AGREGASI TROMBOSIT PADA
SINDROMA KORONER AKUT
T E S I S
Oleh:
S U L I A N T Y
DEPARTEMEN PATOLOGI KLINIK
FAKULTAS KEDOKTERAN USU / RSUP H. ADAM MALIK
M E D A N
AGREGASI TROMBOSIT PADA
SINDROMA KORONER AKUT
T E S I S
Diajukan Untuk Melengkapi Persyaratan Untuk Mencapai Gelar Magister Dalam Bidang Patologi Klinik Pada Fakultas Kedokteran
Universitas Sumatera Utara
Oleh:
S U L I A N T Y
DEPARTEMEN PATOLOGI KLINIKFAKULTAS KEDOKTERAN USU /
RSUP H. ADAM MALIK
KATA PENGANTAR
Puji dan syukur saya ucapkan kepada Bapa yang di Surga oleh karena kasih
karuniaNya, sehingga saya dapat mengikuti Program Pendidikan Dokter spesialis
patologi klinik Fakultas Kedokteran Sumatera utara dan dapat menyelesaikan Karya
tulis (tesis) yang berjudul Agregasi Trombosit pada Sindroma Koroner Akut
Selama saya mengikuti pendidikan dan proses penyelesaian penelitian untuk
karya tulis ini, saya telah banyak mendapat bimbingan, petunjuk, bantuan dan
pengarahan serta dorongan baik moril dan materil dari berbagai pihak sehinggan
saya dapat menyelesaikan pendidikan dan karya tulis ini. Untuk semua itu
perkenankanlah saya menyampaikan rasa hormat dan terimakasih yang tiada
terhingga kepada :
Yth, Prof. Adi Koesoema Aman, SpPK-KH, FISH, sebagai pembimbing
saya yang telah banyak memberikan bimbingan, petunjuk, pengarahan, bantuan dan
dorongan selama dalam pendidikan dan proses penyusunan, sampai selesainya
tesis ini. Saya juga sangat berterimakasih kepada beliau selaku Ketua Departemen
Patologi Klinik yang memberikan kesempatan sebagai peserta Program Pendidikan
Dokter Spesialis Patologi Klinik.
Yth, Dr. Refli Hasan, SpPD,SpJP, pembimbing II dari department Penyait
Dalam yang sudah memberikan banyak bimbingan, petunjuk, pengarahan dan
bantuan mulai dari penyusunan proposal, selama dilaksanakan penelitian sampai
selesainya tesis ini.
Yth, Prof. DR. Dr. Ratna Akbari Ganie, SpPK-KH, FISH dan Dr. Ricke
Loesnihari SpPK-K, sebagai Ketua dan Sekretaris Program Studi di Departemen
membimbing, mengarahkan dan memotivasi sejak awal pendidikan dan
menyelesaikannya.
Yth, Prof. Herman Hariman, PhD, SpPK-KH, FISH, yang memberikan
bimbingan, pengarahan dan masukan selama saya mulai pendidikan sampai
menyelesaikan penulisan tesis ini.
Yth, Prof. Burhanuddin Nasution, SpPK-KN, FISH, yang banyak
memberikan bimbingan dan pengarahan selama pendidikan dan menyelesaikan
penulisan tesis ini
Yth, Prof. Dr. Iman Sukiman, SpPk-KH, FISH, Dr. R. Ardjuna M Burhan,
DMM, SpPK-K (Alm), Dr. Muzahar, DMM, SpPK-K, Dr. Zulfikar Lubis, SpPK-K,
FISH, dr. Tapisari Tambunan, SpPK-KH, Dr. Ozar Sanuddin SpPK-K, Dr. Farida
Siregar, SpPK, Dr. Ulfah Mahidin, SpPK, Dr. Chairul Rahmah, SpPk, Dr. Lina
spPK dan Dr Nelly Elfrida SpPK, semuanya guru-guru saya yang telah banyak
memberikan petunjuk, arahan selama saya mengikuti pendidikan Spesialis Patologi
Klinik dan selama penyelesaian tesis ini. Hormat dan terimakasih saya ucapkan.
Serta ibu Eliyana Ginting yang banyak membantu dalam urusan administrasi
dibagian Patologi Klinik.
Yth, Drs. Abdul jalil Amri Arma, MKes, yang telah memberikan bimbingan,
arahan dan bimbingan di bidang statistik selama saya memulai penelitian sampai
selesainya tesis saya, terimakasih banyak saya ucapkan.
Ucapan terimakasih juga saya ucapkan kepada seluruh teman-teman sejawat
Program Pendidikan Dokter Spesialis Patologi Klinik Fakultas Kedokteran
Universitas Sumatera Utara, para analis dan pegawai, serta semua pihak yang tidak
dapat saya sebutkan satu persatu, atas bantuan dan kerja sama yang diberikan
Hormat dan terimakasih yang sebesar-besarnya kepada Dekan Fakultas
Kedokteran Universitas Sumatera Utara, Rektor Universitas Sumatera Utara,
Direktur rumah Sakit umum Pusat H. Adam Malik yang telah memberikan
kesempatan dan menerima saya untuk mengikuti Program Pendidikan Dokter
Spesialis Patologi Klinik.
Terimakasih yang setulus-tulusnya saya sampaikan kepada ayahanda
Handojo dan ibunda Helen Liana yang telah membesarkan, mendidik serta
memberikan dorongan moril dan materil kepada ananda selama ini. Begitu juga
ucapan terimakasih yang sebesar-besarnya kepada kedua adik saya, Rubianty dan
Widyanty Tandean serta ipar saya, dr.RM Prasojo Soedjatmiko yang
memberikan dorongan, bantuan moril dan materil kepada saya dan keluarga.
Akhirnya terimakasih yang tiada terhingga saya sampaikan kepada suami
saya Dr. Rikimin Tedja,SpKK yang telah mendampingi saya dengan penuh
pengertian, perhatian, memberikan motivasi dan pengorbanan selama saya
mengikuti pendidikan sampai saya dapat menyelesaikan pendidikan ini. Juga untuk
anak-anakku yang tersayang Jonathan Aurelius Tedja dan Yulisha Tedja yang
telah banyak kehilangan perhatian dan kasih sayang selama saya mengikuti
pendidikan.
Semoga tesis ini bermanfaat bagi kita semua. Dan semoga Tuhan Yang
Maha Kuasa memberkati kita semua.
Medan, Maret 2011
Penulis,
DAFTAR ISI
1.2. Perumusan Masalah………...
1.3. Hipotesa Penelitian ………...
1.4. Tujuan Penelitian
1.4.1. Tujuan Umum ………...
1.4.2. Tujuan Khusus ………
1.5. Manfaat Penelitian ………...
BAB 2. TINJAUAN KEPUSTAKAAN………
2.1. Trombosit……….
2.1.1. Produksi Trombosit………
2.1.2. Struktur Trombosit ...
2.1.3. Fungsi Trombosit………...
2.1.4. Pembentukan Sumbat Trombosit Hemostatik Primer…...
2.2. Agregasi Trombosit ...
2.2.3. Obat-obatan Yang Mempengaruhi Agregasi Trombosit...
2.2.4. Pengukuran...
2.2.5. Interpretasi………
2.3. Sindroma Koroner Akut………..
2.3.1.Definisi………
2.3.2.Patofisiologi Sindroma Koroner Akut...
2.3.3.Patogenesis Aterosklerosis………
2.3.4.Diagnosa………
2.4. Trombosit Dalam Sindroma Koroner Akut………..
2.4.1.Disfungsi Endotel……….
2.4.2.Aktivasi dan Agregasi Trombosit………...
2.4.3.Aktivasi Kaskade Koagulasi……….
BAB 3. METODE PENELITIAN………...
3.1. Desain Penelitian ………..
3.2. Tempat dan Waktu Penelitian ………..
3.3. Populasi dan Subyek Penelitian...
3.3.1. Kriteria Inklusi ……….
3.3.2. Kriteria Eklusi ………..
3.4. Perkiraan Besar Sampel……….…
3.5. Bahan dan Cara Kerja……….
3.5.1. Bahan Yang Diperlukan……….………...
3.5.2. Anamnese dan Pemeriksaan Fisik...
3.5.3. Pemeriksaan EKG………...
3.5.4. Pengambilan dan Pengolahan Sampel…………...
3.5.5.1.Pemeriksaan Darah Lengkap………...
3.5.5.2.Pemeriksaan Fungsi Agregasi Trombosit……….
3.6. Pemantapan Kualitas………..
3.7. Ethical Clearance dan Informed Consent………...
3.8. Analisa Data………...
3.9. Batasan Operasional………...
3.10. Perkiraan Biaya Penelitian...
3.11. Jadwal Penelitian...
3.12. Kerangka Konsep …………...
3.13. Kerangka Kerja Operasional ………
BAB 4. HASIL PENELITIAN……….
BAB 5. PEMBAHASAN………..
BAB 6. KESIMPULAN DAN SARAN………
DAFTAR TABEL
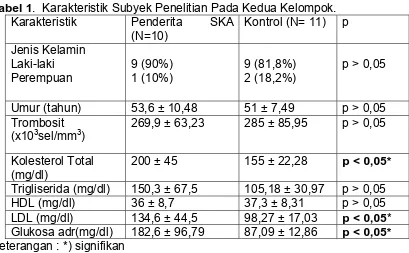
TABEL 1. Karakteristik Subjek Penelitian ...
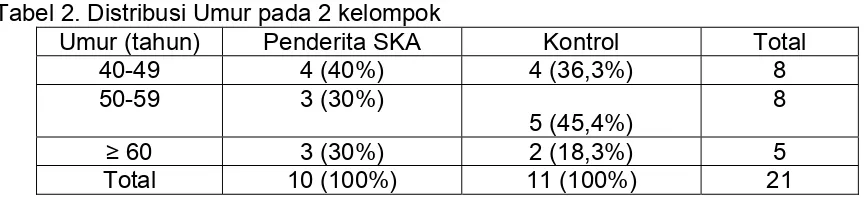
TABEL 2. Distribusi Umur pada 2 kelompok ...
TABEL 3. Hasil pemeriksaan enzim jantung pada pasien SKA ...
TABEL 4. Agregasi Trombosit dengan agonis ADP pada 2 kelompok ...
TABEL 5. Agregasi Trombosit dengan agonis Epinefrin pada 2 kelompok ...
TABEL 6. Agregasi Trombosit dengan atau tanpa dislipidemia ...
TABEL 7. Agregasi Trombosit dengan atau tanpa DM ...
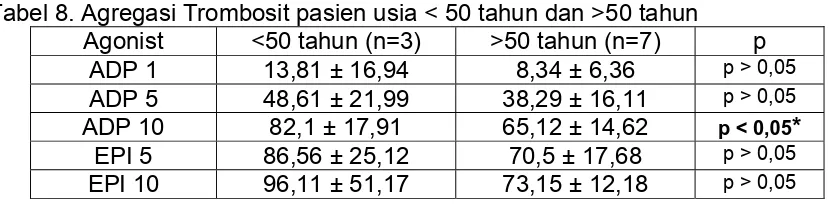
TABEL 8. Agregasi Trombosit pasien usia <50 tahun dan >50 tahun ...
TABEL 9. Agregasi Trombosit dengan atau tanpa hipertensi ... 57
59
60
60
60
61
62
62
DAFTAR LAMPIRAN
Lampiran 1. Status Pasien ...
Lampiran 2. Formulir Persetujuan ...
Lampiran 3. Data Hasil Penelitian ...
Lampiran 4. Lembar Penjelasan Kepada Subjek Penelitian ...
Lampiran 5. Surat Izin Melakukan Penelitian ...
Lampiran 6. Daftar Riwayat Hidup ...
.
77
79
80
82
83
DAFTAR SINGKATAN
cAMP : cyclic 3`,5`- Adenosine Mono Phosphate
SKA : Sindroma Koroner Akut
NO : Nitric Oxide
cGMP : cyclic Guanosine Mono Phosphate
PJK : Penyakit Jantung Koroner
APTS : Angina Pektoris Tak Stabil
NSTEMI : Non ST Elevasi MIokard Infark
STEMI : ST Elevasi MIokard Infark
PRP : Platelet Rich Plasma
PPP : Platelet Poor Plasma
ADP : Adenosine DiPhosphate
CFU : Colony Forming Unit
GP : Glikoprotein
ATP : Adenosine TriPhosphate
PDGF : Platelet Derived Growth Factor
VWF : Von Willebrand Factor
PGG2 : Prostaglandin G2
PGH2 : Prostaglandin H2
EDTA : Etilen Diamine Tetra-acetic Acid
PPACK : d-phenylalanine-proline-arginine chloromethyl ketone
ACD : Acid Citrate Dextrose
ACD-A : Anticoagulant Citrate Dextrose Solution A
PAR : Protease Activated Receptor
FDP : Fibrin Degradation Product
MMLDL : Minimally Modified Low Density Lipoprotein
VCAM : Vascular Cell Adhesion Molecule
LDL : Low Density Lipoprotein
TNF : Tumor Necrosis Factor
LDH : Lactate Dehydogenase
RANTES : Regulated upon activation normal T cells
TGF-β : Transforming Growth Factor-β
EGF : Endothelial Growth Factor
bFGF : basic Fibroblast Growth Factor
PSGL : P Selectin Glycoprotein Ligand
PECAM : Platelet Endothelial Cell Adhesion Molecule
RINGKASAN
Sistem hemostasis merupakan mekanisme tubuh dalam mengontrol respon
terhadap perdarahan atau terjadinya trombosis yang berlebihan sehingga proses
trombogenesis dan proses fibrinolisis dalam keadaan seimbang. Proses hemostasis
pada keadaan normal membantu menghentikan perdarahan dan bila berlebihan
akan menimbulkan oklusi trombotik dan infark sistemik. Trombosis terjadi bila ada
ketidakseimbangan antara faktor trombogenik dan mekanisme proteksi.
Yang termasuk dalam faktor-faktor trombogenik adalah kerusakan dinding
pembuluh darah, rangsangan agregasi trombosit, aktivasi koagulasi darah dan
stasis, sedangkan keadaan-keadaan yang berpengaruh dalam mekanisme proteksi
adalah endotel yang utuh, inhibitor protease dari sistem koagulasi, inaktivasi
koagulasi oleh hati dan sistem fibrinolitik
Sindroma Koroner Akut merupakan istilah terhadap sekumpulan penyakit
arteri koroner yang bersifat trombotik. Sebagai kelainan dasar adalah atersklerosis
yang menyebabkan terbentuknya plak aterom. SKA mencakup angina pectoris tak
stabil (APTS), infark miokard (non ST elevasi miokard infark dan ST elevasi miokard
infark).
Banyak peneliti melaporkan bahwa penyumbatan pembuluh darah otak dan
jantung sering terjadi akibat hiperaktivitas fungsi trombosit. Hiperaktivitas trombosit
dapat meningkatkan agregasi trombosit yang menimbulkan thrombosis, akibatnya
pembuluh darah menjadi tersumbat. Salah satu cara untuk menilai fungsi trombosit
dengan memeriksa agregasi trombosit
Pemeriksaan agregasi trombosit bertujuan mendeteksi abnormalitas fungsi
mikroskopik dan menggunakan analyzer, tetapi yang paling sering dikerjakan
menggunakan analyzer berdasarkan perubahan transmisi cahaya.
Selama periode Oktober sampai Desember 2010 telah dilakukan
suatu penelitian Cross Sectional Study di Departemen Patologi Klinik dan bekerja
sama dengan Departemen Kardiologi dan Departemen Ilmu Penyakit Dalam FK
USU/RSUP.H.Adam Malik Medan. Kelompok kasus adalah pasien sindroma
koroner akut yang memenuhi kriteria inklusi; sebagai kontrol diambil orang normal.
Darah diambil dari vena cubiti dengan Dysposible Syringe 15 cc yang dibagi atas 3
bagian yaitu : 3 cc darah tanpa antikoagulan untuk mendapatkan serum dilakukan
untuk pemeriksaan profil lipid, enzim jantung, kadar gula darah sewaktu. 2 cc
darah EDTA untuk pemeriksaan darah lengkap dan 3,6 cc darah dengan 0,4 cc
antikoagulan Na-Citrat 3,8% untuk mendapatkan plasma kaya trombosit dan
plasma miskin trombosit untuk pemeriksaan agregasi trombosit, dengan memakai
agonis ADP konsentrasi 1, 5 dan 10 µM dan epinefrin dengan konsentrasi 5 dan 10
µM.
Dari penelitian ini didapatkan bahwa ada perbedaan yang bermakna antara
nilai agregasi trombosit pada kelompok Sindroma Koroner Akut dibandingkan
dengan kelompok normal, dengan memakai agonis ADP 1 dan 5 µM (p<0,05).
Juga dijumpai adanya perbedaan bermakna agregasi trombosit dengan agonis
ADP 10 µM pada kelompok pasien Sindroma Koroner Akut dengan usia > 50
tahun.
Orang berusia diatas 50 tahun yang memiliki faktor resiko, sebaiknya
dilakukan pemeriksaan agregasi trombosit.
RINGKASAN
Sistem hemostasis merupakan mekanisme tubuh dalam mengontrol respon
terhadap perdarahan atau terjadinya trombosis yang berlebihan sehingga proses
trombogenesis dan proses fibrinolisis dalam keadaan seimbang. Proses hemostasis
pada keadaan normal membantu menghentikan perdarahan dan bila berlebihan
akan menimbulkan oklusi trombotik dan infark sistemik. Trombosis terjadi bila ada
ketidakseimbangan antara faktor trombogenik dan mekanisme proteksi.
Yang termasuk dalam faktor-faktor trombogenik adalah kerusakan dinding
pembuluh darah, rangsangan agregasi trombosit, aktivasi koagulasi darah dan
stasis, sedangkan keadaan-keadaan yang berpengaruh dalam mekanisme proteksi
adalah endotel yang utuh, inhibitor protease dari sistem koagulasi, inaktivasi
koagulasi oleh hati dan sistem fibrinolitik
Sindroma Koroner Akut merupakan istilah terhadap sekumpulan penyakit
arteri koroner yang bersifat trombotik. Sebagai kelainan dasar adalah atersklerosis
yang menyebabkan terbentuknya plak aterom. SKA mencakup angina pectoris tak
stabil (APTS), infark miokard (non ST elevasi miokard infark dan ST elevasi miokard
infark).
Banyak peneliti melaporkan bahwa penyumbatan pembuluh darah otak dan
jantung sering terjadi akibat hiperaktivitas fungsi trombosit. Hiperaktivitas trombosit
dapat meningkatkan agregasi trombosit yang menimbulkan thrombosis, akibatnya
pembuluh darah menjadi tersumbat. Salah satu cara untuk menilai fungsi trombosit
dengan memeriksa agregasi trombosit
Pemeriksaan agregasi trombosit bertujuan mendeteksi abnormalitas fungsi
mikroskopik dan menggunakan analyzer, tetapi yang paling sering dikerjakan
menggunakan analyzer berdasarkan perubahan transmisi cahaya.
Selama periode Oktober sampai Desember 2010 telah dilakukan
suatu penelitian Cross Sectional Study di Departemen Patologi Klinik dan bekerja
sama dengan Departemen Kardiologi dan Departemen Ilmu Penyakit Dalam FK
USU/RSUP.H.Adam Malik Medan. Kelompok kasus adalah pasien sindroma
koroner akut yang memenuhi kriteria inklusi; sebagai kontrol diambil orang normal.
Darah diambil dari vena cubiti dengan Dysposible Syringe 15 cc yang dibagi atas 3
bagian yaitu : 3 cc darah tanpa antikoagulan untuk mendapatkan serum dilakukan
untuk pemeriksaan profil lipid, enzim jantung, kadar gula darah sewaktu. 2 cc
darah EDTA untuk pemeriksaan darah lengkap dan 3,6 cc darah dengan 0,4 cc
antikoagulan Na-Citrat 3,8% untuk mendapatkan plasma kaya trombosit dan
plasma miskin trombosit untuk pemeriksaan agregasi trombosit, dengan memakai
agonis ADP konsentrasi 1, 5 dan 10 µM dan epinefrin dengan konsentrasi 5 dan 10
µM.
Dari penelitian ini didapatkan bahwa ada perbedaan yang bermakna antara
nilai agregasi trombosit pada kelompok Sindroma Koroner Akut dibandingkan
dengan kelompok normal, dengan memakai agonis ADP 1 dan 5 µM (p<0,05).
Juga dijumpai adanya perbedaan bermakna agregasi trombosit dengan agonis
ADP 10 µM pada kelompok pasien Sindroma Koroner Akut dengan usia > 50
tahun.
Orang berusia diatas 50 tahun yang memiliki faktor resiko, sebaiknya
dilakukan pemeriksaan agregasi trombosit.
BAB I
PENDAHULUAN
1.1. LATAR BELAKANG PENELITIAN
Sel trombosit berbentuk discus dan beredar dalam sirkulasi darah tepi dalam
keadaan tidak mudah melekat (adhesi) terhadap endotel pembuluh darah atau
menempel (agregasi) antar sel-sel trombosit maupun terhadap sel-sel darah yang
lainnya.1,2,3
Peranan sel trombosit pada proses trombogenesis untuk membentuk sumbat
trombosit diawali dengan reaksi adhesi trombosit, kemudian diikuti dengan
perubahan bentuk dan pelepasan isi granula sebagai reaksi sekresi sel trombosit,
selanjutnya terjadi agregasi trombosit untuk membentuk gumpalan dan akhirnya
aktivasi sistem koagulasi oleh membran trombosit.1,2,3
Sistem hemostasis merupakan mekanisme tubuh dalam mengontrol respon
terhadap perdarahan atau terjadinya trombosis yang berlebihan sehingga proses
trombogenesis dan proses fibrinolisis dalam keadaan seimbang. Proses hemostasis
pada keadaan normal membantu menghentikan perdarahan dan bila berlebihan
akan menimbulkan oklusi trombotik dan infark sistemik. Aktivitas sistem hemostasis
akan beradaptasi terhadap umur dan penyakit vaskuler. Sehingga tak dapat
membedakan antara kerusakan pembuluh darah dan kerusakan plak aterosklerotik.
Trombosis juga terjadi bila ada ketidakseimbangan antara faktor trombogenik dan
mekanisme proteksi.3,4,5
Yang termasuk dalam faktor-faktor trombogenik adalah kerusakan dinding
pembuluh darah, rangsangan agregasi trombosit, aktivasi koagulasi darah dan
adalah endotel yang utuh, inhibitor protease dari sistem koagulasi, inaktivasi
koagulasi oleh hati dan sistem fibrinolitik.4,5
Sel trombosit bereaksi terhadap robeknya plak aterosklerotik seperti terhadap
kerusakan traumatik pembuluh darah normal melalui pengendapan sel-sel trombosit
sebagai reaksi adhesi dan agregasi trombosit. Adhesi sel trombosit dimulai dengan
munculnya glikoprotein adhesif pada membran trombosit dan jika trombosit
diaktivasi maka kompleks glikoprotein untuk agregasi akan berikatan dengan
fibrinogen maupun faktor von Willebrand sehingga membentuk agregat. Area yang
kaya akan lipid pada plak yang robek sebagai faktor trombogenik. Aktivasi
glikoprotein untuk agregasi dapat dihambat oleh prostasiklin melalui peningkatan
cAMP didalam trombosit dan NO melalui peningkatan cGMP didalam trombosit yang
dihasilkan oleh endotel yang intak sebagai faktor proteksi.4,5,6
Berdasarkan faktor diatas maka terjadinya aterosklerosis pada pembuluh
darah bukan saja disebabkan oleh penimbunan lemak pada dinding pembuluh darah
tetapi merupakan lingkaran yang kompleks meliputi penimbunan lemak, modulator
sistem imun dan mekanisme trombotik.7,8,9
Terjadinya aterosklerotik pembuluh darah dengan manifestasi stroke dan
penyakit jantung koroner merupakan penyebab utama morbiditas dan mortalitas baik
di negara maju maupun negara berkembang seperti Indonesia. Pada pasien
tersebut yang merupakan akibat komplikasi aterosklerotik, didapatkan agregasi
trombosit yang meningkat dibanding orang sehat.10,11
Hasil pengukuran agregasi trombosit yang meningkat merupakan salah satu
faktor untuk menilai perkembangan thrombosis dan progresivitas aterosklerosis
bertambahnya umur dan hal tersebut berhubungan dengan perkembangan
aterosklerosis.8,9,12
Agregasi trombosit yang stabil oleh karena perubahan fibrinogen menjadi
fibrin membentuk trombi arteri di tempat injuri vaskuler seperti plak atersklerotik yang
robek atau area dimana ada gangguan aliran darah. Trombi tersebut menyebabkan
komplikasi tromboembolik dari aterosklerosis yaitu menimbulkan infark otot jantung,
stroke trombotik dan penyakit pembuluh darah perifer. Beberapa studi melaporkan
reaktivitas trombosit yang bertambah pada keadaan-keadaan tersebut. Untuk
mengetahui agregabilitas trombosit pasien dengan kelainan trombotik arteri akut
melalui pemeriksaan agregasi trombosit secara in vitro maupun aktivasi dan
agregasi trombosit secara in vivo.8,9,13,14,15
Penyakit Jantung Koroner (PJK) merupakan penyebab kematian pertama
pada Negara-negara berkembang. Estimasi pada tahun 2006 di Amerika 700.000
orang mendapatkan serangan baru pertama kali dan kira-kira 500.000 orang dengan
serangan berulang. Serangan pertama kali terjadi rata-rata pada usia 65,8 tahun
pada pria dan 70,4 tahun pada wanita. 50% pada pria dan 63% pada wanita
meninggal tiba-tiba karena PJK tanpa simptom awal. Di Indonesia dari Survei
Kesehatan Rumah Tangga (SKRT) yang dilakukan secara berkala oleh Departemen
Kesehatan menunjukkan bahwa penyakit kardiovaskuler memberikan kontribusi
sebesar 19,8% dari seluruh penyebab kematian pada tahun 1993 dan meningkat
menjadi 24,4% pada tahun 1998.10,16
Penyakit Jantung Koroner (PJK) merupakan penyakit dengan berbagai
keadaan klinis, dari yang asimtomatis, angina stabil maupun sindroma koroner akut
Sindroma Koroner Akut merupakan istilah terhadap sekumpulan penyakit
arteri koroner yang bersifat trombotik. Sebagai kelainan dasar adalah atersklerosis
yang menyebabkan terbentuknya plak aterom. Aterosklerosis adalah suatu kelainan
yang didasari oleh inflamasi. SKA mencakup angina pectoris tak stabil (APTS),
infark miokard (non ST elevasi miokard infark dan ST elevasi miokard infark).16,17
Apabila terdapat pemicu (trigger) yang sering, yaitu aktivitas eksternal yang
berhubungan dengan rangsangan simpatis misalnya stress fisik, stress emosional
atau vasokonstriksi, dapat menyebabkan plak rawan pecah (rupture) sehingga
terjadi kontak antara aliran darah dengan isi trombogenik dari plak atau permukaan
endotel yang terbuka. Sel endotel yang terbuka akan menyebabkan matriks
subendotelial yang sangat trombogenik jadi tidak terlindungi sehingga menimbulkan
pembentukan thrombus secara cepat dan menyebabkan tersumbatnya arteri
koroner.16,17,18
Frekuensi kematian akibat penyumbatan pembuluh darah otak dan miokard di
Indonesia meningkat dari tahun ke tahun. Salah satu faktor penting yang berperan
dalam proses penyumbatan tersebut adalah thrombosis. Banyak peneliti melaporkan
bahwa penyumbatan pembuluh darah otak dan jantung sering terjadi akibat
hiperaktivitas fungsi trombosit. Hiperaktivitas trombosit dapat meningkatkan agregasi
trombosit yang menimbulkan thrombosis, akibatnya pembuluh darah menjadi
tersumbat. Salah satu cara untuk menilai fungsi trombosit dengan memeriksa
agregasi trombosit.8,9,15
Pemeriksaan agregasi trombosit bertujuan mendeteksi abnormalitas fungsi
trombosit. Pemeriksaan dapat dilakukan dengan berbagai cara seperti makroskopik,
mikroskopik dan menggunakan analyzer, tetapi yang paling sering dikerjakan
Tahun 1962 O`Brien dan Born menemukan instrument untuk mengukur
agregasi trombosit yang memakai dasar turbidimetri. Alat ini distandarisasi memakai
plasma kaya trombosit (PRP) sebagai 0% agregasi dan plasma miskin trombosit
(PPP) sebagai 100% agregasi. Dicatat transmisi cahaya yang melalui cuvet berisi
suspensi trombosit yang diaduk pada suhu 37°C. Bila terbentuk aggregate setelah
penambahan agonis, dijumpai peningkatan transmisi cahaya. Respon agregasi
trombosit dihitung dengan membagi jarak dari baseline ke agregasi maksimal
dengan jarak dari baseline ke agregasi 100%. Pola agregasi trombosit dikenal
respon primer terhadap penambahan agonis eksogen seperti ADP, diikuti respon
sekunder dari pelepasan adenine nukleotida yang terdapat dalam granula padat
trombosit. Respon tersebut dikenal sebagai gelombang pertama dan kedua. 19,36,37
Mardiana I membandingkan nilai agregasi trombosit pada penderita angina
pectoris tak stabil dan penderita PJK. Dijumpai perbedaan bermakna antara nilai
agregasi trombosit dengan ADP 5 µg dengan ADP 10 µg antara kelompok angina
pectoris tak stabil dan PJK yaitu 48,26 ± 14,4% vs 37,26 ± 10,2% dan 59,98 ±
15,21% vs 47,64 ± 14,13% (p <0,05). Reversibilitas akan mengurang dengan
bertambahnya usia, yaitu pada penambahan usia 1 tahun akan mengurangi
reversibilitas 0,39%. Keempat factor resiko (hipertensi, merokok, hiperkolesterolemia
dan diabetes mellitus) tidak berperanan pada peningkatan agregasi trombosit.21
Yoshida T pada tahun 1982 melaporkan bahwa pada kelompok pasien angina
pectoris tak stabil (APTS) ditemukan agregasi trombosit yang diinduksi ADP dan
kolagen pada saat istirahat lebih rendah dibandingkan kelompok normal. Tetapi
meningkat setelah latihan. Pada kelompok angina pectoris stabil, tidak ditemukan
Guha S dkk meneliti agregasi trombosit pada 64 orang pasien sindroma
koroner akut 48 jam dan 7 hari setelah pemberian aspirin dan clopidogrel.
Ditemukan peningkatan agregasi trombosit yang diinduksi epinefrin, ADP dan
collagen setelah 48 jam pemberian antitrombosit. Agregasi trombosit meningkat
pada kelompok diabetes dan perokok.23
Hutajulu NC melaporkan bahwa agregasi trombosit meningkat pada hari
pertama infark dan menurun pada hari ketujuh walaupun masih lebih tinggi jika
dibandingkan dengan kelompok kelola. Lokasi infark dan kebiasaan merokok tidak
mempengaruhi peningkatan agregasi trombosit. Tidak terdapat korelasi antara kadar
puncak enzim dan kadar lemak darah dengan peningkatan agregasi trombosit.24
Lakhey M (2005) menjumpai peningkatan agregasi trombosit pada kelompok
pasien penyakit jantung iskemik, dengan berbagai agonis (collagen, ADP, epinefrin
dan thrombin). Konsumsi aspirin berhubungan dengan penurunan agregasi
trombosit pada kelompok pasien iskemik.25.
1.2. Perumusan masalah
Bagaimana perbedaan nilai agregasi trombosit pada penderita sindroma
koroner akut dibandingkan dengan kelompok normal.
1.3. Hipotesa penelitian
Ada perbedaan fungsi agregasi trombosit pada penderita Sindroma Koroner
Akut dibandingkan dengan kelompok normal.
1.4. Tujuan penelitian
1.4.1. Tujuan umum
1.4.2. Tujuan khusus
- Mengetahui perbedaan fungsi agregasi trombosit pada penderita Sindroma
Koroner Akut dibandingkan dengan kelompok normal.
- Mengetahui perbedaan fungsi agregasi trombosit pada penderita Sindroma
Koroner Akut usia diatas 50 tahun dibandingkan dibawah 50 tahun.
1.5. Manfaat penelitian
Dengan pemeriksaan agregasi trombosit, dapat memberi informasi tentang
peranan trombosit dalam patofisiologi terjadinya Sindroma Koroner Akut, sehingga
para klinisi dapat member penatalaksanaan dan pencegahan yang tepat untuk
BAB II
TINJAUAN KEPUSTAKAAN
2.1. TROMBOSIT
2.1.1.Produksi Trombosit
Sel trombosit berasal dari fragmentasi sitoplasma megakariosit sumsum
tulang. Prekursor megakariosit, megakarioblast, muncul melalui proses diferensiasi
dari sel induk hemopoetik. Megakariosit mengalami pematangan dengan replikasi
inti endomitotik yang sinkron, memperbesar volume sitoplasma sejalan dengan
penambahan lobus inti menjadi kelipatan duanya. Pada berbagai stadium dalam
perkembangannya (paling banyak pada stadium inti delapan), sitoplasma menjadi
granular dan trombosit dilepaskan. Produksi trombosit mengikuti pembentukan
mikrovesikel dalam sitoplasma sel yang menyatu membentuk membrane pembatas
trombosit. tiap sel megakariosit menghasilkan 1000-1500 trombosit. Sehingga
diperkirakan akan dihasilkan 35.000/ul trombosit per hari. Interval waktu semenjak
diferensiasi sel induk sampai produksi trombosit berkisar sekitar 10 hari.2,25,26
Jumlah sel trombosit yang bersirkulasi dalam darah tepi sangat tergantung
jumlah sel megakariosit, volume sitoplasma megakariosit, umur trombosit dan
sekuestrasi oleh limpa. Progenitor megakariosit CFU-Mega meningkat atau menurun
sebagai respon terhadap megakariosit26,27
Trombopoetin adalah pengatur utama produksi trombosit, dihasilkan oleh hati
dan ginjal. Trombosit mempunyai reseptor untuk trombopoetin (C-MPL) dan
mengeluarkannya dari sirkulasi, karena itu kadar trombopoetin tinggi pada
trombositopenia akibta aplasia sumsum tulang. Trombopetin meningkatkan jumlah
setelah dimulainya terapi dan tetap tinggi selama 7-10 hari. Interleukin-11 juga dapat
meningkatkan trombosit dalam sirkulasi.2,27,28
Jumlah trombosit normal adalah sekitar 250 x 109/l (rentang 150-400 x 109/l)
dan lama hidup trombosit yang normal adalah 7-10 hari. Hingga sepertiga dari
trombosit produksi sumsum tulang dapat terperangkap dalam limpa yang normal,
tetapi jumlah ini meningkat menjadi 90% pada kasus splenomegali berat.2,27,29
2.1.2.Struktur Trombosit
Glikoprotein permukaan sangat penting dalam reaksi adhesi dan agregasi
trombosit. Adhesi pada kolagen difasilitasi oleh glikoprotein Ia (GP Ia). Glikoprotein
Ib dan IIb/IIIa penting dalam perlekatan trombosit pada von Willebrand factor (VWF)
dan subendotel vascular. Reseptor IIb/IIIa juga merupakan reseptor untuk fibrinogen
yang penting dalam agregasi trombosit.27,28,29
Membran plasma berinvaginasi ke bagian dalam trombosit untuk membentuk
suatu sistem membrane (kanalikular) terbuka yang menyediakan permukaan reaktif
yang luas tempat protein koagulasi plasma diabsorbsi secara selektif. Fosfolipid
membran (faktor trombosit 3) sangat penting dalam konversi faktor X menjadi Xa
dan protrombin (faktor II) menjadi thrombin (faktor IIa).27,28,29
Di bagian dalam trombosit terdapat kalsium, nukleotida (terutama ADP,ATP
dan serotonin) yang terkandung dalam granula padat. Granula alfa mengandung
antagonis heparin, faktor pertumbuhan (PDGF), β-tromboglobulin, fibrinogen, vWF.
Organel spesifik lain meliputi lisosom yang mengandung enzim hifrolitik, dan
peroksisom yang mengandung katalase. Selama reaksi pelepasan, isi granula
2.1.3. Fungsi Trombosit
Fungsi utama trombosit adalah pembentukan sumbat mekanik selama respon
hemostasis normal terhadap cedera vascular. Tanpa trombosit, dapat terjadi
kebocoran darah spontan melalui pembuluh darah kecil. Reaksi trombosit berupa
adhesi, sekresi, agregasi dan fusi serta aktivitas prokagulannya sangat penting
untuk fungsinya.29,30,31
2.1.4.Pembentukan Sumbat Trombosit Hemostatik Primer
Agar dapat terjadi hemostasis primer yang normal, dan agar trombosit
memenuhi tugasnya membentuk sumbat trombosit inisial, maka harus terdapat
trombosit dalam jumlah memadai di dalam sirkulasi, dan trombosit tesebut harus
berfungsi normal. Fungsi hemostasis normal memerlukan peran serta trombosit
yang berlangsung secara teratur, yang penting dalam pembentukan sumbat
hemostatik primer. Hal ini melibatkan, pada awalnya, adhesi trombosit, agregasi
trombosit dan akhirnya reaksi pembebasan trombosit disertai rekrutmen trombosit
lain.1,3,29,31,34,35
2.1.4.1.Adhesi Trombosit
Setelah cedera pembuluh darah, trombosit melekat pada jaringan ikat
subendotel yang terbuka. Trombosit menjadi aktif apabila terpajan ke kolagen
subendotel dan bagian jaringan yang cedera. Adhesi trombosit melibatkan suatu
interaksi antara glikoprotein membrane trombosit dan jaringan yang terpajan atau
cedera. Adhesi trombosit bergantung pada faktor protein plasma yang disebut faktor
von Willebrand, yang memiliki hubungan yang integral dan kompleks dengan faktor
koagulasi antihemofilia VIII plasma dan reseptor trombosit yang disebut glikoprotein
Ib membrane trombosit. Adhesi trombosit berhubungan dengan peningkatan daya
atau jaringan yang cedera. Dengan demikian, terbentuk sumbat hemostatik primer
atau inisial. Pengaktifan permukaan trombosit dan rekrutmen trombosit lain
menghasilkan suatu massa trombosit lengket dan dipermudah oleh proses agregasi
trombosit.30,31,32,33
2.1.4.2.Agregasi
Agregasi adalah kemampuan trombosit melekat satu sama lain untuk
membentuk suatu sumbat. Agregasi awal terjadi akibat kontak permukaan dan
pembebasan ADP dari trombosit lain yang melekat ke permukaan endotel. Hal ini
disebut gelombang agregasi primer. Kemudian, seiring dengan makin banyaknya
trombosit yang terlibat, maka lebih banyak ADP yang dibebaskan sehingga terjadi
gelombang agregasi sekunder disertai rekrutmen lebih banyak trombosit. Agregasi
berkaitan dengan perubahan bentuk trombosit dari discoid menjadi bulat.
Gelombang agregasi sekunder merupakan suatu fenomena ireversibel, sedangkan
perubahan bentuk awal dan agregasi primer masih reversible.30,31,32,33
In vitro, agregasi dapat dipicu dengan reagen ADP, thrombin, epinefrin,
serotonin, kolagen atau antibiotik ristosetin.
Agregasi in vitro juga terjadi dalam dua fase :
1. Agregasi primer atau reversible
2. Agregasi sekunder atau ireversibel.
Pengikatan ADP yang dibebaskan dari trombosit aktif ke membrane trombosit
akan mengaktifkan enzim fosfolipase, yang menghidrolisis fosfolipid di membrane
trombosit untuk menghasilkan asam arakidonat. Asam arakidonat adalah precursor
mediator kimiawi yang sangat kuat baik pada agregasi maupun inhibisi agregasi
yang terlibat dalam jalur prostaglandin. Melalui proses ini, asam arakidonat diubah di
PGG2 dan PGH2. Stimulator kuat untuk agregasi trombosit, senyawa tromboksan
A2, dihasilkan oleh kerja enzim tromboksan sintetase pada berbagai
endoperoksidase siklik ini. Tromboksan A2 adalah senyawa yang sangat aktif, tetapi
tidak stabil yang mengalami penguraian menjadi tromboksan B2 yang stabil dan
inaktif. Tromboksan A2 juga merupakan vasokonstriktor kuat yang akan mencegah
pengeluaran darah lebih lanjut dari pembuluh yang rusak.30,31,32,33
2.1.4.3.Reaksi Pembebasan
Pemajanan kolagen atau kerja thrombin menyebabkan sekresi isi granul
trombosit yang meliputi ADP, serotonin, fibrinogen, enzim lisosom, β-tromboglobulin
dan factor trombosit 4. Kolagen dan thrombin mengaktifkan sintesis prostaglandin
trombosit. Terjadi pelepasan diasilgliserol (yang mengaktifkan fosforilasi protein
melalui protein kinase C) dan inositol trifosfat (menyebabkan pelepasan ion kalsium
intrasel) menyebabkan terbentuknya tromboksan A2.30,31,33
Agregasi primer melibatkan perubahan bentuk trombosit dan disebabkan oleh
kontraksi mikrotubulus. Gelombang agregasi trombosit sekunder melibatkan
terutama pelepasan mediator-mediator kimiawi yang terdapat di dalam granula
padat. Pelepasan ini melengkapi fungsi utama ketiga trombosit, yaitu reaksi
pembebasan. Reaksi pembebasan diperkuat oleh peningkatan kalsium intrasel,
yang semakin mengaktifkan dan meningkatkan pembebasan tromboksan A2.
Tromboksan A2 memperkuat agregasi trombosit serta mempunyai aktivitas
vasokonstriksi yang kuat. Reaksi pelepasan dihambat oleh zat-zat yang
meningkatkan kadar cAMP trombosit, salah satunya adalah prostasiklin (PGI2) yang
disintesis oleh sel endotel vascular. Prostasiklin merupakan inhibitor agregasi
trombosit yang kuat dan mencegah deposisi trombosit pada endotel vascular
2.1.4.4.Aktivitas Prokoagulan Trombosit
Setelah agregasi trombosit dan reaksi pelepasan, fosfolipid membrane yang
terpajan (factor trombosit 3) tersedia untuk 2 jenis reaksi dalam kaskade koagulasi.
Kedua reaksi yang diperantarai fosfolipid ini bergantung pada ion kalsium. Reaksi
pertama (tenase) melibatkan faktor IXa, VIIIa dan X dalam pembentukan faktor Xa.
Reaksi kedua (protrombinase) menghasilkan pembentukan thrombin dari interaksi
factor Xa, Va dan protrombin. Permukaan fosfolipid membentuk cetakan yang ideal
untuk konsentrasi dan orientasi protein-protein tersebut yang penting.3,31,33
2.1.4.5.Agregasi Trombosit Irreversibel
Konsentrasi ADP yang tinggi, enzim yang dilepaskan selama reaksi
pelepasan dan protein kontraktil trombosit menyebabkan fusi yang irreversible pada
trombosit yang beragregasi [ada lokasi cedera vascular. Trombin juga mendorong
terjadinya fusi trombosit, dan pembentukan fibrin memperkuat stabilitas sumbat
trombosit yang terbentuk.3,31,33
2.2.AGREGASI TROMBOSIT
Tahun 1962 O`Brien dan Born menemukan instrument untuk mengukur
agregasi trombosit yang memakai dasar turbidimetri. Alat ini distandarisasi memakai
plasma kaya trombosit (PRP) sebagai 0% agregasi dan plasma miskin trombosit
(PPP) sebagai 100% agregasi. Dicatat transmisi cahaya yang melalui cuvet berisi
suspensi trombosit yang diaduk pada suhu 37°C. Bila terbentuk aggregate setelah
penambahan agonis, dijumpai peningkatan transmisi cahaya. Respon agregasi
trombosit dihitung dengan membagi jarak dari baseline ke agregasi maksimal
dengan jarak dari baseline ke agregasi 100%. Agonis yang berbeda menghasilkan
pola agregasi yang berbeda. Pola agregasi trombosit dikenal respon primer terhadap
adenine nukleotida yang terdapat dalam granula padat trombosit. Respon tersebut
dikenal sebagai gelombang pertama dan kedua. Respon bifasik ini dapat tidak
terlihat pada penambahan agonis konsentrasi tinggi. Dengan agonist kolagen, pola
agregasi menggambarkan adhesi trombosit dengan fibril kolagen diikuti agregasi
trombosit. Aspirin dapat menghambat agregasi trombosit dengan agonis kolagen
dosis rendah, tetapi pada dosis yang lebih tinggi agregasi masih terjadi. 19,36,37
2.2.1.Variabel Pemeriksaa Agregasi trombosit19,36,38,39
2.2.1.1.Venapunksi
Sampel pasien dewasa diambil dengan jarum 18-21 G dan syringe plastic.
Untuk kasus pediatric, dipakai jarum 23-25 G.
2.2.1.2.Antikoagulan
Sitrat
Sodium sitrat (0,102 M, 0,129 M sitrat buffered dan non buffered) dengan
rasio 9 bagian darah dengan 1 bagian antikoagulan merupakn antikoagulan pilihan
untuk pemeriksaan agregasi trombosit. Sebaiknya tidak memakai Vacutainer karena
dikhawatirkan dapat terjadi aktivasi trombosit oleh tekanan shear vakum. Beberap
laboratorium mengkoreksi hematokrit, terutama bila nilai hematokritnya terlalu tinggi
atau rendah. Hardisty dkk menemukan bahwa pada orang dengan nilai hematokrit
yang tinggi, diperlukan lebih banyak agonist oleh karena kurangnya jumlah kalsium
bebas yang terdapat di plasma.
Heparin
Heparin menghambat pembentukan dan aktivitas thrombin melalui ikatan
dengan antitrombin III. Dapat dipakai untuk pemeriksaan trombosit, tetapi pada
dengan adanya heparin, oleh karena itu heparin buka merupakan pilihan untuk
pemeriksaan agregasi trombosit.
EDTA
Agregasi trombosit tergantung adanya kalsium bebas di plasma, EDTA tidak
cocok untuk pemeriksaan agregasi.
PPACK
d-phenylalanine-proline-arginine chloromethyl ketone (PPACK), suatu
antitrombin, mulai dipakai untuk pemeriksaan inhibisi trombosit oleh antagonist Gp
IIb/IIIa.
ACD
Antikoagulan ini menurunkan ph PRP 6,5; karena itu tidak sesuai untuk
pemeriksan agregasi.
ACD-A
Mempertahankan pH PRP 7,3; dapat dipakai untuk pemeriksaan agregasi.
2.2.1.3.Tabung kaca vs plastic
Penyiapan trombosit untuk pemeriksaan agregasi harus dilakukan dengan
memakai tabung plastic atau tabung kaca yang dilapisi silicon. Bila memakai tabung
yang tidak dilapisi, akan terjadi aktivasi trombosit.
2.2.1.4.Koreksi Jumlah Trombosit
Ada berbagai pendapat mengenai perlunya standarisasi jumlah trombosit
PRP. Respon agregasi dapat bervariasi berhubungan dengan jumlah trombosit.
2.2.1.5. Kontaminasi sel darah medah dan lipemia
Pemeriksaan agregasi trombosit berdasarkan transmisi optick adanya bahan
kontaminan seperti sel darah merah atau lemak, dapat mempengaruhi kemampuan
melepaskan ADP, menyebabkan trombosit menjadi refrakter setelah penambahan
ADP eksogen.
2.2.1.6.pH
Agregasi trombosit adalah pH sensitif. Ketika mempersiapkan bahan untuk
pemeriksaan agregasi, pH harus dipertahankan antara 7,2 dan 8,0. Bila pH plasma
dibawah 6,4 tidak terjadi agregasi; dan pada pH diatas 8,0 dapat terjadi agregasi
spontan. Perubahan pH terjadi melaui difusi CO2 dari plasma; karena itu tabung
PRP harus ditutup. Saline isotonic merupakan diluents pilihan utnuk agonist.
2.2.1.7.Temperatur
Pemeriksaan agregasi harus dilakukan pada suhu 37°C agar menyerupai
suasana in vivo.
2.2.1.8.Kecepatan Putaran Agregometer
Supaya terjadi agregasi, trombosit harus kontak satu sama lain. Bila
ditambahkan agonis pada trombosit yang tidak diputar, tidak akan terjadi agregasi.
Kecepatan putaran optimal berdasarkan tinggi kolum PRP, diameter kuvet dan
ukuran stir bar yang dipakai. Tiap pabrik memilki rekomendasi kecepatan putaran
optimal masing-masing.
2.2.1.9. Waktu Pemeriksaan
Sebaiknya pemeriksaan agregasi trombosit dikerjakan dalam 3 jam setelah
sampel diambil.
2.2.2.AGONIST19,36-39
2.2.2.1.ADP
Kadar 1-10 µM ADP sering dipakai pada pemeriksaan agregasi trombosit.
Kadar ADP yang rendah (1-3 µM) menghasilkan kurva tunggal (monofasik) atau
trombosit disagregasi. Kadar ADP yang lebih tinggi (10 atau 20 µM) dapat menutupi
respon bifasik oleh pelepasan ADP endogen. Ini masih dianggap respon bifasik
karena terjadi pelepasan ADP tetapi tidak tampak pada kurva. Aspirin akan
menghambat respon agregasi ADP kadar rendah, karena hambatan jalur
sikooksigenase dan pelepasan isi granul.19,36,39
2.2.2.2.Epinefrin
Biasanya dipakai epinefrin 5-10 µM untuk pemeriksaan agregasi. Dijumpai
gelombang pertama yang kecil, kadang diikuti respon sekunder yang lebih besar.
Gelombang kedua ini dihambat oleh aspirin, obat anti inflamasi non steroid,
antihistamin, beberapa antibiotik.19,36,37
2.2.2.3.Kolagen
Biasanya dipakai kadar 1-5 µg/ml. Kolagen adalah agonist yang paling kuat.
Agregasi trombosit yang diinduksi kolagen menunjukkan lag phase sekitar 1 menit,
dimana pada saat itu trombosit berikatan pada fibril kolagen, mengalami perubahan
bentuk dan reaksi pelepasan. Respon agregasi yang diukur adalah gelombang
kedua setelah aktivasi dan pelepasan trombosit. Pada kadar kolagen yang rendah,
respon agregasi trombosit dapat dihambat aspirin dan obat anti trombosit lain.19,37,38
2.2.2.4.Asam Arakidonat
Dengan siklooksigenase, asam arakidonat diubah menjadi tromboksan A2.
Aspirin menghambat jalur siklooksigenase dan respon agregasi terhadap asam
arakidonat. Pasien yang mengkonsumsi aspirin atau anti trombosit lain, penderita
gangguan pelepasan atau Glanzman tromboastenia akan memberikan hasil
abnormal agregasi trombosit yang diinduksi asam arakidonat. Pasien dengan SPD
2.2.2.5.Ristocetin
Pada trombosit normal, antibiotic ristocetin dengan kadar 1,5 mg/ml,
menyebabkan agregasi trombosit yang trgantung GpIb/VWF. Bila responnya
abnormal, dicurigai penyakit von Willebrand atau sindroma Bernard Soulier (tidak
ada kompleks GpIb-IX-V)19,37
2.2.2.6.Trombin
Trombin adalah agonist trombosit yang sangat poten. Peptida sintetik
Gly-Pro-Arg-Pro (GPRP) menghambat polimerisasi fibrin yang diinduksi thrombin,
sehingga dapat terjadi agregasi trombosit yang diinduksi thrombin. α-trombin dengan
kadar 0,1-0,5 U/ml dapat dipakai untuk mengakivasi trombosit, baik yang washed
atau gel-filtered.19,37,38
2.2.2.7. TRAP
Thrombin receptor activating peptide (TRAP) adalah peptide sintetik yang
berikatan dengan sekuens asam amino N-terminal dari “tethered ligand” yand
dibentuk setelah hidrolisis thrombin protease activatedreceptor (PAR1).
Penambahan TRAP 10 µM menyebabkan aktivasi respon trombin yang sangat kuat
tanpa pemecahan fibrinogen dan pembentukan clot. Pada umumnya trombosit
menunjukkan respon agregasi normal terhadap TRAP kecuali pada Glanzmann
thromboasthenia. Sekarang ini TRAP dipakai untuk memonitor efek farmakodinamik
anti trombosit baru yang menghambat ikatan fibrinogen dengan trombosit atau yang
2.2.3. Obatan-obatan Yang Mempengaruhi Agregasi Trombosit19,36,41,42
2.2.3.1.Antibiotik
Antibiotik yang memiliki struktur gugus β-lactam seperti penisilin dan
sefalosporin, dapat mempengaruhi fungsi trombosit. Mekanismenya diduga akibat
perubahan membrane yang menghambat interaksi reseptor-agonist atau
mempengaruhi influks kalsium.19,36
2.2.3.2.Dipyridamole
Dipyridamole adalah pyrimidopyrimidine yang menghambat uptake adenosine
dalam trombosit, sel endotel dan eritrosit. Hambatan ini menyebabkan peningkatan
lokal kadar adenosine yang menstimulasi adenilat siklase trombosit dan
meningkatkan kadar cyclic 3`,5`-adenosine monophosphate (cAMP). Peningkatan
cAMP mengurangi kemampuan agregasi trombosit.19,36,41
2.2.3.3.Fibrinolitik
Fibrinolisis dan pembentukan fibrin degradation product (FDP) berhubungan
dengan agregasi trombosit. FDP bersaing dengan fibrinogen untuk berikatan dengan
membrane trombosit dan mengganggu agregasi trombosit. Satu penelitian pada
pasien yang mendapat tenecteplase dan alteplase menunjukkan inhibisi bermakna
agregasi trombosit pada pemeriksaan agregasi. Penelitian lain yang
membandingkan reteplase, alteplase dan streptokinase, dijumpai inhibisi agregsi
trombosit pada ketiga kelompok. Pengurangan kadar fibrinogen plasma dan
gangguan ikatan fibrinogen-Gp IIb/IIIa berkorelasi dengan beratnya defek agregasi
trombosit.19,36,41
2.2.3.4.Dextran
Pemeberian dekstran intravena dapat menyebabkan menurunnya fungsi
trombosit spontan dan yang diinduksi agonist serta ekspresi marker aktivasi seperti
P-selectin.19,36,42
2.2.3.5.Anestesi
Anestesi seperti lidokain, dibukain, kokain menyebabkan efek langsung pada
membrane trombosit. Penambahan kokain pada trombosit in vitro menyebabkan
berkurangnya ikatan fibrinogen dengan reseptor Gp IIb-IIIa.19
2.2.3.6.Inhibitor Trombin
Trombin sangat penting dalam patofisiologi sindroma koroner akut. Trombin
memperantarai perubahan fibrinogen menjadi fibrin, mengaktivasi F.XIII yang
membantu stabilisasi clot, dan agonis trombosit yang poten. Generasi terbaru
inhibitor thrombin direk yang bekerja pada antitrombin III dapat menghambat
clot-bound thrombin dan aktivasi trombosit oleh thrombin19,36,41
2.2.3.7.Thienopyridines
ADP berikatan dengan reseptornya P2Y1 dan p2Y12. Reseptor P2Y12
adalah reseptor primer ADP yang memperantarai ikatan fibrinogen dan respon
agregasi. Thienopyridines, ticlopidine dan clopidogrel secara irreversible mengikat
reseptor ini dan menghambat agregasi trombosit.19,36,41
2.2.3.8.Antagonis GpIIb-IIIa
Antagonis GpIIb-IIIa berikatan dengan reseptor GpIIb-IIIa (integrin αIIbβ3) dan
mencegah ikatan fibrinogen atau VWF pada trombosit. Eptifibatide, abciximab dan
tirofiban menghambat agregasi trombosit dengan semua agonis (ADP, kolagen,
2.2.4.Pengukuran
Agregasi trombosit dapat diukur dengan menimbulkan kontak antara plasma
kaya trombosit dengan suatu zat penginduksi agregasi. Sebagian besar zat
penginduksi ini seperti kolagen, epinefrin dan thrombin bekerja melalui efek ADP
yang dibebaskan sendiri oleh trombosit. Penambahan ADP eksogen menyebabkan
agregasi secara langsung. Agregasi dikuantifikasi dengan menentukan apakah
plasma kaya trombosit yang keruh menjadi jernih karena trombosit yang semula
membentuk suspensi merata membentuk agregat berupa gumpalan-gumpalan besar
yang kurang memendarkan cahaya sehingga transmisi sinar melalui tabung lebih
mudah. Agregometer adalah suatu spektrofotometer yang diadaptasi untuk mencatat
perubahan dalam transmisi sinar sementara mempertahankan suhu yang konstan
dan pengocokan perlahan terhadap suspense trombosit.38,39,40
Setelah diperoleh suatu kurva normal transmisi cahaya, trombosit yang
diperiksa dipajankan ke berbagai zat dan berbagai kondisi. Aspirin, obat
antiinflamasi yang lain, dan banyak obat dari golongan fenotiazin sangat
menghambat kemampuan kolagen dan epinefrin menimbulkan agregasi, tetapi tidak
mengganggu efek langsung ADP. Gangguan konstitusional fungsi trombosit berbeda
satu sama lain dalam sifat bahan yang gagal memicu agregasi. Pasien yang
dicurigai mengidap gangguan gangguan ini harus bebas dari semua obat selama
paling tidak 1 minggu sebelum pemeriksaan.36,37,40
Dalam melakukan uji, pungsi vena harus mulus (nontraumatik). Jumlah
trombosit yang digunakan untuk uji harus distandarisasi karena respon agregasi
dipengaruhi oleh jumlah trombosit. Hal inilah yang menyebabkan pasien
trombositopenia sulit dievaluasi. Pemeriksaan agregasi harus dilakukan dalam 3 jam
pendingin karena hal ini menghambat fungsi trombosit; karena itu, uji dilakukan pada
suhu 37°C. Antikoagulan yang digunakan adalah natrium sitrat, dan sampel jangan
dimasukkan ke wadah kaca karena bahan ini akan mengaktifkan trombosit. Sampel
yang mengalami hemolisis atau lipemik dapat mengganggu interpretasi densitas
optis.
2.2.5.Interpretasi
Bahan-bahan penginduksi agregasi yang paling sering digunakan adalah
ADP dengan berbagai konsentasi, kolagen, epinefrin, ristosetin, thrombin dan asam
arakidonat.
ADP konsentrasi rendah memicu agregasi bifasik dengan gelombang primer
dan sekunder. ADP konsentrasi tinggi memicu hanya satu gelombang agregasi.
Pasien dengan gangguan pembebasan trombosit gagal memperlihatkan gelombang
agregasi kedua. Pasien dengan tromboastenia Glanzmann tidak memperlihatkan
agregasi trombosit pada pemberian ADP.
Agregasi dengan kolagen menghasilkan suatu periode laten yang diikuti oleh
sebuah gelombang agregasi. Penurunan agregasi terhadap kolagen terjadi pada
pasien yang mendapat aspirin dan obat anti-inflamasi.
Agregasi dengan epinefrin biasanya bersifat bifasik. Agregasi yang dipicu
oleh epinefrin ini juga terganggu pada pasien yang mendapat aspirin dan obat
anti-inflamasi. Demikian juga, agregasi thrombin bersifat bifasik dan mungkin terganggu
pada defek trombosit intrinsic tertentu.
Walaupun defek kongenital fungsi trombosit jarang dijumpai, banyak penyakit
didapat yang menekan mekanisme pembebasan trombosit. Aspirin jelas merupakan
obat yang paling sering menjadi penyebab, tetapi hanya sedikit pasien yang
trombosit. Pasien dengan uremia, penyakit hati yang parah atau penyakit terkait
alkohol tahap lanjut sering mengalami gangguan perdarahan kompleks yang
mencakup disfungsi trombosit. Ketiga penyakit ini menekan efek kolagen, epinefrin
atau ADP eksogen yang ditambahkan langsung pada pembebasan ADP. Gangguan
gangguan mieloproliferatif dan disproteinemia dapat menimbulkan kelainan serupa.
2.3.SINDROMA KORONER AKUT
2.3.1.Definisi
Sindroma Koroner Akut merupakan istilah terhadap sekumpulan penyakit
arteri koroner yang bersifat trombotik. Sebagai kelainan dasar adalah aterosklerosis
yang menyebabkan terbentuknya plak aterom. Pecahnya plak aterom akan
menyebabkan iskemia sampai nekrosis miokard. SKA mencakup angina pectoris tak
stabil (APTS), infark miokard (non ST Elevasi Mikcard infark dan ST Elevasi Miokard
Infark).
2.3.2.Patofisiologi Sindroma Koroner Akut
SKA dapat terjadi oleh adanya proses thrombosis akut dan proses
vasokonstriksi koroner. Lesi pada arteri koronaria dimulai dengan adanya trauma
minimal yang kronis pada endothelium sehingga mengganggu aliran darah.
Faktor-faktor resiko seperti hipertensi, hiperkolesterolemia, diabetes mellitus, iritasi kronik
dan infeksi menyebabkan disfungsi endotel, terjadi robekan lokal sehingga terjadi
akumulasi lipid dan monosit (makrofag). Lesi aterosklerotik awal disebut fatty streak
yang bersifat vulnerable. Modifikasi faktor resiko akan menyebabkan masukan
lipoprotein berkurang dan menimbulkan parut. Bila masukan lipoprotein meningkat
sehingga menimbulkan oklusi/suboklusi serta mengakibatkan terjadinya angina tak
stabil.
Hubungan waktu dan patofisiologi SKA, jika 10 sampai 20 menit setelah
terjadinya thrombus, dapat terjadi oklusi pembuluh darah temporer. Bila kerusakan
bertambah berat, dapat terjadi oklusi yang persisten yang sapat berlangsung sampai
satu jam (NSTEMI). Bila plak lebih besar dapat terjadi pembentukan yang menetap
sehingga dapat menyebabkan nekrosis transmural (STEMI). Oklusi total pembuluh
darah lebih dari 4-6 jam akan mengakibatkan nekrosis miokard yang ireversibel.
Tindakan reperfusi dalam periode waktu ini akan dapat membantu menyelamatkan
miokardium dan mengurangi morbiditas dan mortalitas.
2.3.3.Patogenesis Aterosklerosis
Pembuluh darah arteri sama seperti organ-organ lain di dalam tubuh yaitu
mengikuti proses umur (ketuaan) dimana terjadi proses yang karakterisktik seperti
penebalan lapisan intima, berkurangnya elastisitas dan bertambahnya diameter
intima.
WHO pada tahun 1958 mendefinisikan aterosklerosis sebagai perubahan
variable intima arteri yang merupakan akumulasi fokal lemak (lipid), kompleks
karbohidrat, darah dan hasil produk darah, jaringan fibrous dan deposit kalsium yang
kemudian diikuti dengan perubahan lapisan media.
2.3.3.1.Mekanisme dasar pembentukan plak.
Pembentukan foam cell
Proses ini diawali adhesi monosit pada permukaan endotel, diikuti migrasi
makrofag. Lipid diambil oleh makrofag, kemudian mengawali pembentukan foam
cell. Perubahan awal ini menghasilkan suatu molekul pro inflamasi yang disebut
minimally modified low density lipoprotein (MMLDL) yang berkontribusi terhadap
ekspresi VCAM pada endotel. Faktor-faktor inflamasi bekerja bersama-sama
menyebabkan migrasi monosit. Perubahan selanjutnya pada molekul LDL mengarah
pada LDL teroksidasi yang dikenali oleh macrophage scavenger receptor. Foam cell
yang terbentuk menghasilkan sitokin-sitokin inflamasi termasuk TNF-α dan
metalloproteinase dan juga factor prokaogulan.
Pembentukan lipid core
Lipid core merupakan ruang dalam matriks jaringan ikat tunika intima yang
terisi dengan debris seluler dan kolesterol. Plak aktif mengandung sejumlah
makrofag berkelompok pada pinggir inti, dengan ekspresi sebagian
metalloproteinase dalam destruksi matriks kolagen.Beberapa lipid ekstrasel yang
berasal dari ikatan LDL terhadap proteoglikans dalam intima, kebanyakan kolesterol
dan ester pada lipid core dilepaskan dari sitoplasma foam cell yang mati. Kehilangan
faktor pertumbuhan akan menginduksi apoptosis terutama bersamaan dengan
adanya TNF-α dalam jumlah besar pada plak. Ekspresi tissue factor oleh makrofag
dalam inti membuat area ini sangat trombogenik.
Proliferasi otot polos dan pembentukan cap
Bagian cap terdiri dari zat kolagen yang mengandung otot polos yang
menghasilkan matriks jaringan ikat. Sel-sel otot polos intima mempunyai
kecenderungan mengalami apoptosis. Migrasi, proliferasi otot polos dan deposisi
kolagen diatur oleh factor pertumbuhan yang dihasilkan oleh tiap sel. Trombosit,
thrombin dan fibrin juga dapat memacu proliferasi sel otot polos bila menumpuk
2.3.3.2.Perkembangan Plak
Menurut American Heart Association (AHA), perkembangan plak aterosklerosis
dapat dibagi 5 tipe yang dapat dihubungkan dengan tampilan klinisnya Yaitu :
1. Lesi awal (tipe 1), berkembang bila monosit melekat pada permukaan endotel
dan bermigrasi dari lumen untuk berakumulasi pada intima.
2. Lesi tipe 2 adalah fatty streak yang terdiri dari akumulasi lipid ekstra seluler
yang berisi foam cell.
3. Lesi tipe 3 seperti lesi tipe 2 yang disertai kelompok-kelompok kecil lipid
ekstraseluler. Meskipun lesi tipe 1-3 merupakan precursor lesi yang lebih
berat, namun belum menimbulkan gejala klinis.
4. Lesi tipe 4, seperti lesi tipe 2 disertai sel-sel otot polos terlihat dalam lesi di
bawah endotel, dan kelompok-kelompok lipid ekstraseluler bersatu
membentuk lipid core. Lesi ini disebut ateroma.
5. Lesi tipe 5a, seperti tipe 4 dengan kapsul fibrous yang tipis disebut juga
fibroateroma. Lesi tipe 5b adalah ateroma dengan kalsifikasi berat didalam
lipid core. Lesi 5c adalah fibrous ateroma atau pembentukan thrombus mural
dengan komponen lipid yang minimal. Lesi tipe 4 dan 5 biasanya asimtomatik,
namun dapat juga berupa angina stabil. Lesi tipe 5b dan 5c biasanya dengan
angina tak stabil
6. Lesi tipe 6 merupakan lesi yang berkomplikasi dengan thrombosis, dengan
tampilan klinis sindroma koroner akut. LEsi tipe 4 dan 5 disebut plak tidak
2.3.3.3.Disrupsi Plak
Disrupsi plak memegang peranan penting untuk terjadinya Sindroma Koroner
Akut. Resiko terjadinya ruptur plak tergantung dari kerentanan atau ketidakstabilan
plak, bukan adari ukuran atau derajat penyempitannya.
Faktor-faktor yang mempengaruhi instabilitas dan ruptur plak
Faktor Eksternal :
1. Sistemik : faktor hemodinamik dan farmakologik
2. Faktor intrinsik dari plak : besarnya plak, lokasi plak, kepadatan dan
ketebalan lipid dan ketebalan kap yang menyelimuti plak.
Faktor Internal :
1. Aktifitas sel inflamasi
2. Infeksi
3. Disfungsi endotel
4. Proliferasi sel otot polos
2.3.3.4.Trombosis Plak
Lebih dari 75% trombus yang ditemukan pada SKA terletak di tempat dimana
plak mengalami ruptur. Bila plak yang tidak stabil mendapat pencetus maka cap
yang tipis tersebut akan koyak dan terjadi pembentukan trombus yang dimulai dari
fisura atau robekan kap tadi.
Mula-mula terjadi akumulasi platelet di tempat koyakan, dengan adanya fibrin
akan membentuk gumpalan dini yang disebut white thrombus yang secara langsung
berusaha menutupi semua permukaan yang robek. Kemudian eritrosit menutupi
mengakibatkan oklusi koroner dan vasokonstriksi, sehingga akhirnya menimbulkan
tampilan klinis yang disebut dengan Sindroma Koroner Akut.
2.3.4.DIAGNOSA
2.3.4.1.Anamnesis
Nyeri dada tipikal merupakan gejala kardinal pasien infark miokard akut.
Lokasi nyeri substernal, retrosternal dan prekordial. Sifat nyeri : rasa sakit seperti
ditekan, rasa terbakar, tertindih benda berat, seperti ditusuk dan rasa diperas.
Penjalaran biasanya ke lengan kiri, leher, rahang bawah, gigi, punggung, perut dan
lengan kanan. Neri tidak membaik dengan istirahat atau minum obat nitrat. Pada
APS, rasa nyeri berkurang dengan istirahat atau obat-obatan dan nyeri dada < 20
menit.
2.3.4.2.Pemeriksaan Laboratorium
Identifikasi dini pada penderita SKA adalah dengan pemeriksaan petanda
cedera miokard seperti LDH, CK-MB, myoglobin dan troponin jantung (Troponin T
atau Troponin I). LDH meningkat setelah 24-48 jam bila ada infark, mencapai
puncak 3-6 hari dan kembali normal dalam 8-14 hari. CK-MB meningkat setelah 3
jam pasca infark, mencapai puncak dalam 10-36 jam dan kembali normal dalam 3-4
hari. Myoglobin dapat dideteksi 1 jam setelah infark dan mencapai puncaknya dalam
4-8 jam.
Troponin (cTnT dan cTnI) meningkat setelah 2 jam paska infark, mencapai
puncak dalam 10-24 jam dan cTnT masih dapat dideteksi setelah 5-14 hari, cTnI
masih dapat dideteksi setelah 5-10 hari. Troponin T dan I spesifik untuk kerusakan
2.3.4.3.Elektrokardiografi
Pemeriksaan EKG 12 sadapan harus dilakukan pada pasien dengan nyeri
dada. Pemeriksaan ini harus segera dilakukan 10 menit setelah pasien sampai di
IGD. Perubahan EKG pada STEMI adalah ST elevasi yang diikuti terbentuknya
gelombang Q patologis. Perubahan ini harus ditemui minimal pada 2 sandapan yang
berdekatan.
Gambaran EKG pada NSTEMI adalah depresi segmen ST > 0,05 Mv, inverse
gelombang T ditandai dengan >0,2 Mv dan inverse gelombang T yang simetris di
sandapan prekordial. Pada NSTEMI 1-6% dengan gambaran EKG normal.
Pemeriksaan EKG pada angina pectoris tak stabil adalah adanya depresi
segmen ST atau tanpa inverse gelombang T. Pada angina pectoris tak stabil 4%
penderita dengan gambaran EKG normal.
Diagnosa dilakukan berdasarkan kriteria WHO yaitu : terpenuhinya minimal 2
dari 3 kriteria berikut ini : nyeri dada iskemik yang khas, perubahan EKG dan
peningkatan enzim-enzim jantung.
2.4.Trombosit dalam Sindroma Koroner Akut
2.4.1.Disfungsi Endotel
Aterosklerosis koroner adalah suatu proses inflamasi kronis yang dapat
menjadi akut dengan rupturnya plak dan thrombosis arteri. Trombosit memegang
peran penting dalam oklusi vaskuler pada plak aterosklerotik koroner yang ruptur,
menimbulkan Sindroma Koroner Akut, terdiri dari miokardial infark (MI), non ST
segmen elevasi miokardial infark (NSTEMI) dan angina pectoris tak stabil (APTS).
arteri koroner akut mendukung adanya peranan trombosit dalam Sindroma Koroner
Akut.
Dalam keadaan normal, trombosit bersirkulasi dalam pembuluh darah tanpa
interaksi dengan sel lain. Sel endotel pembuluh darah normal mencegah perlekatan
maupun aktivasi trombosit dengan produksi bahan antitrombotik antara lain
prostasiklin (prostaglandin I2 atau PGI2) dan nitric oxide (NO), ekspresi
ecto-ADPase pada permukaan endotel. Adanya faktor resiko (merokok, diabetes,
hipertensi, kadar LDL yang tinggi, tekanan tinggi pada stenosis arteri, vasoaktif
amine, radikal bebas dan infeksi mikroorganisme) menyebabkan disfungsi endotel.
Disfungsi endotel yang ditandai dengan penurunan bioavaibilitas NO, mencetuskan
serangkaian proses pembentukan lesi aterosklerosis. Jalur NO memliki interaksi
sinergistik dengan pembentukan/degradasi nukleotida siklik dan fosforilasi protein
pada trombosit dan sel otot polos, yang mengatur fungsi kardiovaskular (tonus
vaskular, inhibisi agregasi trombosit serta adhesi leukosit, dan pencegahan
proliferasi sel otot polos).
Terganggunya permeabilitas sawar endotel memperantarai rekrutmen
monosit yang bersirkulasi dan plasma lipid ke dinding arteri, juga deposisi trombosit
pada endotel yang terluka. Dengan pelepasan faktor mitogenik, memperantarai
migrasi dan proliferasi sel otot polos, bersama peningkatan akumulasi lipid dan
sintesa jaringan ikat membentuk plak ateromatous tipikal. Proses yang terus
berlanjut menyebabkan hiperplasia lapisan intima-media pembuluh darah dan
perkembangan plak aterosklerotik.Plak yang rentan terdiri dari :
1. Inti lipid nekrotik yang luas (meliputi >40% total volume plak)
4. Berkurangnya kolagen dan sel otot polos
5. Materi trombotik dengan deposisi trombosit dan fibrin
2.4.2.Aktivasi dan Agregasi Trombosit
Trombosit yang pertama kali menuju vaskuler yang trauma dimana trombosit
dapat langsung melekat pada endotel, kolagen yang terekspos dan atau makrofag.
Terjadinya perlekatan trombosit ke dinding arteri dan aktivasinya tidak harus mutlak
dibutuhkan gangguan endotel. Trombosit dapat juga diaktivasi pada stadium awal
aterosklerosis. Hal ini diduga oleh karena :
1. Berkurangnya mekanisme antitrombotik endotel
2. Terbentuknya oksigen reaktif dari factor resiko aterosklerosis (adanya
hipertensi, hiperkolesterolemia, merokok dan diabetes berhubungan
dengan meningkatnya jumlah trombosit teraktivasi)
3. Meningkatnya mediator protrombotik dan proinflamasi di sirkulasi atau di
Trombosit yang teraktivasi melepaskan faktor kemotaktik (RANTES, platelet
factor-4), factor pertumbuhan (PDGF, TGF-β, EGF, bFGF) yang merangsang
migrasi, akumulasi, proliferasi sel otot polos dan leukosit menuju lapisan intima.
Pada aterosklerosis awal, mikrotrombi di permukaan luminal dapat mempotensiasi
perkembangan aterosklerosis melalui paparan dinding pembuluh darah dengan
faktor-faktor mitogen, sedangkan pada stadium akhir aterosklerosis, thrombosis
mural berhubungan dengan pertumbuhan plak aterosklerotik dan oklusi luminal
progresif.
Perlekatan awal trombosit pada endotel yang trauma diperantarai ikatan
glikoprotein (GP) Ibα dengan von Willebrand factor (VWF) dan molekul adhesi
endotel P-selectin. P-selectin terdapat dalam granul α trombosit dan badan
Weibel-Palade endotel. Bila sel teraktivasi, P-selectin dengan cepat menuju permukaan sel.
P-selectin sekarang dianggap pertanda aktivasi trombosit, pada aterosklerosis
dijumpai ekspresi P-selectin trombosit yang lebih tinggi. Ada banyak ligan P-selectin
yand diekspresikan trombosit (sulfatides, Gp Ib, PSGL-1), mucosal vascular
addressin cell adhesion molecule 1, di leukosit (PSGL-1) serta di endotel
(GlyCAM-1, CD34). Sel endotel yang mengalami disfungsi juga mengekspresikan VCAM-(GlyCAM-1,
vitronectin receptor αvβ3 dan PECAM-1, yang menyebabkan adhesi trombosit ke
dinding vascular.
Ikatan VWF – Gp Ib/V/IX bersifat kurang stabil. Reseptor Gp VI yang
langsung berikatan dengan kolagen dan menginduksi aktivasi reseptor adhesif lain
seperti integrin αIIbβ3 (Gp IIb/IIIa) dan α2β1. Keduanya menyebabkan ikatan
trombosit dengan permukaan endotel yang kuat, stabil dan irreversibel.
Agonist yang bersirkulasi seperti epinefrin, thrombin, serotonin, tromboxane
teraktivasi, trombosit mengalami perubahan bentuk dan terjadi peningkatan kadar
kalsium bebas dalam sitosol, menyebabkan pelepasan komponen granul trombosit,
disebut degranulasi trombosit. Komponen yang dilepaskan antara lain ADP. ADP
yang dilepaskan adri trombosit memiliki efek autokrin, menyebabkan agregasi
trombosit yang stabil melalui interaksi dengan reseptor spesifik (P2Y1 dan P2Y12);
juga memliki efek parakrin dengan berikatan dengan reseptor ADP pada trombosit
yang berdekatan,sehingga terjadi penguatan aktivasi trombosit. Aktivasi trombosit
akan menginduksi aktivasi fosfolipase A2 yang memicu metabolisme asam
arakidonat. COX-1 trombosit mengkatalis perubahan asam arakidonat menjadi
Prostaglandin G2/H2. PGH2 akan diubah oleh enzim tromboksan sintetase menjadi
tromboksan A2 (TxA2), dilepas ke sirkulasi dan berikatan dengan reseptornya,
memperkuat aktivasi trombosit dan vasokonstriksi.
2.4.3.Aktivasi Kaskade Koagulasi
Setelah vaskular terganggu terjadi aktivasi koagulasi. Diduga kuat faktor
jaringan (tissue factor TF) diekspresikan oleh sel busa, faktor trombogenik yang
mengaktivasi kaskade koagulasi. Endotel berubah menjadi prokoagulan sedangkan
permukaan trombosit mengkatalisasi pembentukan thrombin dari protrombin.
Aktivasi kaskade koagulasi menghasilkan trombin. Sinyal trombin melalui protease
activated receptors (PARs) menyebabkan hiperplasia intima, inflamasi,
BAB III
METODE PENELITIAN
3.1. Desain penelitian
Penelitian dilakukan dengan observasional analitik dengan cara cross
sectional (potong lintang). Pengambilan sampel dengan cara consecutive sampling,
dimana jumlah sampel dibatasi minimal sesuai perkiraan jumlah sampel atau sampai
batas waktu pengumpulan sampel yang ditetapkan. Pengukuran variabel dilakukan
hanya satu kali.
3.2. Tempat dan waktu penelitian
Penelitian dilakukan di Departemen Patologi Klinik FK USU/RSUP. H. Adam
Malik Medan bekerja sama dengan Departemen Ilmu Penyakit Dalam dan
Departemen Kardiologi FK USU/RSUP. H. Adam Malik Medan, mulai Oktober
sampai dengan Desember 2010.
3.3. Populasi dan subyek penelitian
Kelompok kasus adalah pasien-pasien sindroma koroner akut yang dirawat di
IGD dan ruang CVCU RSUP. H. Adam Malik Medan. Sebagai kelompok
pembanding adalah pasien yang datang ke laboratorium Patologi Klinik RSUP H.
Adam Malik Medan untuk memeriksakan darahnya yang secara anamnesa dan
pemeriksaan fisik tidak pernah terkena sindroma koroner akut berdasarkan jenis
kelamin dan umur yang sesuai dengan penderita sindroma koroner akut.
Kriteria inklusi