PENENTUAN PROFIL FARMAKOKINETIKA
DIKLOFENAK PADA KELINCI JANTAN
Skripsi
Oleh : Sofia Rahmi Nim : 050804084
FAKULTAS FARMASI
UNIVERSITAS SUMATERA UTARA
MEDAN
PENENTUAN PROFIL FARMAKOKINETIKA
DIKLOFENAK PADA KELINCI JANTAN
SKRIPSI
Diajukan untuk melengkapi salah satu syarat untuk memperoleh gelar Sarjana Farmasi pada Fakultas Farmasi
Universitas Sumatera Utara
Oleh : Sofia Rahmi Nim : 050804084
FAKULTAS FARMASI
UNIVERSITAS SUMATERA UTARA
MEDAN
PENGESAHAN SKRIPSI
PENENTUAN PROFIL FARMAKOKINETIKA
DIKLOFENAK PADA KELINCI JANTAN
Oleh :
SOFIA RAHMI NIM 050804084
Dipertahankan dihadapan Panitia Penguji Fakultas Farmasi
Universitas Sumatera Utara Pada tanggal : 05 Juni 2010
Pembimbing I, Panitia Penguji,
Dr.Edy Suwarso,S.U.,Apt. Dr.Karsono,Apt
NIP 130935857 NIP 131415891
Pembimbing II,
Prof.Dr.rer.nat.Effendi De Lux Putra,S.U.Apt Dr.Edy Suwarso, S.U.,Apt.
NIP 195206191983031001 NIP130935857
Drs.Muchlisyam,Msi., Apt.
NIP 195006221980021001
Drs.Saiful Bahri, M.S.,Apt.
NIP 131285999
Medan, Juni 2010 Fakultas Farmasi Universitas Sumatera Utara
Dekan,
Prof.Sumadio Hadisahputra, Apt.
KATA PENGANTAR
Puji dan syukur kehadirat Allah SWT yang telah melimpahkan rahmat, karunia,
dan ridho-Nya sehingga penulis dapat menyelesaikan skripsi yang berjudul “Penentuan
Profil Farmakokinetika Diklofenak pada Kelinci Jantan”. Skripsi ini diajukan sebagai
salah satu syarat untuk memperoleh gelar Sarjana Farmasi pada Fakultas Farmasi
Universitas Sumatera Utara.
Tujuan penelitian ini adalah untuk mengetahui profil farmakokinetika baku dari
Natrium Diklofenak (senyawa kimia yang larut dalam air) sebagai dasar dalam penelitian
selanjutnya untuk obat jadi (sediaan) Natrium Diklofenak.
Pada kesempatan ini penulis menyampaikan terima kasih yang sebesar-besarnya
kepada bapak Dr.Edy Suwarso,S.U.,Apt. dan bapak Prof.Dr.rer.nat.Effendi De Lux Putra,
S.U.,Apt. yang telah membimbing dengan penuh kesabaran, tulus dan ikhlas selama
penelitian dan penulisan skripsi ini berlangsung. Ucapan terima kasih juga disampaikan
kepada bapak Dekan Fakultas Farmasi Universitas Sumatera Utara, Prof.Dr.Sumadio
Hadisahputra, Apt. yang telah memberikan bantuan dan fasilitas selama masa pendidikan.
Penulis juga tidak lupa mengucapkan terima kasih dan penghargaan yang tulus
kepada kedua orang tua Drs.Azhary Tambusai, M.A. dan Dra.Khairina Nasution, M.S.
atas dorongan dan pengorbanan baik moril maupun materil dalam penyelesaian skripsi
ini. Serta ucapan terima kasih kepada teman-teman yang telah membantu dalam
penelitian dan penyelesaian skripsi ini.
Medan, 2010
Penulis,
PENENTUAN PROFIL FARMAKOKINETIKA
DIKLOFENAK PADA KELINCI JANTAN
Abstrak
Fase
farmakokinetik
berkaitan dengan masuknya zat aktif ke dalam
tubuh. Pemasukan
in vivo
tersebut secara keseluruhan merupakan fenomena
fisiko-kimia yang terpadu di dalam organ penerima obat. Penelitian ini
bertujuan untuk mengetahui bagaimana profil farmakokinetika dari Natrium
diklofenak.
Untuk mendapatkan keadaan yang optimal, penelitian ini dilakukan
dengan cara memberikan larutan Natrium diklofenak baku kepada 6 ekor
hewan kelinci jantan yang beratnya sekitar 1,5-2 kg dengan pemberian
secara oral. Dan untuk mendapatkan kadar obat dalam darah pada
masing-masing kelinci jantan maka darah diambil dengan selang waktu 0,25 jam;
0,5 jam; 0,75 jam; 1,25 jam; 1,5 jam; 2,5 jam; 3,5 jam; 4,5 jam; 5,5 jam.
Lalu divortex dengan menggunakan TCA dan disentrifuge. Pengukuran
kadar obat Natrium diklofenak dalam plasma kelinci jantan dilakukan
dengan menggunakan KCKT (Kromatografi Cair Kinerja Tinggi). Fase
gerak yang digunakan untuk mengukur kadar Natrium diklofenak dalam
plasma kelinci jantan adalah MeOH : buffer asetat (Na asetat 6,8 g / l
sesuaikan sampai pH 4,2 dengan HCl
(p)) dengan perbandingan (90 : 10) dan
laju alir 1,4 ml / menit.
Dari hasil penentuan parameter farmakokinetika Natrium diklofenak
diketahui nilai rata-rata ± SD dari K
a0,963 ± 0,422 jam
-1; AUC
0-∞2,831 ±
0,710 mcg / L jam; C
maks0,073 ± 3,157x10
-3
mcg / L; T
maks2,445 ± 0,343
jam; Vd 24,027 ± 4,197 L; K
el0,030 ± 6,06x10
-3
jam
-1; t
1/224,323 ± 6,298
jam; Klirens 0,703 ± 0,201 L / jam.
Dari hasil di atas dapat disimpulkan bahwa laju absorpsi Natrium
Diklofenak adalah 0,963 ± 0,422 jam
-1, dan laju eliminasinya adalah 0,030
± 6,06x10
-3jam
-1.
THE DETERMINATION OF THE PHARMACOKINETIC
PARAMETER OF DICLOFENAC IN MALE RABBIT
Abstract
Pharmacokinetic phase refers to the entrance of active substance to the
body. The in
vivo
influx, overall, is a harmonious physico-chemical
phenomenon in the drug receiving organ. This research objective is to know
and assess the pharmacokinetic form of diclofenac in combination with its
sodium salt.
To obtain optimal condition, this research was done by giving
standard diclofenac sodium solution to six male rabbits, weight about 1,5-2
kg orally. And to get the value of drug blood level in each rabbit, then the
blood was extracted in fixed time range 0,25 hour; 0,5 hour; 0,75 hor; 1,25
hour; 1,5 hour; 2,5 hour; 3,5 hour; 4,5 hour; 5,5 hour. Then put in a vortex
by using TCA and centrifuged. The determination of the Diclofenac sodium
in male rabbit’s plasma was done using HPLC. The mobile phase used in
measuring the level of diclofenac sodium in male rabbit plasma was MeOH :
acetate buffer (Sodium acetate 6,8 g / l fixed until pH 4,2 with concentrated
HCl
(p)ratio (90:10) and flow rate 1,4 ml / minute.
From the result of the determination of the pharmacokinetic parameter of
Diclofenac Sodium mean is known ± deviation Standard (SD) from K
a0,963
± 0,422 hour
-1; AUC
0-∞2,831 ± 0,710 mcg / L hour; C
maks0,073 ± 3,157x10
-3mcg / L; T
maks2,445 ± 0,343 hour; Vd 24,027 ± 4,197 L; K
el0,030 ±
6,06x10
-3hour
-1; t
1/224,323 ± 6,298 hour; Klirens 0,703 ± 0,201 L / hour.
From the result can be conclude that the rate absorption is 0,963 ±
0,422 jam
-1, and the rate elimination is 0,030 ± 6,06x10
-3jam
-1.
DAFTAR ISI
Halaman
JUDUL
LEMBAR PENGESAHAN KATA PENGANTAR
ABSTRAK... i
ABSTRACT... ii
DAFTAR ISI... iii
DAFTAR GAMBAR... iv
DAFTAR TABEL... v
DAFTAR LAMPIRAN... vi
BAB I PENDAHULUAN... 1
1.1 Latar Belakang ... 1
1.2 Perumusan Masalah ... 2
1.3 Hipotesis ... 3
1.4 Tujuan Penelitian ... 3
1.5 Manfaat Penelitian ... 3
BAB II TINJAUAN PUSTAKA... 4
2.1 Diklofenak... 4
2.1.1 Rumus Bangun... 4
2.1.2 Sifat Fisiko Kimia ... 4
BAB III METODOLOGI PERCOBAAN... 18
3.1 Alat... 18
3.2 Bahan ... 18
3.3 Pembuatan Suspensi CMC 1% ... 18
3.4 Pembuatan Suspensi Natrium Diklofenak 0,5%... 18
3.5 Pembuatan larutan Induk Baku Natrium Diklofenak... 19
3.6 Pembuatan Buffer Asetat ... 19
3.7 Pembuatan Fase Gerak... 19
3.8 Penyiapan Alat Kromatografi Cair Kinerja Tinggi... 19
3.9 Hewan Percobaan... 20
3.10 Pengambilan Sampel Darah Untuk Kurva Baku ... 20
3.11 Penentuan Profil Farmakokinetika dari Natrium diklofenak ... 20
BAB IV HASIL DAN PEMBAHASAN... 22
BAB V KESIMPULAN DAN SARAN... 29
5.1 Kesimpulan ... 29
5.2 Saran ... 29
DAFTAR GAMBAR
GAMBAR HALAMAN
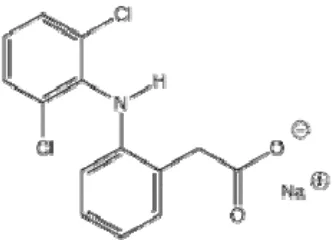
1. Struktur Kimia Diklofenak Natrium ... 4
4.1 Kromatogram hasil penyuntikan larutan Natrium Diklofenak BPFI dengan konsentrasi 500 mcg / ml fase gerak
MeOH : buffer asetat (90:10) waktu tambat 2,6 menit ... 22
4.2 Kurva baku Natrium Diklofenak ... 23
4.3 Konsentrasi Rata-Rata (log c) vs Waktu (t) Natrium Diklofenak
DAFTAR TABEL
TABEL HALAMAN
4.1 Nilai Konsentrasi dan Luas Area Natrium Diklofenak
Dalam Plasma ... 23
4.2 Nilai Konsentrasi Rata-Rata ± SD (Standard Deviasi)
Seluruh Hewan Percobaan Terhadap Waktu (t)... 25
4.3 Nilai Rata-Rata dan Satuan Parameter Farmakokinetika Natrium Diklofenak Dalam Plasma dengan menggunakan
DAFTAR LAMPIRAN
LAMPIRAN HALAMAN
1. Sertifikat Pengujian Natrium Diklofenak BPFI ... 33
2. Hasil Orientasi Gambar dengan Menggunakan Alat KCKT ... 34
2.1 Kromatogram hasil penyuntikan larutan Natrium Diklofenak BPFI dengan konsentrasi 500 mcg / ml, fase gerak MeOH : buffer asetat (68 :32) waktu tambat 6,3 menit... 34
2.2 Kromatogram hasil penyuntikan larutan Natrium Diklofenak BPFI dengan konsentrasi 500 mcg / ml, fase gerak MeOH : buffer asetat (70 :30), waktu tambat 2,3 menit... 34
2.3 Kromatogram hasil penyuntikan larutan Natrium Diklofenak BPFI dengan konsentrasi 500 mcg / ml, fase gerak MeOH : buffer asetat (80 : 20), waktu tambat 3,1 menit... 35
3. Natrium Diklofenak Dalam Plasma ... 36
3.1 Natrium Diklofenak dalam Plasma 0,25 jam... 36
3.2 Natrium Diklofenak dalam Plasma 0,5 jam... 36
3.3 Natrium Diklofenak dalam Plasma 0,75 jam... 36
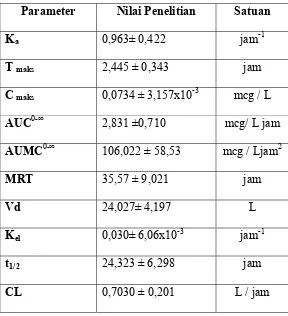
3.4 Natrium Diklofenak dalam Plasma 1,25 jam... 36
3.5 Natrium Diklofenak dalam Plasma 1,5 jam... 37
3.6 Natrium Diklofenak dalam Plasma 2,5 jam... 37
3.7 Natrium Diklofenak dalam Plasma 3,5 jam... 37
3.8 Natrium Diklofenak dalam Plasma 4,5 jam... 37
3.9 Natrium Diklofenak dalam Plasma 5,5 jam... 38
4. Perhitungan Persamaan Regresi dari Kurva Kalibrasi Natrium Diklofenak BPFI yang Diperoleh Secara KCKT... 39
5. Koversi Perhitungan Dosis Antar Jenis Hewan ... 41
6. Perhitungan Dosis yang Diberikan Kepada Masing-Masing Hewan Percobaan... 22
7.1 Konsentrasi Obat Pada Masing-Masing Hewan Percobaan... 46
8. Contoh Perhitungan Parameter Farmakokinetika Metode
Stripe Secara Komputerisasi ... 48
9. Contoh Perhitungan Parameter Farmakokinetika Secara Manual ... 50
10. Alat Ultrasonic Cleaner dan Penyaring... 56
10.1 Alat Ultrasonic Cleaner... 56
10.2 Penyaring ... 56
11. Alat KCKT dan Syringe 100µl ... 57
11.1 Alat KCKT (shimadzu)... 57
11.2 Syringe 100 µl (SCE)... 57
12. Kelinci, Alat Vorteks dan Sentrifuge... 58
12.1 Kelinci... 58
12.2 Alat Vorteks ... 58
PENENTUAN PROFIL FARMAKOKINETIKA
DIKLOFENAK PADA KELINCI JANTAN
Abstrak
Fase
farmakokinetik
berkaitan dengan masuknya zat aktif ke dalam
tubuh. Pemasukan
in vivo
tersebut secara keseluruhan merupakan fenomena
fisiko-kimia yang terpadu di dalam organ penerima obat. Penelitian ini
bertujuan untuk mengetahui bagaimana profil farmakokinetika dari Natrium
diklofenak.
Untuk mendapatkan keadaan yang optimal, penelitian ini dilakukan
dengan cara memberikan larutan Natrium diklofenak baku kepada 6 ekor
hewan kelinci jantan yang beratnya sekitar 1,5-2 kg dengan pemberian
secara oral. Dan untuk mendapatkan kadar obat dalam darah pada
masing-masing kelinci jantan maka darah diambil dengan selang waktu 0,25 jam;
0,5 jam; 0,75 jam; 1,25 jam; 1,5 jam; 2,5 jam; 3,5 jam; 4,5 jam; 5,5 jam.
Lalu divortex dengan menggunakan TCA dan disentrifuge. Pengukuran
kadar obat Natrium diklofenak dalam plasma kelinci jantan dilakukan
dengan menggunakan KCKT (Kromatografi Cair Kinerja Tinggi). Fase
gerak yang digunakan untuk mengukur kadar Natrium diklofenak dalam
plasma kelinci jantan adalah MeOH : buffer asetat (Na asetat 6,8 g / l
sesuaikan sampai pH 4,2 dengan HCl
(p)) dengan perbandingan (90 : 10) dan
laju alir 1,4 ml / menit.
Dari hasil penentuan parameter farmakokinetika Natrium diklofenak
diketahui nilai rata-rata ± SD dari K
a0,963 ± 0,422 jam
-1; AUC
0-∞2,831 ±
0,710 mcg / L jam; C
maks0,073 ± 3,157x10
-3
mcg / L; T
maks2,445 ± 0,343
jam; Vd 24,027 ± 4,197 L; K
el0,030 ± 6,06x10
-3
jam
-1; t
1/224,323 ± 6,298
jam; Klirens 0,703 ± 0,201 L / jam.
Dari hasil di atas dapat disimpulkan bahwa laju absorpsi Natrium
Diklofenak adalah 0,963 ± 0,422 jam
-1, dan laju eliminasinya adalah 0,030
± 6,06x10
-3jam
-1.
THE DETERMINATION OF THE PHARMACOKINETIC
PARAMETER OF DICLOFENAC IN MALE RABBIT
Abstract
Pharmacokinetic phase refers to the entrance of active substance to the
body. The in
vivo
influx, overall, is a harmonious physico-chemical
phenomenon in the drug receiving organ. This research objective is to know
and assess the pharmacokinetic form of diclofenac in combination with its
sodium salt.
To obtain optimal condition, this research was done by giving
standard diclofenac sodium solution to six male rabbits, weight about 1,5-2
kg orally. And to get the value of drug blood level in each rabbit, then the
blood was extracted in fixed time range 0,25 hour; 0,5 hour; 0,75 hor; 1,25
hour; 1,5 hour; 2,5 hour; 3,5 hour; 4,5 hour; 5,5 hour. Then put in a vortex
by using TCA and centrifuged. The determination of the Diclofenac sodium
in male rabbit’s plasma was done using HPLC. The mobile phase used in
measuring the level of diclofenac sodium in male rabbit plasma was MeOH :
acetate buffer (Sodium acetate 6,8 g / l fixed until pH 4,2 with concentrated
HCl
(p)ratio (90:10) and flow rate 1,4 ml / minute.
From the result of the determination of the pharmacokinetic parameter of
Diclofenac Sodium mean is known ± deviation Standard (SD) from K
a0,963
± 0,422 hour
-1; AUC
0-∞2,831 ± 0,710 mcg / L hour; C
maks0,073 ± 3,157x10
-3mcg / L; T
maks2,445 ± 0,343 hour; Vd 24,027 ± 4,197 L; K
el0,030 ±
6,06x10
-3hour
-1; t
1/224,323 ± 6,298 hour; Klirens 0,703 ± 0,201 L / hour.
From the result can be conclude that the rate absorption is 0,963 ±
0,422 jam
-1, and the rate elimination is 0,030 ± 6,06x10
-3jam
-1.
BAB I
PENDAHULUAN
1.1Latar Belakang
Fase farmakokinetik berkaitan dengan masuknya zat aktif ke dalam tubuh.
Pemakaian in vivo tersebut secara keseluruhan merupakan fenomena fisiko-kimia yang
terpadu di dalam organ penerima obat. Fase farmakokinetik ini merupakan salah satu
unsur penting yang menentukan profil keberadaan zat aktif pada tingkat biofase dan
selanjutnya menentukan aktivitas terapetik obat (Aiache, 1993).
Secara umum dan kualitatif kejadian in vivo untuk setiap zat aktif tertentu dan
untuk setiap jenis reseptor tertentu selalu tetap; namun secara kuantitatif keadaan ini
beragam tergantung pada sifat fisiko-kimia, zat aktif, keadaan fisiologi subjek penerima
serta keadaan fisio-patologis subjek yang sama. Penelitian farmakokinetik suatu zat aktif
merupakan penelitian identifikasi dan kuantifikasi perjalanan obat dalam tubuh (Aiache,
1993).
Aktivitas serta toksisitas suatu obat tergantung pada lama keberadaan dan
perubahan zat aktif di dalam tubuh. Penelitian tentang nasib obat dalam tubuh merupakan
rangkaian penyidikan yang harus dilakukan untuk memahami suatu obat serta untuk
memilih bentuk sediaan yang sesuai agar diperoleh efek terapi yang dikehendaki (Aiache,
1993).
Dalam praktek terapetik, suatu obat harus mencapai tempat kerja yang diinginkan
setelah masuk ke dalam tubuh dengan jalur yang terbaik. Obat harus diabsorpsi ke dalam
memberikan efek harus dikeluarkan dengan kecepatan tertentu melalui inaktifasi
metabolik, melalui ekskresi dari tubuh atau gabungan kedua proses ini (Katzung, 1998).
Keefektifan suatu obat diperoleh apabila suatu obat dapat diabsorpsi dan
mencapai konsentrasi yang efektif di dalam darah, jaringan dan tempat kerjanya.
Pemberian informasi tentang perjalanan obat dalam tubuh dipengaruhi oleh efek terapetik
maupun efek toksik (Nawaz, 2004).
Natrium diklofenak merupakan derivat sederhana fenil asetat yang termasuk
Non-Steroidal Anti-Inflammatory Drug (NSAID) yang terkuat anti radangnya, tetapi
mempunyai efek samping pada pemakaian sediaan obat dalam jangka waktu yang lama,
yaitu dapat menyebabkan pendarahan pada saluran cerna (Goodman and Gilman, 1996).
Natrium diklofenak mempunyai aktivitas anti- rematik, anti-radang dan
analgetik-antipiretik. Digunakan terutama untuk mengurangi rasa nyeri akibat peradangan pada
berbagai keadaan rematik dan kelainan degeneratif pada sistem otot rangka. Diklofenak
diserap secara cepat dan sempurna di dalam lambung, kadar plasma tertinggi dicapai 2
jam setelah pemberian oral, dengan waktu paruh antara 1-3 jam (Anonim, 2007).
1.2Perumusan Masalah
Berdasarkan latar belakang di atas, muncul perumusan masalah dalam penelitian ini
adalah :
1.3Hipotesis
Berdasarkan perumusan masalah di atas, maka yang menjadi hipotesis dalam
penelitian ini adalah :
Profil farmakokinetika Natrium diklofenak mengikuti orde satu dan model satu
kompartemen terbuka.
1.4Tujuan Penelitian
Berdasarkan hipotesis di atas, maka tujuan penelitian ini adalah :
Untuk mengetahui profil farmakokinetika dari Natrium diklofenak.
1.5 Manfaat Penelitian
Manfaat dari penelitian di atas adalah dengan diketahuinya profil farmakokinetika
baku Natrium Diklofenak (senyawa kimia yang larut dalam air) tersebut dapat sebagai
BAB II
TINJAUAN PUSTAKA
2.1 Diklofenak
2.1.1 Rumus Bangun
Gambar 1. Struktur kimia Diklofenak Natrium
2.1.2 Sifat Fisikokimia
Rumus Molekul : C14H10Cl2NO2Na
Berat Molekul : 318,3
Nama Kimia : Natrium{0-[2,6 dikofenil aminofenil} asetat
Pemerian : Serbuk kristal, putih atau agak kekuningan, agak hi
groskopis (USP Pharmacopiea, 2007).
Diklofenak merupakan derivat fenil asetat dan termasuk NSAID yang terkuat
daya anti-radangnya dengan efek samping yang lebih kecil dibandingkan dengan obat
lainnya. Obat ini sering digunakan untuk segala macam nyeri, juga pada migrain dan
encok. Lagi pula secara parenteral sangat efektif untuk menanggulangi rasa nyeri hebat
Diklofenak adalah turunan asam fenilasetat sederhana yang merupakan
penghambat COX yang kuat dengan efek anti-inflamasi, analgesik, dan antipiretik. Obat
ini cepat diabsorpsi setelah pemberian oral dan mempunyai waktu paruh yang pendek.
Obat ini dianjurkan untuk kondisi peradangan kronis seperti artritis rematoid dan
osteoartritis serta untuk pengobatan nyeri otot rangka akut. Efek samping terjadi kira-kira
20% penderita dan meliputi distres saluran cerna, perdarahan saluran cerna dan tukak
lambung (Payan,1998). Inhibisi sintesis prostaglandin dalam mukosa saluran cerna sering
menyebabkan kerusakan gastrointestinal (dispepsia, mual dan gastritis). Efek samping
yang paling serius adalah perdarahan gastrointestinal dan perforasi (Neal, 2006).
Diklofenak resorpsinya dari usus cepat dan lengkap, tetapi ketersediaan hayatinya
rata-rata 55% akibat FPE.. Efek analgetiknya dimulai setelah 1 jam, secara rektal dan
intramuskuler lebih cepat, masing-masing setelah 30 dan 15 menit. Penyerapan garam K
(Cataflam) lebih pesat dari pada garam Na. Ikatan obat dengan protein plsma di atas 99%,
waktu paruhnya 1 jam. Ekskresi melalui kemih berlangsung 60% sebagai metabolit dan
20% dalam empedu dan tinja, sisanya dalam bentuk tidak berubah (Tjay, 2002).
Profil keberadaan bahan obat dalam darah sebagai fungsi dari waktu
menggambarkan interaksi antara fase ketersediaan zat aktif dan fase disposisinya. Selain
itu profil tersebut juga mengungkapkan nasib obat di dalam tubuh. Oleh karena fenomena
penyerapan zat aktif dari darah menuju jaringan dapat terjadi secara bolak-balik
(reversible), maka selalu terjadi hubungan dinamik antara konsentrasi zat aktif dalam
jaringan dan konsentrasi zat aktif dalam darah (Aiache, 1993).
Absorpsi sistemik suatu obat dari saluran cerna atau tempat ekstravaskuler yang
seperti luas permukaan dinding usus, kecepatan pengosongan lambung, pergerakan
saluran cerna dan aliran darah ke tempat absorpsi, semuanya mempengaruhi laju dan
jumlah absorpsi obat (Shargel, 2005).
Absorpsi merupakan suatu fenomena yang memungkinkan zat aktif melewati
jalur pemberian obat menuju sistem peredaran darah, dan penyerapan obat terjadi secara
langsung dengan mekanisme perlintasan membran. Tanpa mengabaikan masalah
ketersediaan hayati, maka harus dibahas pentingnya bentuk sediaan, perlunya zat aktif
berada dalam bentuk yang sesuai agar dapat menembus membran dan pentingnya
kelarutan atau keterlarutan zat aktif padat (Aiache, 1993).
Diklofenak adalah golongan obat non steroid dengan aktivitas anti inflamasi,
analgetik-antipiretik. Aktivitas diklofenak dengan jalan menghambat enzim
siklo-oksigenase sehingga pembentukan prostaglandin terhambat (Altaher, 2005).
NSAID berkhasiat analgetis, antipiretik serta anti radang, dan sering sekali
digunakan untuk menghalau gejala penyakit rematik, seperti artrosis dan spondylosis.
Obat ini efektif unruk peradangan lain akibat trauma (pukulan, benturan, kecelakaan),
juga misalnya setelah pembedahan, atau pada memar akibat olahraga. Obat ini dipakai
pula untuk mencegah pembengkakan bila diminum sedini mungkin dalam dosis yang
cukup tinggi (Tjay, 2002).
Cara kerja NSAID untuk sebagian besar berdasarkan hambatan sintesis
prostaglandin, dimana kedua jenis cyclo-oxygenase (COX) di blokir. NSAID ideal
hendaknya hanya menghambat COX-2 (peradangan) dan tidak COX-1 (perlindungan
mukosa lambung), lagi pula menghambat lipo-oxygenase (pembentukan leukotrien)
COX-2 dibanding COX-1. Diklofenak menghambat biosintesa prostaglandin, dan juga
mengurangi pembentukan leukotrien, yang dapat memberikan kontribusi kepada aktivitas
anti-inflamasi. Obat ini waktu paruhnya pendek pada sebagian besar spesies, termasuk
manusia, tetapi terakumulasi di situs peradangan, dimana mencapai konsentrasi yang
lebih tinggi di non-peradangan jaringan, dan sama dengan yang dicapai dalam plasma
(Veterinaria, 2006).
Absorpsi obat ini melalui saluran cerna berlangsung cepat dan lengkap. Obat ini
terikat 99% pada protein plasma dan mengalami efek metabolisme lintas pertama (first
pass effect = FPE). Walaupun waktu paruhnya singkat yakni sekitar 1-3 jam, Na
diklofenak diakumulasi di cairan sinovial yang menjelaskan efek terapi di sendi jauh
lebih panjang dari waktu paruh obat tersebut (Altaher, 2005).
Informasi tentang kecepatan dan tingkat absorpsi obat jarang mempunyai
kepentingan klinis. Namun, absorpsi biasanya terjadi selama dua jam pertama setelah
dosis obat dan bervariasi menurut asupan makanan, posisi tubuh dan aktivitas. Oleh
karena itu tidak boleh mengambil darah sebelum absorpsi lengkap (kira-kira 2 jam
setelah dosis oral) (Holford, 1998).
Proses-proses absorpsi, distribusi dan eliminasi (metabolisme dan ekskresi) yang
dialami oleh hampir semua obat pada dosis terapi mengikuti kinetika orde pertama (first
order), artinya kecepatan proses-proses tersebut sebanding dengan jumlah obat yang ada
(yang tinggal). Jadi jumlah obat yang dibasorpsi, distribusi dan dieliminasi persatuan
waktu makin lama makin sedikit, sebanding dengan jumlah obat yang masih belum
Absorpsi obat adalah perpindahan obat dari tempat pemberian menuju ke darah
dan target aksinya. Untuk memasuki aliran sistemik (darah), obat harus dapat melintasi
membran (barier) yang merupakan faktor terpenting bagi obat untuk mencpai tempat
aksinya (misalnya otak, jantung, dan anggota badan yang lain). Obat harus dapat
melewati berbagai membran sel (misalnya sel usus halus, pembuluh darah, sel gilia di
otak, dan sel saraf) (Shargel, 2005).
Penyebaran zat aktif tergantung pada berbagai parameter, terutama sifat
fisiko-kimia molekul obat. Dengan demikian proses penyerapan zat aktif terjadi apabila
sebelumnya sudah dibebaskan dari sediaan dan sudah melarut dalam cairan biologi
setempat. Tahap pelepasan dan pelarutan zat aktif merupakan tahap penentu pada proses
penyerapan zat aktif, baik dalam hal jumlah yang diserap maupun laju penyerapannya
(Aiache, 1993).
Jumlah obat yang masuk ke tubuh tergantung kepada kecepatan dan tingkat
transfer obat dari tempat pemberian ke dalam darah. Kelebihan dosis atau kekurangan
dosis yang relatif terhadap dosis yang diresepkan sering dapat diketahui dengan
pengukuran konsentrasi. Variasi-variasi tingkat ketersediaan hayati lebih sering
disebabkan oleh adanya metabolisme selama absorpsi, walaupun kadang-kadang dapat
pula disebabkan oleh kesalahan pembuatan formulasi obat tertentu (Holford, 1998).
Pada distribusi khususnya melalui peredaran darah, obat yang telah melalui hati
bersamaan dengan metabolitnya disebarkan secara merata ke seluruh jaringan tubuh.
Melalui kapiler dan cairan ekstra sel (yang mengelilingi jaringan) obat diangkut ke
tempat kerjanya di dalam sel (cairan intra-sel), yaitu organ atau otot yang sakit. Tempat
melakukan aktivitasnya bila konsentrasi setempatnya cukup tinggi selama waktu yang
cukup lama. Seringkali distribusi obat tidak merata akibat beberapa gangguan, yaitu
adanya rintangan, misalnya rintangan darah-otak, terikatnya obat pada protein darah atau
jaringan dan lemak (Tjay, 2002).
Pada tahap distribusi ini penyebarannya sangat peka terhadap berbagai pengaruh
yang terkait dengan tahap penyerapan dan tahap yang terjadi sesudahnya yaitu peniadaan,
serta terkait pula dengan komposisi biokimia serta keadaan fisiopatologi subyeknya,
disamping itu perlu diingat kemungkinan adanya interaksi dengan molekul lainnya. Pada
tahap ini merupakan fenomena dinamik, yang selalu terdiri dari fase peningkatan dan
penurunan kadar zat aktif. Pengertian akumulasi dan penimbunan terutama penimbunan
bahan toksik, harus dijajaki dari sudut pandang dinamik, maksudnya melihat perbedaan
antara kecepatan masuk dan kecepatan keluar. Sebenarnay penimbunan bahan toksik
merupakan efek racun atau hasil fatal sebagai akibat lambat atau sangat lambatnya laju
pengeluaran dibandingkan laju penyerapan. Pengertian tentang waktu paruh biologik
suatu zat aktif, seringkali diartikan dengan waktu setengah peniadaan dan bertumpu pada
kinetik, maka pengurangan laju peniadaan obat yang terbaca merupakan penjumlahan
aljabar dari laju peniadaan murni dan laju kembalinya zat aktif dari jaringan menuju
darah (distribusi inversi) (Aiache, 1993).
Bila obat diberikan per oral, maka availabilitas sistemiknya kurang dari 1 dan
besarnya bergantung pada jumlah obat yang dapat menembus dinding saluran cerna
(jumlah obat yang dibasorpsi) dan jumlah obat yang mengalami eliminasi presistemik
Obat yang digunakan secara oral akan melalui lever (hepar) sebelum masuk ke
dalam darah menuju ke daerah lain dari tubuh (misalnya otak, jantung, paru-paru dan
jaringan lainnya). Di dalam lever terdapat enzim khusus (yaitu sitokrom P-450) yang
akan mengubah obat menjadi bentuk metabolitnya. Metabolit umumnya menjadi lebih
larut dalam air (polar) dan akan dengan cepat diekskresi ke luar tubuh melalui urin, feses,
keringat, dan lain-lain. Hal ini akan secara dramatik mempengaruhi kadar obat dalam
plasma dimana obat yang mengalami first pass metabolism akan kurang
bioavailabilitasnya sehingga efek yang dihasilkan juga berkurang (Hinz, 2005).
Efektivitas suatu senyawa obat pada pemakaian klinik berhubungan dengan
farmakokinetikanya. Efek obat terhadap tubuh pada dasarnya merupakan akibat interaksi
obat dengan reseptornya; maka secara teoretis intensitas efek obat baik efek terapi
maupun efek toksik tergantung dari kadar obat di tempat reseptor atau tempat kerjanya.
Oleh karena kadar obat di tempat kerja belum dapat diukur, maka sebagai gantinya
diambil kadar obat dalam plasma / serum yang umum dalam keseimbangan dengan
kadarnya di tempat kerja (Setiawati, 2005).
Farmakokinetika menggunakan model matematik untuk menguraikan
proses-proses absorpsi, distribusi, biotranformasi dan ekskresi. Dengan memperkirakan besarnya
kadar obat dalam plasma sebagai fungsi dari besarnya dosis terhadap waktu pengambilan
sampel darah dalam penetapan kadar obat dalam darah tersebut (Setiawati, 2005).
Aktivitas serta toksisitas obat tergantung pada lama keberadaan dan perubahan zat
aktif di dalam tubuh. Penelitian tentang nasib obat dalam tubuh merupakan rangkaian
penyidikan yang harus dilakukan untuk mengethui kapan obat tersebut menunjukkan
diberikan akan memberikan efek terapi atau efek toksik dengan melihat nilai ambang
terapi dari obat tersebut. (Aiache, 1993).
Pada umumnya zat aktif suatu obat akan menunjukkan efek farmakologik pada
titik-tangkap jaringan bila bahan tersebut telah mencapai tempat tersebut dengan
perantaraan darah. Peredaran darah bagaikan “lempeng berputar” dari perjalanan obat.
Fenomena penyerapan sebagai tahap awal farmakokinetika, ditentukan oleh penembusan
zat aktif ke dalam darah yang selanjutnya oleh darah dihantarkan menuju sasaran kerja
farmakologik, mengalami perubahan hayati dan selanjutnya ditiadakan (Aiache, 1993).
Efek samping yang lazim ialah mual, gastritis, eritema kulit dan sakit kepala sama
seperti semua obat NSAID. Pemakaian obat ini harus berhati-hati pada penderita tukak
lambung. Peningkatan enzim transaminasi dapat terjadi pada 15% pasien dan umumnya
kembali ke normal. Pemakaian selama kehamilan tidak dianjurkan (Wilmana, 2005).
Diklofenak merupakan non-steroid anti-inflamasi (NSAID) yang telah digunakan
dalam farmakoterapi selama bertahun-tahun. Diklofenak diindikasikan untuk pengobatan
berbagai peradangan dan pasca trauma gangguan degeneratif, serta perawatan
pra-operasi untuk katarak-ekstraksi (Veterinaria, 2006).
Diklofenak mempunyai durasi kerja singkat.. Obat ini bisa memberikan analgesia
pasca operasi yang cukup dan tidak menyebabkan depresi napas. Efek analgesik NSAID
yang terdapat pada diklofenak digunakan baik di perifer maupun di sentral, tetapi efek
perifernya lebih banyak. Efek analgesik biasanya berhubungan dengan efek antiinflamasi
dan diakibatkan oleh inhibisi sistesis prostaglandin dalam jaringan yang meradang.
Prostaglandin mempotensiasi nyeri yang disebabkan mediator inflamasi lain. Pada
vaskuler. Akan tetapi, inhibisi sistesis prostaglandin oleh NSAID mengurangi inflamasi
daripada menghilangkannya karena obat ini tidak menghambat mediator inflamasi
lainnya. Meskipun demikian, pada sebagian besar pasien dengan artritis reumatoid, efek
anti-inflamasi relatif ringan untuk mengurangi nyeri, kekakuan dan pembengkakan.
Namun, tidak mengubah perjalanan penyakit (Neal, 2006).
Tipe metabolisme dibedakan menjadi dua bagian yaitu Nonsynthetic Reactions (Reaksi
Fase I) dan Synthetic Reactions (Reaksi fase II). Reaksi fase I terdiri dari oksidasi,
reduksi, hidrolisa, alkali, dan dealkilasi. Metabolitnya bisa lebih aktif dari senyawa
asalnya. Umumnya tidak dieliminasi dari tubuh kecuali dengan adanya metabolisme lebih
lanjut. Reaksi fase II berupa konjugasi (glukoronidasi dan sulfatasi) yaitu penggabungan
suatu obat dengan suatu molekul lain. Metabolitnya umumnya lebih larut dalam air dan
mudah diekskresikan (Hinz, 2005).
Faktor yang mempengaruhi metabolisme obat yaitu induksi enzim yang dapat
meningkatkan kecepatan biotransformasi dirinya sendiri, atau obat lain yang
dimetabolisme oleh enzim yang sama yang dapat menyebabkan toleransi. Selain itu
inhibisi enzim yang merupakan kebalikan dari induksi enzim, biotransformasi obat
diperlambat, menyebabkan bioavailabilitasnya meningkat, menimbulkan efek menjadi
lebih besar dan lebih lama. Kompetisi (interaksi obat) juga berpengaruh terhadap
metabolisme dimana terjadi oleh obat yang dimetabolisir oleh sistem enzim yang sama
(contoh alkohol dan barbiturat). Perbedaan individu juga berpengaruh terhadap
metabolisme karena adanya genetic polymorphism, dimana seseorang mungkin memiliki
Metabolit umumnya merupakan suatu bentuk yang lebih larut dalam air
dibandingkan molekul awal. Perubahan sifat fisiko-kimia ini paling sering dikaitkan
dengan penyebaran kuantitatif metabolit yang dapat sangat berbeda dari zat aktifnya
dengan segala akibatnya. Jika metabolit ini merupakan mediator farmakologik, maka
akan terjadi perubahan, baik berupa peningkatan maupun penurunan efeknya (Aiache,
1993).
Obat akan dieliminasi dari dalam tubuh dalam bentuk metabolitnya. Organ
ekskresi utama adalah ginjal yang menghasilkan urin. Namun bisa juga melalui
paru-paru, keringat, air liur, feses dan asi (Hinz, 2005).
Obat dan metabolitnya yang terlarut dalam plasma melintasi dinding glomeruli
secara pasif dengan ultrafiltrat. Ekskresi dapat diperlancar dengan memperkuat disosiasi
obat yang kebanyakan bersifat asam atau basa lemah dengan derajat ionisasi yang agak
ringan (Tjay, 2002).
Untuk dapat menilai suatu obat secara klinis, menetapkan dosis dan skema
penakarannya yang tepat, perlu adanya sejumlah keterangan farmakokinetik. Khususnya
mengenai kadar obat di tempat tujuan kerja (target site) dan dalam darah, serta perubahan
kadar ini dalam waktu tertentu. Pada umumnya besarnya efek obat tergantung pada
konsentrasinya di target site dan ini berhubungan erat dengan konsentrasi plasma
(Waldon, 2008).
Turunnya kadar plasma obat dan lama efeknya tergantung pada kecepatan
metabolisme dan ekskresi. Kedua faktor ini menentukan kecepatan eliminasi obat yang
dinyatakan dengan pengertian plasma half-life eliminasi (waktu paruh, t1/2) yaitu rentang
Kecepatan eliminasi obat dan plasma t1/2-nya tergantung dari kecepatan biotransformasi
dan ekskresi. Obat dengan metabolisme cepat half life-nya juga pendek. Sebaliknya zat
yang tidak mengalami biotrasformasi atau yang diresorpsi kembali oleh tubuli ginjal,
dengan sendirinya t1/2-nya panjang (Waldon, 2008).
AUC (Area Under Curve) adalah permukaan di bawah kurva (grafik) yang
menggambarkan naik turunnya kadar plasma sebagai fungsi dari waktu. AUC dapat
dihitung secara matematis dan merupakan ukuran untuk bioavailabilitas suatu obat.
AUC dapat digunakan untuk membandingkan kadar masing-masing plasma obat bila
penentuan kecepatan eliminasinya tidak mengalami perubahan. Selain itu antara kadar
plasma puncak dan bioavailabilitas terdapat hubungan langsung (Waldon, 2008).
Plasma half-life merupakan ukuran untuk lamanya efek obat, maka t1/2 bersama
grafik kadar-waktu penting sekali sebagai dasar untuk menentukan dosis dan frekuensi
pemberian obat yang rasional, dengan kata lain berapa kali sehari sekianmg. Dosis yang
terlalu tinggi atau terlalu frekuen dapat menimbulkan efek toksis, sedangkan dosis
terlampau rendah atau terlalu jarang tidak menghasilkan efek, bahkan pada
kemoterapeutika dapat menimbulkan resistensi kuman (Waldon, 2008).
Obat dengan half-life panjang, lebih dari 24 jam pada umumnya cukup diberikan
dosis satu kali sehari dan tidak perlu sampai 2 atau 3 kali. Kecuali bila obat sangat terikat
pada protein, sedangkan kadar plasma tinggi diperlukan untuk efek terapeutiknya.
Sebaliknya, obat yang dimetabolisasi cepat dan t1/2-nya pendek, perlu diberikan sampai
3-6 kali sehari agar kadar plasmanya tetap tinggi (Waldon, 2008).
Waktu konsentrasi plasma mencapai puncak dapat disamakan dengan waktu yang
tmaks absorpsi obat adalah terbesar, dan laju absorpsi obat sama dengan laju eliminasi
obat. Absorpsi masih berjalan setelah tmaks tercapai, tetapi pada laju yang lebih lambat.
Harga tmaks menjadi lebih kecil (berarti sedikit waktu yang diperlukan untuk mencapai
konsentrasi plasma puncak) bila laju absorpsi obat menjadi lebih cepat (Shargel, 2005).
Konsentrasi plasma puncak menunjukkan konsentrasi obat maksimum dalam
plasma setelah pemberian secara oral. Untuk beberapa obat diperoleh suatu hubungan
antara efek farmakologi suatu obat dan konsentrasi obat dalam plasma. Konsentrasi
plasma puncak memberi suatu petunjuk bahwa obat cukup diabsorpsi secara sistemik
untuk memberi suatu respons terapetik. Selain itu konsentrasi plasma puncak juga
memberi petunjuk dari kemungkinan adanya kadar toksik obat (Shargel, 2005).
Volume distribusi (vd) menunjukkan volume penyebaran obat dalam tubh dengan
kadar plasma atau serum. Vd tidak perlu menunjukkan volume penyebaran obat yang
sesungguhnya ataupun volume secara anatomik, tetapi hanya volume imajinasi dimana
tubuh dianggap sebagi 1 kompartemen yang terdiri dari plasma atau serum, dan Vd
menghubungkan jumlah obat dalam tubuh dengan kadarnya dalam plasma atau serum
(Setiawati, 2005).
Besarnya Vd ditentukan oleh ukuran dan komposisi tubuh, fungsi kardiovaskular,
kemampuan molekul obat memasuki berbagai kompartemen tubuh, dan derajat ikatan
obat dengan protein plasma dan dengan berbagai jaringan. Obat yang tertimbun dalam
jaringan sehingga kadar dalam plasma rendah sekali, sedangkan obat yang terikat dengan
kuat pada protein plasma sehingga kadar dalam plasma cukup tinggi mempunyai vd
Volume distribusi yang diperoleh mencerminkan suatu keseimbangan antara
ikatan pada jaringan, yang mengurangi konsentrasi plasma dan membuat nilai distribusi
lebih besar, dengan ikatan pada protein plasma, yang meningkatkan konsentrasi plasma
dan membuat volume distribusi menjadi lebih kecil. Perubahan-perubahan dalam ikatan
dengan jaringan ataupun dengan plasma dapat mengubah volume distribusi yang
ditentukan dari pengukuran-pengukuran konsentrasi plasma (Holford, 1998).
Klirens obat adalah suatu ukuran eliminasi obat dari tubuh tanpa
mempermasalahkan mekanisme prosesnya. Umumnya, jaringan tubuh atau organ
dianggap sebagai suatu kompartemen cairan dengan volume terbatas (volume distribusi)
dimana obat terlarut di dalamnya (Shargel, 2005).
Untuk beberapa obat rute pemakaian mempengaruhi kecepatan metabolismenya.
Obat-obat yang diberikan secara oral diabsorpsi secara normal dalam duodenal dari usus
halus dan ditanspor melalui pembuluh mesenterika menuju vena porta hepatik dan
kemudian ke hati sebelum ke sirkulasi sistemik. Obat-obat yang dimetabolisme dalam
jumlah besar oleh hati atau oleh sel-sel mukosa usus halus menunjukkan availabilitas
sistemik yang jelek jika diberikan secara oral. Metabolisme secara oral sebelum mencapai
BAB III
METODOLOGI PERCOBAAN
3.1 Alat
Alat-alat yang digunakan adalah politube, beaker glass, vortex, waterbath,
sentrifuge, labu tentukur, pH meter, gelas ukur, neraca analitik (Baeco Germany), pipet
volume, sarung tangan, animal box, spuit, pisau cukur, perangkat KCKT( Shimadzu), dan
alat lain yang dibutuhkan.
3.2Bahan
Bahan yang digunakan dalam penelitian ini adalah BPFI Na-diklofenak, TCA,
heparin (PT. Pratapa Nirmala), akuabides (PT.Ikapharmindo Putramas).
3.3 Pembuatan Suspensi CMC 1%
Air suling dipanaskan hingga mendidih. Sebanyak 1000 mg CMC ditimbang.
Setelah mendidih dimasukkan air suling ke dalam cawan porselen sebanyak 1/3 dari
bagian air. Ditaburkan CMC ke dalam porselen secara perlahan dan diamkan 30 menit.
Diaduk hingga membentuk massa yang transparan. Kemudian ditambahkan air suling
yang masih panas sampai 1000 ml dan dihomogenkan kemudian dimasukkan ke dalam
3.4 Pembuatan Suspensi Natrium Diklofenak 0,5 %
Ditimbang 50 mg Natrium Diklofenak, digerus dan dilarutkan dalam suspensi 1%.
Digerus kembali hingga homogen dan dimasukkan ke dalam labu 100 ml. Dibilas dan ad
kan sampai garis tanda.
3. 5 Pembuatan Larutan Induk Baku Natrium Diklofenak
Timbang seksama sejumlah 25,0 mg diklofenak BPFI, dimasukkan ke dalam labu
tentukur 50 ml. dicukupkan dengan fase gerak hingga garis tanda. Dikocok sampai
homogen sehingga diperoleh larutan dengan konsentrasi 500 mcg / ml.
3.6 Pembuatan Buffer Asetat
Sebanyak 6,8 g Na asetat ditimbang pada neraca analitik, dimasukkan ke dalam
labu 1000 ml lalu ditambahkan sedikit demi sedikit akuabides sampai Na asetat terlalut
sempurna. Lalu dimasukkan ke dalamnya HCl (p) dan disesuaikan pHnya sampai pH 4,2
dengan menggunakan alat pH meter. Lalu dicukupkan dengan akuabides sampai garis
tanda.
3.7 Pembuatan Fase Gerak
Fase gerak yang digunakan adalah campuran antara MeOH dan buffer asetat
dengan perbandingan 90:10, disaring dengan menggunakan membran filter PTFE 0,5 µm
3.8 Penyiapan Alat Kromatografi Cair Kinerja Tinggi
Alat kromatografi dihidupkan, pengukuran dilakukan dengan menggunakan
kolom Agilent tipe TC-C18, laju alir 1,4 ml / menit, detektor UV pada panjang
gelombang 273 nm. Pompa yang digunakan mode aliran tetap dengan sistem elusi
isokratik.
3.9 Hewan Percobaan
Hewan yang digunakan adalah kelinci jantan dengan berat 1,5-2 kg, yang telah
dikondisikan selama 1 minggu dan diberi makanan kangkung segar selama penelitian
berlangsung.
3.10 Pengambilan Sampel Darah Untuk Kurva Baku
Ambil darah kelinci jantan kira-kira 5 ml, dimasukkan ke dalam tabung yang sudah berisi
2 tetes heparin. Siapkan 4 buah tabung dan masing-masing tabung masukkan Larutan
Induk Baku Natrium diklofenak dengan konsentrasi 80 mcg / ml; 90 mcg / ml; 105,6 mcg
/ ml; 120 mcg / ml kemudian di ad kan dengan darah masing-masing ad 1 ml, kemudian
ditambahkan TCA sebanyak 1 ml lalu divorteks dan disentrifuge untuk diambil
plasmanya, dan diukur kadarnya dengan menggunakan alat KCKT (Kromatografi Cair
Kinerja Tinggi) dengan menyuntikannya sebanyak 20 µl (3 kali).
3.11 Penentuan Profil Farmakokinetika Dari Natrium Diklofenak
Enam ekor kelinci jantan diambil darahnya masing-masing 1 ml (untuk blanko)
kelinci tersebut diberikan suspensi Natrium Diklofenak dengan dosis yang telah
dikonversikan (dosis manuasia ke dosis kelinci) terhadap dosis lazim 25 mg. Lalu
diambil darahnya kira-kira 1 ml dengan rentang waktu yang telah ditetapkan. Rentang
waktunya berkisar : 0,25 jam; 0,5 jam; 0.75 jam; 1,25 jam; 1,5 jam; 2,5 jam; 3,5 jam; 4,5
jam; 5,5 jam. Dan kemudian ditambahkan TCA sebanyak 1 ml lalu divorteks dan
disentrifuge untuk diambil plasmanya. Setelah itu diukur kadarnya dengan menggunakan
BAB IV
HASIL DAN PEMBAHASAN
Sebelum dilakukan penetapan kadar Natrium diklofenak terlebih dahulu
ditentukan perbandingan fase gerak dengan menggunakan metode KCKT melalui
orientasi fase geraknya berupa MeOH : buffer asetat dengan panjang gelombang 273 nm.
Orientasi digunakan untuk mengetahui perbandingan fase gerak, laju alir, waktu tambat
dan tekanan kolom yang optimal dengan cara menyuntikkan larutan Natrium diklofenak
pada konsentrasi 500 mcg / ml sebanyak 20µl ke dalam sistem KCKT dengan
perbandingan fase gerak MeOH : buffer asetat (90:10) dan laju alir yang tetap yaitu 1,4
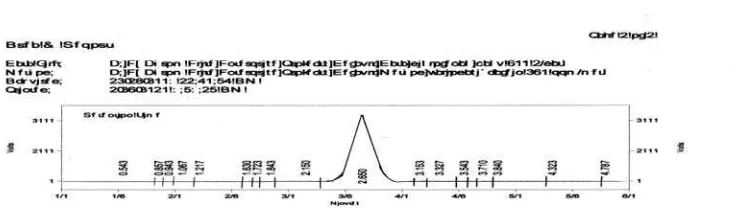
[image:34.612.107.478.356.461.2]ml / menit diperoleh waktu tambat yaitu 2,6 menit, seperti yang tertera pada Gambar 4.1.
Gambar 4.1 Kromatogram hasil penyuntikan larutan Natrium Diklofenak BPFI dengan
konsentrasi 500 mcg / ml, fase gerak MeOH : buffer asetat (90:10)
Dari Gambar 4.1 lebih lanjut dapat diketahui nilai konsentrasi dan luas area
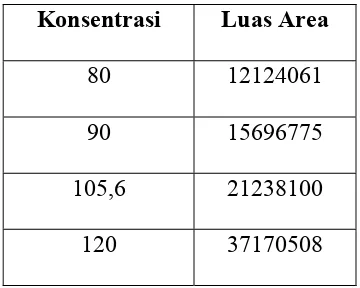
Tabel 4.1 Nilai Konsentrasi dan Luas Area Natrium Diklofenak dalam Plasma
Konsentrasi Luas Area
80 12124061
90 15696775
105,6 21238100
120 37170508
Dari Tabel 4.1 lebih lanjut digambarkan kurva baku Natrium Diklifenak seperti
yang tertera pada Gambar 4.2.
Gambar 4.2 Kurva Baku Natrium Diklofenak
Kadar sampel dapat dihitung menggunakan persamaan rumus berikut Y =
601980,46X-37978506,5 yaitu mensubstitusikan Y dengan luas puncak sampel. Hasil
[image:35.612.89.462.406.522.2]Berdasarkan kromatogram dan kurva kalibrasi hasil penyuntikan Natrium
diklofenak BPFI di atas, selanjutnya dilakukan penyuntikan dari plasma 6 ekor kelinci
jantan yang mengandung Natrium diklofenak untuk penentuan nilai parameter
farmakokinetiknya.
Dari data yang diperoleh pada lampiran 4, makadapat diketahui nilai konsentrasi
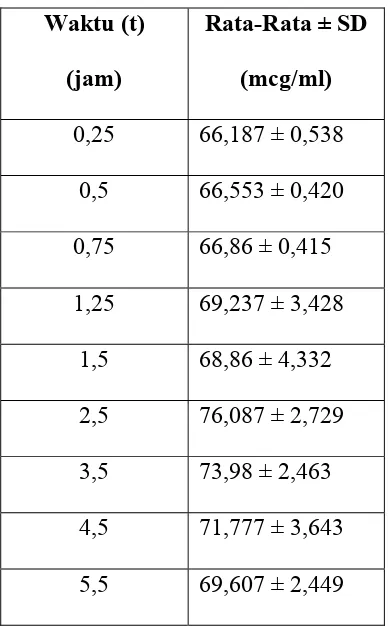
Tabel 4.2 Nilai Konsentrasi Rata-Rata ± SD (Standard Deviasi) Seluruh Hewan
Percobaan Terhadap Waktu
Waktu (t)
(jam)
Rata-Rata ± SD
(mcg/ml)
0,25 66,187 ± 0,538
0,5 66,553 ± 0,420
0,75 66,86 ± 0,415
1,25 69,237 ± 3,428
1,5 68,86 ± 4,332
2,5 76,087 ± 2,729
3,5 73,98 ± 2,463
4,5 71,777 ± 3,643
5,5 69,607 ± 2,449
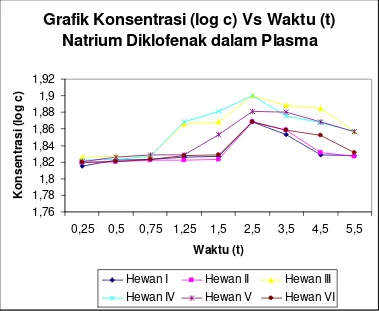
Dari Tabel 4.2 di atas dapat digambarkan konsentrasi rata-rata (log c) VS waktu
Konsentrasi Rata-Rata (log c) Vs Waktu (t) Natrium Diklofenak dalam
Plasma
1,74 1,76 1,78 1,8 1,82 1,84 1,86 1,88 1,9 1,92
0,25 0,5 0,75 1,25 1,5 2,5 3,5 4,5 5,5
Waktu (t)
K
o
n
s
e
n
tr
a
s
i R
a
ta
-R
a
ta
(l
o
g
[image:38.612.94.422.74.272.2]c)
Gambar 4.3 Konsentrasi Rata-Rata (log c) VS Waktu (t) Natrium Diklofenak dalam
Plasma
Dari Tabel 4.2 juga dapat diketahui nilai parameter farmakokinetika natrium
diklofenak dalam plasma pada hasil penelitian dengan menggunakan metode stripe secara
Tabel 4.3 Nilai Rata-Rata dan Satuan Parameter Farmakokinetika Natrium
Diklofenak dalam Plasma dengan Menggunakan Metode Stripe Secara
Komputerisasi
Parameter Nilai Penelitian Satuan
Ka 0,963± 0,422 jam-1
T maks 2,445 ± 0,343 jam
C maks 0,0734 ± 3,157x10-3 mcg / L
AUC0-∞ 2,831 ±0,710 mcg/ L jam
AUMC0-∞ 106,022 ± 58,53 mcg / Ljam2
MRT 35,57 ± 9,021 jam
Vd 24,027± 4,197 L
Kel 0,030± 6,06x10-3 jam-1
t1/2 24,323 ± 6,298 jam
CL 0,7030 ± 0,201 L / jam
Dari data di atas diperoleh nilai Ka 0,963 ± 0,422 jam-1disebabkan karena hewan
percobaan berupa 6 ekor kelinci jantan yang diberikan Na-diklofenak secara per oral
harus melewati sistem ELP (Efek Lintas Pertama) dimana obat akan mengalami
metabolisme pada membran usus sebelum mencapai sirkulasi sistemik sehingga jumlah
obat yang dihasilkan lebih sedikit dan obat yang diabsorpsi juga sedikit. Nilai Tmaks yang
serta nilai AUC0-∞2,831 ±0,710 mcg/ L jam dan nilai MRT 35,57 ± 9,021 jam karena
untuk mencapai waktu maksimum dibutuhkan waktu yang lama, disebabkan oleh obat
harus mengalami proses metabolisme pada membran usus sebelum mencapai sirkulasi
sistemik sehingga waktu yang dibutuhkan obat di dalam tubuh (MRT) lebih lama serta
keberadaan obat dalam tubuh (AUC0-∞)semakinsedikit dan konsentrasi maksimum yang
diperoleh juga kecil.
Nilai Vd (volume distribusi yang diperoleh 24,027± 4,197 L dan nilai Kel 0,030±
6,06x10-3 jam-1 disebabkan karena faktor kondisi tubuh hewan percobaan yang tidak
memiliki banyak lemak sehingga volume cairan tubuh yang membawa obat di dalam
tubuh menuju ke sirkulasi sistemik lebih banyak dan menyebabkan proses eliminasi yang
dihasilkan juga semakin cepat. Nilai t1/2 24,323 ± 6,298 jam dan nilai CL (klirens) 0,7030
± 0,201 L / jam disebabkan karena semakin kecil laju eliminasi maka separuh jumlah
obat dalam tubuh akan semakin besar dan keberadan obat dalam tubuh akan semakin
kecil sehingga proses pembuangan (nilai klirens) akan semakin bertambah besar. Ini
BAB V
KESIMPULAN DAN SARAN
5.1 Kesimpulan
1. Waktu retensi kromatogram Natrium diklofenak muncul pada penggunaan
KCKT menggunakan kolom Agilent tipe TC-C18, laju alir 1,4 ml / menit, fase gerak
MeOH : buffer asetat (90 : 10) dengan waktu tambat 2,6 menit.
2. Profil farmakokinetika Natrium diklofenak yang diperoleh memiliki laju
absorpsi yaitu (0,963 ± 0,422 jam-1). Tmaks yang diperoleh menunjukkan waktu yang
relatif lama (2,445 ± 0,343 jam), konsentrasi maksimum (Cmaks) yang diperoleh (0,073 ±
3,157x10-3 mcg / L). Selain itu AUC0-∞ (2,831 ± 0,710 mcg / L jam). Volume distribusi
yang diperoleh (24,027 ± 4,197 L), serta laju eliminasi yang diperoleh (0,030 ± 6,06x10-3
jam-1). Waktu paruhnya (24,323 ± 6,298 jam), serta pembuangan (Klirens) (0,7030 ±
0,201 L / jam).
5.2 Saran
1. Disarankan agar melanjutkan penelitian dengan melakukan pemberian
perjalanan oba yang berbeda.
2. Disarankan melakukan penelitian dengan senyawa lain yang berinteraksi dengan
DAFTAR PUSTAKA
Aiache, J.M. (1993).Farmasetika 2-Biofarmasi. Edisi Kedua. Surabaya : Penerbit
Airlangga University Press. Hal. 7-11, 39.
Altaher, A.Y. (2005). Pharmacokinetics Of Diclofenac In Sheep Following Intra Venous
and Intramuscular Administration.Vol 13, No.2-3.Saudi Pharmaceutical Journal.
Pages 107-108.
Anonim. (2007). Diklofenak. ( http://library@lib.unair.ac.id)
Goodman, Gilman, A., Hardman J. G., Limbird L. E. (1996). Goodman and Gilman’s
Pharmacologycal Basis of Therapeutics. Ninth Edision. C. Graw Hill Company:
Page. 617- 635.
Hinz, B. (2005). Bioavailability of Diclofenac Pottassium at Low Doses. Germany :
Department of Experimental and Clinical Pharmacology and Toxicology,
Friedrich Alexander University Erlangen-Nurnberg, Fahrstrasse 17, D-91054
Erlangen. Pages 80-81.
Holford, N.H. (1998). Farmakokinetik dan farmakodinamik : Pemilihan Dosis yang
Rasional dan waktu Kerja Obat. Farmakologi Dasar dan Klinik. Edisi VI. Jakarta.
Hal. 36-38.
Katzung, B. (1998). Farmakologi dasar dan klinik. Edisi VI. Jakarta. Hal. 36.
Nawaz, R. (2004). Kinetics Of Diclofenac Sodium.Singel Oral Dose Disposition in Male
Volunteers.Faisalabad : Department of Chemistry University of Agriculture. Page
2.
Neal, M .J.(2006). Farmakologi Medis. Edisi Kelima. Jakarta : Penerbit Erlangga Hal.
Rizki, M.A.R. (2008). Optimasi Fase Gerak Metanol-Air Untuk Analisa Kuantitatif
Campuran Teofilin dan Efedrin dalam sample Tablet Dengan Metode KCKT.
Skripsi. Fakultas Farmasi Unair.
Rohman, A. (2007). Kimia Farmasi Analisis. Yogyakarta : Penerbit Pustaka Pelajar. Hal.
378- 415.
Setiawati, A. (2005). Farmakokinetik Klinik. Farmakologi dan Terapi. Edisi 4. Jakarta :
Bagian Farmakologi Fakultas Kedokteran Universitas Indonesia. Hal. 811-815.
Shargel, L. (2005). Biofarmasetika dan Farmakokinetika Terapan.Edisi Kedua.
Surabaya : Airlangga University Press. Hal. 137, 167, 201.
Soewandhi, S. N. Reni, I. et.all. (2007). Polimorfisme Diklofenak Natrium. Bandung :
Kelompok Keilmuan Farmasetika. Sekolah Farmasi ITB. Hal. 1-8.
Sumitrapura, Y.C. Herwanto. S. (1997). Profil Farmakokinetik dan Ketersediaan Hayati
Tiga Sediaan tablet Natrium Diklofenak salut Enterik. Vol. 2. No. 2. Bandung :
Jurusan Farmasi Fakultas MIPA ITB, Bagian Penelitian dan Pengembangan PT.
Sanbe Farma. Hal. 47- 49.
Tjay T.H. & Kirana R. (2002). Obat-Obat Penting. Edisi Kelima. Jakarta : PT. Elex
Media Komputindo. Hal. 296, 309, 313.
USP Pharmacopiea. (2007). The National Formulary. Edition 30. The United States
Veterinaria, A. Zorica et.all. (2006).Farmakikinetika Diklofenak Pada Babi Setelah
Pemberian Intramuskular Dosis Tunggal. Vol 56. N0.4. Beograd. Hal. 323-325.
Waldon, D.J. (2008). Pharmacokinetics and Drug Metabolism. Cambridge : Amgen,
Inc., One Kendall Square, Building 1000, USA.
Wilmana, P.F. (2005). Analgesik-Antipiretik Anti Inflamasi Non steroid.Farmakologi dan
Terapi. Edisi 4. Jakarta : Bagian Farmakologi Fakultas Kedokteran Universitas
Lampiran 2. Hasil Orientasi dengan Menggunakan Alat KCKT
2.1 Kromatogram hasil penyuntikan larutan Natrium Diklofenak BPFI dengan
konsentrasi 500 mcg / ml, fase gerak MeOH : buffer asetat (68:32), waktu tambat
6,3 menit
2.2 Kromatogram hasil penyuntikan larutan Natrium Diklofenak BPFI dengan
konsentrasi 500 mcg / ml, fase gerak MeOH : buffer asetat (70:30), waktu tambat 2,3
2.3 Kromatogram hasil penyuntikan larutan Natrium Diklofenak BPFI dengan
konsentrasi 500 mcg / ml, fase gerak MeOH : buffer asetat (80:20), waktu tambat
Lampiran 3. Natrium Diklofenak Dalam Plasma
[image:48.612.95.477.317.420.2]Gambar 3.1 Natrium diklofenak dalam plasma 0,25 jam
Gambar 3.2 Natrium diklofenak dalam plasma 0,5 jam
[image:48.612.101.477.486.578.2]Gambar 3.4 Natrium diklofenak dalam plasma 1,25 jam
Gambar 3.5 Natrium diklofenak dalam plasma 1,5 jam
Gambar 3.6 Natrium diklofenak dalam plasma 2,5 jam
[image:49.612.103.483.351.432.2] [image:49.612.93.483.496.569.2]Gambar 3.8 Natrium diklofenak dalam plasma 4,5 jam
[image:50.612.96.480.330.408.2]Lampiran 4. Perhitungan Persamaan Regresi Dari Kurva Kalibrasi Natrium
Diklofenak BPFI yang Diperoleh Secara KCKT
No Konsentrasi
(mcg/ ml)
X
Luas Puncak
Y XY X
2
Y2
1. 80 12124061 969924880 6400 1,5x1014
2. 90 15696775 1412709750 8100 2,5x1014
3. 105,6 21238100 2242743360 11151,36 4,5x1014
4. 120 37170508 4460460960 14400 1,4x1015
Σ 395,6 86229444 9085838950 40051,36 2,25x1015
Rata-Rata
98,9 21557361 227145974 10012,84 5,6x1014
Y = aX + b
a = n(Σxy)- (Σx)( Σy)
a = 4(9085838950)-(395,6)(86229444)
4(40051,36)-(395,6)2
a = 601980,46
b = Y - aX
b = 21557361- 601980,46 (98,9)
b = -37978506,5
Y = aX + b
Sehingga diperoleh persamaan regresi Y = 601980,46X-37978506,5
Untuk mencari hubungan kadar (X) dengan luas puncak (Y) digunakan pengujian
koefisien korelasi (r)
r = n(Σxy)-(Σx)(Σy)
[n(x2)(x)2][n(y2)(y)2]
r = 4(9085838950)-(395.6)(86229444)
] ) 86229444 (
) 10 25 , 2 ( 4 ][ ) 6 , 395 ( ) 36 , 40051 ( 4
[ 2 x 15 2
Lampiran 5. Konversi Perhitungan Dosis Antar Jenis Hewan
Perlakuan Mencit 20g
Tikus
200g
Marmot
400g
Kelinci
1,5kg
Kera
4kg
Anjing
12kg
Manusia
70kg
Mencit
20g 1,0 7,0 12,25 27,8 64,1 124,2 387,9
Tikus
200g 0,14 1,0 1,74 3,9 9,2 17,8 56,0
Marmot
400g 0,08 0,57 1,0 2,25 5,2 10,2 31,5
Kelinci
1,5kg 0,04 0,25 0,44 1,0 2,4 4,5 14,2
Kera
4kg
0,016 0,11 0,19 0,42 1,0 1,9 6,1
Anjing
12kg 0,008 0,06 0,10 0,22 0,52 1,0 3,1
Manusia
Lampiran 6. Perhitungan Dosis yang Diberikan Kepada Masing-Masing Hewan
Percobaan
1. Hewan I
Suspensi Natrium Diklofenak 0,5%
Dosis lazim = 25 mg
Berat hewan = 1,94 kg
Konversi pada hewan kelinci = 0,07
Dosis konversi = 25 x 0,07 = 1,75 mg
Dosis dari perkiraan berat per kg BB = 1000 g x 1,75 mg = 1,17 mg / kg BB
1500 g
Dosis = 1940 g x 1,17 mg = 2,3 mg
1000 g
2. Hewan II
Suspensi Natrium Diklofenak 0,5%
Dosis lazim = 25 mg
Berat hewan = 1,64 kg
Konversi pada hewan kelinci = 0,07
Dosis konversi = 25 x 0,07 = 1,75 mg
Dosis dari perkiraan berat per kg BB = 1000 g x 1,75 mg = 1,17 mg / kg BB
1500 g
Dosis = 1640 g x 1,17 mg = 1,9 mg
1000 g
Volume dosis yang diberikan =
mg mg
5 , 0
9 , 1
x 1 ml = 3,8 ml
3. Hewan III
Suspensi Natrium Diklofenak 0,5%
Dosis lazim = 25 mg
Berat hewan = 1,19 kg
Konversi pada hewan kelinci = 0,07
Dosis konversi = 25 x 0,07 = 1,75 mg
Dosis dari perkiraan berat per kg BB = 1000 g x 1,75 mg = 1,17 mg / kg BB
1500 g
Dosis = 1190 g x 1,17 mg = 1,4 mg
1000 g
Volume dosis yang diberikan =
mg mg
5 , 0
4 , 1
4. Hewan IV
Suspensi Natrium Diklofenak 0,5%
Dosis lazim = 25 mg
Berat hewan = 1,54 kg
Konversi pada hewan kelinci = 0,07
Dosis konversi = 25 x 0,07 = 1,75 mg
Dosis dari perkiraan berat per kg BB = 1000 g x 1,75 mg = 1,17 mg / kg BB
1500 g
Dosis = 1540 g x 1,17 mg = 1,8 mg
1000 g
Volume dosis yang diberikan =
mg mg
5 , 0
8 , 1
x 1 ml = 3,6 ml
5. Hewan V
Suspensi Natrium Diklofenak 0,5%
Dosis lazim = 25 mg
Berat hewan = 1,84 kg
Konversi pada hewan kelinci = 0,07
Dosis konversi = 25 x 0,07 = 1,75 mg
Dosis dari perkiraan berat per kg BB = 1000 g x 1,75 mg = 1,17 mg / kg BB
1500 g
Dosis = 1840 g x 1,17 mg = 2,15 mg
1000 g
Volume dosis yang diberikan =
mg mg
5 , 0
15 , 2
6. HewanVI
Suspensi Natrium Diklofenak 0,5%
Dosis lazim = 25 mg
Berat hewan = 1,64 kg
Konversi pada hewan kelinci = 0,07
Dosis konversi = 25 x 0,07 = 1,75 mg
Dosis dari perkiraan berat per kg BB = 1000 g x 1,75 mg = 1,17 mg / kg BB
1500 g
Dosis = 1640 g x 1,17 mg = 1,9 mg
1000 g
Volume dosis yang diberikan =
mg mg
5 , 0
9 , 1
Lampiran 7.
7.1 Konsentrasi Obat Pada Masing-Masing Hewan Percobaan
Waktu
(t)
Hewan
I
Hewan
II
Hewan
IIi
Hewan
IV
Hewan
V
Hewan
VI
Rata-Rata ± SD
0,25 65,39 66,07 67,04 66,42 66,13 66,07 66,187 ± 0,538
0,5 66,42 66,13 67,07 66,53 67,04 66,13 66,553 ± 0,420
0,75 66,55 66,40 67,25 67,07 67,36 66,53 66,86 ± 0,415
1,25 67,04 66,42 73,39 73,88 67,44 67,25 69,237 ± 3,428
1,5 67,07 66,53 73,88 76,07 71,26 67,36 68,86 ± 4,332
2,5 73,88 73,88 79,45 79,45 75,98 73,88 76,087 ± 2,729
4,5 67,44 67,84 76,65 73,66 73,88 71,19 71,777 ± 3,643
5,5 67,24 67,07 71,83 72,83 71,83 67,84 69,607 ± 2,449
Grafik Konsentrasi (log c) Vs Waktu (t)
Natrium Diklofenak dalam Plasma
1,76 1,78 1,8 1,82 1,84 1,86 1,88 1,9 1,92
0,25 0,5 0,75 1,25 1,5 2,5 3,5 4,5 5,5
Waktu (t)
K
ons
e
nt
ra
s
i (
log c
)
Hewan I Hewan II Hewan III
Hewan IV Hewan V Hewan VI
Lampiran 8. Contoh Perhitungan Parameter Farmakokinetika Metode Stripe
University of Illinois at Chicago-College of Pharmacy Department of
Pharmacodynamics
Hewan I
Time Concentration Calculated % Difference
0.25 65.39 65.69 -0.453
0.50 66.42 66.14 0.424
0.75 66.55 66.50 0.075
1.25 67.04 67.01 0.048
1.50 67.07 67.16 -0.119
2.50 73.88 67.21 9.037
3.50 71.25 66.60 6.535
4.50 67.44 65.54 2.819
5.50 67.24 64.18 4.551
A (1) = -14,872 B (1) = -0,342 jam-1
A (2) = 80,013 B (2) = -0,034 jam-1
N (1) = 5 r (1) = -0,996
N (2) = 4 r (2) = -0,957
AIC = 47, 37 SS = 79,418
There is no lag time
Absorption half life = -2,027 jam
Half life = 20,513 jam
AUC (0-Tn) = 362, 99
AUC (Tn-inf) is 84, 57 % of AUC (0-inf)
AUMC = 70876, 91 mcg / ml jam2
MRT = 30, 12 jam
Vd (ss) = %29449, 359 ml
= 29, 45 L
Total clearance = 977, 57355 ml / jam
= 0,978 L / jam
Assumed fraction absorbed = 1.000
Calculated c max = 67, 28 mcg / ml
T max = 2, 09 jam
Lampiran 9. Contoh Perhitungan Parameter Farmakokinetika Secara Manual
(jam) (mcg / ml)
0,25 65,39 79,383 /-13,992/
0,5 66,42 78,711 /-12,291/ Ln R = LnB-Ka.t
0,75 66,55 78,045 /-11,491/ =2,60268-0,2248t
1,25 67,04 76,729 /-9,691/ R = B.e-Ka.t
1,5 67,07 76,079 /-9,004/ = 13,500e-0,2248t
2,5 73,88 Ln CE = Ln A-Kel.t r =- 0,8105
3,5 71,25 = 4,38279-0,03401t
4,5 67,44 CE = A.e-Kel.t
5,5 67,24 = 80,061e-0,03401 t
r = -0,946
Waktu (t) Konsentrasi (C) Ln C X2 Y2 XY (jam) (mcg / ml)
(X) (Y)
2,5 73,88 4,302 6,25 18,507 10,755
3,5 71,25 4,266 12,25 18,199 14,931
4,5 67,44 4,211 20,25 17,733 18,9495
5,5 67,24 4,208 30,25 17,707 23,144
ΣX = 16 ΣY=16,987 ΣX2=69 ΣY2=72,146 ΣXY = 67, 77795
X
= 4 Y= 4, 24675a = 2
16 ) 69 ( 4 ) 987 , 16 ( 16 ) 7795 , 67 ( 4
b = Y- aX
= 20 6802 , 0
b = 4, 24675-(-0,03401x 4)
= -0, 3401 = 4, 38279
r = n(Σxy)-(Σx)(Σy)
= ] ) 987 , 16 ( ) 146 , 72 ( 4 ][ ) 16 ( ) 69 ( 4 [ ) 987 , 16 )( 16 ( ) 77795 , 67 ( 4 2 2 = -0,946
Waktu (t) R Ln R X2 Y2 XY
(jam)
(X) (Y)
0,25 /-13,992/ 2,638 0,625 6,959 0,6595
0,5 /-12,291/ 2,509 0,25 6,295 1,2545
0,75 /-11,491/ 2,442 0,5625 5,963 1,8315
1,25 /-9,691/ 2,271 1,5625 5,157 2,83875
1,5 /-9,004/ 2,198 2,25 4,831 3,297
ΣX = 4,25 ΣY= 12,058 ΣX2 = 5,25 ΣXY = 9,88125 X = 0,85 Y= 2,4116
a =
2 2 ) ( ) ( ) )( ( ) ( x x n y x xy n
b = Y- aX
a =
2 ) 25 , 4 ( ) 25 , 5 ( 5 ) 058 , 12 25 , 4 ( ) 88125 , 9 ( 5 x
= 2,4116-(-0,2248 x0,85)
= -0,2248 = 2,60268
r = n(Σxy)-(Σx)(Σy)
[n(x2)(x)2][n(y2)(y)2]
= ] ) 058 , 12 ( ) 205 , 29 ( 5 ][ ) 25 , 4 ( ) 25 , 5 ( 5 [ ) 058 , 12 )( 25 , 4 ( ) 88125 , 9 ( 5 2 2
= -0,8105
Kel = 0, 03401 jam-1 T1/2 el =
03401 , 0 693 , 0
= 20, 376 jam
Ka = 0,2248 jam-1
AUC0-t = { 2 1 1xt C } + { 2 ) ( )
(C2 C1 x t2 t1
} + {
2
) (
)
(Cn Cn1 x tn tn1
}
AUC0-5,5={
2 25 , 0 391 , 65 x }+{ 2 ) 25 , 1 5 , 1 )( 038 , 67 075 , 67 ( }+ { 2 ) 25 , 0 5 , 0 )( 391 , 65 420 , 66 ( }+{ 2 ) 5 , 0 75 , 0 )( 420 , 66 554 , 66 ( } +{ 2 ) 75 , 0 25 , 1 )( 554 , 66 038 , 67 ( }+{ 2 ) 5 , 1 5 , 2 )( 075 , 67 884 , 73 ( } +{ 2 ) 5 , 2 5 , 3 )( 884 , 73 252 , 71 ( }+{ 2 ) 5 , 3 5 , 4 )( 252 , 71 436 , 67 ( }+ { 2 ) 5 , 4 5 , 5 )( 436 , 67 236 , 67 ( }
= 371,152 mcg / ml jam
AUC5,5-∞ = 03401 , 0 236 , 67
= 1976,948 mcg / ml jam
AUC0-∞ = AUC0-5,5 + AUC5,5-∞
= 371,152 + 1976,948
= 2348,1 mcg / jam
c. AUMC
t Cxt
0,25 16,348
0,5 33,21 0,75 49,9155 1,25 83,7975 1,5 100,6125 2,5 184,71 3,5 249,382 4,5 303,462 5,5 369,798
AUMC0-t={ }+{
2
) )( (C2t2 C1t1 t2 t1
}+{
2
) )(
(Cntn Cn1tn1 tn tn1
}
AUMC0-5,5={ 2 ) 348 , 16 25 , 0 ( x }+{ 2 ) 25 , 0 5 , 0 )( 348 , 16 21 , 33 ( }+ { 2 ) 5 , 0 75 , 0 )( 21 , 33 9155 , 49 ( }+ { 2 ) 75 , 0 25 , 1 )( 9155 , 49 7975 , 83 ( }+ { 2 ) 25 , 1 5 , 1 )( 7975 , 83 6125 , 100 ( }+{ 2 ) 5 , 1 5 , 2 )( 6125 , 100 71 , 184 ( }+ { 2 ) 5 , 2 5 , 3 )( 71 , 184 382 , 249 ( }+{ 2 ) 5 , 3 5 , 4 )( 382 , 249 462 , 303 ( }+ { 2 ) 5 , 4 5 , 5 )( 462 , 303 798 , 369 ( }
= 1047,868 mcg / ml jam2
AUMC5,5-∞ = 03401 , 0 798 , 396
= 10873,214 mcg / ml jam2
AUMC0-∞ = AUMC0-5,5 + AUMC5,5-∞
= 1047,868 + 10873,214
= 11921,082 mcg / ml jam2
MRT =
0 0 AUC AUMC = 1 , 2348 082 , 11921
= 5,08 jam
Cmaks = .max
) (
. ket
e Ke Ka Vd Dosis Ka
= . 0,03401 9,9
) 03401 , 0 2248 , 0 ( 794 , 28800 2300000 2248 , 0 x e x