yang diujikan adalah metanol-plasma dengan perbandingan 1 : 1 dan 4 : 1, kemudian dilakukan perngamatan kromatogram plasma blangko dengan melihat apakah pada daerah waktu retensi N-(hidroksietil)-p-metoksi sinamamida terdapat pengotor plasma atau tidak. Hasil yang diperoleh dari kedua komposisi metanol yang diujikan untuk mengendapkan protein adalah tidak satupun komposisi pelarut yang menghasilkan puncak pengotor pada waktu retensi N-(hidroksietil)-p-metoksi sinamamida saat analisis dilakukan. Gambar dapat dilihat pada Gambar 4.5 dan 4.7.
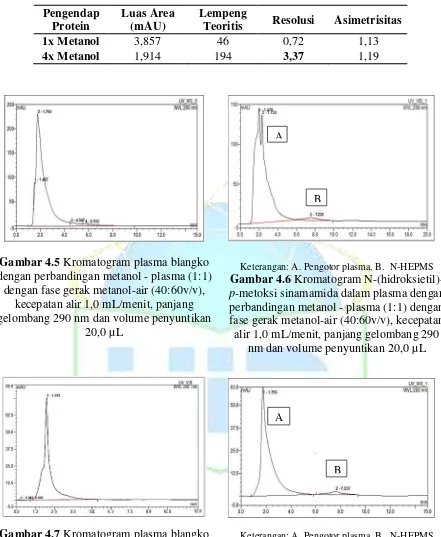
Tabel 4.3 Hasil optimasi pengendapan protein
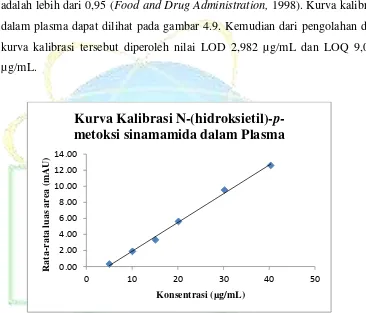
Gambar 4.5 Kromatogram plasma blangko dengan perbandingan metanol - plasma (1:1)
dengan fase gerak metanol-air (40:60v/v), kecepatan alir 1,0 mL/menit, panjang gelombang 290 nm dan volume penyuntikan
20,0 µL
Gambar 4.7 Kromatogram plasma blangko dengan perbandingan metanol - plasma (4:1)
dengan fase gerak metanol-air (40:60v/v), kecepatan alir 1,0 mL/menit, panjang gelombang 290 nm dan volume penyuntikan
20,0 µL
Keterangan: A. Pengotor plasma, B. N-HEPMS
Gambar 4.6 Kromatogram
N-(hidroksietil)-p-metoksi sinamamida dalam plasma dengan perbandingan metanol - plasma (1:1) dengan fase gerak metanol-air (40:60v/v), kecepatan alir 1,0 mL/menit, panjang gelombang 290
nm dan volume penyuntikan 20,0 µL
Keterangan: A. Pengotor plasma, B. N-HEPMS
Gambar 4.8 Kromatogram
N-(hidroksietil)-p-metoksi sinamamida dalam plasma dengan perbandingan metanol - plasma (4:1)
dengan fase gerak metanol-air (40:60v/v), kecepatan alir 1,0 mL/menit, panjang gelombang 290 nm dan volume penyuntikan
20,0 µL
A
B A
38
UIN Syarif Hidayatullah Jakarta
Dari data diatas, dengan membandingkan kedua komposisi metanol yang digunakan untuk mengendapkan protein plasma dapat dilihat bahwa pada penambahan metanol 4 kali volume plasma memberikan pemisahan yang paling baik dengan pengotor dalam plasma, yaitu dengan nilai resolusi 3,37 dimana telah
memenuhi persyaratan resolusi ≥1,5, serta menghasilkan puncak senyawa dengan kriteria puncak yang paling baik. Hal ini dapat dilihat dari nilai resolusi yang lebih besar, nilai lempeng teoritis yang lebih besar, serta asimetrisitas yang kecil bila dibandingkan dengan penambahan metanol 1 kali volume plasma.
Nilai resolusi yang besar menyatakan metode ekstraksi menggunakan metanol 4 kali volume plasma dapat memisahkan puncak pengotor plasma yang muncul pada waktu retensi sekitar 1,753 dengan puncak N-(hidroksietil)-p-metoksi sinamamida yang muncul pada waktu retensi sekitar 7,237 dengan pemisahan yang paling baik.
4.5 Validasi Metode Analisis N-(hidroksietil)-p-metoksi sinamamida dalam Plasma secara In Vitro
4.5.1 Pengukuran Batas Kuantifikasi Terendah(LLOQ)
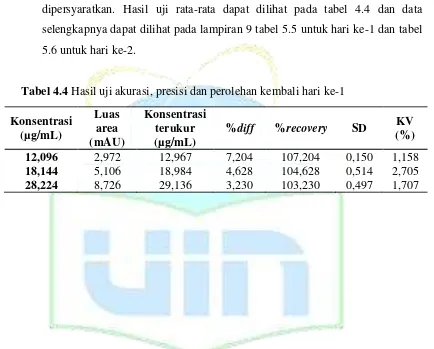
Pengukuran sebanyak 5 konsentrasi larutan N-(hidroksietil)-p-metoksi sinamamida dalam plasma dengan konsentrasi 10,08 µg/mL; 15,12 µg/mL; 20,16 µg/mL; 30,24 µg/mL; dan 40,32 µg/mL menghasilkan persamaan regresi y = 0,361x - 1,7941 dengan linieritas koefisien korelasi 0,9953. Dari hasil pengolahan data diperoleh LOQ 9,675 µg/mL.
Kemudian LLOQ dibuat dengan cara mengencerkan ½ konsentrasi LOQ. LLOQ merupakan standar terendah pada kurva kalibrasi yang dapat diterima (Food and Drug Administration, 2001). Pada penelitian ini, konsentrasi LLOQ yang dibuat adalah 5,04 µg/mL.
4.5.2 Pembuatan Kurva Kalibrasi dan Uji Linieritas dalam Plasma secara In Vitro
40,32 µg/mL. Persamaan garis kurva kalibrasi yang diperoleh adalah y = 0,3559x - 1,6435 dengan koefisien korelasi 0,9962; dimana x adalah konsentrasi senyawa dan y adalah luas area senyawa. Koefisien korelasi ini mendekati persyaratan nilai koefisien korelasi yang ideal sehingga dapat dapat disimpulkan bahwa metode analisis N-(hidroksietil)-p-metoksi sinamamida dalam plasma dengan konsentrasi 5,04 – 40,32 µg/mL memenuhi kriteria uji linieritas dan dapat diterima untuk suatu metode analisis yang valid. Pada analisis senyawa dalam plasma, nilai koefisien korelasi yang dapat diterima adalah lebih dari 0,95 (Food and Drug Administration, 1998). Kurva kalibrasi dalam plasma dapat dilihat pada gambar 4.9. Kemudian dari pengolahan data kurva kalibrasi tersebut diperoleh nilai LOD 2,982 µg/mL dan LOQ 9,037 µg/mL.
Gambar 4.9 Kurva Kalibrasi N-(hidroksietil)-p-metoksi sinamamida dalam plasma
4.5.3 Uji Selektivitas
Selektivitas adalah kemampuan metode analisis untuk membedakan dan mengukur secara kuantitatif analit dengan adanya komponen lain di dalam sampel (Food and Drug Administration, 2001). Pada percobaan ini komponen lain tersebut adalah pengotor plasma. Uji selektivitas ini dilakukan terhadap enam plasma manusia dari sumber yang berbeda pada konsentrasi LLOQ yaitu 5,04 µg/mL, diperoleh nilai koefisien variasi 2,123% dan % diff antara 8,795%
40
UIN Syarif Hidayatullah Jakarta
sampai 15,045% serta tidak terdapat puncak pengotor plasma yang muncul pada kromatogram. Data hasil uji selektivitas dapat dilihat pada lampiran 8 tabel 5.4.
4.5.4 Uji Akurasi, Presisi dan Perolehan Kembali (% recovery)
Uji akurasi bertujuan memperoleh data kedekatan nilai rata-rata hasil uji yang diperoleh dengan nilai konsentrasi analit sebenarnya. Untuk analisis dalam matriks biologis, selisih hasil analisis dengan nilai konsentrasi yang sebenarnya adalah + 15%, kecuali jika pengukuran dilakukan pada kadar LLOQ maka tidak boleh melebihi 20% dan nilai dari uji perolehan kembali berada dalam rentang 80-120% (Food and Drug Administration, 2001). Perolehan kembali analit tidak harus 100% namun tingkat perolehan kembali analit harus konsisten, presisi, dan reprodusibel (Food and Drug Administration, 2001).
Uji presisi bertujuan memperoleh data kedekatan hasil uji yang satu dengan yang lainnya. Pengukuran presisi pada setiap level konsentrasi, harus memiliki nilai koefesien variasi (KV) tidak lebih dari 15%, kecuali LLOQ dimana nilai KV tidak boleh lebih dari 20% (Food and Drug Administration, 2001). Pada penelitian ini, uji akurasi dan presisi dilakukan selama 2 hari.
Adapun pada uji perolehan kembali hari ke-1, diperoleh rata-rata
%recovery untuk konsentrasi rendah sebesar 107,204%, konsentrasi sedang 104,628% dan konsentrasi tinggi 103,320%. Adapun pada hari ke-2, diperoleh rata-rata %recovery untuk konsentrasi rendah sebesar 105,295%, konsentrasi sedang 106,597% dan konsentrasi tinggi 107,974%. Dari hasil percobaan ini, uji akurasi, presisi, dan perolehan kembali analisis N-(hidroksietil)-p-metoksi sinamamida dalam plasma yang dilakukan sudah memenuhi kriteria yang dipersyaratkan. Hasil uji rata-rata dapat dilihat pada tabel 4.4 dan data selengkapnya dapat dilihat pada lampiran 9 tabel 5.5 untuk hari ke-1 dan tabel 5.6 untuk hari ke-2.
Tabel 4.4 Hasil uji akurasi, presisi dan perolehan kembali hari ke-1
Konsentrasi (µg/mL)
Luas area (mAU)
Konsentrasi terukur (µg/mL)
%diff %recovery SD KV
(%)
42 UIN Syarif Hidayatullah Jakarta
BAB V
KESIMPULAN DAN SARAN
5.1 Kesimpulan
1. Kondisi optimum untuk analisis N-(hidroksietil)-p-metoksi sinamamida menggunakan KCKT dapat dilakukan dengan detektor DAD, kolom Acclaim® Polar Advantage II C18 dengan panjang kolom 4,6 x 150 mm ukuran partikel 3 µm; fase gerak metanol akuabides (40:60); laju alir 1,0 mL/menit; volume injeksi 20 µL; waktu akuisisi 15 menit dan dideteksi pada panjang gelombang 290 nm.
2. N-(hidroksietil)-p-metoksi sinamamida dalam plasma diekstraksi dengan cara pengendapan protein dengan mencampurkan metanol pada perbandingan metanol-plasma 4:1, lalu di vortex selama 20 detik kemudian disentrifugasi pada kecepatan 3000 rpm selama 10 menit.
3. Dari hasil validasi diperoleh nilai LLOQ sebesar 5,04 µg/mL, pada rentang konsentrasi 5,04 – 40,32 µg/mL dihasilkan kurva kalibrasi N-(hidroksietil)-p-metoksi sinamamida yang linier dengan koefisien korelasi (r) 0,9962, akurasi (%diff) dari metode ini antara -2,961% sampai 11,954% dengan presisi (KV) antara 0,439% sampai 4,306%. Hasil validasi metode tersebut menunjukkan bahwa metode analisis telah memenuhi kriteria linieritas, presisi, dan akurasi sesuai ketentuan yang berlaku sehingga dapat digunakan untuk analisis N-(hidroksietil)-p-metoksi sinamamida dalam plasma.
5.2 Saran
1. Disarankan untuk dapat dilakukan pengembangan metode validasi analisis N-(hidroksietil)-p-metoksi sinamamida dalam plasma secara
in vitro yang lebih lengkap.
2. Dapat dilakukan analisis -(hidroksietil)-p-metoksi sinamamida secara
DAFTAR PUSTAKA
Anonim. (1995). Farmakope Indonesia edisi IV. Jakarta: Departemen Kesehatan RI.
Barus, Rosbina. (2009). Amidasi Etil p-Metoksi Sinamat yang Diisolasi dari Kencur (Kaempferia galanga, L) melalui Amidasi dengan Dietanolamin.
Skripsi. Medan: Universitas Sumatera Utara.
Effendy. (2004). Kromatografi Cair Kinerja Tinggi dalam Bidang Farmasi. Sumatera Utara: FMIPA USU.
Evans, G. (2004). A Handbook of Bioanalysis and Drug Metabolism. USA: CRC Press.
Food and Drug Administration. (2001). Bioanalytical Method Validation. Rockville: Center for Veterinary Medicine.
Food and Drug Administration. 1988. Guidance for Industry: Bioanalytical Methods Validation for Human Studies. Rockville: Center for Drug Evaluation and Research (CDER).
Gandjar, I. G. dan Abdul Rohman. (2007). Kimia Farmasi Analisis. Yogyakarta: Pustaka Pelajar.
Ganong, W.F. (2001). Fisiologi kedokteran Edisi 20. Terj. Djauhari W, Dewi I, dan Minarma S. Jakarta: ECG.
Harahap, Y. (2010). Peran Bioanalisis dalam Penjaminan Kualitas Obat dan Peningkatan Kualitas Hidup Pasien. Depok: UI Press.