METHOD COMPARATION AND VERIFICATION OF TOTAL CARBOHYDRATE
ANALYSIS WITH LUFF-SCHOORL AND ANTHRONE SULFURIC ACID
Manikharda, Hanifah Nuryani Lioe and Dian Herawati
Department of Food Science and Technology, Faculty of Agricultural Technology, Bogor Agricultural University, IPB Darmaga Campus, PO Box 16680, Bogor, West Java,
Indonesia.
Phone +62852 13 374 396, E-mail : [email protected]
ABSTRACT
Carbohydrate plays crucial role in food industry. Therefore an accurate, direct and reliable carbohydrate analysis is needed. Among many colorimetric methods for carbohydrate determination, the Anthrone-sulfuric acid is the most commonly used. The Anthrone-sulfuric method for carbohydrate analysis is simple and sensitive. However, the SNI official method for carbohydrate analysis employing the Luff-Schoorl method which is time consuming, difficult for untrained staff and the reduction reactions are seldom stoichiometric. Therefore a new candidate method employing Anthrone sulfuric acid was proposed to replace the SNI 01-2891-1992 total carbohydrate analysis.
In this research both methods were compared using three matrices which represent general food matrices in liquid form based on AOAC proposed triangle scheme. Samples from the low, medium and high content of carbohydrate from the triangle scheme were selected. The selected samples were coconut milk, soy sauce and sweet soy sauce. Based on the comparation result, Anthrone method as a new proposed method proved ineligible to replace the SNI 01-2891-1992. Thus the next step taken was to verify the SNI 01-2891-1992 method through its repeatability and accuracy. Accuracy was accessed using reference material and standard addition. The repeatability showed acceptable precision. But the standard addition exhibited poor recovery value in SNI 01-2891-1992 method of total carbohydrate.
I.
PENDAHULUAN
1.1.
Latar Belakang
Karbohidrat merupakan komponen yang sering kita jumpai dalam bahan pangan. Karbohidrat dalam pangan ada dalam berbagai macam bentuk dari glukosa sederhana hingga bentuk polisakarida yang kompleks. Contoh bahan pangan yang banyak mengandung karbohidrat diantaranya serealia dan umbi-umbian. Karbohidrat berkontribusi besar dalam menyusun produk pangan pada umumnya (Fennema 1996) dan merupakan salah satu makronutrien yang dibutuhkan oleh tubuh. Lebih dari 70% kebutuhan energi manusia dipenuhi dengan karbohidrat (BeMiller 2010). Sifat fungsional karbohidrat yang penting dalam proses pengolahan pangan, menyebabkan keberadaan karbohidrat menjadi komponen yang perlu diperhatikan dan dianalisis.
Analisis total karbohidrat telah lama dilakukan pada berbagai sampel seperti ekstrak tanaman (Yemm dan Willis 1954), tanah (Safarik dan Satruckova 1992), feses (Ameen and Powell 1985), produk farmasi (Leyva et al 2007) dan produk pangan (BeMiller 2009). Jumlah karbohidrat dalam produk pangan perlu diketahui, antara lain untuk: standardisasi identitas pangan, label nutrisi, deteksi adanya adulterasi dan untuk pengembangan suatu produk pangan. Peran karbohidrat yang signifikan terutama dalam produk pangan menjadikan analisis total karbohidrat penting.
Pengukuran karbohidrat sejak dahulu hingga sekarang masih dilakukan adalah menggunakan metode by difference dalam sistem analisis proksimat Weende yaitu dengan mengurangi kadar air, kadar protein, kadar lemak dan kadar abu dari total bahan pangan yang diujikan (Southgate 1976). Akan tetapi pada metode by difference terdapat kelemahan yaitu dapat menyebabkan hasil yang kurang akurat. Hasil yang kurang akurat diakibatkan oleh akumulasi dari kesalahan pada metode yang digunakan untuk menganalisis komponen lain, seperti protein dan lemak, sehingga nilai yang didapat semakin jauh dari nilai sebenarnya. Selain itu juga ada kemungkinan komponen nonkarbohidrat seperti asam organik, lignin dan tanin ikut terhitung sebagai karbohidrat.
metode refraktometri, gravimetri, polarimetri, titrimetri dan kolorimetri kondensasi (Southgate 1976). Banyaknya metode analisis yang dikembangkan tentu dapat menimbulkan kebingungan karena setiap metode dapat menghasilkan nilai yang berbeda. Dengan demikian, perlu ditetapkan persetujuan untuk menggunakan satu metode.
Metode yang digunakan untuk analisis total karbohidrat langsung yang ditetapkan oleh BSN (Badan Standardisasi Nasional) melalui SNI 01-2891-1992, yaitu tentang cara uji makanan dan minuman, adalah Metode Luff-Schoorl. Namun terdapat kelemahan pada Metode Luff-Schoorl karena dapat menimbulkan hasil yang kurang konsisten (Faulks dan Timms 1985) sehingga tingkat kepercayaan terhadap hasil kurang. Selain itu Metode Luff-Schoorl juga membutuhkan pekerjaan yang tidak sederhana dan lebih banyak memakan waktu dibanding metode analisis kolorimetri.
Beberapa metode yang digunakan untuk menganalisis total karbohidrat secara langsung selain Luff–Schoorl, yaitu Metode Anthrone sulfat, fenol sulfat, orsinol dan resorsinol. Metode Anthrone sulfat adalah yang paling umum digunakan (Leyva et al 2008) dengan menggunakan instrument spektofotometer UV-Visible. Metode Anthrone ini memiliki banyak keunggulan antara lain kesederhanaan ujinya, spektrumnya yang luas dan sensitifitasnya yang cukup baik (Koehler 1952).
Analis pangan sampai sekarang masih terikat dengan prosedur analisis yang telah ditetapkan oleh peraturan yaitu SNI (Standard Nasional Indonesia) 01-2891-1992. Penggunaan metode yang baku merupakan hal yang penting untuk menjamin bahwa hasil yang diperoleh sesuai dengan persyaratan yang ditetapkan oleh pemerintah (Nielsen, 2010). Beberapa metode analisis pangan bersifat empiris yaitu metode itu masih digunakan hingga saat ini karena memang metode itu yang sudah digunakan sejak dulu dan hasil yang didapat cukup konsisten (Sawyer 1984). Begitu pula halnya dengan Metode Luff-Schoorl yang dijadikan metode standard dalam SNI 01-2891-1992 karena sifatnya yang empiris.
Metode analisis total karbohidrat dengan menggunakan Metode Anthrone sulfat bukan merupakan metode standard, maka perlu divalidasi sebelum digunakan. Selain itu, validasi metode terutama untuk matriks pangan yang spesifik penting untuk menjamin ketepatan dari metode yang digunakan (Nielsen, 2010). Dengan adanya validasi, kita dapat mengetahui bahwa hasil dari analisis itu dapat dipercaya pada matriks pangan yang dianalisis.
pangan cair dan belum diketahui validitas Metode Anthrone sulfat dengan hidrolisis asam untuk menganalisis karbohidrat total secara langsung terutama pada matriks pangan cair untuk dapat menggantikan Metode Luff-Schoorl. Oleh karena itu tujuan dari penelitian ini adalah membandingkan kedua metode pada matriks pangan cair dengan tingkat karbohidrat rendah, sedang dan tinggi dan menentukan metode mana yang lebih baik untuk digunakan dalam analisis rutin dan melakukan validasi Metode Anthrone atau verifikasi metode yang sudah baku yaitu Luff Schoorl berdasarkan hasil perbandingan metode.
1.2.
Tujuan
1.2.1. Tujuan Umum
Tujuan umum dari penelitian ini adalah menentukan metode yang lebih baik untuk analisis total karbohidrat antara metode SNI 01-2891-1992 secara titrimetri dan metode kandidat dengan Anthrone sulfat secara spektrofotometri.
1.2.2.
Tujuan Khusus
Tujuan khusus dari penelitian ini adalah:
1. Melakukan perbandingan hasil analisis total karbohidrat dengan menggunakan dua metode berbeda yaitu metode SNI 01-2891-1992 secara titrimetri dengan metode kandidat yang menggunakan Anthrone sulfat secara spektrofotometri.
2. Melakukan validasi Metode Anthrone sulfat atau verifikasi metode SNI berdasarkan hasil yang diperoleh dari perbandingan metode pada berbagai matriks.
1.3.
Manfaat Penelitian
Manfaat dari penelitian ini adalah:
1. Mendapatkan informasi mengenai metode analisis mana yang lebih baik untuk digunakan pada analisis total karbohidrat secara rutin.
2. Mendapatkan informasi mengenai tingkat validitas metode yang digunakan
1.4.
Hipotesis
II.
TINJAUAN PUSTAKA
2.1.
Karbohidrat
Kebanyakan ahli kimia kesulitan dalam mengelompokkan bahan apa saja yang termasuk ke dalam karbohidrat. Definisi klasik karbohidrat berdasarkan asal katanya yaitu carbo dari bahasa Latin dan hydros dari bahasa Yunani adalah ‘hidrat dari karbon’ yang mengandung hidrogen dan oksigen dengan perbandingan 2:1 (Southgate 1978) atau elemen yang terdiri dari air dan karbon dengan perbandingan 1:1 (Kennedy dan White 1988). Karbohidrat adalah senyawa organik yang mengandung karbon, hidrogen dan oksigen baik dalam bentuk molekul sederhana maupun kompleks (Christian dan Vaclavik 2003).
Karbohidrat telah menjadi sumber energi utama untuk metabolisme pada manusia dan sarana untuk memelihara kesehatan saluran pencernaaan manusia. Karbohidrat adalah penyumbang utama dari komponen yang membentuk produk pangan baik sebagai komponen alami maupun bahan yang ditambahkan. Karbohidrat meliputi lebih dari 90% dari berat kering tanaman. Karbohidrat banyak tersedia dan murah. Penggunaannya sangat luas dan jumlah penggunaannya cukup besar (Fennema 1996) baik untuk pemanis, pengental, penstabil, gelling agents dan fat replacer (Christian dan Vaclavik 2003). Karbohidrat dapat dimodifikasi baik secara kimia dan biokimia dan modifikasi itu digunakan untuk memperbaiki sifat dan memperluas penggunaannya.
2.1.1
Struktur karbohidrat
Karbohidrat digunakan dalam kimia untuk senyawa dengan formula Cm(H2O)n, tetapi kini rumus molekul itu tidak secara kaku digunakan untuk mendefinisikan karbohidrat (Kennedy dan White 1988). Sebelumnya beberapa ahli kimia memasukkan formaldehid dan glikoaldehid sebagai karbohidrat, namun sekarang istilah karbohidrat dalam biokimia, tidak mengikutsertakan senyawa yang kurang dari tiga atom karbon. Southgate (1978) menggunakan definisi karbohidrat sebagai senyawa yang tersusun oleh polihidroksi aldehid, keton, alkohol, asam dan turunan sederhananya serta polimernya yang memiliki ikatan polimer tipe asetal.
2.1.2. Monosakarida
Monosakarida terdiri dari tiga sampai delapan karbon atom, tetapi umumnya hanya lima atau enam yang biasa ditemukan. Biasanya monosakarida digolongkan berdasarkan jumlah atom karbonnya, misalnya triosa (C3H6O3), tetrosa (C4H8O3), pentosa (C5H10O5) dan heksosa (C6H12O6). Dari golongan tersebut dapat dibagi lagi berdasarkan gugus fungsional yang ada, misalnya dari golongan heksosa ada aminoheksosa (C6H13O5N), deoksiheksosa (C6H12O5) dan asam heksuronat (C6H10O7). Contoh monosakarida adalah glukosa dan fruktosa.
2.1.3. Oligosakarida
Oligosakarida terdiri dari beberapa monosakarida (2-10) yang saling terikat oleh ikatan glikosidik. Tetapi ada juga yang mengklasifikasikan sendiri karbohidrat dengan dua gugus gula sebagai disakarida. Menurut Christian dan Vaclavik (2003) disakarida terdiri dari dua molekul monosakarida yang bergabung dengan ikatan glikosidik. Contoh disakarida di pangan adalah maltosa, selubiosa, dan sukrosa. Oligosakarida yang memiliki lebih dari tiga gugus gula contohnya adalah rafinosa dan stakiosa.
2.1.4. Polisakarida
Polisakarida merupakan polimer dari gula sederhana yang tersusun atas lebih dari sepuluh monomer gula sederhana. Contoh polisakarida di makanan adalah pati, pektin dan gum. Ketiganya adalah polimer karbohidrat kompleks dengan sifat yang berbeda, tergantung unit gula penyusunnya, tipe ikatan glikosidik dan derajat percabangan molekul.
2.2. Pentingnya Analisis Total Karbohidrat
2.3. Total Karbohidrat dalam Bahan Pangan dan Metode Analisisnya
2.3.1. Definisi total karbohidrat
Total karbohidrat atau total karbohidrat menurut Badan Pengawasan Obat dan Makanan (2005) meliputi gula, pati, serat pangan dan komponen karbohidrat lain. Pernyataan jumlah total karbohidrat dalam gram penyajian yang dinyatakan dengan nilai gram terdekat, jika penyajian kurang dari 0,5 gram, jumlah kadarnya dapat dinyatakan sebagai nol dan jika penyajian lebih dari 0,5 gram dibulatkan ke kelipatan 1 gram terdekat. Total karbohidrat dapat dinyatakan dengan total karbohidrat by difference.
Total karbohidrat dalam pengukuran karbohidrat dengan metode langsung dinyatakan dalam bentuk persen yang setara dengan glukosa. Satuan glukosa (glucose equivalent) juga dapat diganti dengan larutan gula lain yang dijadikan sebagai larutan standar.
2.3.2. Metode analisis total karbohidrat
Sejumlah teknik analisis telah dikembangkan untuk mengukur jumlah dan tipe karbohidrat yang ada di bahan pangan. Kadar karbohidrat di bahan pangan dapat diketahui dengan menghitung persentase yang tersisa setelah semua komponen lain telah diukur (total carbohydrate by difference), yaitu dengan persamaan (1.1) (SNI 01-2891-1992):
(1.1)
Metode by differenceini masih digunakan oleh FDA, tetapi metode ini dapat menghasilkan nilai yang salah karena ada kemungkinan terjadi akumulasi kesalahan dari metode-metode yang digunakan untuk mengukur komponen lain, dan kemungkinan adanya komponen non karbohidrat yang terukur sebagai karbohidrat menyebabkan penyimpangan yang lebih besar. Pengukuran kadar karbohidrat secara langsung lebih baik karena didapat hasil lebih yang akurat.
2.3.2.1. Analisis karbohidrat langsung
Uji karbohidrat yang resmi ditetapkan oleh BSN dalam SNI 01-2891-1992 yaitu analisis total karbohidrat dengan menggunakan metode Luff Schoorl. Pada tahun 1936 International Commission for Uniform Methods of Sugar Analysis mempertimbangkan Metode Luff-Schoorl sebagai salah satu metode yang digunakan untuk menstandarkan analisis gula pereduksi karena metode Luff Schoorl saat itu menjadi metode yang resmi dipakai di pulau Jawa, di samping nominator lainnya yaitu metode Lane-Eynon. Tetapi pada saat itu metode kolorimetri belum banyak berkembang dan dalam catatan komisi itu terdapat agenda untuk melakukan penyeragaman analisis gula dengan metode kolorimetri.
Berikut ini adalah beberapa jenis analisis total karbohidrat langsung:
2.3.2.1.1. Analisis total karbohidrat dalam SNI 01-2891-1992
Seluruh senyawa karbohidrat yang ada dipecah menjadi gula-gula sederhana (monosakarida) dengan bantuan asam yaitu HCl dan panas. Monosakarida yang terbentuk kemudian dianalisis dengan Metode Luff-Schoorl. Prinsip analisis dengan Metode Luff-Schoorl yaitu reduksi Cu2+ menjadi Cu 1+ oleh monosakarida. Monosakarida bebas akan mereduksi larutan basa dari garam logam menjadi bentuk oksida atau bentuk bebasnya. Kelebihan Cu2+ yang tidak tereduksi kemudian dikuantifikasi dengan titrasi iodometri (SNI 01-2891-1992).
Reaksi yang terjadi (1.2):
Karbohidrat kompleks→ gula sederhana (gula pereduksi) Gula pereduksi+ 2 Cu2+→ Cu2O(s)
2 Cu2+(kelebihan) + 4 I-→ 2 CuI2→ 2 CuI-+ I2 I2+ 2S2O32-→ 2 I-+ S4O6
2-(1.2) Osborne dan Voogt (1978) mengatakan bahwa Metode Luff-Schoorl dapat diaplikasikan untuk produk pangan yang mengandung gula dengan bobot molekuler yang rendah dan pati alami atau modifikasi.
2.3.2.1.2.
Analisis total karbohidrat dengan Metode Anthrone sulfat
Penggunaan Metode Anthrone untuk analisis total karbohidrat mulai berkembang sejak penggunaan pertama kali oleh Dreywood pada tahun 1946 untuk uji kualitatif. Dasar dari reaksi ini adalah kemampuan karbohidrat untuk membentuk turunan furfural dengan keberadaan asam dan panas, yang kemudian diikuti dengan reaksi dengan anthrone yang menghasilkan warna biru kehijauan (Sattler dan Zerban 1948) dalam Brooks et al (1986).
Anthrone, C6H4COC6H4CH2, adalah turunan dari anthraquinone. Senyawa ini diproduksi oleh reduksi katalitik dari anthraquinone oleh asam hidroklorat dengan keberadaan logam timah. Senyawa ini mungkin ada dalam bentuk keto atau enol, yang masing-masing dikenal dengan nama anthrone and anthranol. Reaksinya dapat dilihat pada persamaan (1.3):
(1.3)
Mekanisme pembentukan warna anthrone dengan gula telah diteliti. Hurd dan Isenhour (1932) dan Wolfrom et al (1948) mempostulasikan bahwa karbohidrat dan turunannya mengalami pembentukan cincin dalam keberadaan asam kuat dari mineral, seperti yang ditunjukkan untuk glukosa (1.4):and Zerban (1948) menyarankan bahwa pembentukan warna hijau pada reaksi anthrone tergantung oleh keberadaan 5-(hidroksimetil)-2-furaldehid, atau senyawa furfural yang mirip, yang dibentuk oleh reaksi asam sulfat pada karbohidrat.
Momose et al. (1957) melakukan kromatografi pada ekstrak benzene dari pewarna terhadap alumina dan menunjukkan bahwa bagian yang dapat larut dari benzene-terdiri dari beberapa pewarna yang memberikan pewarnaan yang berbeda dengan asam sulfat. Mereka menentukan berat molekul dari salah satu pewarna utama yaitu kurang lebih 530, dan mempostulasikan formula dari pewarna itu (C47H30O3). Mereka menyimpulkan bahwa 3 mol anthrone bereaksi dengan 1 mol glukosa, yang digambarkan dalam persamaan (1.5):
3C
14H
10O + C
6H
12O
6
C
47H
3O
30+ 5H
2O + CH
2O
(1.5)
Dari data analisis dan spektrum inframerah dari pewarna, dan mekanisme reaksinya dipertimbangkan, mereka menduga struktur yang mungkin adalah 1,2,5,- atau 1,3,5,-trianthronylidenepentane.
Ludwig dan Goldberg (1956) melaporkan adaptasi dari Metode Anthrone kolorimetri untuk analisis total karbohidrat secara kuantitatif pada pangan. Metode yang digunakan relatif cepat dan akurat serta lebih baik daripada metodologi analisis karbohidrat sebelumnya, yaitu metode Somogyi-Shaffer-Hartmann yang menggunakan teknik teknik iodometri dan prinsip gula pereduksi. Mereka menunjukkan bahwa persiapan hidrolisis dan deproteinisasi tidak perlu dilakukan ketika teknik anthrone digunakan.
Kekurangan dari Metode Anthrone adalah ketidakstabilan dari reagen (anthrone yang dilarutkan dalam asam sulfat), sehingga perlu dilakukan persiapan reagen yang baru setiap hari. Dreywood (1946) memperhatikan bahwa panas yang dihasilkan oleh pelarutan asam sulfat merupakan bagian yang penting dalam uji. Morris (1948) melihat signifikansi dari panas pada reaksi anthrone dan menunjukkan bahwa pada sejumlah karbohidrat yang diberikan, intensitas warna bervariasi dengan jumlah panas yang dihasilkan. Oleh karena itu kurva standar juga perlu dibuat setiap hari.
Nilai total karbohidrat tidak dapat dinyatakan dalam persen karbohidrat, tetapi lebih baik dinyatakan dengan istilah glucose equivalents per cent, karena kepekatan warna yang dihasilkan dari reaksi anthrone bervariasi dengan tipe gula yang ada. Kepekatan warna yang sama contohnya, ditunjukkan oleh 100 µg. glukosa, 105 µg. maltosa, dan 111 µg glikogen. Gula murni lain selain glukosa dapat dikalkulasi dengan faktor konversi. Tetapi jika terdapat campuran karbohidrat yang tidak diketahui pada bahan pangan faktor konversi itu tidak dapat digunakan, dan hasilnya bukan persentase karbohidrat absolut, melainkan ekuivalen glukosa, yang dapat bervariasi dari nilai persentasi karbohidrat yang sebenarnya dengan jumlah yang tidak dapat ditentukan. Keganjilan ini tidak signifikan ketika nilai glucose equivalents per cent digunakan hanya sebagai basis untuk mengkonversi nilai total karbohidrat menjadi nilai total pangan (Beck dan Bibby 1961). Untuk tujuan ini glucose equivalents per cent hanya sebagai indeks dari persentasi absolute dari masing-masing karbohidrat dalam pangan.
2.4. Validasi dan Verifikasi Metode
Metode analisis memiliki beberapa atribut, seperti ketepatan, ketelitian, spesifisitas, sensitivitas, kemandirian, dan kepraktisan, yang harus dipertimbangkan ketika akan digunakan (Garfield et al. 2000). Informasi yang digunakan untuk mengambil keputusan harus seimbang dengan pertimbangan praktis seperti biaya, waktu, risiko, kesalahan, dan tingkat keahlian yang diperlukan. Selain itu suatu laboratorium yang akan menerapkan suatu metode perlu mempertimbangkan apakah data validasi yang ada mengenai metode tersebut cukup memadai atau apakah masih membutuhkan tindakan validasi ulang sebelum metode itu digunakan. Selanjutnya jika data validasi telah cukup memadai, laboratorium perlu mengetahui apakah level performa yang ditunjukkan oleh data validasi tersebut mampu dilaksanakan. Untuk mencapai level performa itu dibutuhkan analis yang kompeten serta peralatan dan fasilitas yang memadai (Jelita 2011).
oleh laboratorium lain atau metode yang telah dipublikasi tetapi belum menjadi metode baku. Ketika data validasi yang ada telah memadai, yaitu seperti pada metode yang telah divalidasi oleh organisasi terstandarisasi seperti AOAC (Association of Official Analytical Chemists) Internasional, laboratorium umumnya hanya menjaga performa data dengan cara melakukan verifikasi metode.
Validasi metode analisis adalah suatu tindakan penilaian terhadap parameter tertentu, berdasarkan percobaan laboratorium, untuk membuktikan bahwa parameter tersebut memenuhi persyaratan untuk penggunaannya (Harmita, 2004). Berdasarkan Harvey (2000), validasi merupakan suatu proses evaluasi kecermatan dan keseksamaan yang dihasilkan oleh suatu prosedur dengan nilai yang dapat diterima. Sebagai tambahan, validasi memastikan bahwa suatu prosedur tertulis memiliki detail yang cukup jelas sehingga dapat dilaksanakan oleh analis atau laboratorium yang berbeda dengan hasil yang sebanding. Menurut AOAC (2002) validasi metode menunjukkan apakah suatu metode sesuai dengan tujuan yang diinginkan. Dalam praktiknya, memungkinkan untuk merancang percobaan yang akan dilakukan sehingga karakteristik validasi yang sesuai dapat diterapkan untuk mendapatkan hasil yang cukup dan menyeluruh mengenai kemampuan suatu prosedur analisis, seperti: spesifisitas, linearitas, rentang, akurasi (kecermatan), dan presisi (keseksamaan) (EMA, 1995).
Verifikasi metode adalah suatu tindakan validasi metode tetapi hanya pada beberapa beberapa karakteristik performa saja. Laboratorium harus menentukan karakteristik performa yang dibutuhkan. Spesifikasi analisis dapat menjadi acuan untuk merancang proses verifikasi. Rancangan yang baik akan menghasilkan informasi yang dibutuhkan serta meminimalisir tenaga, waktu, serta biaya. Pemilihan parameter validasi atau verifikasi tergantung pada beberapa faktor seperti aplikasi, sampel uji, tujuan metode, dan peraturan lokal atau internasional.
Adapun beberapa parameter analisis yang harus dipertimbangkan dalam validasi metode analisis :
2.4.1. Akurasi
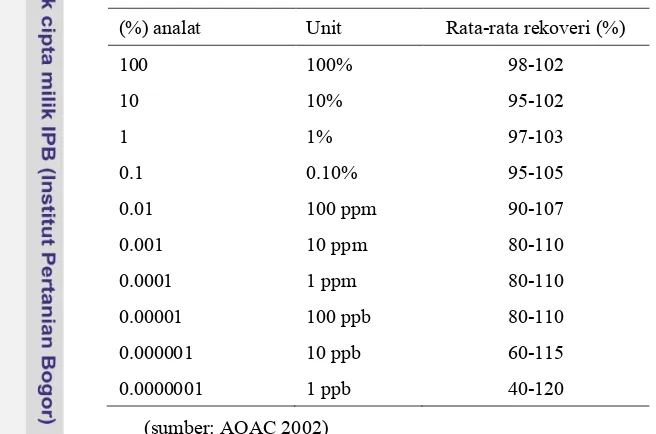
hasilnya dengan hasil yang dilakukan oleh lab lain (Smith, 2010) atau dengan menggunakan metode referen (Walton 2001). Akurasi juga dapat diketahui dengan melakukan uji rekoveri (Walton 2001). Hasil uji ini akurasi dapat dinyatakan sebagai persen perolehan kembali (recovery) analat yang ditambahkan pada sampel. Sampel ditambahkan (spiking) dengan standar yang telah diketahui jumlah dan kadarnya (EMA, 1995). Rentang nilai penerimaan kecermatan suatu metode akan bervariasi sesuai kebutuhannya (FAO, 1998). Adapun AOAC menetapkannya seperti dalam Tabel 1.
Tabel 1 Persentase rekoveri yang dapat diterima sesuai dengan konsentrasi analat
(%) analat Unit Rata-rata rekoveri (%)
100 100% 98-102
10 10% 95-102
1 1% 97-103
0.1 0.10% 95-105
0.01 100 ppm 90-107
0.001 10 ppm 80-110
0.0001 1 ppm 80-110
0.00001 100 ppb 80-110
0.000001 10 ppb 60-115
0.0000001 1 ppb 40-120
(sumber: AOAC 2002)
2.4.2. Presisi
Presisi dalam hal ripitabilitas diukur dengan menghitung relative standard deviation atau simpangan baku relatif (RSD) dari beberapa ulangan dengan menggunakan rumus (1.6):
(1.6)
Standar deviasi ripitabilitas bervariasi tergantung pada konsentrasi (AOAC 2002). Oleh karena itu hasil yang didapat dari perhitungan dibandingkan hasilnya dengan nilai yang ada di Tabel 2.
Tabel 2 Nilai presisi (RSD) sesuai dengan konsentrasi analat (%) analat Konsentrasi RSD (%)
100 100% 1
10 10% 1.5
1 1% 2
0.1 0.10% 3
0.01 100 ppm 4
0.001 10 ppm 6
0.0001 1 ppm 8
0.00001 10 ppb 15
(sumber: AOAC 2002)
Nilai yang didapat juga dapat dibandingkan atau dengan menggunakan rumus (1.7):
(1.7)
dengan C adalah konsentrasi yang didapat dari rataan.
Nilai yang dapat diterima untuk ripitabilitas adalah antara 1/2 dan 2 kali dari nilai yang dijadikan sebagai pembanding. Ada juga yang menggunakan RSD Horwitz sebagai nilai pembanding, RSD Horwitz dihitung dengan rumus (1.8):
(1.8)
2.4.3. Spesifisitas
Spesifisitas dari metode analitik tertentu berarti metode itu hanya mendeteksi komponen yang diinginkan. Metode analitis dapat bersifat sangat spesifik untuk komponen tertentu atau pada beberapa kasus dapat menganalisis spektrum komponen yang luas (Smith, 2010).
Spesifisitas suatu metode diuji dengan membandingkan hasil dari sampel yang mengandung pengotor dengan hasil sampel yang tidak mengandung pengotor. Pada dasarnya, spesifisitas dapat diuji secara langsung atau tidak langsung. Pendekatan secara tidak langsung ditinjau dari penerimaan parameter akurasi. Pendekatan secara langsung ditinjau dari keberadaan komponen pengganggu (Ermer dalam Ermer dan Miller, 2005). Cara yang terakhir dilakukan dengan menambahkan sejumlah tertentu komponen pengganggu pada larutan standar murni. Jika diperkirakan tidak adanya komponen pengganggu pada sampel, spesifisitas dapat ditunjukkan dengan membandingkan hasil uji sampel dengan standar (EMA, 1995).
2.4.4. Limit Deteksi dan Limit Kuantitasi
Limit deteksi atau Limit of Detection(LOD) suatu metode analisis adalah jumlah terkecil dari analat yang dapat dideteksi namun jumlah ini belum tentu dapat dikuantisasi dengan presisi yang baik oleh metode tersebut. Limit kuantitasi atau Limit of Quantitation (LOQ) yang disebut juga limit determinasi adalah konsentrasi terendah dari analat yang dapat ditentukan secara kuantitatif dengan presisi dan akurasi yang dapat diterima (Ermer dalam Ermer dan Miller, 2005).
Giese (2004) menyatakan bahwa terdapat dua cara untuk menentukan LOD dan LOQ, yaitu dengan menentukan kurva kalibrasi menggunakan sepuluh level konsentrasi, atau melakukan analisis blanko berulang. Tetapi ada masalah dalam pendekatan menggunakan blanko karena seringkali sulit diukur dan variasinya sangat tinggi. Lebih lanjut, nilai yang didapat dengan pendekatan seperti ini tidak bergantung dari analat (AOAC 2002).
Limit deteksi hanya berguna untuk mengontrol ketidakmurnian yang tidak diinginkan yang konsentrasinya harus tidak lebih dari level tertentu dan mengontrol kontaminan dengan konsentrasi rendah, sedangkan materi yang bermanfaat harus ada pada konsentrasi yang cukup tinggi agar dapat menjadi fungsional. Limit deteksi dan determinasi seringkali bergantung pada kemampuan instrumen (AOAC 2002).
2.4.5. Linieritas
dengan konsentrasi analat dalam sampel pada range tertentu (Leyva et al 2008). Linieritas dapat diuji secara informal dengan membuat plot residual yang dihasilkan oleh regresi linier pada respon konsentrasi dalam satu seri kalibrasi (Thompson et al. 2002).
Linieritas harus dievaluasi dengan pemeriksaan visual terhadap plot absorbansi yang merupakan fungsi dari konsentrasi analat. Jika hubungannya linier, hasil uji dievaluasi lebih lanjut secara statistik dengan perhitungan garis regresi. Dalam penentuan linieritas, sebaiknya menggunakan minimum lima konsentrasi (EMA, 1995). Rentang penerimaan linieritas tergantung dari tujuan pengujian. Pada kondisi yang umum, nilai koefisien regresi (r2) ≥ 0,99.
2.5. Matriks Sampel
Suatu metode harus dapat menunjukkan rekoveri dan ripitabilitas yang dapat diterima pada konsentrasi dan matriks yang mewakili kelompok sampel dimana metode itu hendak diterapkan (AOAC 2002). Suatu metode yang hendak diterapkan pada “pangan” secara umum, metode tersebut perlu diujikan pada jenis pangan yang dianggap mewakili kelompok pangan secara umum. Sampel yang yang dianggap mewakili dapat dipilih berdasarkan skema segitiga atau
Gambar 1. Matriks pangan berdasarkan kadar protein, lemak dan karbohidrat (Nielsen 2010).
Nielsen (2010) mengatakan bahwa kemampuan suatu metode analisis dipengaruhi oleh matriks pangan (misalnya komponen dari pangan tersebut terutama lemak, protein dan karbohidrat). Matriks pangan merupakan tantangan terbesar bagi para analis pangan. Makanan dengan kadar lemak tinggi dan kadar gula tinggi dapat menghasilkan interferensi yang berbeda dengan makanan dengan kadar lemak rendah dan kadar gula rendah. Prosedur digesti dan tahap ekstraksi sangat penting bagi hasil analisis yang akurat. Hal ini tergantung pada matriks pangan. Kompleksitas dari berbagai sistem pangan seringkali membutuhkan lebih dari satu teknik dan prosedur untuk komponen spesifik tertentu, termasuk pengetahuan mengenai teknik mana yang sesuai untuk matriks pangan yang spesifik.
Metode analitik yang umum harus dapat menganalisis kesembilan kombinasi yang ada, menggantikan metode yang spesifik pada matriks tertentu (matrix dependent method). Misalnya dengan menggunakan metode yang dipengaruhi oleh matriks, kita mungkin dapat menggunakannya untuk menganalisis bahan yang rendah protein, dengan karbohidrat dan lemak sedang seperti coklat dan keripik kentang. Tetapi untuk bahan dengan protein tinggi, lemak rendah dan karbohidrat tinggi seperti susu rendah lemak, harus digunakan metode analisis yang lain. Hal ini cukup merepotkan dan kemungkinan nilai yang didapat dari hasil analisis kedua metode perlu dievaluasi (Nielsen 2010).
mengonfirmasi komposisi dari sampel. Berikut data mengenai sampel yang akan digunakan dalam perbandingan metode:
2.5.1. Kecap manis
Kecap manis merupakan produk olahan kedelai, yang teksturnya kental dan berwarna coklat kehitaman (Suprapti 2005). Komposisi kimia kecap manis dapat dilihat pada Tabel 3.
Tabel 3 Komposisi kimia kecap manis, kecap asin dan santan
Komponen
Kadar (%)
Kecap manis Kecap asin Santan
Air 29,61a 63, 84a 54,9c
Protein kasar 1,46a 6,55a 4,20b
Lemak 0,14a 0,35a 34,30b
Abu 7,64a 18,48a 1-1,3c
Karbohidrat 61,15a 10,78a 5,60b
Garam (NaCl) 6,27a 18,43a (tidak ada informasi)
Sumber: aJudoamidjojo (1987) , bDirektorat Gizi (1967), c Woodroof (1979)
Kandungan gula dan viskositas yang tinggi dari produk ini disebabkan karena penambahan gula dalam proses pembuatannya. Komponen terbesar kecap manis adalah karbohidrat, terutama sukrosa, glukosa dan fruktosa (Kusumadewi, 2011). Kandungan gula kecap manis, yaitu 26-61%, lebih banyak dari kecap asin yang hanya 4-19% (Judoamidjojo 1987). Kandungan asam amino yang cukup tinggi dari kecap manis karena salah satu bahan yang digunakan untuk membuatnya adalah kedelai yang memiliki kandungan protein yang tinggi (Santoso 1994). Rincian jenis asam amino kecap manis dapat dilihat pada Tabel 4.
Tabel 4. Kandungan asam amino kecap asin dan kecap manis (g/100g) Asam amino Kecap Asin Kecap Manis
Asam aspartat 0,42 0,03
Treonin 0,21 0,01
Serin 0,29 0,01
Glutamat 0,63 0,10
Prolin 0,16 0,01
Glisin 0,15 0,00
Alanin 0,30 0,02
Valin 0,30 0,02
Metionin 0,08 0,00
Isoleusin 0,29 0,02
Leusin 0,41 0,02
Tirosin 0,15 0,02
Fenilalanin 0,24 0,02
Lisin 0,27 0,01
Histidin 0,09 0,00
Arginin 0,27 0,00
Triptofan 0,00 0,00
Sistein 0,00 0,00
Sumber: Judoamidjojo et al (1985)
2.5.2. Kecap kedelai asin
Kecap kedelai asin atau yang biasa dikenal dengan nama kecap asin merupakan hasil fermentasi dari kedelai. Menurut definisi SNI 01-3543-994 kecap kedelai adalah produk cair yang diperoleh dari hasil fermentasi dan atau cara kimia (hidrolisis) kacang kedelai (Glycine max. L) dengan atau tanpa penambahan bahan makanan lain dan bahan tambahan makanan yang diizinkan. Warna dari kecap asin adalah coklat gelap. Tetapi warna ini bergantung pada proses penuaan atau
agingnya. Kecap asin mirip dengan kecap manis, hanya tanpa penambahan gula. Komposisi kimia dari kecap kedelai dapat dilihat dari Tabel 3 dan kandungan asam aminonya dapat dilihat pada Tabel 4.
2.5.3. Santan
distabilisasi secara alamiah oleh protein (globulin dan albumin) dan fosfolipida (Tangsuphoom dan Coupland, 2008). Senyawa δ-C8-laktone, δ-C10-laktone, dan n-oktanol merupakan komponen volatil utama dan memberikan karakteristik aroma pada santan kelapa (Lin dan Wilkens 2006),
Adapun komposisi kimia santan dapat dilihat di Tabel 3. Tetapi komposisi kimianya masih bervariasi tergantung pada varietas lokasi tumbuh, cara budi daya, kematangan buah, dan metode ekstraksi seperti jumlah penambahan air dan suhu ekstraksi. Menurut Seow dan Gwee (1997), komposisi kimia santan kelapa yang diekstraksi dengan tanpa penambahan air terdiri atas protein 2.6-4.4%; lemak 32-40%; air 50-54%; dan abu 1-1.5%.
2.5.4. Bahan Acuan
Semua metode instrumental membutuhkan bahan acuan, sekalipun untuk metode yang mengukur analat yang empiris. Analat yang empiris adalah analat yang nilainya tidak seperti senyawa kimia yang stoikiometris yang bersifat tetap. Analat empiris merupakan hasil dari penerapan prosedur yang biasa digunakan untuk mengukurnya, contohnya untuk kadar air, kadar abu, kadar lemak, kadar karbohidrat (by difference) dan kadar serat (AOAC 2002).
Bahan acuan memainkan peranan penting untuk mengetahui akurasi dalam melakukan validasi. Bahan acuan disini dapat diartikan sebagai bahan atau zat yang memiliki sifat-sifat tertentu yang cukup homogen dan stabil, yang telah ditetapkan untuk dapat digunakan dalam pengukuran atau dalam pengujian suatu contoh. Bahan acuan dapat digunakan untuk mengontrol presisi pengukuran walaupun bahan acuan tersebut tidak memiliki nilai acuan(assigned value), sedangkan untuk kalibrasi atau untuk mengontrol kebenaran pengukuran hanya bahan acuan yang memiliki nilai acuan yang dapat digunakan (Dara 2010). Kalibrasi dan pengontrolan analisis sangat penting, karena menyangkut kehandalan hasil pengujian. Untuk pengambilan keputusan yang krusial diperlukan hasil pengujian yang dapat dipercaya (Nuryatini 2010). Bahan acuan ini dapat diperoleh dari berbagai produsen bahan acuan seperti Puslit Kimia LIPI yang telah mengembangkan beberapa bahan acuan (in-house reference materials) khususnya untuk pengujian dalam bidang lingkungan dan pangan (Dara 2010).
penggunaan CRM masih dianjurkan, tetapi dengan disertai dengan pengujian tambahan. Jika diperlukan dan dapat dilakukan, sejumlah CRM yang sesuai dengan matriks dan konsentrasi analit sebaiknya diujikan (Thompson et al 2002).
III.
METODOLOGI PENELITIAN
3.1.
Bahan dan Alat
3.1.1
Bahan
Seluruh bahan kimia yang digunakan memiliki grade analitik. Asam sulfat terkonsentrasi (H2SO498%), reagen anthrone, KI, HCl 37%, Na2CO3, asam sitrat, standar glukosa, CH3COOH 100%, Na2S2O3.5H2O, heksana, HgO dan indikator pati berasal dari Merck, Jerman. Kalium dikromat (K2CrO7), Cu2SO4.5H2O, H3BO3,K2SO4dan NaOH berasal dari CICA, Jepang. Standar amilosa (potato amylose) berasal dari Sigma-Aldrich. Es, indikator fenolftalein, kapas bebas lemak dan air distilasi. Sampel matriks pangan cair yang digunakan untuk penelitian perbandingan metode analisis yaitu kecap asin, kecap manis dan santan. Selain itu juga untuk verifikasi digunakan sampel berupa bahan acuan tepung kedelai dan tepung kacang hijau yang diperoleh dari LIPI Kimia Bandung dan bahan acuan susu bubuk dari BBIA Bogor.
3.1.2. Alat
Alat yang digunakan pada penelitian ini adalah hot plate(Cimarec 3 Thermolyne USA), oven vakum (V0-7-3 Ogawa Seiki Japan), tanur (4800 Furnace Barnstead Thermolyne USA),
waterbath(Type 1008, GFL Gesselschaft fur Labortechnik mbH D-30938 Burgwedel Germany), kertas saring, alat ekstraksi soxhlet (kondensor dan pemanas listrik), labu lemak, desikator berisi bahan pengering, batang pengaduk, tabung reaksi, tabung reaksi bertutup, gelas piala, labu takar, baskom plastik, sudip, batang pengaduk, pipet tetes, pipet ukur, pH meter (Orion model 210 A, Thermo Electron Corp. USA), erlenmeyer, neraca analitik (Precisa XT 220A, Swiss), bulb, vortex, spektrofotometer (UV Mini 1240, UV-Vis Spectrophotometer, Shimadzu Japan), stopwatch, buret (volume 25 mL), cawan porselen, cawan alumunium dan labu Kjeldahl.
3.2. Metode Penelitian
3.2.1.
Penentuan matriks sampel
Penentuan matriks sampel dilakukan untuk mendapatkan sampel yang mewakili segitiga pangan. Selain itu juga digunakan untuk mendapatkan informasi mengenai komponen lain yang terdapat pada sampel yang akan digunakan.
3.2.1.1. Pemilihan sampel untuk uji perbandingan metode berdasarkan studi
literatur
Studi literatur dilakukan untuk memetakan beberapa sampel berdasarkan ke dalam skema segitiga matriks pangan. Dari hasil pemetaan akan dipilih sampel yang dapat mewakili matriks dengan kadar karbohidrat rendah, sedang dan tinggi.
3.2.1.2. Analisis proksimat
Hasil pemilihan sampel berdasarkan literatur dikonfirmasi komposisinya dengan analisis proksimat. Selain untuk konfirmasi, analisis proksimat juga berfungsi untuk identifikasi komponen yang ada dalam sampel. Analisis proksimat yang dilakukan meliputi kadar air, kadar lemak, kadar abu, kadar protein dan kadar karbohidrat menggunakan metode dari SNI 01-2891-1992 (Cara Uji Makanan dan Minuman).
3.2.2. Perbandingan metode
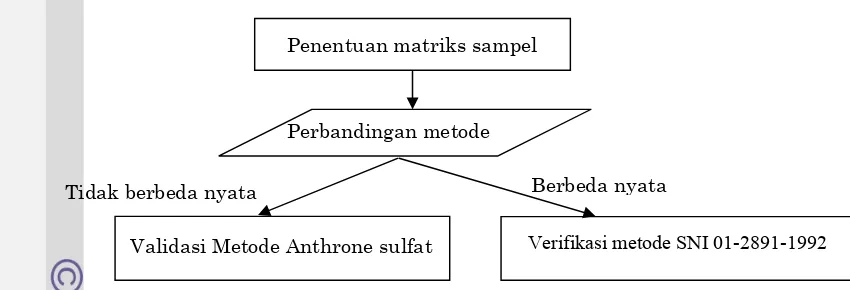
Perbandingan metode dilakukan untuk mengevaluasi sejauh mana kedua metode yang diperbandingkan menghasilkan kesesuaian nilai. Hasil dari perbandingan metode dapat digunakan untuk melihat apakah metode yang baru (metode kandidat) dapat menggantikan metode yang digunakan sebelumnya.
Sebanyak tiga kali ulangan dilakukan menggunakan metode kandidat dan metode SNI 01-2891-1992 pada tiga matriks yang telah ditentukan. Setelah itu hasil dari metode kandidat dan metode SNI 01-2891-1992 dibandingkan dan disesuaikan dengan data analisis proksimat.
Gambar 2.Tahapan penelitian validasi metode analisis karbohidrat
Validasi Metode Anthrone sulfat Verifikasi metode SNI 01-2891-1992
Tidak berbeda nyata Berbeda nyata
Penentuan matriks sampel
Perbandingannya meliputi uji varian (uji F), independent student t-testdan korelasi kedua metode dengan regresi linear. Jika hasil analisis metode kandidat tidak berbeda nyata dengan hasil analisis metode SNI 01-2891-1992 serta sesuai dengan hasil uji proksimat, maka akan dilakukan validasi metode kandidat. Jika hasil yang didapatkan berbeda jauh, maka akan dilakukan verifikasi pada metode SNI 01-2891-1992. .
3.2.3. Validasi Metode Anthrone sulfat
Validasi dilakukan pada matriks sampel yang terpilih yaitu sampel yang mewakili kadar karbohidrat rendah, kadar karbohidrat sedang dan kadar karbohidrat tinggi dan bahan acuan. Sampel dari matriks karbohidrat rendah, sedang dan tinggi diukur kadar karbohidratnya untuk mengetahui tingkat validitas dari Metode Anthrone sulfat. Penentuan tingkat validasi ini meliputi:
3.2.3.1. Presisi
Ripitabilitas merupakan salah satu aspek presisi yang menggambarkan keseragaman nilai yang diperoleh dari rangkaian pengukuran berulang terhadap analat dengan menggunakan prosedur analisis yang sama (Leyva et al 2008). Sebanyak 7 kali ulangan dengan prosedur yang sama, hari yang sama dan analis yang sama dilakukan pada sampel kemudian dihitung RSDnya. Besarnya RSD dalam satuan % menunjukkan ripitabilitas. Keberterimaan RSD analisis ditentukan sebesar 2/3 RSD Horwitz (Garfield 2000) atau 1/2 sampai 2 kali RSD AOAC (AOAC 2002). Reprodusibilitas diukur dengan melakukan analisis yang sama setelah dua bulan sejak dilakukan analisis pertama. Hasil analisis dibandingkan lalu diuji secara statistik untuk melihat apakah hasil berbeda signifikan atau tidak.
3.2.3.2. Akurasi
Akurasi dilaksanakan dengan mengggunakan bahan acuan tepung kedelai dan tepung kacang hijau dari LIPI Kimia Bandung dan bahan acuan susu bubuk dari BBIA Bogor. Selain itu uji rekoveri juga dilakukan.
Tujuan uji rekoveri adalah memeriksa adanya interferensi kompetitif dan efek dari matriks sampel (Koch dan Peter 1999; Cembrowski dan Sullivan 1992). Uji rekoveri dilakukan dengan menggunakan sampel yang dispike (ditambahkan) standard glukosa. Percobaan spiking
(2.1)
3.2.3.3. Linieritas
Linieritas dari metode analitis yang menggambarkan kemampuan suatu metode untuk hasil analisis yang proporsional dengan konsentrasi analat pada sampel dalam range tertentu baik secara langsung maupun melalui transformasi matematik (Leyva et al 2008). Untuk mengetahui linieritas metode, sebanyak tujuh kali ulangan dilakukan pada standar glukosa dengan 6-8 konsentrasi. Kemudian tiap kali ulangan dihitung rataan, SD1dan RSD1. Selain itu tiap ulangan diplotkan persamaan garis dari kurva kalibrasi dan dihitung koefisien korelasinya (r2). Selanjutnya ditabulasikan nilai y yang baru berdasarkan persamaan garis yang ada. Dari nilai y yang baru dihitung rataan, standar deviasinya (yang kemudian disebut SD2) dan RSDnya. Uji F digunakan untuk mengetahui apakah ada perbedaan signifikan pada variansi kurva pada tiap kelompok konsentrasi.
3.2.4. Verifikasi metode SNI 01-2891-1992
Verifikasi dilakukan dengan mengukur kadar karbohidrat matriks sampel yang terpilih yaitu sampel yang mewakili kadar karbohidrat rendah, kadar karbohidrat sedang dan kadar karbohidrat tinggi dan beberapa sampel yang telah diketahui nilainya yaitu bahan acuan (reference material). Verifikasi ini meliputi atribut presisi (ripitabilitas) dan akurasi (dengan bahan acuan uji rekoveri).
3.2.4.1. Presisi
3.2.4.2. Akurasi
Akurasi dilaksanakan dengan mengggunakan bahan acuan tepung kedelai dan tepung kacang hijau dari LIPI Kimia Bandung dan bahan acuan susu bubuk dari BBIA Bogor. Selain itu uji rekoveri juga dilakukan. Tujuan uji rekoveri adalah memeriksa adanya interferensi kompetitif dan efek dari matriks sampel (Koch dan Peter 1999; Cembrowski dan Sullivan 1992). Uji rekoveri dilakukan dengan menggunakan sampel yang dispike standard glukosa. Spiking dilakukan sebanyak tujuh ulangan pada sampel bahan acuan. Sebelumnya juga dilakukan uji terhadap sampel yang tidak dispiking. Akurasi dilihat dari nilai rekoveriyang diperoleh. Recovery dihitung dengan rumus (2.2):
IV.
HASIL DAN PEMBAHASAN
4.1.
Pemilihan Matriks Sampel
Matriks pangan sangat mempengaruhi performa suatu metode, terutama komponen mayor seperti protein, karbohidrat, dan lemak, oleh karena itu beberapa sampel pangan cair dari hasil studi literatur dipilih berdasarkan tiga kriteria karbohidratnya yaitu mewakili matriks sampel dengan kadar karbohidrat rendah, sedang dan tinggi menurut skema segitiga yang disusun oleh AOAC International seperti pada Gambar 1. Penempatan sampel menurut studi literatur dapat dilihat pada Gambar 3. Sampel kecap manis dimasukkan pada kelompok pangan dengan karbohidrat tinggi, sampel kecap asin dimasukkan pada kelompok pangan dengan karbohidrat sedang, lemak rendah dan protein sedang serta santan dimasukkan pada kelompok pangan dengan karbohidrat rendah, protein rendah dan lemak tinggi. Kemudian dilakukan analisis proksimat dengan menggunakan metode SNI 01-2891-1992 untuk melakukan konfirmasi terhadap komposisi dan identitasnya. Hasil analisis proksimat dapat dilihat pada Tabel 5. Hasil analisis proksimat sesuai dengan penempatan yang dilakukan berdasarkan studi literatur.
Tabel 5. Komposisi proksimat matriks sampel cair yang terpilih untuk uji perbandingan metode analisis total karbohidrat (N=2)
No Sampel Kadar Air
(g/100g)
Kadar Abu (g/100g)
Kadar Protein (g/100g)
Kadar Lemak (g/100g)
Kadar Karbohidrat by difference(g/100g)
1 Kecap Manis 27.92 5.37 1,45 0,30 64,96
2 Kecap Asin 72.50 19.01 4.78 0,06 3,65
3 Santan 53.15 0.52 3,55 41,78 1,00
4.2.
Perbandingan metode
Hasil analisis total karbohidrat dengan menggunakan Metode Luff-Schoorl dan Metode Anthrone sulfat pada tiga matriks sampel pangan cair (kecap manis, kecap asin dan santan), yang mewakili skema segitiga matriks pangan, diuji statistik dengan SPSS 17.0 dengan menggunakan uji F menunjukkan bahwa varian kedua metode tidak berbeda nyata pada tingkat kepercayaan 95% untuk sampel kecap asin, kecap manis, dan santan. Hasil uji F dapat dilihat pada Lampiran 2. Hal ini menunjukkan tidak ada perbedaan signifikan dalam segi presisi dari Metode Luff-Schoorl dengan Metode Anthrone sulfat untuk sampel kecap manis dan kecap asin dan santan.
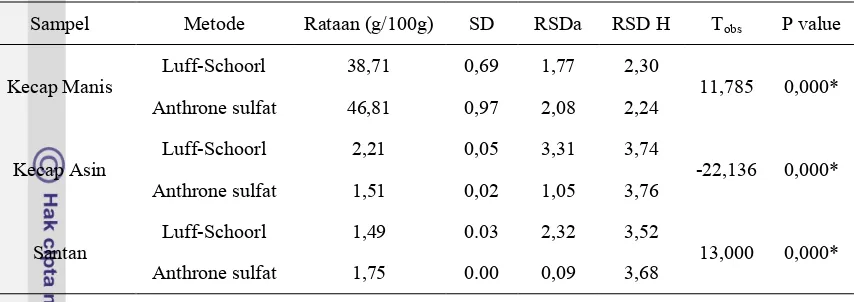
Tabel 6. Perbandingan Metode Anthrone sulfat dan Luff-Schoorl untuk analisis karbohidrat total pada 3 matriks sampel pangan cair (N=3)
Sampel Metode Rataan (g/100g) SD RSDa RSD H Tobs P value
Kecap Manis
Luff-Schoorl 38,71 0,69 1,77 2,30
11,785 0,000*
Anthrone sulfat 46,81 0,97 2,08 2,24
Kecap Asin
Luff-Schoorl 2,21 0,05 3,31 3,74
-22,136 0,000*
Anthrone sulfat 1,51 0,02 1,05 3,76
Santan
Luff-Schoorl 1,49 0.03 2,32 3,52
13,000 0,000*
Anthrone sulfat 1,75 0.00 0,09 3,68
*berbeda nyata
Berdasarkan uji F dan uji t pada hasil analisis total karbohidrat dengan menggunakan metode Luff-Schoorl dan Metode Anthrone sulfat terlihat adanya bias. Varian kedua metode tidak berbeda signifikan sedangkan hasil analisis kedua metode menunjukkan adanya perbedaan signifikan. Oleh karena itu, dilakukan uji korelasi dengan regresi linear untuk mengestimasi kesalahan sistematis (systematic error) diantara kedua metode.
Gambar 4. Perbandingan hasil analisis karbohidrat total pada tiga matriks sampel pangan cair ditambah dengan tiga matriks sampel pangan padat (N=18) dengan metode SNI (Luff-Schoorl) dan Metode Anthrone sulfat
Tepung beras
Kecap manis
Sarden
Kecap asin Santan
Perbandingan antara kedua metode dilakukan dengan menggunakan suplemen data dari penelitian Novitri (2011). Hasil regresi linier dapat dilihat pada Gambar 4; dengan koefisien korelasi (r2) dari kurva regresi (y=1.1873x-1.6264) menunjukkan nilai yang memuaskan yaitu 0.9797 (n=18). Nilai ini menunjukkan bahwa range konsentrasi yang digunakan memadai untuk analisis regresi sederhana, tetapi nilai ini tidak digunakan untuk menentukan apakah suatu metode akurat, relatif terhadap metode baku (Walton 2001; Westgard 1998), yang dalam hal ini adalah Luff Schoorl. Slope kurva regresi (1.1873) memperlihatkan bahwa kurva sedikit lebih curam dibandingkan kurva regresi yang ideal yaitu 1:1. Hal ini menunjukkan adanya proportional systematic error diantara metode yang digunakan (Walton 2001) dan terlihat bahwa Metode Anthrone sulfat sedikit lebih sensitif dibandingkan metode Luff-Schoorl. Dari intercept kurva regresi (-1.6264) kita dapat melihat bahwa Metode Anthrone menghasilkan nilai analisis 1.63% lebih rendah dibanding metode Luff-Schoorl pada sampel dengan nilai karbohidrat terendah (intercept1.6264 pada nilai total karbohidrat Metode Anthrone= 0). Nilai ini juga menunjukkan
estimated constant errordiantara kedua metode (Walton 2001). Dari penjelasan ini menunjukkan bahwa, meskipun korelasi cukup baik, terdapat mutual biasdiantara kedua metode. Tetapi karena konsentrasi dari populasi sampel kurang mewakili seluruh populasi matriks pangan secara umum, kesimpulan regresi linear pada perbandingan metode ini belum dapat dijadikan landasan yang kokoh. Regresi ini hanya memberikan gambaran sepintas dari populasi yang diujikan yaitu kecap manis, kecap asin, santan, sarden, susu bubuk dan tepung beras.
Hasil analisis menggunakan uji F, independent student t-test dan regresi linear sederhana tehadap perbandingan hasil analisis menggunakan metode Luff-Schoorl dan Metode Anthrone sulfat pada tiga sampel matriks pangan cair, menunjukkan bahwa dengan presisi yang tidak berbeda nyata, nilai hasil yang didapat oleh kedua metode berbeda nyata. Oleh karena itu penyebab bias dari kedua metode dianalisis.
lagi bahwa nilai analisis metode by difference dapat mengandung akumulasi kesalahan, oleh karena itu nilai yang ada hanya dijadikan perbandingan.
Tabel 7. Karbohidrat total dari tiga sampel matriks pangan cair dengan beberapa metode
Sampel Kadar karbohidrat (g/100g)
by difference Luff-Schoorl Anthrone sulfat
Kecap manis 64,96 38,71 46,81
Kecap asin 3,65 1,57 1,51
Santan 1,00 1,49 1,75
Dilihat dari Tabel 7 pada sampel kecap manis dan kecap asin, hasil metode pengukuran karbohidrat secara langsung yaitu baik Luff-Schoorl maupun Metode Anthrone sulfat, nilainya lebih kecil dibandingkan metode by difference. Metode by difference dapat memiliki kesalahan positif karena metode ini tidak dapat membedakan komponen non karbohidrat seperti asam organik, tanin dan lignin. Baik kecap asin dan kecap manis merupakan produk hasil fermentasi oleh kapang, oleh karena itu produk samping hasil metabolit, seperti asam organik, dapat terkandung dalam kecap manis dan kecap asin.
Adapun nilai analisis sampel santan baik metode by difference dan Luff-Schoorl menunjukkan nilai yang hamper sama, yaitu jika dibulatkan nilainya 1%. Adapun Metode Anthrone nilainya sedikit lebih besar dibandingkan metode by difference maupun Luff-Schoorl. Hal ini dapat disebabkan karena kandungan gula sederhana (terutama dalam bentuk glukosa dan fruktosa) yang ada pada santan tidak sebanyak pada kecap manis maupun kecap asin, sehingga pengaruh degradasi gula sederhana pada tahap hidrolisis asam tidak terlalu terlihat. Selain itu komponen non karbohidrat yang dapat terhitung sebagai karbohidrat oleh metode by difference, seperti asam organik, tidak terlalu banyak terdapat pada sampel santan yang digunakan.
Metode by differencetidak dapat dijadikan sebagai acuan karena metode ini tidak lepas dari banyak bias. Perbedaan nilai antara metode by difference dengan metode lainnya menunjukkan bahwa ada kemungkinan nilai yang didapat baik oleh Metode Anthrone sulfat maupun Metode Luff-Schoorl, terutama untuk sampel kecap asin dan kecap manis, bukanlah nilai kadar total karbohidrat karena serat kasar seperti selulosa juga tidak dapat dihidrolisis dengan asam kuat encer saja (Southgate 1976) dan juga tidak dapat dikatakan sebagai nilai total available
karbohidrat juga karena sulit untuk memisahkan fraksi pati dari karbohidrat struktural (Loomys dan Shull 1937). Nilai yang didapat lebih cocok jika disebut sebagai nilai total karbohidrat yang dapat terhidrolisis oleh asam (Weinmann 1946).
Pengaruh faktor konversi yang digunakan juga dapat berdampak pada perbedaan nilai yang didapat antara metode kandidat, Luff-Schoorl dan metode by difference. Tanpa melihat jenis karbohidrat yang banyak terkandung pada matriks, faktor konversi 0.9 diterapkan untuk semua matriks. Adapun dalam perbandingan metode ini pengaruh komponen lain seperti lemak dan protein belum dapat diketahui melalui penelitian ini.
Luff-Schoorl menunjukkan nilai yang sedikit lebih besar dibandingkan Metode Anthrone sulfat (selisih rataan 0.06%). Ada juga kemungkinan interferensi komponen pereduksi yang bukan gula yang menyebabkan kesalahan positif pada metode Luff Schoorl.
Tiap metode memang memiliki keterbatasan. Metode Anthrone sulfat rentan terhadap interferensi non spesifik (Faulks dan Timms 1985) salah satunya keberadaan ion halida (Fales et al 1961) terutama ion Cl yang berasal dari tahap hidrolisis dengan HCl. Intensitas warna yang dihasilkan oleh reaksi Anthrone juga berbeda-beda untuk gula yang berbeda (Yemm dan Willis 1954). Selain itu reaksi senyawa Anthrone cenderung lebih baik untuk senyawa heksosa dan reaksi dengan pentose kurang menghasilkan warna yang stabil (Koehler 1952; Southgate 1976).
Penggantian suatu metode dengan metode lain dapat dilakukan jika kedua metode memiliki kesesuaian hasil yang dapat diterima. Meski presisi kedua metode tidak berbeda nyata berdasarkan uji F, uji T yang dilakukan menunjukkan Metode Anthrone sulfat dan Metode Luff-Schoorl menghasilkan nilai yang berbeda nyata pada aplikasinya untuk sampel kecap manis, kecap asin dan santan yang mewakili matriks pangan cair. Karena kedua metode berbeda nyata dan tidak ada acuan bahwa Metode Anthrone sulfat memiliki nilai yang lebih akurat dibanding metode yang telah baku (Luff-Schoorl dalam SNI 01-2891-1992), maka Metode Anthrone sulfat dianggap tidak dapat menggantikan Metode Luff-Schoorl, sehingga tahap selanjutnya yang dilakukan adalah verifikasi Metode Luff-Schoorl yang telah baku. Selain karena Metode Anthrone pada tahap yang telah dilakukan dianggap tidak dapat menggantikan Metode Luff-Schoorl, keputusan untuk melakukan verifikasi ini diambil karena Metode Luff-Schoorl merupakan metode yang telah baku (ditetapkan dalam SNI 01-2891-1992).
diterima. Sampai saat ini uji profisiensi lab untuk pemenuhan persyaratan SNI 19-17025-2000 masih menggunakan nilai konsensus dari peserta lab uji, maka penggunaan metode baku manual SNI masih menjadi alternatif yang lebih baik untuk mendapatkan hasil analisis dengan performa yang memenuhi standard. Oleh karena itu, tahap validasi Metode Anthrone tidak dilakukan dan dan hanya dilakukan verifikasi terhadap metode baku yaitu Luff-Schoorl.
4.3.
Verifikasi metode SNI 01-2891-1992
Tingkat validasi tergantung status dari suatu metode pada struktur analitik (AOAC 2002), yang dimaksud disini adalah validasi seperti apakah yang harus diterapkan pada suatu metode tergantung status metode itu sendiri. Metode yang telah baku hanya memerlukan verifikasi dari kemampuan suatu laboratorium untuk mencapai karakteristik performa yang ditetapkan, sedangkan di sisi lain untuk metode yang masih baru atau aplikasi suatu metode pada matriks yang baru memerlukan validasi (AOAC 2002). Karena Metode Luff-Schoorl dalam SNI 01-2891-1992 sudah baku maka hanya dilakukan verifikasi. Karakteristik yang akan dinilai dalam verifikasi adalah aspek presisi dan akurasi
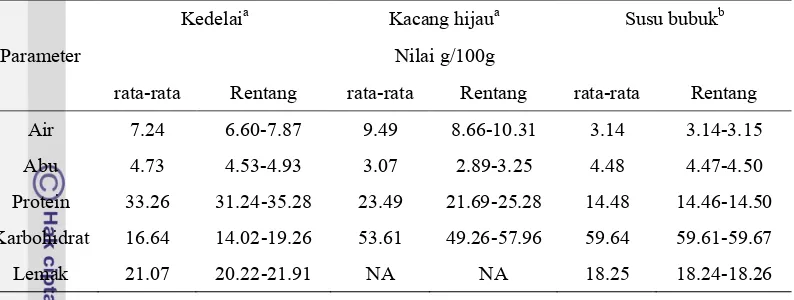
Tabel 8 Komposisi proksimat bahan acuan yang digunakan dalam verifikasi metode karbohidrat total SNI 01-2891-1992
Parameter
Kedelaia Kacang hijaua Susu bubukb
Nilai g/100g
rata-rata Rentang rata-rata Rentang rata-rata Rentang
Air 7.24 6.60-7.87 9.49 8.66-10.31 3.14 3.14-3.15
Abu 4.73 4.53-4.93 3.07 2.89-3.25 4.48 4.47-4.50
Protein 33.26 31.24-35.28 23.49 21.69-25.28 14.48 14.46-14.50 Karbohidrat 16.64 14.02-19.26 53.61 49.26-57.96 59.64 59.61-59.67
Lemak 21.07 20.22-21.91 NA NA 18.25 18.24-18.26
a
berdasarkan nilai yang tercantum pada bahan acuan LIPI Kimia
b
berdasarkan hasil analisis proksimat Lab Kimia LD-ITP
Bahan acuan yang dipakai jika dimasukkan ke dalam matriks segitiga pangan akan terbagi menjadi dua kelas matriks dalam segitiga pangan. Kedelai masuk ke dalam kelas dengan kadar karbohidrat rendah, lemak rendah dan protein sedang (yang ditandai dengan nomor 8 pada matriks segitiga pangan di Gambar 1). Sedangkan kacang hijau dan susu bubuk akan masuk ke dalam kelas protein rendah, lemak rendah dan karbohidrat sedang (yang ditandai dengan nomor 6 pada matriks segitiga pangan di Gambar 1). Sebelumnya pada perbandingan metode digunakan sampel yang mewakili tiga kelas matriks dalam segitiga pangan (Gambar 3). Sehingga kalau dijumlah sampel dan bahan acuan yang digunakan telah mewakili 5 dari 9 matriks segitiga pangan yang ada.
4.3.1. Aspek presisi
Walton (2001) merekomendasikan evaluasi terhadap presisi sebagai langkah pertama dalam validasi metode. Jika presisi metode sudah tidak baik, maka sulit untuk mendapatkan hasil yang dapat dipercaya. Salah satu aspek yang umum digunakan dalam verifikasi adalah ripitabilitas (Mullins 2003). Tetapi dalam pengujian presisi metode untuk validasi satu lab (single laboratory validation) dapat berupa ripitabilitas dan reprodusibilitas intralab.
4.3.1.1. Ripitabilitas bahan acuan
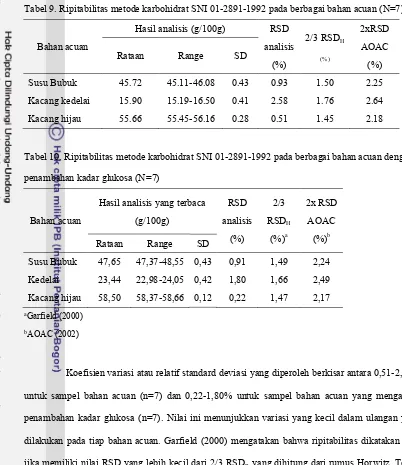
Tabel 9. Ripitabilitas metode karbohidrat SNI 01-2891-1992 pada berbagai bahan acuan (N=7)
Bahan acuan
Hasil analisis (g/100g) RSD analisis (%) 2/3 RSDH (%) 2xRSD AOAC (%)
Rataan Range SD
Susu Bubuk 45.72 45.11-46.08 0.43 0.93 1.50 2.25
Kacang kedelai 15.90 15.19-16.50 0.41 2.58 1.76 2.64
Kacang hijau 55.66 55.45-56.16 0.28 0.51 1.45 2.18
Tabel 10. Ripitabilitas metode karbohidrat SNI 01-2891-1992 pada berbagai bahan acuan dengan penambahan kadar glukosa (N=7)
Bahan acuan
Hasil analisis yang terbaca (g/100g) RSD analisis (%) 2/3 RSDH (%)a 2x RSD AOAC (%)b
Rataan Range SD
Susu Bubuk 47,65 47,37-48,55 0,43 0,91 1,49 2,24
Kedelai 23,44 22,98-24,05 0,42 1,80 1,66 2,49
Kacang hijau 58,50 58,37-58,66 0,12 0,22 1,47 2,17
a
Garfield (2000)
b
AOAC (2002)
Koefisien variasi atau relatif standard deviasi yang diperoleh berkisar antara 0,51-2,58% untuk sampel bahan acuan (n=7) dan 0,22-1,80% untuk sampel bahan acuan yang mengalami penambahan kadar glukosa (n=7). Nilai ini menunjukkan variasi yang kecil dalam ulangan yang dilakukan pada tiap bahan acuan. Garfield (2000) mengatakan bahwa ripitabilitas dikatakan baik jika memiliki nilai RSD yang lebih kecil dari 2/3 RSDRyang dihitung dari rumus Horwitz. Tetapi AOAC (2002) mengatakan bahwa nilai yang dapat diterima untuk ripitabilitas adalah antara 0,5 sampai 2 kali dari nilai yang terhitung berdasarkan rumus atau di Tabel 2. Bahkan nilai RSD di bawah 5% dapat diterima, meskipun terkadang batas itu tergantung tipe dari analisis (Smith 2010).
dalam range yang dapat diterima. Begitupula jika mengikuti acuan Smith (2010), yaitu RSD masih di bawah 5%.
Nilai RSD kedelai cenderung lebih besar dibanding kacang hijau dan susu bubuk baik pada bahan acuan dengan penambahan glukosa maupun bahan acuan tanpa penambahan glukosa. Hal ini dapat disebabkan oleh konsentrasi karbohidrat pada kedelai yang lebih kecil dibandingkan susu bubuk dan kacang hijau. Akan tetapi jika dilihat dari nilai standard deviasi(SD)nya sendiri, kedelai memiliki SD yang hampir sama bahkan cenderung lebih kecil dibandingkan susu bubuk. Hal ini mengindikasikan bahwa konsentrasi karbohidrat yang lebih kecil (hingga pada range lebih dari ±15,90 gram karbohidrat setara glukosa/100 gram sampel) bukan berarti menyebabkan keterulangan yang lebih buruk dibandingkan konsentrasi karbohidrat yang lebih tinggi. Adanya kecenderungan bahwa nilai SD susu bubuk lebih besar dari kedelai lebih besar dari kacang hijau perlu diteliti lebih lanjut untuk mengetahui komponen apa dari tiap bahan acuan yang mungkin dapat menyebabkan variasi yang ada. Dalam penelitian ini, range konsentrasi ±15,90-58.50 gram karbohidrat setara glukosa/100 gram sampel pada sampel kacang hijau, kedelai dan susu bubuk masih memiliki kerterulangan (ripitabilitas) yang dapat diterima terutama pada lab tempat penelitian dilaksanakan telah dikonfirmasi.
4.3.1.2. Reprodusibilitas bahan acuan dan matriks sampel
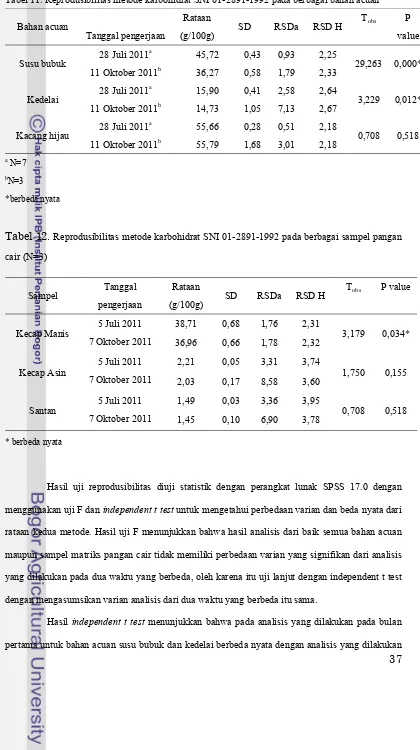
Tabel 11. Reprodusibilitas metode karbohidrat SNI 01-2891-1992 pada berbagai bahan acuan
Bahan acuan
Tanggal pengerjaan
Rataan
(g/100g) SD RSDa RSD H
Tobs P
value
Susu bubuk 28 Juli 2011 a
45,72 0,43 0,93 2,25
29,263 0,000* 11 Oktober 2011b 36,27 0,58 1,79 2,33
Kedelai 28 Juli 2011 a
15,90 0,41 2,58 2,64
3,229 0,012* 11 Oktober 2011b 14,73 1,05 7,13 2,67
Kacang hijau 28 Juli 2011 a
55,66 0,28 0,51 2,18
0,708 0,518 11 Oktober 2011b 55,79 1,68 3,01 2,18
a
N=7
b
N=3
*berbeda nyata
Tabel 12. Reprodusibilitas metode karbohidrat SNI 01-2891-1992 pada berbagai sampel pangan cair (N=3)
Sampel Tanggal
pengerjaan
Rataan
(g/100g) SD RSDa RSD H
Tobs P value
Kecap Manis
5 Juli 2011 38,71 0,68 1,76 2,31
3,179 0,034*
7 Oktober 2011 36,96 0,66 1,78 2,32
Kecap Asin
5 Juli 2011 2,21 0,05 3,31 3,74
1,750 0,155
7 Oktober 2011 2,03 0,17 8,58 3,60
Santan
5 Juli 2011 1,49 0,03 3,36 3,95
0,708 0,518
7 Oktober 2011 1,45 0,10 6,90 3,78
* berbeda nyata
Hasil uji reprodusibilitas diuji statistik dengan perangkat lunak SPSS 17.0 dengan menggunakan uji F dan independent t testuntuk mengetahui perbedaan varian dan beda nyata dari rataan kedua metode. Hasil uji F menunjukkan bahwa hasil analisis dari baik semua bahan acuan maupun sampel matriks pangan cair tidak memiliki perbedaan varian yang signifikan dari analisis yang dilakukan pada dua waktu yang berbeda, oleh karena itu uji lanjut dengan independent t test dengan mengasumsikan varian analisis dari dua waktu yang berbeda itu sama.
pada bulan kedua yang berselang lebih dari dua bulan sejak analisis pertama, sedangkan untuk bahan acuan kacang hijau tidak berbeda nyata. Adapun hasil independent t testpada analisis yang dilakukan pada bulan pertama untuk sampel matriks pangan cair yaitu kecap asin dan santan tidak berbeda nyata dengan analisis yang dilakukan pada bulan kedua, sedangkan untuk sampel kecap manis berbeda nyata. Nilai yang berbeda nyata ini mengindikasikan reprodusibilitas yang buruk.
Jumlah total karbohidrat yang ada pada bahan acuan seharusnya tidak akan banyak berubah karena lingkungan. Jika diasumsikan bahwa bahan acuan cenderung bersifat stabil, maka perubahan atau ketidakkonsistenan dapat berasal dari analis, reagen, atau lingkungan yang mempengaruhi performa metode itu sendiri. Meskipun reagen seperti natrium tiosulfat dan reagen lain disiapkan segar, reagen Luff yang digunakan untuk analisis pada bulan kedua sama dengan yang digunakan pada bulan pertama karena diasumsikan reagen ini bersifat stabil. Tetapi ternyata hasil analisis menunjukkan adanya ketidakkonsistenan dalam ripitabilitas dan reprodusibilitas, sehingga ada kemungkinan jika reagen kurang stabil dalam penyimpanan lebih dari 2 bulan. Hal ini juga dapat menyebabkan bias. Adapun ketidakkonsistenan dari analis dan perubahan kondisi pada lingkungan juga dapat mempengaruhi performa metode.
Koefisien variasi atau relatif standard deviasi yang diperoleh untuk analisis yang dilakukan pada bulan pertama cenderung lebih baik dibandingkan hasil analisis yang dilakukan pada bulan kedua. Hal ini juga yang dapat menunjukkan bahwa adanya ketidakkonsistenan pada analisis yang dilakukan pada bulan kedua. Hal ini kemungkinan besar dapat disebabkan karena adanya perubahan pada reagen, matriks, analis dan lingkungan. Reagen dapat mengalami perubahan seperti yang disebutkan sebelumnya. Dari segi analis, metode yang memiliki tahapan yang panjang dan melelahkan dapat menyebabkan performa metode kurang konsisten. Selain itu perubahan dari matriks sampel (dalam hal ini terutama matriks sampel pangan cair) baik secara biologis atau kimia dapat menyebabkan hasil kurang konsisten baik untuk ripitabilitas maupun reprodusibilitas. Dari sini dapat dilihat juga bahwa reprodusibilitas metode dipengaruhi oleh matriks sampel yang dianalisis.
reprodusibilitasnya buruk (analisis yang dilakukan dalam selang waktu dua bulan hasilnya berbeda nyata).
4.3.2. A
spek akurasi
Akurasi dari metode SNI 01-2891-1992 dilakukan dengan menggunakan bahan acuan dan uji rekoveri. Hasil analisis terhadap bahan acuan dapat dilihat pada Tabel 13, dan uji rekoveri dengan menggunakan standard glukosa dapat dilihat pada Tabel 14.
Tabel 13. Akurasi metode karbohidrat total SNI 01-2891-1992 pada berbagai bahan acuan (N=7)
Bahan acuan Rentang bahan acuan(g/100g)
Hasil analisis (g/100g)
Rataan Range SD
Susu Bubuk 59,61-59,67a 45,72 45,11-46,08 0,43
Kedelai 14,02-19,26b 15,90 15,19-16,50 0,41
Kacang hijau 49,26-57,96b 55,66 55,45-56,16 0,28 a
berdasarkan hasil analisis by differenceLab Kimia LD-ITP
b
berdasarkan nilai yang tercantum pada bahan acuan LIPI Kimia (analisis Luff-Schoorl)
4.3.2.1. Akurasi berdasarkan bahan acuan
Bahan acuan yang digunakan bukanlah Certified Reference Material (CRM), melainkan hanya bahan acuan yang nilai (assigned value) komposisinya berdasarkan konsensus beberapa lab dan digunakan untuk uji profisiensi. Sekalipun demikian, bahan acuan seperti ini masih dapat digunakan untuk mengetahui adanya bias (Thompson et al 2002). Hasil analisis terhadap bahan acuan menunjukkan nilai yang masih dalam rentang yang tercantum pada bahan acuan, kecuali untuk bahan acuan susu bubuk. Khusus untuk susu bubuk rentangnya masih sempit karena nilai yang ditampilkan merupakan hasil uji dari satu lab saja dan itupun masih menggunakan metode by difference.
dimaksud di sini adalah bahan acuan yang nilai komposisinya merupakan hasil konsensus beberapa lab, bukan bahan acuan yang nilainya tetap seperti senyawa kimia standard.
4.3.2.2. Akurasi berdasarkan uji rekoveri
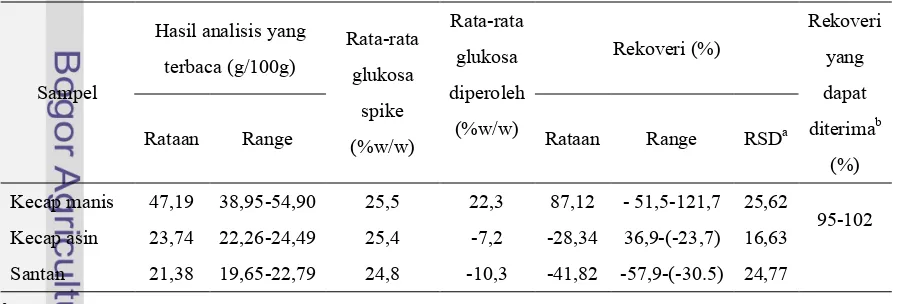
Bias yang terlihat dari perbandingan metode dapat dijelaskan dengan uji rekoveri (Lumsden 2000). Berdasarkan perbandingan metode yang telah dilakukan sebelumnya, diperkirakan adanya proportional error. Proportional systematic errordapat diperkirakan dengan uji rekoveri (Lumsden 2000; Koch dan Peter 1999). Selain itu uji rekoveri dapat digunakan untuk mendukung studi yang menggunakan bahan acuan (Thompson et al 2002). Rekoveri yang dilakukan pada penelitian ini dilakukan pada bahan acuan dan pada matriks sampel pangan cair. Baik pada bahan acuan maupun matriks sampel pangan cair hanya menggunakan satu level konsentrasi, yaitu dengan menggunakan glukosa sebanyak 10% dari berat total sampel untuk bahan acuan dan sebanyak ±25% dari berat total sampel untuk matriks bahan pangan cair . Hasil uji rekoveri dapat dilihat pada Tabel 14 dan Tabel 15.
Tabel 14. Hasil uji rekoveri pada berbagai bahan acuan dengan spike glukosa (N=7)
Bahan acuan
Hasil analisis yang terbaca (g/100g) Rata-rata glukosa spike (%w/w) Rata-rata glukosa diperoleh (%w/w) Rekoveri (%) Rekoveri yang dapat diterimab (%)
Rataan Range Rataan Range RSDa
Susu Bubuk 47,65 47,37-48,55 10,0 6,7 65,0 62,2-74,1 6,68
95-102
Kedelai 23,44 22,98-24,05 10,0 9,1 91,0 86,3-96,9 4,64
Kacang hijau 58,50 58,37-58,66 10,0 8,4 84,0 82,6-85,5 1,48
[image:41.595.84.533.556.708.2]aRSD analisis dari rekoveri bmenurut AOAC(2002)
Tabel 15. Hasil uji rekoveri pada berbagai sampel pangan cair dengan spike glukosa (N=7)
Sampel
Hasil analisis yang terbaca (g/100g) Rata-rata glukosa spike (%w/w) Rata-rata glukosa diperoleh (%w/w) Rekoveri (%) Rekoveri yang dapat diterimab (%)
Rataan Range Rataan Range RSDa
Kecap manis 47,19 38,95-54,90 25,5 22,3 87,12 - 51,5-121,7 25,62
95-102 Kecap asin 23,74 22,26-24,49 25,4 -7,2 -28,34 36,9-(-23,7) 16,63
Santan 21,38 19,65-22,79 24,8 -10,3 -41,82 -57,9-(-30.5) 24,77
a
RSD analisis dari rekoveri
b
4.3.2.2.1.
Rekoveri dengan bahan acuan
Uji rekoveri dengan spiking glukosa untuk susu bubuk pada Tabel 13 menunjukkan rata-rata rekoveri 65,0%, untuk kedelai didapatkan rata-rata rekoveri 91,0% dan untuk kacang hijau didapatkan rata-rata rekoveri 84,0%. Uji rekoveri dengan spiking glukosa untuk sampel kecap manis pada Tabel 14 menunjukkan rata-rata rekoveri 87,12%; untuk sampel kecap asin didapatkan rata-rata rekoveri -28,34%, dan untuk sampel santan didapatkan rata-rata rekoveri -41,82%. Berdasarkan uji rekoveri tidak ada hasil yang menunjukkan nilai rekoveri yang dapat diterima berdasarkan batas yang ditetapkan oleh AOAC (2002). Meski nilai rekoveri yang baik belum tentu menandakan bahwa nilai analisis merupakan nilai yang sebenarnya karena efek dari analat yang ditambahkan dengan analat dalam bentuk alaminya mungkin berbeda, tetapi nilai rekoveri yang buruk jelas menunjukkan adanya bias dari nilai yang sebenarnya (Thompson et al 2002).
Nilai rekoveri sampel kedelai (91,03%) lebih besar daripada kacang hijau (83,95%) dan lebih besar daripada susu bubuk (65,0%). Hal ini menunjukkan bahwa efek matriks yang dapat mengganggu analisis paling besar terlihat pada bahan acuan susu bubuk. Selain itu nilai rekoveri yang kurang dari 60-70% perlu pemeriksaan yang mengarah pada perbaikan (AOAC 2002) karena kemungkinan nilai rekoveri ini menunjukkan bahwa ada kesalahan sistematis akibat adanya komponen matriks lain yang menganggu dalam analisis seperti maltodekstrin yang digunakan sebagai bahan pengisi pada susu bubuk. Courtin et al (2000) mengatakan bahwa nilai yang dihasilkan oleh analisis maltodekstrin dengan metode gula pereduksi cenderung lebih kecil dibandingkan dengan metode kolorimetri dan adanya komponen lain yang memiliki kemampuan mereduksi dapat mempengaruhi gula pereduksi yang ada. Nilai rekoveri rata-rata untuk bahan acuan susu bubuk adalah 65%, sehingga jika Metode Luff-Schoorl seperti dalam prosedur SNI 01-2891-1992 diaplikasikan sampel yang komposisinya mirip seperti pada bahan acuan susu bubuk diperkirakan ada kemungkinan kesalahan sistematis dapat terjadi.
4.3.2.2.2.
Rekoveri dengan sampel matriks uji