UIN SYARIF HIDAYATULLAH JAKARTA
Validasi Metode Analisis Etil p-metoksisinamat dalam
Plasma secara In Vitro menggunakan Kromatografi Cair
Kinerja Tinggi (KCKT)
SKRIPSI
ANI KURNIAWATI
1112102000042
PROGRAM STUDI FARMASI
FAKULTAS KEDOKTERAN DAN ILMU KESEHATAN
UIN SYARIF HIDAYATULLAH JAKARTA
UIN SYARIF HIDAYATULLAH JAKARTA
Validasi Metode Analisis Etil p-metoksisinamat dalam
Plasma secara In Vitro menggunakan Kromatografi Cair
Kinerja Tinggi (KCKT)
SKRIPSI
Diajukan sebagai salah satu syarat memperoleh gelar Sarjana Farmasi
ANI KURNIAWATI
1112102000042
PROGRAM STUDI FARMASI
FAKULTAS KEDOKTERAN DAN ILMU KESEHATAN
UIN SYARIF HIDAYATULLAH JAKARTA
v ABSTRAK
Nama : Ani Kurniawati Program Studi : Farmasi
Judul Skripsi : Validasi Metode Analisis Etil p-metoksisinamat dalam Plasma secara In Vitro Menggunakan Kromatografi Cair Kinerja Tinggi (KCKT).
Etil p-metoksisinamat (EPMS) merupakan salah satu metabolit sekunder yang terdapat pada tanaman kencur (Kaempferia galanga. L) dari family Zingiberaceae. Senyawa ini memiliki aktivitas farmakologinya sebagai antiinflamasi dan analgesik. Tujuan penelitian ini adalah memperoleh optimasi kondisi analisis dan metode yang tervalidasi untuk analisis EPMS dalam plasma secara in vitro
menggunakan KCKT dengan Diode Array Detector (DAD). EPMS diekstraksi dari plasma dengan metode pengendapan protein menggunakan metanol. Metanol dimasukkan ke dalam plasma dengan perbandingan 4:1 kemudian divortex selama 20 detik, dan disentrifugasi pada kecepatan 3000 rpm selama 10 menit. Analisis dilakukan dengan isokratik pada kolom C18 fase terbalik Acclaim® Polar
Advantage II (4,6 x 150 mm, 3µm) dan fase gerak metanol-aquabidestilata (60:40v/v) pada laju alir 1,0 mL/menit. Deteksi dilakukan pada panjang gelombang 308 nm dan volume penyuntikan 20 µL. Pada rentang konsentrasi 2,02-40,4 µg/mL dihasilkan kurva kalibrasi yang linier dengan koefesien korelasi (r) sebesar 0,9989. Nilai %diff akurasi metode ini berada diantara -12,4507% dan 10,5212% dengan nilai presisi (KV) antara 0,1417% dan 7,5913% serta uji perolehan kembali antara 87,5493% dan 110,5212%.
vi ABSTRACT
Name : Ani Kurniawati Programme Study : Farmasi
Title : Analytical Method Validation of Ethyl P-methoxycinnamate Plasma In Vitro using High Performance Liquid Chromatography
Ethyl p-metoxycinnamate (EPMC) is one of secondery metabolite which is found in kencur (Kampferia galangal Linn) family Zingiberaceae. These compounds have biological activity as anti-inflammatory and analgetic. The aim of this study was to obtin the optimum conditions and validated methods for analysis of EPMC in plasma in vitro. The method of analysis using High Performance Liquid Chromatography (HPLC) with Diode Array Detector. EPMC was extracted from plasma by protein deproteination method using methanol. A mixture of plasma and methanol (1:4 (v/v)) is shaked with vortex for 20 seconds and centrifuged on 3000 rpm for 10 minutes. The analysis was done by using isocratic technique on reverse phase C18 column Acclaim® Polar Advantage II (4,6 x 150 mm, 3µm) and
mobile phase consisted methanol-aquabidest (60:40v/v) at flow rate of 1,0 mL/min. Samples were detected at a wavelength of 308 nm and injection volume is 20 µL. Linierity was established for range concentration of 2,02-40,4 µg/mL with correlation (r) of 0,9989. Accuracy (%diff) of this method is between -12,4507% to 10,5212% with precision between 0,1417% to 7,5913% and test relative recovery between 87,5493% to 110,5212%.
Keywords: Ethyl p-metoxycinnamate, High Performance Liquid Chromatography. protein deproteination, method validation.
vii
KATA PENGANTAR
Alhamdulillah, puji dan syukur senantiasa penulis panjatkan kehadirat Allah SWT, karena berkat rahmat dan hidayah-Nya yang telah dilimpahkan serta segala anugerah-Nya yang telah diberikan sehingga penulis dapat menyelesaikan skripsi ini. Salawat serta salam senantiasa penulis curahkan kepada Nabi Muhammdad SAW, keluarga, sahabat, dan para pengikutnya. Skripsi ini berjudul Validasi Metode Analisis Etil p-metoksisinamat dalam plasma secara In Vitro
menggunakan Kromatografi Cair Kinerja Tinggi yang telah diajukan sebagai persyaratan untuk memperoleh gelar Sarjana Farmasi Program Studi Farmasi FKIK UIN Syarif Hidayatullah Jakarta.
Dalam penyelesaian skripsi ini, penulis mendapatkan bantuan dari berbagai pihak berupa ilmu pengetahuan, pengarahan, semangat, bantuan materi, dan motivasi. Oleh karena itu, dengan tulus penulis ingin mengucapkan banyak terimakasih kepada nama-nama yang telah terlulis dibawah ini, semoga Allah senantiasa melimpahkan rahmat dan karunia-Nya kepada:
1. Bapak Supandi, M.Si., Apt selaku Pembimbing I dan Ibu Zilhadia, M.Si., Apt selaku Pembimbing II yang telah meluangkan waktunya untuk memberikan bimbingan, saran, bantuan dan dukungan kepada penulis serta kesabaran dan kepercayaanya selama penelitian dan penyusunan skripsi ini.
2. Dr. H. Arif Sumantri, SKM, M.Kes, selaku Dekan Fakultas Kedokteran dan Ilmu Kesehatan Universitas Islam Negeri Syarif Hidayatullah Jakarta yang telah memberikan banyak motivasi dan bantuan.
3. Ibu Dr.Nurmeilis, M.si.,Apt selaku Ketua Program Studi Farmasi Falkutas Kedokteran dan Ilmu Kesehatan UIN Syarif Hidayatullah Jakarta.
x
DAFTAR ISI
Halaman
HALAMAN JUDUL ... i
HALAMAN PERSYARATAN ORISINILITAS ... ii
HALAMAN PERSETUJUAN PEMBIMBING ... iii
HALAMAN PENGESAHAN ... iv
ABSTRAK ... v
ABSTRACT ... vi
KATA PENGANTAR ... vii
HALAMAN PERSETUJUAN PUBLIKASI KARYA ILMIAH ... ix
DAFTAR ISI ... x
DAFTAR TABEL……….. xiii
DAFTAR GAMBAR……….. xiv
DAFTAR LAMPIRAN……….. xvi
BAB I PENDAHULUAN ... 1
1.1LatarBelakang ... 1
1.2PerumusanMasalah ... 2
1.3TujuanPenelitian ... 3
1.4Manfaat Penelitian ... 3
BAB II TINJAUAN PUSTAKA ... 4
2. 1. Etil p-metoksisinamat... 4
2. 2. Plasma ... 5
2. 3.Analisis Obat dalam Plasma... 6
2. 4.Kromatografi Cair Kinerja Tinggi ... 8
2.4.1 Pendahuluan……….8
2.4.2 Keuntungan KCKT………..9
2.4.3 Cara Kerja ... 9
2.4.4 Instrumentasi KCKT ... 10
2.4.4.1Wadah Fase Gerak pada KCKT ... 11
2.4.4.2 Fase Gerak KCKT ... 11
xi
2.4.4.4 Penyuntikan (injector) sampel pada KCKT ... 12
2.4.4.5 Kolom pada KCKT ... 12
2.4.4.6 Fase Diam KCKT ... 13
2.4.4.7 Detektor KCKT ... 14
2.4.4.8 Intergrator ... 16
2.4.5 Penggunaan KCKT dalam Analisis Farmasi ... 16
2.4.6 Analasis Kuantitatif ... 16
2.4.6.1 Metode Baku Eksternal ... 16
2.4.6.2 Metode Baku Internal ... 17
2.4.6.3 Metode Presentase Tinggi/Lebar Puncak atau Metode Normalisasi Internal ... 17
2. 5.Validasi Metode Analisis ... 18
2.5.1 Ketepatan (Akurasi) ... 19
2.5.2 Presisi ... 20
2.5.3 Perolehan Kembali (%Recovery) ... 20
2.5.4 Kurva Kalibrasi ... 21
2.5.4.1 Linieritas ... 21
2.5.4.2 Batas Deteksi (limit of detection, LOD) ... 21
2.5.4.3 Batas Kuantifikasi (limit of quantification, LOQ) ... 22
2.5.5 Selektivitas ... 22
2.5.6 Uji Kesesuaian Sistem... 23
2.5.7 Stabilitas ... 23
2.5.7.1 Stabilitas Beku dan Cair ... 23
2.5.7.2 Stabilitas Suhu Jangka Pendek ... 24
2.5.7.3 Stabilitas Jangka Panjang ... 24
2.5.7.4 Stabilitas Larutan Stok ... 24
2.5.7.5 Stabilitas Post-Preparatif ... 25
BAB III METODE PENELITIAN ... 26
3.1Tempat dan Waktu Penelitian ... 26
3.2Bahan dan Alat ... 26
3.2.1 Bahan ... 26
xii
3.3 Prosedur Kerja ... 26
3.3.1 Pembuatan Larutan Induk EPMS ... 26
3.3.2 Pembuatan Fase Gerak ... 27
3.3.3 Tahapan Optimasi ... 27
3.3.3.1 Penentuan Panjang Gelombang Maksimum ... 27
3.3.3.2 Pemilihan Komposisi Fase Gerak ... 27
3.3.3.3 Uji Kesesuaian Sistem ... 28
3.3.3.4 Penetapan Metode Ekstraksi ... 28
3.3.4 Validasi Metode Analisis EPMS dalam Plasma ... 28
3.3.4.1 Pengukuran LLOQ ... 28
3.3.4.2 Pembuatan Kurva Kalibrasi ... 29
3.3.4.3 Selektivitas ... 29
3.3.4.4 Uji Akurasi ... 30
3.3.4.5 Uji Presisi ... 30
BAB IV HASIL DAN PEMBAHASAN ... 31
4.1 Penetapan Panjang Gelombang Analisis ... 31
4.2 Optimasi Kondisi Analisis ... 32
4.2.1 Pemilihan Komposisi Fase Gerak ... 32
4.2.2 Uji Kesesuaian Sistem... 34
4.2.3 Penetapan Metode Ekstraksi ... 34
4.3 Validasi Metode Analisis EPMS dalam Plasma In Vitro ... 36
4.3.1 Pengukuran Limit Kuantitasi Terendah (LLOQ) ... 36
4.3.2 Pembuatan Kurva Kalibrasi ... 37
4.3.3 Uji Selektivitas ... 38
4.3.4 Uji Akurasi ... 39
4.3.5 Uji Presisi ... 40
BAB V KESIMPULAN DAN SARAN ... 41
5.1 Kesimpulan ... 41
5.2 Saran ... 42
DAFTAR PUSTAKA ... 43
xiii
DAFTAR TABEL
Tabel Halaman
Tabel 3.1 Komposisi Fase Gerak ...27
Tabel 4.1 Hasil Penetapan Komposisi Fase Gerak ...33
Tabel 4.2 Hasil Uji Rata-rata Kesesuain Sistem EPMS ...34
Tabel 4.3 Hasil Optimasi Pengendapan Protein Plasma ...35
Tabel 4.4 Data Hasil Uji Selektivitas ...39
Tabel 5.1 Uji Kesesuaian Sistem ...48
Tabel 5.2 Data Hasil Pengukuran Limit Kuantitasi Terendah (LLOQ) ...49
Tabel 5.3 Data Hasil Uji Linieritas ...50
Tabel 5.4 Data Hasil Perhitungan Batas Deteksi (LOD) dan Batas Kuantitasi (LOQ) ...52
Tabel 5.5 Data Hasil Uji Akurasi dan Presisi Hari ke-1 ...53
Tabel 5.6 Data Hasil Uji Akurasi dan Presisi Hari ke-2 ...53
xiv
DAFTAR GAMBAR
Gambar Halaman
Gambar 2.1 Struktur Kimia EPMS ...4
Gambar 2.2 Diagram Alir KCKT ...10
Gambar 4.1 Spektrum Serapan Gelombang Maksimum EPMS ...31
Gambar 4.2 Kromatogram EPMS Murni pada Fase Gerak Metanol 100% ...33
Gambar 4.3 Kromatogram EPMS Murni pada Fase Gerak Metanol-Air (80:20 v/v) ...33
Gambar 4.4 Kromatogram EPMS Murni pada Fase Gerak Metanol-Air (70:30 v/v) ...33
Gambar 4.5 Kromatogram EPMS Murni pada Fase Gerak Metanol-Air (60:40 v/v) ...33
Gambar 4.6 Kromatogram Blangko Plasma dengan Perbandingan Metanol- Plasma (1:1) ...36
Gambar 4.7 Kromatogram EPMS pada Perbandingan Metanol dalam Plasma (1:1) ...36
Gambar 4.8 Kromatogram Blangko Plasma dengan Perbandingan Metanol- Plasma (4:1) ...36
Gambar 4.9 Kromatogram EPMS pada Perbandingan Metnaol dalam Plasma (4:1) ...36
Gambar 4.10 Kurva Kalibrasi EPMS dalam Plasma ...37
Gambar 5.1 Kromatogram Sampel Plasma Kosong (Blangko) ...47
Gambar 5.2 Kromatogram EPMS dalam Sampel Plasma ...47
Gambar 5.3 Alat Kromatografi Cair Kinerja Tinggi ...55
Gambar 5.4 Overlay dari Beberapa Kromatogran pada Kurva Kalibrasi ...56
Gambar 5.5 Plasma ...57
Gambar 5.6 EPMS dalam Plasma Setelah Divortex ...57
Gambar 5.7 Vial KCKT ...57
Gambar 5.8 Pot Penyimpanan Sampel EPMS ...57
xv
Gambar 5.10 Kolom KCKT ...57
Gambar 5.11 Sentrifugator ...58
Gambar 5.12 Sonikator ...58
xvi
DAFTAR LAMPIRAN
Lampiran Halaman
Lampiran 1 Alur Penelitian ...46
Lampiran 2 Kromatogram Hasil Analisa ...47
Lampiran 2 Uji Kesesuaian Sistem ...48
Lampiran 3 Pengukuran Limit Kuantitasi Terendah (LLOQ) ...49
Lampiran 4 Uji Linearitas dan Pembuatan Kurva Kalibrasi ...50
Lampiran 5 Uji Batas Deteksi dan Batas Kuantitasi ...52
Lampiran 6 Uji Akurasi dan Presisi ...53
Lampiran 7 Rumus-rumus ...54
Lampiran 8 Alat Kromatografi Cair Kinerja Tinggi ...55
Lampiran 9 Overlay Kromatogram Kurva Kalibrasi ...56
BAB I PENDAHULUAN
1.1 Latar Belakang
Etil p-metoksisinamat merupakan senyawa hasil isolat dari kencur (Kaempferia galanga. L) yang termasuk kedalam spesies tumbuhan dari suku zingiberaceae, bagian yang sering digunakan ialah rimpangnya. Senyawa Etil p-metoksisinamat (80,05%) merupakan kandungan utama yang terdapat didalam ekstrak kencur (Umar et al., 2012).
Etil p- metoksisinamat telah dilaporkan memiliki aktivitas sebagai anti-tuberkulosis, nematisidal, penolak nyamuk, larvasidal, antineoplastik dan potensi sebagai antimikroba (Umar et al., 2014). Selain itu, Etil p-metoksisinamat telah terbukti berperan penting dalam efek farmakologinya sebagai antiinflamasi. Dalam studi in vitro, etil p-metoksisinamat secara non-selektif mampu menghambat aktivitas COX-1 dan COX-2, dengan masing-masing 1,12 µM dan 0,83 µM. Hasil ini memvalidasi aktivitas antiinflamasi kencur yang dihasilkan oleh penghambatan COX-1 dan COX-2 (Umar et al., 2012). Hasil penelitian baru-baru ini yang dilakukan oleh Umar et al, (2014) menunjukkan bahwa EPMS memiliki efek analgesik dan antiinflamasi melalui mekanisme penghambatan sintesis sitokin pro-inflamasi meliputi TNF-α dan IL-1 secara in vivo dan in vitro. Efek ini juga melibatkan penghambatan vital sel endogen seperti proliferasi, sistesis dan migrasi dari vascular endotel growth factor.
Aktivitas antiinflamasi EPMS dapat digunakan untuk pengembangan obat baru. Dalam pengembangan obat tradisional, kandidat obat tersebut akan melalui serangkaian uji yaitu uji praklinik dan uji klinik sebelum diresmikan sebagai obat oleh badan pemberi izin. Uji praklinik merupakan persyaratan uji untuk memperoleh informasi tentang efikasi (efek farmakologi), profil farmakokinetik dan toksisitas kandidat obat. Setelah dinyatakan memenuhi persyaratan uji praklinik, suatu senyawa kandidat obat selanjutnya akan diuji pada manusia (uji klinik) (Sukandar., 2006).
2
UIN Syarif Hidayatullah Jakarta
yang diperoleh dalam bioanalisis akan menjadi pedoman untuk studi klinis dan studi keamanan obat (Harahap., 2010). Analisis dapat dilakukan apabila metode yang digunakan telah divalidasi sehingga didapatkan metode bioanalisis yang valid berdasarkan pada Food and Drug Administration, Guidance for Industry: Bioanalytical Method Validation, 2001. Selain itu, validasi perlu dilakukan agar hasil analisis yang diperoleh terpercaya, cermat, handal dan dapat dipertanggung jawabkan secara ilmiah (Ganjar & Rohman., 2007). Parameter-parameter penting dalam validasi metode bioanalisis adalah linearitas, limit of detection (LOD), limit of quantification (LOQ), selektivitas, akurasi dan presisi.
Metode analisis yang dipilih adalah Kromatografi Cair Kinerja Tinggi (KCKT) dimana telah digunakan secara luas untuk penemuan dan pengembangan metode analisis baru (Evans., 2004). Salah satu keuntungan dari KCKT adalah dapat menghitung sampel dengan kadar yang sangat rendah sehingga dapat digunakan untuk konsentrasi obat dalam plasma yang diukur mencapai level mikrogram sampai nanogram atau pikogram (Johnson & Stevenson., 1991). Cara ini ideal untuk analisis beragam obat dalam cairan biologis karena selektif, sederhana dan kepekaannya tinggi (Parwa., 1991).
Pada penelitian ini, akan dilakukan validasi metode analisis EPMS dalam plasma secara in vitro menggunakan KCKT dimana pemisahan pada kromatografi menggunakan fase diam C18 (Oktadesil silika atau ODS). Fase diam tersebut
paling banyak digunakan karena mampu memisahkan senyawa-senyawa dengan kepolaran yang rendah, sedang, maupun tinggi (Gandjar & Rohman., 2007). Penyiapan sampel plasma dilakukan dengan menggunakan metanol. Fase gerak yang digunakan adalah metanol dan akuabidestilata yang dilakukan secara isokratis. Pemilihan fase gerak didasarkan pada senyawa EPMS yang mampu larut dalam beberapa pelarut dengan kepolaran bervariasi seperti metanol, air, etanol, etil asetat dan heksana (Taufikhurohmah, 2008). Detektor yang digunakan adalah DAD (Diode Array Detector).
1.2 Perumusan masalah
3
b. Bagaimanakah metode ekstraksi untuk analisis EPMS dalam plasma menggunakan KCKT?
c. Apakah analisis EPMS dalam plasma secara in vitro menggunakan KCKT memiliki nilai validitas yang sesuai dengan persyaratan?
1.3 Tujuan Penelitian
a. Mendapatkan optimasi kondisi analisis EPMS menggunakan KCKT. b. Mendapatkan metode ekstraksi untuk analisis EPMS dalam plasma
menggunakan KCKT.
c. Memperoleh metode yang tervalidasi untuk analisis EPMS dalam plasma secara in vitro menggunakan KCKT.
1.4Manfaat penelitian
4 UIN Syarif Hidayatullah Jakarta BAB II
TINJAUAN PUSTAKA
2.1 Etil p-metoksisinamat (EPMS)
EPMS merupakan senyawa hasil isolat dari rimpang kencur (Kaempferia galanga. L). EPMS merupakan senyawa yang mampu larut dalam beberapa pelarut dengan kepolaran bervariasi seperti metanol, air, etanol, etil asetat dan heksana. Hal ini disebabkan karena karena EPMS bersifat non polar yang merupakan suatu ester yang mengandung cincin benzene dan gugus metoksi. Selain itu, bersifat agak polar karena mengandung gugus karbonil yang mengikat etil (Taufikhurohmah, 2008).
Gambar 2.1 Struktur kimia Etil p-metoksisinamat
[www.chemicalbook.com]
EPMS memiliki rumus molekul C12H14O3 dan berat molekul 206,4 g/mol
yang berbentuk kristal berwarna putih, berbau aromatik khas lemah dan memiliki titik lebur 49oC (Umar et al., 2012).
5
Mekanisme kerja EPMS sebagai antiinflamasi melalui penghambatan non-selektif pada aktivitas COX-1 dan COX-2 secara in vitro dimana enzim ini berguna dalam pembentukan mediator inflamasi yaitu prostaglandin (Umar etal., 2012). Hasil penelitian baru-baru ini yang dilakukan oleh Umar et al. (2014) menunjukkan bahwa EPMS memiliki efek analgesik dan antiinflamasi melalui mekanisme penghambatan sintesis sitokin pro-inflamasi meliputi TNF-α dan IL-1 secara in vivo dan in vitro. Efek ini juga melibatkan penghambatan vital sel endogen seperti proliferasi, sistesis dan migrasi dari vaskular endotel growth factor.
2.2 Plasma
Darah membentuk sekitar 8% dari berat badan tubuh total dan memiliki volume rata-rata 5 liter pada wanita dan 5,5 liter pada pria. Darah terdiri dari tiga jenis unsur sel khusus yang terendam dalam cairan kompleks plasma, yaitu eritrosit dalam jumlah yang besar, leukosit dalam jumlah yang relatif sangat sedikit 2 permil dari jumlah eritrosit dan trombosit yang memiliki fungsi penting pada penggumpalan darah (Sherwood.,1996).
Apabila suatu sampel darah utuh ditaruh dalam sebuah tabung reaksi dan diberi zat antikoagulan, unsur-unsur sel yang lebih berat akan secara perlahan mengendap di dasar dan plasma yang lebih ringan naik kebagian atas. Proses ini dipercepat oleh pemusingan (sentrifugasi), yang dengan cepat menyebabkan sel-sel mengendap di dasar tabung (Sherwood.,1996). Bobot jenis darah bervariasi antara 1,054-1,060, sedangkan bobot jenis plasma darah sekitar 1,024-1,028 (Poedjaji., 1994)
6
UIN Syarif Hidayatullah Jakarta 2.3 Analisis Obat dalam Plasma
Penentuan kadar obat dalam sampel biologis merupakan hal yang sangat penting dalam evaluasi dan interpretasi data farmakokinetika. Cairan biologis yang umum digunakan untuk analisis adalah darah, plasma (serum), dan urin.
Analisis konsentrasi obat dalam darah biasanya tidak digunakan dalam farmakokinetik karena darah merupakan sistem fisik kompleks yang mengandung sel darah merah, sel darah putih dan platelet yang tersuspensi dalam plasma. Darah dengan elemen selular yang telah dihilangkan dengan sentifugasi (plasma) atau pembekuan (serum) lebih disukai (Rosenbaun., 2011). Sampel biologis yang paling umum digunakan adalah plasma karena hubungan konsentrasi obat dalam plasma dengan efek terapetik yang ditimbulkan baik (Kelly., 1992).
Pemeriksaan kadar obat dalam plasma merupakan suatu metode yang sesuai untuk pemantauan pengobatan dan memungkinkan untuk penyesuaian dosis obat dan mengoptimasi terapi (Shargel., 2005). Salah satu keuntungan dari KCKT adalah dapat menghitung sampel dengan kadar yang sangat rendah sehingga dapat digunakan untuk konsentrasi obat dalam plasma yang diukur mencapai level mikrogram sampai nanogram atau pikogram (Johnson & Stevenson., 1991).
Pada matriks biologis seperti plasma mengandung sejumlah besar komponen endogen yang dapat menggangu analisis dimana sebagian besar obat akan berikatan dengan protein plasma sehingga harus dibebaskan terlebih dahulu. Oleh karena itu, diperlukan preparasi sampel dengan tujuan agar dapat memisahkan atau mengisolasi obat yang akan ditentukan dari komponen endogen plasma yang dapat mengganggu analisis, membebaskan obat dari sisi pengikatan protein dan memekatkan obat agar diperoleh analisis yang sensitif. Kegiatan preparasi sampel merupakan hal penting dalam bioanalisis (Harahap., 2010). Beberapa teknik preparasi sampel yang digunakan untuk mengisolasi obat dari matriks biologis antara lain:
a. Pengendapan protein
7
sangat efesien untuk mengendapkan protein. Penambahan larutan yang berisi ion logam berat atau penambahan pelarut organik seperti metanol, etanol, asetonitril dan aseton kedalam sampel biologis telah digunakan secara luas dalam bioanalisis karena kompatibilitasnya dengan fase gerak KCKT, meskipun memiliki efesiensi yang relatif rendah dalam memisahkan protein. Pelarut organik akan mengendapkan protein bedasarkan prinsip polaritas dan menurunkan solubilitas protein. (Evans., 2004; Harahap., 2010)
b. Ultrafiltrasi
Larutan bebas protein dapat diperoleh melalui proses penyaringan dengan melewatkan larutan pada suatu membran semipermeabel yang selektif dengan menggunakan tekanan hidrostatik (1-10 atm) untuk memberikan dorongan dalam proses pemisahan. (Harahap, 2010)
c. Ekstraksi cair-cair (Liquid-liquid Extraction)
Ekstraksi cair-cair adalah proses pemindahan atau pemisahan suatu komponen dari satu fase ke fase lainnya yang tidak saling bercampur satu sama lain, proses ini disebut partisi atau distribusi. Salah satu fasenya yaitu fase
aqueous dan fase lainnya merupakan pelarut organik. Larutan aqueous yang dapat digunakan adalah air, larutan yang bersifat asam/basa, garam dan lainnya. Pelarut organik yang dapat digunakan adalah heksan, etil asetat, toluene dan lainnya. Pelarut organik non polar untuk mengekstraksi senyawa yang bersifat lipofil, sedangkan senyawa hidrofil lebih mudah larut dalam pelarut organik yang relatif polar.
Metode ekstraksi dengan pelarut organik merupakan cara yang paling umum digunakan untuk pemisahan parsial. pH fase air harus dioptimasi agar diperoleh bentuk tidak terionisasi, karena obat dapat terekstraksi dalam pelarut organik apabila dalam bentuk tidak terionisasi. Optimasi dapat dilakukan dengan menghitung atau menentukan pKa obat.
8
UIN Syarif Hidayatullah Jakarta
penyaringan dapat menghilangkan air dari fase organik. Selanjutnya hasil penguapan direkonstitusi menggunakan pelarut yang sesuai. Kelemahan dari metode yaitu terjadinya pembentukan emulsi dan tidak dapat diaplikasikan kesemua analit, contohnya metode ini sulit digunakan untuk analit yang bersifat sangat polar (Evans 2004.; Harahap., 2010)
d. Ekstraksi fase padat (Solid Phase Extraction)
Prinsip mekanisme pemisahan dan isolasi yang digunakan dalam pemisahan fase padat yaitu fase terbalik, fase normal dan ion exchange sama seperti yang digunakan dalam KCKT. Metode ekstraksi fase padat ini berdasarkan prinsip kromatografi. Prinsip umum dari ekstraksi fase padat yaitu adsorpsi obat dari larutan kedalam adsorben atau fase diam. Partikel silika berukuran 40-60 µm merupakan adsorben yang sering digunakan berkaitan dengan membentuk fase hidrokarbon. Adsorben yang paling baik kapasitasnya dalam mengadsorbsi analit adalah C18.
Pada metode ini, digunakan kolom berukuran kecil (catridge) dengan adsorben yang memiliki sifat mirip dengan sifat analit yang diperiksa. Ekstraksi fase padat adalah suatu teknik yang dapat mengatasi beberapa masalah yang ditemui pada ekstraksi cair-cair. Secara umum SPE menggunakan 5 tahap yaitu pengkondisian, penyeimbangan fase diam, memasukkan sampel, pencucian untuk menghilangkan senyawa pengganggu, dan elusi sampel (Evans., 2004; Harahap., 2010).
2.4 Kromatografi Cair Kinerja Tinggi
2.4.1 Pendahuluan
9
KCKT paling sering digunakan untuk: menetapkan kadar senyawa-senyawa tertentu seperti asam-asam amino, asam-asam nukleat, dan protein-protein dalam cairan fisiologis; menentukan kadar senyawa-senyawa aktif obat, produk hasil samping proses sintesis, atau produk-produk degradasi dalam sediaan farmasi; memonitor sampel-sampel yang berasal dari lingkungan; memurnikan senyawa dalam suatu campuran; memisahkan polimer dan menentukan distribusi berat molekulnya dalam suatu campuran; kontrol kualitas; dan mengikuti jalnnya reaksi sintesis.
Keterbatasan metode KCKT adalah untuk identifikasi senyawa, kecuali jika KCKT dihubungkan dengan spektrofotometer massa (MS) dan apabila sampelnya sangat kompleks, maka resolusi yang baik sulit diperoleh. (Gandjar & Rohman., 2007)
2.4.2 Keuntungan KCKT
KCKT termasuk metode analisis terbaru yaitu suatu teknik kromatografi dengan fase gerak cairan dan fase diam cairan atau padat. Adapun keuntungan dari KCKT, yaitu : cepat, daya pisah baik, peka, detektor unik, pemilihan kolom dan eluen sangat bervariasi, dapat menghitung sampel dengan kadar yang sangat rendah, kolom dapat dipakai kembali, ideal untuk molekul besar dan ion, mudah memperoleh kembali cuplikan, mampu memisahkan molekul-molekul dari suatu campuran, mudah melaksanakannya, dapat dihindari terjadinya dekomposisi atau kerusakan bahan yang dianalisis, dan resolusi yang baik (Johnson & Steveson., 1991; Effendy., 2004).
2.4.3 Cara kerja KCKT
10
UIN Syarif Hidayatullah Jakarta
gerak dan fase diam. Penggunaan kromatografi cair secara sukses terhadap suatu masalah yang dihadapi membutuhkan penggabungan secara tepat dari berbagai macam kondisi operasional seperti jenis kolom, fase gerak, panjang dan diameter kolom, kecepatan alir fase gerak, suhu kolom, dan ukuran sampel (Gandjar & Rohman., 2007).
2.4.4 Instrumentasi KCKT
Instrumentasi KCKT pada dasarnya terdiri atas delapan komponen pokok yaitu: wadah fase gerak, alat untuk memasukkan sampel, kolom, detektor, wadah penampung buangan fase gerak, tabung penghubung, dan suatu komputer atau integrator atau perekam (Gandjar & Rohman., 2007).
.
Keterangan: 1 = Tempat fase gerak + penyaring; 2 = saluran penghubung dengan frit; 3 = pompa
(dengan monometer); 4 = injector sampel (autosampler); 5 = kolom (dengan thermostat) ; 6 =
detektor; 7 = pembuangan; 8 = pengolah data
Gambar 2.3 Diagram Alir KCKT
11
2.4.4.1 Wadah Fase Gerak pada KCKT
Wadah fase gerak harus bersih dan lembam (inert). Wadah pelarut kosong ataupun labu laboratorium dapat digunakan sebagai wadah fase gerak. Wadah yang digunakan dapat menampung fase gerak antara 1 sampai 2 liter. Fase gerak sebelum digunakan harus dilakukan
degassing (penghilangan gas) yang ada pada fase gerak, sebab adanya gas akan berkumpul dengan komponen lain terutama di pompa dan detektor sehingga akan mengganggu analisis (Gandjar & Rohman., 2007).
2.4.4.2 Fase gerak pada KCKT
Fase gerak atau eluen biasanya terdiri atas campuran pelarut yang dapat bercampur yang secara keseluruhan berperan dalam daya elusi dan resolusi. Daya elusi dan resolusi ini ditentukan oleh polaritas keseluruhan pelarut, polaritas fase diam, dan sifat komponen-komponen sampel. Untuk fase normal (fase diam lebih polar daripada fase gerak), kemampuan elusi meningkat dengan meningkatnya polaritas pelarut. Sementara untuk fase terbalik (fase diam kurang polar dari fase gerak), kemampuan elusi menurun dengan meningkatnya polaritas pelarut.
Elusi dapat dilakukan dengan cara isokratik (komposisi fase gerak tetap selama elusi) atau dengan cara bergradien (komposisi fase gerak berubah-ubah selama elusi). Elusi bergradien digunakan untuk meningkatkan resolusi campuran yang kompleks terutama jika sampel mempunyai kisaran polaritas yang luas.
12
UIN Syarif Hidayatullah Jakarta
Fase gerak adalah salah satu variabel yang mempengaruhi pemisahan. Oleh karena itu setidaknya fase gerak harus murni (tidak terdapat kontaminan), tidak bereaksi dengan wadah (packing), sesuai dengan detektor, melarutkan sampel, memiliki viskositas rendah, bila
diperlukan memudahkan “sample recovery”, dan diperdagangan dapat
diperoleh dengan harga murah (reasonable price) (Johnson & Steveson., 1991).
2.4.4.3 Pompa pada KCKT
Pompa berfungsi untuk menggerakan fase gerak melalui kolom (Johnson & Steveson., 1991). Tujuan penggunaan pompa adalah untuk menjamin proses penghantaran fase gerak berlangsung secara tepat, reprodusibel, konstan, dan bebas dari gangguan. Pompa yang cocok untuk KCKT adalah pompa harus inert terhadap fase gerak. Bahan yang umum dipakai untuk pompa adalah gelas, baja tahan karat, telfon, dan batu nilam. Ada 2 jenis pompa dalam KCKT yaitu : pompa dengan tekanan konstan, dan pompa dengan aliran fase gerak yang konstan.
Pompa yang digunakan sebaiknya mampu memberikan tekanan sampai 5000 psi dan mampu mengalirkan fase gerak dengan kecepatan alir 3 mL/menit. Untuk tujuan preparatif, pompa yang digunakan harus mampu mengalirkan fase gerak dengan kecepatan 20 mL/menit (Gandjar & Rohman., 2007).
2.4.4.4 Penyuntikan (injektor) sampel pada KCKT
13
2.4.4.5 Kolom pada KCKT
Kolom merupakan jantung kromatograf. Keberhasilan atau kegagalan analisis bergantung pada pilihan kolom dan kondisi kerja yang tepat. Kolom berfungsi untuk memisahkan masing-masing komponen. Kolom dapat dibagi menjadi dua kelompok, yaitu (Johnson & Steveson., 1991) :
1. Kolom analitik : Diameter dalam 2-6 mm. Panjang kolom tergantung pada jenis material pengisi kolom. Untuk kemasan pellicular, panjang yang digunakan adalah 50-100 cm. Untuk kemasan poros mikropartikulat, 10-30 cm.
2. Kolom preparatif: umumnya memiliki diameter 6 mm atau lebih besar dan panjang kolom 25 -100 cm.
2.4.4.6 Fase Diam pada KCKT
Kebanyakan fase diam pada KCKT berupa silika yang dimodifikasi secara kimiawi, silika yang tidak dimodifikasi, polimer-polimer stiren dan divinilbenzen. Permukaan silika adalah polar dan sedikit asam karena adanya residu gugus silanol (Si-OH).
Silika dapat dimodifikasi secara kimiawi dengan menggunakan reagen-reagen seperti klorosilan yang akan bereaksi dengan gugus silanol dan menggantinya dengan gugus-gugus fungsional yang lain. Hasil reaksi yang diperoleh disebut dengan silika fase terikat yang stabil terhadap hidrolisis karena terbentuk ikatan-ikatan siloksan (Si-O-O-Si). Silika yang dimodifikasi ini mempunyai karakteristik kromatografik dan selektifitas yang berbeda jika dibandingkan dengan silika yang tidak dimodifikasi.
Oktadesil silika (ODS atau C18) merupakan fase diam yang
14
UIN Syarif Hidayatullah Jakarta
2.4.4.7Detektor KCKT
Suatu detektor dibutuhkan untuk mendeteksi adanya komponen sampel di dalam kolom (analisis kualitatif) dan menghitung kadamya (analisis kuantitatif) (Johnson & Steveson., 1991). Detektor pada KCKT dikelompokkan menjadi 2 golongan yaitu : detektor universal (yang mampu mendeteksi zat secara umum, tidak bersifat spesifik, tidak bersifat selektif) seperti detektor indeks bias dan detektor spektrofotometri massa; dan golongan detektor yang spesifik yang hanya akan mendeteksi analit secara spesifik dan selektif, seperti detektor UV-Vis, detektor flouresensi, dan elektrokimia.
Idealnya, suatu detektor harus mempunyai karakteristik sebagai berikut (Gandjar & Rohman., 2007) :
1. Mempunyai respon terhadap solut yang cepat dan reprodusibel, 2. Mempunyai sensitifitas yang tinggi, yakni mampu mendeteksi solut
pada kadar yang sangat kecil, 3. Stabil dalam pengoperasiannya,
4. Mempunyai sel volume yang kecil sehingga mampu meminimalkan pelebaran pita.
5. Signal yang dihasilkan berbanding lurus dengan konsentraasi solut pada kisaran yang luas (kisaran dinamis linier), dan
6. Tidak peka terhadap perubahan suhu dan kecepatan alir fase gerak. Detektor KCKT yang umum digunakan adalah detektor UV 254 nm. Variabel panjang gelombang dapat digunakan untuk mendeteksi banyak senyawa dengan rentang yang lebih luas. Detektor indeks refraksi juga digunakan secara luas, terutama pada kromatografi eksklusi, tetapi umumnya kurang sensitif jika dibandingkan dengan detektor UV (Johnson & Steveson, 1991). Beberapa detektor yang sering digunakan pada KCKT (Gandjar & Rohman, 2007) :
a. Detektor Spektofotometri UV-Vis
15
gugus-gugus kromofik. Detektor spektrofotometri UV-Vis dapat berupa detektor dengan panjang gelombang tetap (merupakan detektor yang paling sederhana) serta detektor dengan panjang gelombang bervariasi.
b. Detektor Photodiode-array (PDA)
Detektor PDA meruapakan detektor UV-Vis dengan berbagai keistimewaan. Detektor ini mampu memberikan kumpulan kromatogram secara simultan pada panjang gelombang yang berbeda pada sekali proses (single run). Selama proses berjalan, suatu kromatogram pada panjang gelombang yang diinginkan (biasanya antara 190-400) dapat ditampilkan.
c. Detektor Flouresensi
Flouresensi merupakan fenomena luminisensi yang terjadi ketika suatu senyawa menyerap sinar UV atau visible lalu mengemisikannya pada panjang gelombang yang lebih besar. Tidak semua senyawa obat mempunyai sifat flouresen sehingga detektor flouresensi ini sangat spesifik. Disamping itu, detektor ini juga sangat sensitif dibandingkan dengan detektor UV.
d. Detektor Indeks Bias
Detektor ini akan merespon setiap perbedaan indeks bias antara analit (zat terlarut) dengan pelarutnya (fase geraknya). Penggunaan detektor ini terutama untuk senyawa-senyawa yang tidak mempunyai gugus kromofor.
e. Detektor Elektrokimia
16
UIN Syarif Hidayatullah Jakarta
cukup untuk mengoperasikannya supaya didapatan garis dasar (baseline) yang stabil.
2.4.4.8 Intergrator
Alat ini akan mengukur sinyal elektronik yang dihasilkan oleh detektor lalu mem-plotkannya sebagai suatu kromatogram yang selanjutnya dievaluasi oleh analisis (pengguna). Alat pengumpul data seperti komputer, integrator, atau rekorder, dihubungkan dengan detektor (Gandjar & Rohman, 2007).
2.4.5 Penggunaan KCKT dalam Analisis Farmasi
Kromatografi Cair Kinerja Tinggi (KCKT) merupakan suatu metoda pemisahan canggih dalam analisis farmasi yang dapat digunakan sebagai uji identitas, uji kemurnian dan penetapan kadar. Titik beratnya adalah untuk analisis senyawa-senyawa yang tidak mudah menguap dan tidak stabil pada suhu tinggi, yang tidak bisa dianalisis dengan Kromatografi Gas (Effendy., 2004). Metode KCKT merupakan metode yang sangat populer untuk menetapkan kadar senyawa obat baik dalam bentuk sediaan maupun dalam sampel hayati. Hal ini disebabkan KCKT merupakan metode yang memberikan sensitifitas dan spesifitas yang tinggi (Gandjar & Rohman., 2007).
2.4.6 Analisis Kuantitatif
Mengukur luas puncak dari suatu komponen zat yang dianalisis merupakan dasar perhitungan kuantitatif. Beberapa metode yang dapat digunakan, yaitu :
2.4.6.1Metode Baku Eksternal
17
kromatogram senyawa tertentu yang ada dalam sampel. Sampel yang mengandung senyawa tertentu yang akan ditetapkan konsentrasinya dan telah disiapkan, selanjutnya diinjeksikan dan dianalisis dengan cara yang sama (Gandjar dan Rohman., 2007).
2.4.6.2Metode Baku Internal
Sejumlah baku internal ditambahkan pada sampel dan standar. Baku internal dapat menghilangkan pengaruh karena adanya perubahan-perubahan pada ukuran sampel atau konsentrasi karena variasi instrumen. Salah satu alasan utama digunakan baku internal adalah jika suatu sampel memerlukan suatu perlakuan sampel yang sangat signifikan sehingga dapat mengakibatkan berkurangnya sampel. Jika baku internal ditambahkan pada sampel sebelumnya dilakukan preparasi sampel, maka baku internal dapat mengoreksi hilangnya sampel-sampel ini. Syarat-syarat suatu senyawa dapat digunakan sebagai baku internal adalah (Gandjar dan Rohman., 2007):
1. Terpisah dengan baik dari senyawa yang dituju atau puncak-puncak yang lain.
2. Mempunyai waktu retensi yang hampir sama dengan analit. 3. Tidak terdapat dalam sampel.
4. Memiliki kemiripan sifat-sifat dengan analit dalam tahapan-tahapan penyiapan sampel.
5. Tidak mempunyai kemiripan sifat kimiawi dengan analit. 6. Tersedia dalam perdagangan dengan kemurnian yang tinggi. 7. Stabil dan tidak reaktif dengan sampel atau dengan fase gerak.
8. Mempunyai respon detektor yang hampir sama dengan analit pada konsentrasi yang digunakan.
2.4.6.3Metode Presentase Tinggi/Lebar Puncak atau Metode Normalisasi Internal
18
UIN Syarif Hidayatullah Jakarta
yang menghasilkan puncak. Dalam metoda yang paling sederhana diukur lebar atau tinggi puncak, yang kemudian dinormalisasi (ini berarti bahwa setiap lebar atau tinggi puncak diekspresikan sebagai suatu persentase dari total). Hasil normalisasi dari lebar atau tinggi puncak memberikan komposisi dari campuran yang dianalisis.
Ada dua masalah dengan pendekatan ini, yaitu: Kita harus yakin bahwa kita telah menghitung semua komponen, yang tiap-tiap komponen muncul sebagai suatu puncak yang terpisah pada kromatogram. Komponen-komponen dapat berkoelusi, atau ditahan di dalam kolom, atau terelusi tanpa terdeteksi. Kemudian, Kita harus mengasumsi bahwa kita memperoleh respons detektor yang sama untuk setiap komponen. Untuk mengatasi kesulitan ini, maka kalibrasi detektor diperlukan (Gandjar dan Rohman., 2007).
2.5 Validasi Metode Analisis
Validasi metode analisis adalah suatu tindakan penilaian terhadap parameter tertentu, berdasarkan percobaan laboratorium, untuk membuktikan bahwa parameter tersebut memenuhi persyaratan untuk penggunaannya (Harmita., 2004).
Suatu metode analisis harus divalidasi untuk melakukan verifikasi bahwa parameter-parameter kinerjanya cukup mampu untuk mengatasi problem analisis, karenanya suatu metode harus divalidasi, ketika (Gandjar dan Rohman., 2007) :
a. Metode baru dikembangkan untuk mengatasi problem analisis tertentu.
b. Metode yang sudah baku direvisi untuk menyesuaikan perkembangan atau karena munculnya suatu problem yang mengarahkan bahwa metode baku tersebut harus direvisi.
c. Penjaminan mutu yang mengindikasikan bahwa metode baku telah berubah seiring dengan berjalannya waktu.
19
e. Untuk mendemonstrasikan kesetaraan antar 2 metode, seperti antara metode baru dan metode baku.
Pada validasi metode bioanalisis terdapat tiga tipe dan tingkatan validasi, yaitu (Food and Drug Administration, Guidance for Industry: Bioanalytical Method Validation., 2001) :
a. Validasi lengkap (full validation)
Validasi lengkap ini sangat penting apabila ingin mengembangkan metode dan mengimplementasikan metode bioanalisis untuk pertama kalinya. Validasi ini penting untuk obat baru dan untuk penentuan metabolitnya.
b. Validasi parsial (partial validation)
Validasi parsial merupakan modifikasi dari metode bioanalisis yang yang sudah divalidasi.
c. Validasi silang (cross validation)
Validasi silang dilakukan dengan membandingkan parameter-parameter validasi apabila digunakan dua atau lebih metode bioanalisis untuk mendapatkan data pada studi yang sama atau pada studi yang berbeda. Pada validasi ini digunakan metode validasi yang original sebagai pembanding dan metode bioanalisis lainnya sebagai komparator.
Validasi metode analisis yang dilakukan dalam matriks biologi biasanya disebut sebagai validasi metode bioanalisis. Validasi metode bioanalisis ini digunakan pada studi farmakologi klinis, pengujian bioavaibilitas (BA) dan bioekuivalensi (BE), serta uji farmakokinetika (PK). Metode analisis yang selektif dan sensitif untuk evaluasi obat dan metabolitnya (analit) secara kuantitatif sangat berpengaruh terhadap kesuksesan studi farmakologi pre-klinik dan klinik. Parameter-parameter penting dalam validasi metode bioanalisis adalah akurasi, presisi, selektivitas, sensitivitas, reprodusibilitas, dan stabilitas. Pengembangan metode analisis meliputi evaluasi selektivitas, akurasi, presisi, uji perolehan kembali (%recovery), kurva kalibrasi, dan stabilitas (Harahap, 2010).
2.5.1 Ketepatan (Akurasi)
20
UIN Syarif Hidayatullah Jakarta
sampel yang mengandung analit dalam jumlah yang diketahui. Akurasi dilakukan minimal lima kali pengukuran untuk setiap konsentrasi. Minimal dari tiga konsentrasi didalam rentang konsentrasi yang direkomendasikan, yaitu pada konsentrasi rendah, sedang dan tinggi. Pengukuran akurasi memenuhi syarat jika nilai rata-rata berada pada 15% nilai sebenarnya, kecuali jika pengukuran dilakukan pada kadar LLOQ maka tidak boleh melebihi 20%. Perbedaan nilai rata-rata dan nilai yang sebenarnya menentukan akurasi (US FDA., 2001; Harahap., 2010)
2.5.2 Presisi
Presisi menggambarkan kedekatan hasil pengukuran individual analit yang satu dengan yang lainnya. Presisi diukur menggunakan sedikitnya lima kali pengukuran untuk setiap konsentrasi dan menggunakan minimal tiga kosentrasi yaitu rendah, sedang, tinggi. Pengukurannya dapat dilakukan intra assay (dalam satu kali analisis) dan inter assay (dilakukan analisis selama 5 hari). Pengukuran presisi pada setiap level konsentrasi, harus memiliki nilai koefesien variasi (KV) tidak lebih dari 15%, kecuali LLOQ dimana nilai KV tidak boleh lebih dari 20% (US FDA., 2001; Harahap., 2010)
2.5.3 Perolehan kembali (%Recovery)
21
2.5.4 Kurva Kalibrasi
Kurva kalibrasi merupakan hubungan antara respon instrumen dengan konsentrasi analit yang diketahui. Kurva kalibrasi harus terdiri dari 1 sampel blanko (matriks tanpa baku dalam), 1 sampel zero (matriks dengan baku dalam) dan 6-8 sampel non-zero yang mencakup kisaran konsentrasi pengukuran (termasuk konsentrasi pada LLOQ).
Kurva kalibrasi dapat diterima jika memenuhi persyaratan berikut: 20% simpangan dari nilai LLOQ, 15 % simpangan dari standar selain LLOQ dan setidaknya empat dari enam sampel non zero memenuhi persyaratan tersebut, termasuk LLOQ dan kosentrasi tinggi pada kurva kalibrasi.
2.5.4.1 Linearitas
Linearitas suatu metode bioanalisis harus diuji untuk mengetahui adanya hubungan yang linier antara kadar zat dengan respon detektor. Liniearitas diperoleh dari koefesien korelasi (r) pada analisis regresi linear yang didapat dari kurva kalibrasi. Dengan dilakukan uji ini, maka dapat diketahui batas-batas konsentrasi dari analit yang memberikan respon detektor yang linier. Analisis harus dilakukan pada konsentrasi yang termasuk batas-batas linier dari konsentrasi yang telah dilakukan (Harahap., 2010)
2.5.4.2 Batas Deteksi (limit of detection, LOD)
Batas deteksi didefinisikan sebagai konsentrasi analit terendah dalam sampel yang masih dapat dideteksi, meskipun tidak selalu dapat dikuantifikasi. Definisi batas deteksi yang paling umum digunakan dalam kimia analisis adalah bahwa batas deteksi merupakan kadar analit yang memberikan respon sebesar respon blangko (yb) ditambah dengan 3
simpangan baku blangko (3Sb).
22
UIN Syarif Hidayatullah Jakarta
kurva baku pada level yang mendekati LOD sesuai dengan rumus, LOD = 3 (SD/S). Standar deviasi respon dapat ditentukan berdasarkan pada standar deviasi blanko, pada standar deviasi residual dari garis regresi, atau standar deviasi intersep y pada garis regresi (Gandjar dan Rohman., 2007).
2.5.4.3 Batas Kuantifikasi (limit of quantification, LOQ)
Batas kuantifikasi didefinisikan sebagai kosentrasi analit terendah dalam sampel yang dapat ditentukan dengan presisi dan akurasi yang dapat diterima pada kondisi operasional metode yang digunakan. ICH menggunakan 2 metode untuk menentukan LOQ yaitu: metode non instrumental visual dan metode perhitungan bedasarkan pada standar deviasi respon (SD) dan kemiringan (slope, S) kurva baku sesuai dengan rumus, LOQ = 10 (SD/S). Standar deviasi respon dapat ditentukan berdasarkan pada standar deviasi blanko, pada standar deviasi residual dari garis regresi, atau standar deviasi intersep y pada garis regresi (Gandjar dan Rohman., 2007).
Batas kuantifikasi terendah (Lower limit of quantification, LLOQ) adalah analit terkecil yang dapat ditentukan dengan ketelitian dan akurasi tertentu (Harahap, 2010). Standar terendah pada kurva kalibrasi harus diterima sebagai LLOQ jika: respon analit pada LLOQ minimal lima kali respon sampel blanko dan puncak analit dapat diidentifikasi, tidak ada gangguan, reprodusibel dengan presisi 20% dan akurasi 80-120% (US FDA.,2001).
2.5.5 Selektivitas
23
interferensi dan selektivitas pada lower limit of quantification (LLOQ) (US FDA,2001).
2.5.6 Uji Kesesuaian Sistem
Uji kesesuaian sistem didefinisikan sebagai serangkaian uji untuk menjamin bahwa metode tersebut dapat menghasilkan akurasi dan presisi yag dapat diterima. Seorang analisis harus memastikan bahwa sistem dan prosedur yang digunakan harus mampu memberikan data yang dapat diterima.
United State Pharmacopeia (USP) menentukan parameter yang dapat digunakan untuk menetapkan kesesuaian sistem sebelum analisis. Parameter-parameter yang digunakan meliputi: bilangan lempeng teori (N), faktor tailing,
kapasitas (k’ atau α) dan nilai standar deviasi relative (RSD) tinggi puncak dan
luas puncak dari serangkaian injeksi. Pada umumnya ada 2 kriteria yang biasanya dipersyaratkan untuk kesesuaian sistem suatu metode yaitu jika nilai RSD < 1% untuk 5 kali injeksi larutan baku pada pengujian komponen yang jumlahnya banyak (komponen mayor) dan untuk senyawa-senyawa dengan kadar sekelumit, nilai RSD dapat diterima jika antara 5-15% (Gandjar dan Rohman., 2007).
2.5.7 Stabilitas
Stabilitas obat dalam cairan biologis adalah fungsi dari kondisi penyimpanan, sifat kimia obat, matriks dan sistem penyimpanan. Penyimpanan dapat membuat obat yang ada didalam matriks biologis dapat terurai sehingga tidak dapat terdeteksi sewaktu sampel dianalisis.
24
UIN Syarif Hidayatullah Jakarta
2.5.7.1Stabilitas Beku dan Cair (Freeze and Thaw Stability)
Stabilitas analit harus ditentukan setelah tiga siklus beku dan cair. Setidaknya tiga aliquot dari masing-masing kosentrasi rendah dan tinggi disimpan pada suhu penyimpanan yang diinginkan selama 24 jam dan dicairkan pada suhu kamar. Ketika mencair seluruhnya, sampel dibekukan kembali selama 12-24 jam pada kondisi yang sama. Siklus beku cair diulang sebanyak dua kali, kemudian analisis dilakukan pada siklus ketiga. Jika analit tidak stabil pada suhu yang diinginkan, maka uji dapat dilakukan dengan menyimpan sampel pada suhu -70OC selama tiga siklus beku dan cair.
2.5.7.2Stabilitas Suhu Jangka Pendek (Short-Term Temperature Stability)
Tiga aliquot dari masing-masing konsentrasi rendah dan tinggi harus diberikan pada suhu kamar dan disimpan pada suhu ini selama 4-24 jam (atau berdasarkan durasi yang diinginkan dimana sampel akan dijaga pada temperature ruang sesuai dengan uji yang diinginkan) kemudian dianalisis.
2.5.7.3Stabilitas Jangka Panjang (Long-Term Stability)
Waktu penyimpanan pada evaluasi stabilitas jangka panjang harus melebihi waktu antara pengumpulan sampel pertama kali dan sampel terakhir dianalisis. Stabilitas jangka panjang ditentukan dengan menyimpan sedikitnya tiga aliquot dari masing-masing konsentrasi rendah dan tinggi pada kondisi yang sama seperti uji sampel. Konsentrasi dari semua sampel harus dibandingkan dengan rata-rata nilai perolehan kembali yang sesuai dengan konsentrasi standar dari hari pertama uji stabilitas jangka panjang.
2.5.7.4Stabilitas Larutan Stok (Stock Solution Stability)
25
selama periode tertentu, stabilitas harus terdokumentasi. Setelah tercapainya waktu penyimpanan yang diinginkan, stabilitas harus diuji dengan membandingkan respon instrument terhadap larutan baru yang telah disiapkan.
2.5.7.5Stabilitas Post-Preparatif
26 UIN Syarif Hidayatullah Jakarta BAB III
METODE PENELITIAN
3.1 Tempat dan Waktu Penelitian
Penelitian dilaksanakan di Laboratorium Farmakognosi dan Fitokimia, Laboratorium Kimia Obat, Laboratorium Penelitian 1 dan Laboratorium Penelitian 2 Program Studi Farmasi FKIK Universitas Islam Negeri Syarif Hidayatullah Jakarta pada Februari 2016 sampai Juni 2016.
3.2 Bahan dan Alat
3.2.1 Bahan
Etil p-metoksisinamat (hasil isolasi), metanol (Merck), akuabides (Otsuka), plasma (PMI DKI Jakarta)
3.2.2 Alat
Kromatografi Cair Kinerja Tinggi (Dionex UltiMate® 3000) yang terdiri dari; pompa, autosampler, kolom Acclaim® Polar Advantage II (C18; 3 µm; 4,6 x 150 mm), detektor DAD (Diode Array Detector), program komputer PC (Chormeleon®). Spektrofotometer Ultraviolet-Visibel (Hitachi U-2910), vortex, sentrifugator (Eppendorf Centrifuge 5417R) dengan tabung sentrifugasi, syringe filter (Sartorius, RC 0,45 µm), pompa vakum (Welch®), timbangan analitik (AND-6H202), Suntikan (Terumo), mikropipet (Rainin), tabung vacutainer, lemari pendingin, dan sonikator (Elmasonic 5), alat-alat gelas.
3.3 Prosedur Kerja
3.3.1 Pembuatan Larutan Induk EPMS Konsentrasi 1000 µg/mL
27
3.3.2 Pembuatan Fase Gerak
Berbagai perbandingan komposisi fase gerak dibuat dengan mencampurkan metanol dan akuabides. Fase gerak yang telah dibuat kemudian disaring menggunakan vakum dan filter 0,45 µm. Gas yang terdapat di dalam larutan dihilangkan menggunakan sistem penghilang gas (degasser). Perbandingan komposisi fase gerak dapat dilihat pada Tabel 3.1.
Tabel 3.1 Komposisi Fase Gerak
No. Metanol Akuabides
1 100 -
2 80 20
3 70 30
4 60 40
3.3.3 Tahapan Optimasi
3.3.3.1 Penentuan Panjang Gelombang Maksimum untuk Analisis
Larutan induk EPMS diencerkan hingga diperoleh konsentrasi 5,05 µg/mL dengan metanol. Masing-masing larutan tersebut diukur nilai serapannya pada panjang gelombang 200-400 nm menggunakan. Spektrofotometri UV-Visibel, ditentukan panjang gelombang maksimumnya.
3.3.3.2 Pemilihan Komposisi Fase Gerak
28
UIN Syarif Hidayatullah Jakarta
3.3.3.3 Uji Kesesuaian Sistem
Larutan EPMS dengan konsentrasi 10,1 µg/mL diinjeksikan sebanyak 20 µL ke alat KCKT dengan kondisi analisis terpilih, diulangi sebanyak lima kali. Kemudian dihitung jumlah plat teoritis, HETP (Height Equivalent Theoritical Plate), asimetrisitas dan %RSD (Relative Standard Deviation).
3.3.3.4 Penetapan Metode Ekstraksi (Polson, 2002)
Kedalam tabung sentrifus dimasukkan 0,5 ml plasma dan dicampur metanol dengan perbandingan 1:1 dan 1:4. Kemudian dikocok dengan vortex selama 20 detik. Setelah itu, disentrifugasi pada 3000 rpm selama 10 menit. Lalu supernatan diambil dan disaring menggunakan
syringe filter 0,45 µm. Kemudian diinjeksikan supernatan sebanyak 20 µL ke alat KCKT. Kemudian dianalisis kromatogram dari masing-masing perbandingan untuk mengetahui kondisi kromatogram blanko plasma.
Setelah itu, dibuat larutan dalam plasma yang mengandung larutan EPMS dengan konsentrasi 10,1 µg/mL. Kemudian diambil sebanyak 0,5 mL dari larutan tersebut, diekstraksi menggunakan metanol dalam plasma dengan perbandingan 1:1 dan 1:4. Sebanyak 20 µL diinjeksikan ke alat KCKT kemudian dicatat waktu retensi dan luas puncak, asimetrisitas dan resolsinya.
3.3.4 Validasi Metode Analisis EPMS dalam Plasma
3.3.4.1 Pengukuran Limit Kuantitasi Terendah (LLOQ)
29
EPMS dalam plasma dari masing-masing konsentrasi dan dibuat kurva kalibrasinya. Dari data pengukuran kemudian dihitung nilai LOQ.
Setelah nilai batas kuantitasi (LOQ) diperoleh, nilai LLOQ diperoleh dari konsentrasi setengah atau seperempat nilai LOQ.
3.3.4.2 Pembuatan Kurva Kalibrasi dan Uji Linearitas dalam Plasma In Vitro
Dibuat larutan blangko plasma dan 7 sampel yaitu larutan EPMS dengan konsentrasi 2,02; 5,05; 10,1; 15,15; 20,2; 25,25; 40,4 µg/mL dimana dalam rentang konsentrasi tersebut terdapat LLOQ didalamnya, Kemudian dipreprasi sesuai prosedur. Lalu supernatan masing-masing sebanyak 20 µL diinjeksikan ke alat KCKT pada kondisi analisis terpilih. Setelah itu dianalisis regresi perbandingan luas puncak terhadap konsentrasi EPMS dalam plasma dari masing-masing konsentrasi dan dibuat kurva kalibrasi dengan persamaan garis regresi linier (y=a + bx). Dihitung koefesien korelasi (r) dari kurva tersebut. Kemudian dihitung LOQ (limit batas kuantitasi) dan LOD (limit batas deteksi).
LOQ (limit batas kuantitasi) dihitung melalui persamaan garis regresi linier dari kurva kalibrasi, dengan rumus :
LOQ = �
�
Sedangkan nilai batas deteksi (LOD) diperoleh dengan rumus :
LOD =3
30
UIN Syarif Hidayatullah Jakarta
berbeda. Kemudian dihitung nilai % KV (Koefesien Variasi) dengan nilai < 20% dan akurasinya (%diff) dengan nilai + 20%.
3.3.4.4 Uji Akurasi
Dibuat larutan EPMS dengan 3 kosentrasi. Konsentrasi rendah (3 kali LLOQ) yaitu 6,06 µg/mL; konsentrasi sedang yaitu 18,18 µg/mL dan konsentrasi tinggi yaitu 30,3 µg/mL bedasarkan kurva kalibrasi. Setelah itu dipreparasi sesuai prosedur. Supernatan sebanyak 20 µL diinjeksikan ke alat KCKT dengan kondisi terpilih, yang diulangi sebanyak 3 kali. Kemudian dihitung presentase akurasi (%diff) dan perolehan kembali (% recovery) dari masing-masing konsentrasi larutan tersebut. Nilai rata-rata %diff disyaratkan + 15% dari nilai sebenarnya. Sedangkan nilai perolehan kembali dihitung dengan cara membandingkan konsentrasi EPMS dalam darah yang diperoleh dari hasil ekstraksi dengan konsentrasi EPMS yang sebenarnya dikalikan dengan 100%. Perolehan kembali tidak harus 100%, tetapi tingkat perolehan kembali analit dan baku dalam harus konsisten, presisi, dan reprodusibel.
3.3.4.5 Uji Presisi
BAB IV
HASIL DAN PEMBAHASAN
4.1 Penetapan Panjang Gelombang Analisis
Pada penelitian ini, pemilihan panjang gelombang analisis dilakukan menggunakan alat spektrofotometer UV-Vis. Senyawa aktif EPMS pada konsentrasi 5,05 µg/mL diukur pada panjang gelombang 200 hingga 400 nm, dimana senyawa EPMS memiliki gugus kromofor di dalam molekulnya sehingga dapat diperoleh spektrum serapannya. Spektrum serapan yang dihasilkan menunjukkan bahwa EPMS berada panjang gelombang sinar UV yaitu 308 nm. Sehingga didapatkan panjang gelombang maksimum untuk analisis yaitu 308 nm. Hal ini sesuai dengan literatur, dimana analisis EPMS dideteksi pada panjang gelombang 308 nm (Salkar,2014). Pemilihan panjang gelombang analisis ini berguna untuk meningkatkan selektivitas dan sensitifitas analisis dari sampel yang digunakan. Data spektrum serapan senyawa zat aktif EPMS dapat dilihat pada Gambar 4.1 dibawah ini.
32
UIN Syarif Hidayatullah Jakarta
4.2 Optimasi Kondisi Analisis
4.2.1 Pemilihan Komposisi Fase Gerak
Analisis EPMS dalam plasma secara in vitro menggunakan kromatografi cair kinerja tinggi dilakukan pada kondisi optimum dengan laju alir 1 mL/menit, panjang gelombang analisis 308 nm, volume penyuntikan 20 µL dan menggunakan kolom Acclaim® (C18) 150 mm. Sistem kromatografi yang digunakan adalah sistem isokratik dengan komposisi fase gerak metanol dan air dalam berbagai perbandingan. Fase gerak tersebut bersifat polar sehingga dapat memisahkan senyawa EPMS yang memiliki molekul-molekul bersifat polar.
Komposisi fase gerak sebagai kondisi awal yang digunakan adalah metanol 100%. Pada fase gerak metanol 100% diperoleh kromatogram senyawa EPMS yang memiliki waktu retensi (tR) 2,077 menit. Kemudian komposisi fase gerak diubah dengan penambahan air dalam perbandingan metanol-air (80:20 v/v); (70:30 v/v) dan (60:40 v/v). Dimana pada komposisi fase gerak tersebut menghasilkan waktu retensi secara berurutan yaitu (tR) 3,393 menit, 6,440 menit dan 10,037 menit. Pengubahan fase gerak metanol 100% dengan penambahan air pada komposisi fase gerak dilakukan untuk menghasilkan puncak EPMS pada waktu retensi diatas menit kedua, karena pengotor dalam plasma umumnya muncul pada menit kedua sehingga nantinya dapat mengganggu puncak dari senyawa EPMS.
33
Tabel 4.1 Hasil penetapan komposisi fase gerak pada konsentrasi 10,1µg/mL, kecepatan alir 1 mL/menit, panjang gelombang 308 nm, dan volume penyuntikan 20 µL.
Fase gerak (v/v) TR (menit)
Luas puncak (mAU)
N HETP Asimetrisitas
Metanol 2,077 23,4096 165 0,9090 1,52 Metanol-Air (80:20) 3,393 22,7340 86 1,7442 0,98 Metanol-Air (70:30) 6,440 20,4664 129 1,1628 2,16 Metanol-Air (60:40) 10,037 21,4820 245 0,6122 1,86
Gambar 4.2 Kromatogram EPMS murni pada
konsentrasi 10,1µg/mL dengan fase gerak
metanol 100%, kecepatan alir 1 mL/menit, panjang gelombang 308 nm dan volume
penyuntikan 20 µL.
Gambar 4.3 Kromatogram EPMS murni pada
konsentrasi 10,1 µg/mLdengan komposisi fase
gerak metanol-air (80:20 v/v), kecepatan alir 1 mL/menit, panjang gelombang 308 nm dan
volume penyuntikan 20 µL.
Gambar 4.4 Kromatogram EPMS murni pada konsentrasi 10,1 µg/mL dengan komposisi fase gerak metanol-air (70:30 v/v), kecepatan alir 1
mL/menit, panjang gelombang 308 nm dan volume penyuntikan 20 µL.
Gambar 4.5 Kromatogram EPMS murni pada konsentrasi 10,1 µg/mL dengan komposisi fase gerak metanol-air (60:40 v/v), kecepatan alir 1
34
UIN Syarif Hidayatullah Jakarta
4.2.2 Uji Kesesuaian Sistem
Sebelum metode analisis yang terpilih akan digunakan perlu dilakukan uji kesesuaian sistem. Dimana uji kesesuaian sistem ini dilakukan untuk memastikan bahwa sistem dan prosedur yang digunakan harus mampu memberikan data yang dapat diterima. Dalam uji kesesuaian sistem terdapat beberapa parameter-parameter, yaitu plat teoritis (N) dengan syarat > 2500, asimetrisitas dengan syarat < 2,5 dan syarat utama yaitu nilai koefesien variasi luas puncak dengan syarat %RSD < 2 % dari pengulangan injeksi. Suatu metode dapat diterima apabila dari uji kesesuaian sistem terdapat minimal dua parameter yang memenuhi persyaratan (Gandjar & Rohman., 2007).
Dari hasil analisis 5 kali penyuntikan, didapatkan luas puncak dari zat aktif EPMS. Nilai koefesien variasi yang diperoleh dari luas puncak adalah sebesar 1,2491% dan nilai koefesien variasi yang diperoleh dari waktu retensi adalah sebesar 0,8924 %. Uji kesesuaian sistem yang telah dilakukan telah memenuhi persyaratan yang ditetapkan, kecuali pada parameter plat teoritis (N). Keefesienan kolom dapat dilihat beberapa diantaranya pada nilai bilangan plat teoritis, HETP dan asimetrisitas. Dari data yang diperoleh plat teoritis yang dihasilkan tidak memenuhi persyaratan hal ini menunjukkan bahwa kondisi kolom yang digunakan kurang baik. Data dapat dilihat pada Tabel 4.2 dan data selengkapnya tercantum dalam Lampiran 2 Tabel 5.1.
Tabel 4.2 Hasil uji rata-rata kesesuaian sistem EPMS pada konsentrasi 10,1 µg/mL dengan komposisi fase gerak metanol-air (60:40 v/v) pada kecepatan alir 1 ml/menit, panjang
gelombang 308 nm dan volume penyuntikan 20 µL.
Parameter Syarat Hasil yang diperoleh Kesimpulan %RSD waktu retensi < 2% 0,8924% √
%RSD Luas puncak < 2% 1,2491% √ Lempeng Teoritis (N) > 2500 153 X
Asimetrisitas < 2,5 0,934 √
4.2.3 Penetapan Metode Ekstraksi
35
cara pengendapan protein menggunakan pelarut organik untuk mendenaturasi dan mengendapkan protein. Pelarut organik akan mengendapkan protein bedasarkan prinsip polaritas dan menurunkan solubilitas protein. Penambahan volume pelarut organik perlu dioptimasi agar EPMS terekstraksi dengan baik. Didalam prosedur penetapan metode ekstraksi adanya perlakuan vortex adalah untuk mencampurkan plasma yang telah ditambahkan dengan pelarut organik dan perlakuan sentrifugasi bertujuan untuk memisahkan pelarut pengestrak dengan protein-protein plasma setelah pengocokan. Pelarut organik yang digunakan adalah metanol dengan perbandingan metanol dalam plasma 1:1 dan 4:1. Setelah dilakukan pengamatan, dari dua perbandingan tersebut, tidak ada puncak pengotor protein yang mengganggu pada waktu retensi EPMS yang dilihat dari kromatogram blangko plasma. Gambar dapat dilihat pada Gambar 4.6 dan 4.8.
Kemudian dilakukan pengamatan pada kromatogram plasma yang mengandung EPMS dengan melakukan metode ekstraksi yang sama. Data hasil analisis menunjukkan bahwa komposisi metanol sebanyak 4 kali bagian plasma merupakan metode pengendapan protein yang baik, dikarenakan menghasilkan asimetrisitas yang baik dan nilai resolusi yang paling besar sehingga dapat memisahkan puncak pengotor plasma dengan EPMS jika dibandingkan dengan perbandingan komposisi metanol sebanyak 1 kali bagian plasma. Data dapat dilihat pada Tabel 4.3 dan Gambar 4.7 dan 4.9.
Tabel 4.3 Hasil optimasi pengendapan protein plasma
Perbandingan Pelarut Metanol dengan plasma
Luas puncak (mAu)
N Resolusi Asimetrisitas
36
UIN Syarif Hidayatullah Jakarta
4.3 Validasi Metode Analisis EPMS dalam Plasma In Vitro
4.3.1 Pengukuran Limit Kuantitasi Terendah (LLOQ)
Validasi diawali dengan pengukuran batas kuantitasi (LOQ) dan batas kuantitasi terendah (LLOQ). Rentang konsentrasi dalam plasma yang digunakan adalah 5,05-40,4 µg/mL. Dari rentang konsentrasi tersebut didapatkan persamaan regresi linear Y=0,4215x-0,4924. Kemudian dihitung nilai LOQ, nilai LOQ yang diperoleh adalah 5,3767 µg/mL. data perhitungan dapat dilihat selengkapnya pada Lampiran 3 Tabel 5.2. Kemudian dihitung nilai LLOQ dengan melakukan pengenceran konsentrasi LOQ menjadi
Gambar 4.6 Kromatogram blangko plasma pada perbandingan metanol dalam plasma 1:1
dengan fase gerak metanol-air (60:40v/v), kecepatan alir 1 mL/menit, panjang gelombang
308 nm dan volume penyuntikan 20 µL.
Gambar 4.8 Kromatogram blangko plasma pada perbandingan metanol dalam plasma 4:1
dengan fase gerak metanol-air (60:40v/v), kecepatan alir 1 mL/menit, panjang gelombang
308 nm dan volume penyuntikan 20 µL.
Keterangan: A. Pengotor plasma dan B. EPMS Gambar 4.9 Kromatogram EPMS pada perbandingan metanol dalam plasma 4:1 dengan fase gerak metanol-air (60:40v/v), kecepatan alir 1 mL/menit, panjang gelombang 308 nm dan
volume penyuntikan 20 µL.
Keterangan: A. Pengotor plasma dan B. EPMS Gambar 4.7 Kromatogram EPMS pada perbandingan metanol dalam plasma 1:1 dengan fase gerak metanol-air (60:40v/v), kecepatan alir 1 mL/menit, panjang gelombang 308 nm dan
volume penyuntikan 20 µL.
A
B A
37
setengah atau seperempatnya, sehingga nilai LLOQ yang diperoleh dari setengah atau seperempat LOQ adalah sekitar 2,02 µg/mL. Setelah nilai LLOQ diperoleh, maka dilakukan pembuatan kurva kalibrasi dan uji linearitas EPMS dalam plasma dengan rentang konsentrasi 2,02-40,4 µg/mL dimana didalamnya terdapat konsentrasi LLOQ.
4.3.2 Pembuatan Kurva Kalibrasi
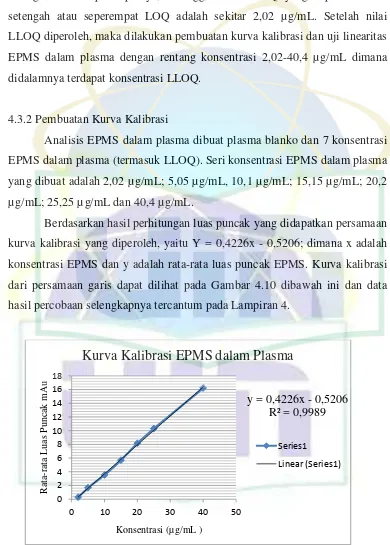
Analisis EPMS dalam plasma dibuat plasma blanko dan 7 konsentrasi EPMS dalam plasma (termasuk LLOQ). Seri konsentrasi EPMS dalam plasma yang dibuat adalah 2,02 µg/mL; 5,05 µg/mL, 10,1 µg/mL; 15,15 µg/mL; 20,2 µg/mL; 25,25 µg/mL dan 40,4 µg/mL.
Berdasarkan hasil perhitungan luas puncak yang didapatkan persamaan kurva kalibrasi yang diperoleh, yaitu Y = 0,4226x - 0,5206; dimana x adalah konsentrasi EPMS dan y adalah rata-rata luas puncak EPMS. Kurva kalibrasi dari persamaan garis dapat dilihat pada Gambar 4.10 dibawah ini dan data hasil percobaan selengkapnya tercantum pada Lampiran 4.
Gambar 4.10 Kurva kalibrasi EPMS dalam plasma
Uji Linearitas diperoleh dari kurva kalibrasi EPMS dalam plasma. Linieritas EPMS dalam plasma dapat dilihat pada Gambar 4.10. Metode analisis EPMS dalam plasma dengan rentang konsentrasi 2,02-40,4 µg/mL
y = 0,4226x - 0,5206
Kurva Kalibrasi EPMS dalam Plasma
Series1
38
UIN Syarif Hidayatullah Jakarta
memenuhi kriteria uji linearitas dan dapat diterima untuk suatu metode analisis yang valid dikarenakan dalam pembuatan kurva kalibrasi, diperoleh persamaan regresi linier Y = 0,4226x-0,5206 dengan koefesien korelasi r = 0,9989. Dimana kriteria linearitas untuk sediaan dalam matriks biologis adalah r = 0,95 atau lebih (FDA, 1998). Maka dapat disimpulkan bahwa metode telah memenuhi persyaratan kurva kalibrasi yang baik.
Dengan menggunakan kurva kalibrasi diatas, kemudian dihitung Limit Deteksi (LOD) dan Limit Kuantifikasi (LOQ). LOD adalah konsentrasi analit terendah dalam sampel yang masih dapat dideteksi, meskipun tidak selalu dapat dikuantifikasi. LOQ adalah kosentrasi analit terendah dalam sampel yang dapat ditentukan dengan presisi dan akurasi yang dapat diterima pada kondisi operasional metode yang digunakan. Nilai LOD yang didapatkan adalah 1,4511 µg/mL dan nilai LOQ yang didapatkan adalah 4,8370 µg/mL. Data perhitungan nilai LOD dan LOQ dapat dapat dilihat selengkapnya pada Lampiran 5.
4.3.3 Uji Selektivitas
Selektivitas dilakukan untuk mengetahui bahwa metode yang ditetapkan dapat membedakan dan mengukur kadar analit dengan adanya komponen-komponen lain dalam sampel (cairan biologis). Persyaratan untuk uji selektivitas yaitu %KV dengan nilai tidak lebih dari 15%, kecuali LLOQ dimana nilai KV tidak boleh lebih dari 20% dan akurasi nya (%diff) dengan nilai + 15% dari nilai sebenarnya (FDA, 2001).