Improvement of Farm Animal Breeding by
DNA Sequencing
G. Darshan Raj
Department of Animal Husbandry and Veterinary Services, Government of Karnataka,
India
1. Introduction
Animal breeds with specific qualities like adaptability to local climatic conditions, farming situations , resistance to diseases and those meet the requirements of local needs and perform under available feeds and fodder are valuable and present requirement for the world. Main farm animals cattle, sheep, goat and pigs were domesticated 9,000–11,000 years ago, whereas the dog was domesticated earlier, ~14,000 years ago, and the chicken ~4,500 years ago. Most domestic animals have a much broader genetic basis. Molecular studies have shown that the two main forms of domestic cattle, European–African (Bos Taurus taurus) and Asian (Bos taurus indicus), originate from two different subspecies of the wild ancestor (Loftus et al., 1994). There are thirty well recognized cattle breeds in India which are highly adoptive to local conditions and each breed has distinctive characters. The world population of buffalo is about 158M in comparison with around 1.3B cattle, 1B sheep and 500-600M goats. There are two types of domestic water buffalo, the River Buffalo (Bubalus bubalis) which are more widely spread globally, and are the predominant type found in the west from India to Europe. The second type, the Swamp Buffalo (Bubalus carabanesis), is found more frequently in the east from India to the Philippines. The two major types of buffalo can be distinguished by their different karyotypes, the Swamp Buffalo has 48 chromosomes, whereas the River buffalo has 50. However, the two types crossbreed to produce fertile hybrid progeny and so cannot be considered as truly different species.
In India and Pakistan there are well defined breeds, each with their own individual qualities. In South Asia 18 River buffalo breeds are recognized, which can be placed into 5 major groups designated as the Murrah, Gujarat, Uttar Pradesh, Central Indian and South Indian breeds. The Mediterranean Buffalo is of the River type and has been strongly selected for milk production, particularly in Italy. It has been genetically isolated for a long time and as a result has become genetically distinct from other breeding populations. The high level of milk production of the Mediterranean Buffalo has resulted in it been exported to many countries worldwide.
identified in India. There are nearly 102 breeds of goats in the world, of which 20 breeds are in India. Goats are reared for two purposes i.e. meat & milk. Meat and Milk production is the main objective. The domestic goat (Capra aegagrus hircus) is a subspecies of goat domesticated from the wild goat of southwest Asia and Eastern Europe. The goat is a member of the Bovidae family and is closely related to the sheep as both are in the goat-antelope subfamily Caprinae. Goats are one of the oldest domesticated species. Goats have been used for their milk, meat, hair, and skins over much of the world. In the twentieth century they also gained in popularity as pets.
Farm animal populations harbor rich collections of mutations with phenotypic effects that have been purposefully enriched by breeding. Farm animals are of particular interest for identifying genes that control growth, energy metabolism, development, appetite, reproduction and behavior, as well as other traits that have been manipulated by breeding. Genome research in farm animals will add to our basic understanding of the genetic control of these traits and the results will be applied in breeding programmes to reduce the incidence of disease and to improve product quality and production efficiency (Leif Andersson, 2001).
2. Evaluation of animal traits
Many of the traits of interest in animal production are quantitative traits. Evaluation of genetic merit of animals is still essentially based on the application of the theory of quantitative genetics. Traditionally, the genetic improvement of livestock breeds has been based on phenotypic selection. The past century was characterized by the development of quantitative theory and methodology towards the accurate selection and prediction of genetic response (Walsh, 2000).This resulted in the selection of a number of economically important genetic traits in cattle, sheep, pigs and poultry. The conceptual basis of this theory is the polygenic model, which assumes that quantitative traits result from the action (and interaction) of a large number of minor genes, each with small effect. The resulting effects are then predicted using powerful statistical methods (animal model), based on pedigree and performance recording of traits from the individual animal and its relatives (Yahyaoui, 2003). Most of morphological markers are sex limited, age dependent, and are significantly influenced by the environment. Biochemical markers show low degree of polymorphism. The various genotypic classes are indistinguishable at the phenotypic level because of the dominance effect of the marker and low genome coverage (Montaldo and Meza-Herrera, 1998).
2.1 Genome mapping
The advances in molecular genetics technology in the past two decades, particularly Nucleic acid-based markers, has had a great impact on gene mapping, allowing identification of the underlying genes that control part of the variability of these multigenic traits. Broadly, two experimental strategies have been developed for this purpose: linkage studies and candidate gene approach (Jeffreys et al., 1985).
2.1.1 Linkage studies
markers (generally micro satellites) and the trait being analyzed. Markers that tend to co-segregate with the analyzed trait provide approximate chromosomal location of the underlying gene (or genes) involved in part of the trait variability determinism (Yahyaoui, 2003).
2.1.2 Candidate gene approach
The candidate gene approach studies the relationship between the traits and known genes that may be associated with the physiological pathways underlying the trait (Liu et al., 2008). In other words, this approach assumes that a gene involved in the physiology of the trait could harbor a mutation causing variation in the trait. The gene or part of gene, are sequenced in a number of different animals, and any variation found in the DNA sequences, is tested for association with variation in the phenotypic trait (Koopaei and Koshkoiyeh, 2011). This approach has had some success. For example a mutation was discovered in the estrogen receptor locus (ESR) which results increased litter size in pig. There are 2 problems with the candidate gene approach. Firstly, there are usually a large number of candidate genes affecting the trait, so many genes must be sequenced in several animals and many association studies carried out in a large sample of animals. Secondly, the causative mutation may lie in a gene that would not have been regarded prior as an obvious candidate for this particular trait. Candidate gene approach is performed in 5 steps: 1) collection of resource population. 2) Phenotyping of the traits. 3) Selection of gene or functional polymorphism that potentially could affect the traits. 4) Genotyping of the resource population for genes or functional polymorphism. Lastly, one is statistical analysis of phenotypic and genotypic data (Da, 2003). This is an effective way to find the genes associated with the trait. So far a number of genes have been investigated. Candidate gene approach has been ubiquitously applied for gene disease research, genetic association studies, biomarker and drug target selection in many organisms from animals (Tabor et al., 2002). The traditional candidate gene approach is largely limited by its reliance on existing knowledge about the known or presumed biology of the phenotype under investigation, unfortunately the detailed molecular anatomy of most biological traits remain unknown (Zhu and Zhao, 2007).
A candidate gene approach has been already successfully applied to identify several DNA markers associated with production traits in livestock The principle is based on the fact that variability within genes coding for protein products involved in key physiological mechanisms and metabolic pathways directly or indirectly involved in determining an economic trait (e.g. feed efficiency, muscle mass accretion, reproduction efficiency, disease resistance, etc.) might probably explain a fraction of the genetic variability for the production trait itself. The first step is the identification of mutations in candidate genes that can be analyzed in association studies in specific designed experiments. For the roles they play, growth hormone (GH) and myostatin (MSTN) genes can be considered candidate genes for meat production traits and casein genes in milk production traits (Rothschild and Soller, 1997).
2.1.2.1 Candidate gene polymorphism and DNA sequencing technique
number of human genes was initially estimated at around 50,000-1, 00,000. However, based on the recent complete sequencing of the human genome in the 2002 year , this number has been revised to 25000-30000 only. A similar estimate has been reported for most of the farm animal species. These genes prescribe the development of the living species and contribute to the biological biodiversity of our planet. A feature of the genome that has confused geneticists for years is that the coding sequences (genes) only represent some 3to 5 percent on the genome. Whereas the rest is sequences (genes) unknown functions including different types of repetitive DNA, introns, 5’ and 3’ untranslated regions and pseudogenes. The sizes of genes vary from less than 100 bp to over 2000kb (Fields et al., 1994).
Recent developments in DNA technologies have made it possible to uncover a large number of genetic polymorphisms at the DNA sequence level and to use them as markers for evaluation of the genetic basis for observed phenotypic variability. These markers posses unique genetic properties and methodological advantages that make them more usefull and amendable for genetic analysis and other genetic markers. The possible applications of molecular markers in livestock industry are by conventional breeding programme to transgenic breeding technologies. In conventional breeding strategies molecular markers have several short range or immediate applications viz., parentage determination , genetic distance estimation , determination of twin zygosity and free martinism, sexing preimplation embryo and identification of genetic disease carrier and long range applications viz ., Genome mapping and marker assisted selection. In transgenic breeding molecular markers can be used as reference points for identification of the animals carrying the trasgenes. The progress in the genetic markers suggests their potential use for genetic improvement in the livestock species.
The progress in the DNA recombinant technology , gene cloning and High throughput Sequencing technique during last two decades have brought revolutionary changes in the field of basic as well as applied genetics which provide several new approaches for genome analysis with greater genetic resolution . it is now possible to uncover a large number of genetic polymorphisms at DNA sequence level and to use them as markers for evaluation of the genetic basis for observed phenotypic variability. The first demonstration of DNA level polymorphism, known as restriction fragment polymorphism (RFLP) an almost unlimited number of molecular markers have accumulated. Currently, more powerful and less laborious techniques to uncover new types of DNA makers are steadily being introduced. The introduction of polymerase chain reaction (PCR) in conjunction with the constantly increasing DNA sequence data also represents a mile stone in this endeavor.
suggest that an animal is heterozygous. However animal may be misclassified as homozygote if there is polymorphism in the primer sequence, which prevents the allele from being amplified and therefore not being detected on the electrophoresis gel referred to as null allele. A null allele will often be detected when misparentage routinely found for a marker system. An animal might also be misclassified, if another, non allelic form was amplified with the PCR primers and digestion of the PCR product results in a different sized fragments. A Non allelic form is revealed by sequencing the fragments contained within the electrophoresis bands, which is recommended step when establishing marker system (Jiang and Ott, 2010).
However new technologies have significantly advanced our ability to identify SNP’s and then explore multiple candidate genes at one time at a much lower cost/polymorphism than the PCR-RFLP method. The identification SNP’s within a gene or genetic region is now relatively easy To do this , genomic DNA of key animals within a population are sequenced using high throughput automatic sequencing and then compared with the other sequences within the population or to sequences which are publically available databases .
This approach is already being employed with regard to bovine leukocyte adhesion deficiency (BLAD; Shuster et al., 1992) and genes with major effects, such as the halothane locus in swine (Rempel et al., 1993) and alpha S1-casein in goat (Manfredi et al.,
1995).Currently, direct sequencing is one of the high throughput methods for mutation detection, and is the most accurate method to determine the exact nature of a polymorphism. Sanger dideoxy-sequencing can detect any type of unknown polymorphism and its position, when the majority of DNA contains that polymorphism. Fluorescent sequencing can have variable sensitivity and specificity in detecting heterozygotes because of the inconsistency of base-calling of these sites (Yan et al., 2000). Thus, it has only limited utility when the polymorphism is present in a minor fraction of the total DNA (for example in pooled samples of DNA or in solid tumors) due to low sensitivity. DNA sequencing is usually used as a second step to confirm and identify the exact base altered in the target region previously identified as polymorphic by using scanning methods.
2.1.2.1.1 Different methods of DNA sequencing
thousand (Roche) to 40 million (Illumina) target DNAs and sequencing these fragments simultaneously. Furthermore, these new technologies allow the direct sequencing of DNA or cDNA without any cloning step and at decreasing cost per sequenced base . Although all of these new technologies produced extremely short reads when they initially entered the market, the performance of (NGS) platforms have increased substantially. As an example: the first nextgeneration sequencer released by 454 (GS20) had an average read-length of 110 bp. A second improved sequencer has since then been introduced, the GS-FLX, which is able to obtain average read lengths of 250 bases and is able to perform mate-paired reads. A more recent release is the 454 FLX Titanium which has an average read length of 400 bp
Technology Approach Maxthrougput
Table 1. Accuracy of different DNA sequencing methods: Kerstens, (2010).
Throughput is compared by giving the maximum number of bases per second a platform generates at it's optimal read length. Paired-end sequencing is for some platforms restricted to a read length. Sequencing cost is compared by indicating the price per mega base and only includes the costs of reagents and costs to perform one sequencing run.
1. Applied Biosystems http://www.appliedbyosystems.com, 2. Roche Applied Science http://www.roche-applied-science.com, 3. Illumina, Inc. http://www.illumina.com
4. Applied Biosystems http://www.appliedbyosystems.com 5. Helicosbio http://www.helicosbio.com (Kerstens, 2010)
from 50 to 400 bp. Further, it is promised that new, third generation single molecule sequencing will soon be available and should deliver complete genome sequence that is in time and pricing affordable for every research group (Perez-Enciso and Ferretti, 2010)
NGS technologies allow cost effective sequencing of multiple individuals which is beneficial for SNP detection. SNP discovery through parallel pyrosequencing (454) of an individual human genome identified 3.32 million SNPs, with 606,797 of those as novel SNPs (Wheeler et al ., 2008) which is comparable with the shotgun-sequenced Venter genome (cost $70 million) that had 3.47 million SNPs, with 647,767 of those being novel (Wheeler et al ., 2008) .These authors stated that at least a 20X genome coverage is required to call 99% of heterozygous bases correctly within a single individual, resulting in a sequencing cost of 2 million dollar. For the Illumina method similar results (3.07 million SNPs of which 420 thousand novel SNPs) can be obtained by sequencing an individual genome with 36X coverage for less than half a million dollars (Wang et al ., 2008) or ~ 4 million SNPs of which 26% are novel SNPs at 30X coverage for a quarter of a million dollar. (Bentley et al ., 2008). More recent developments using single molecule sequencing further reduce the sequencing costs. For example, the discovery of 2.8 million SNPs by whole genome resequencing at 28X coverage using this technology reduces the costs to less than 50 thousand dollars (Pushkarev et al ., 2009). These developments indicate that extremely high throughput sequencing machines that produce relatively short reads are favorable for SNP discovery in a whole genome resequencing approach. In addition, simulations suggest that 85% of 35 bp reads can be placed uniquely on the human genome whereas 95% of 35 bp paired end reads with 200 bp insert sizes have unique placement (Li et al. ,2008). However SNP discovery in species with limited public genomic resources benefits from longer read lengths. It has been demonstrated that reads produced with pyrosequencing technology can be assembled de novo into reasonably long contigs that subsequently can serve as a genome reference on which short reads of other individuals can be mapped to detect SNPs (Novaes et al., 2008). At present whole genome sequence assembly by alignment of relative short NGS fragments without the availability of a reference genome is tedious, but possible for less complex mammalian sized genomes (Li et al ., 2009). Besides the consistent pattern of non-uniform sequence coverage each NGS platform generates (Harismendy et al., 2009), which is substantially lower in AT-rich repetitive sequences, repetitive regions are hard to reconstruct by short-read sequence assembly. Possible NGS SNP detection strategies circumventing the requirement of a sequenced reference genome are (ultra short read) sequencing of more than one genotype and alignment of that data by using: (1) genome or transcriptome sequence data from model species closely related to the species of interest, (2) whole transcriptome or reduced representative genome sequence data for the species of interest, based on Roche/454 sequence technology (Wiedmann et al ., 2008).
farm animals, as many disease carriers are removed from breeding populations by purifying selection. By studying diverse phenotype over time, researchers can now monitor mutations that occur as wild species become domesticated (Fadiel et al., 2005).
2.2 Animal genome projects
According to Wikipedia, Genome projects are scientific endeavours that ultimately aim to determine the complete genome sequence of an organism (be it an animal, a plant, a fungus, a bacterium, anarchaean, a protist or a virus) and to annotate protein-coding genes and other important genome-encoded features. The genome sequence of an organism includes the collective DNA sequences of each chromosome in the organism. The release of the first draft of the chicken genome in March 2004 spawned the current boom in chicken genomic research (Antin and Konieczka, 2005) evolutionary standpoint; investigation of the chicken genome will provide significant information needed to understand the vertebrate genome evolution, since the chicken is between the mammal and fish on the evolutionary tree. Furthermore, the chicken remains significant as a food animal which comprises 41% of the meat produced in the world and serves as a reliable model for the study of diseases and developmental biology (Dequeant and Pourquie, 2005). With this sequenced genome, chicken breeders will have a framework for investigating polymorphisms of informative quantitative traits to continue their directed evolution of these species (Fadiel et al., 2005)
The sequencing of the pig genome generated an invaluable resource for advancements in enzymology, reproduction, endocrinology, nutrition and biochemistry research (Wernersson, et al., 2005 and Rothschild, et al., 2003) Since pigs are evolutionarily distinct to both humans and rodents, but have co-evolved with these species, the diversity of selected phenotypes make the pig a useful model for the study of genetic and environmental interactions with polygenic traits (Blakesley et al., 2004). The sequencing of the pig genome is also instrumental in the improvement of human health. Clinical studies in areas such as infectious disease, organ transplantation, physiology, metabolic disease, pharmacology, obesity and cardiovascular disease have used pig models (Rothschild, 2004). In the near future, the sequencing of the porcine genome will allow gene markers for specific diseases to be identified, assisting breeders in generating pig stocks resistant to infectious diseases (Klymiuk, and Aigner, 2005 and Fadiel et al ., 2005)
Cattle is of great interest since it represents a group of eutherian mammals phylogenetically distant from primates (Larkin, et al., 2003 and Kumar, and Hedges, 1998). Working with the cow species, B.taurus, is significant because the cow is such an economically important animal. This form of livestock makes up the beef and milk production industry. The identification of numerous single-nucleotide polymorphisms (SNPs) makes it possible for geneticists to find associations between certain genes and cow traits that will eventually lead to the production of superior-quality beef (Adam, 2002).
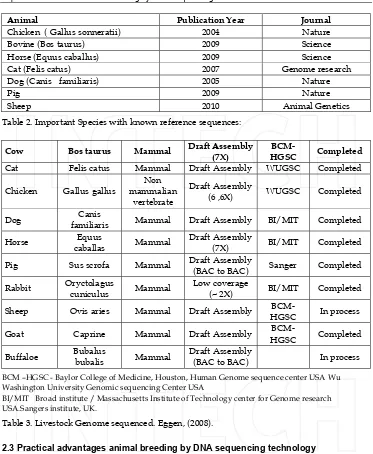
Animal Publication Year Journal
Chicken ( Gallus sonneratii) 2004 Nature
Bovine (Bos taurus) 2009 Science
Horse (Equus caballus) 2009 Science
Cat (Felis catus) 2007 Genome research
Dog (Canis familiaris) 2005 Nature
Pig 2009 Nature
Sheep 2010 Animal Genetics
Table 2. Important Species with known reference sequences:
Cow Bos taurus Mammal Draft Assembly (7X)
BCM-HGSC Completed
Cat Felis catus Mammal Draft Assembly WUGSC Completed
Chicken Gallus gallus
familiaris Mammal Draft Assembly BI/MIT Completed
Horse Equus
caballas Mammal
Draft Assembly
(7X) BI/MIT Completed
Pig Sus scrofa Mammal Draft Assembly
(BAC to BAC) Sanger Completed
Rabbit Oryctolagus
cuniculus Mammal
Low coverage
(~ 2X) BI/MIT Completed
Sheep Ovis aries Mammal Draft Assembly
BCM-HGSC In process
Goat Caprine Mammal Draft Assembly
BCM-HGSC Completed
Buffaloe Bubalus
bubalis Mammal
Draft Assembly
(BAC to BAC) In process
BCM –HGSC - Baylor College of Medicine, Houston, Human Genome sequence center USA Wu Washington University Genomic sequencing Center USA
BI/MIT Broad institute / Massachusetts Institute of Technology center for Genome research USA.Sangers institute, UK.
Table 3. Livestock Genome sequenced. Eggen, (2008).
2.3 Practical advantages animal breeding by DNA sequencing technology
and many millions of animals. Genetic markers had limited use during the century after Mendel’s principles of genetic inheritance were rediscovered because few major QTL were identified and because marker genotypes were expensive to obtain before 2008. Genomic evaluations implemented in the last two years for dairy cattle have greatly improved reliability of selection, especially for younger animals, by using many markers to trace the inheritance of many QTL with small effects. More genetic markers can increase both reliability and cost of genomic selection. Genotypes for 50,000 markers now cost <US$200 per animal for cattle, pigs, chickens, and sheep. Lower cost chips containing fewer (2,900) markers and higher cost chips with more (777,000) markers are already available for cattle, and additional genotyping tools will become available for cattle and other species VanRaden et al., (2011). Livestock selection has used estimated breeding values (EBV) based on phenotypic data and pedigree records for more than 40 years. More recently, advances in molecular genetic techniques, in particular DNA sequencing, have led to the discovery of regions of the genome that influence traits in livestock. However utilizing both sources of data in genetic evaluation schemes, such as BREEDPLAN, has been a challenge due to the heterogeneity of data sources, the multi-trait nature of the evaluations, and unknown effects of the marker information on all traits in the evaluation. The SmartGene for Beef project identified significant effects of the Catapult Genetics GeneSTAR tenderness markers on meat tenderness as recorded by the objective measure of shear force These results have been used to further develop methods for combining EBVs (i.e. phenotypic and pedigree data) and gene marker information into a single marker-assisted EBV called an EBVM. Flight time is an objective measure of an animal’s temperament which has been shown to be heritable and moderately genetically correlated with SF, thus representing a potential genetic indicator trait for meat tenderness (Johnston and Graser 2009).
2.3.1 Cattle
2.3.1.1 Dairy animals
An Example for milk trait, Schild and Geldermann (1996), Koczan et al. (1991), and Bleck et al. (1996) identified mutations in the 5' flanking regions of bovine casein genes. In their experiments nine of these mutations were screened by DNA sequencing to see whether they might change affinity to nuclear proteins– transcription factors affect expression of relevant genes and quantitatively influence composition of milk proteins. Eighty-one Polish Black and White (BW) and 195 Polish Red (PR) cows were screened by DNA sequencing for polymorphism. Both breeds were found to differ significantly in the distribution of various genotypes in the 5'-flanking regions of S1- and S2-casein genes. No polymorphism was
found in the 5'-flanking region of the -casein gene. However, preferential associations were found between individual promoter genotypes and protein variants of S1- and -caseins. It
provided strong evidences for the existence of specific haplotype combinations, including both coding and regulatory sequences at the casein locus in the Polish cattle breeds. These results showed that nucleotide sequence variations in the promoter regions of bovine casein genes might change the affinity of these regions to nuclear proteins – transcription factors – and thus affect the expression of relevant genes that quantitatively influence the composition of milk proteins (Martin et al., 2002).
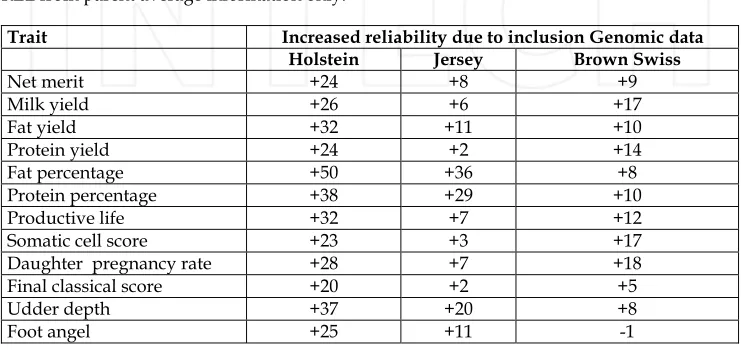
the BovinSNP50 BeadChip (VanRaden et al., 2009; Cole et al., 2009). Genotypic and phenotypic data of these bulls were used to estimate the effects of 38,416 SNP markers (after discarding markers with low minor allele frequency and markers that were in complete linkage disequilibrium with adjacent markers) on production, type, longevity, udder health, and calving ability. Next, the estimated SNP effects were used to compute the genomic PTA of each of 2,035 young Holstein bulls born from 2000-2003 that had no progeny. Finally, the 2009 PTA of each bull in the latter group, which was based on information from its progeny, was compared with the traditional PA and the genomic PTA computed from 2004 data. The same process was repeated in the Jersey breed (1,361 older animals and 388 young bulls) and the Brown Swiss breed (512 older animals and 150 young bulls). Results in Table IV show the increase in reliability (REL) due to genomic information, as compared with the REL from parent average information only.
Trait Increased reliability due to inclusion Genomic data
Holstein Jersey Brown Swiss
Net merit +24 +8 +9
Milk yield +26 +6 +17
Fat yield +32 +11 +10
Protein yield +24 +2 +14
Fat percentage +50 +36 +8
Protein percentage +38 +29 +10
Productive life +32 +7 +12
Somatic cell score +23 +3 +17
Daughter pregnancy rate +28 +7 +18
Final classical score +20 +2 +5
Udder depth +37 +20 +8
Foot angel +25 +11 -1
Table 4. Reliability changes due to the inclusion of genomic data in national genetic evaluations VanRaden et al.,(2009).
2.3.1.2 Beef animals
growth, and may lead to the development of new molecular indicators of tenderness or marbling. Some of these genes are specifically regulated by genetic and nutritional factors or differ between different beef cuts (Hocquette et al ., 2007).
An Example for meat trait, a visibly distinct muscular hypertrophy (mh), commonly known as double muscling, occurs with high frequency in the Belgian Blue and Piedmontese cattle breeds. The autosomal recessive mh locus causing double-muscling condition in these cattle maps to bovine chromosome 2 within the same interval as myostatin, a member of the
super family of genes. Because targeted disruption of myostatin in mice results in a muscular phenotype very similar to that seen in double-muscled cattle, Kambadur et al ., (1997) have evaluated this gene as a candidate gene for double-muscling condition by cloning the bovine myostatin cDNA and examining the expression pattern and sequence of the gene in normal and double-muscled cattle. The analysis demonstrated that the levels and timing of expression do not appear to differ between Belgian Blue and normal animals, as both classes show expression initiating during fetal development and being maintained in adult muscle. Moreover, sequence analysis reveals mutations in heavy-muscled cattle of both breeds.
2.3.2 Goat
The International Goat Genome Consortium (IGGC) was created in March 2010 in Shenzhen and is coordinated by Professor Wen Wang, Zhang Wenguang (China) and Gwenola Tosser-Klopp (INRA). The initial activity of the consortium has focused on coordination in three areas, the results of which should benefit the planned activity in 3SR, namely: de novo goat genome assembly and resequencing; development of a Goat Radiation Hybrid Panel and Mapping; and production of a high-density SNP chip. Assembly of the goat genome is now underway in the Beijing Genome Institute (BGI). The average depth of sequencing is around 60X and optical mapping has been used to increase the scaffold size. Collection of samples for resequencing from around the world is now underway and once completed may provide information on domestication events (Zhang, 2011).
2.3.3 Sheep
The recent availability of the OvineSNP50 Beadchip (Illumina) opens promising perspectives on using molecular information in the management of breeding schemes of dairy and meat sheep populations. The large amount of information provided by this tool may have a strong impact on studies aimed at verifying the feasibility of Marker (or Gene) Assisted Selection or Genome-Wide Selection programs. The OvineSNP50 Beadchip has been recently used by the International Sheep Genomics Consortium (ISGC; www.sheephapmap.org) to genotype samples from 64 different sheep breeds. The sheep breed population may have great advantages by introducing molecular information in the selection scheme. Indeed the possibility to predict breeding values by genomic data might lead to reduce costs of phenotype recording and increase the number of selection objectives (Usai et al., 2010).
2.3.4 Pig
processes. It has the potential to increase genetic progress as well as offering an insight into pig disease and immunity traits and will assist in efforts to preserve the global heritage of rare, endangered and wild pigs. It also will be important for the study of human health because pigs are very similar to humans in their physiology, behavior and nutritional needs. To date, researchers have identified several genes or DNA regions that are associated with traits of economic importance including reproduction, growth, lean and fat quantity, meat quality traits and disease resistance. A number of gene and marker tests are now available commercially from genotyping service companies. Examples are CAST (meat quality), ESR and EPOR (litter size), FUT1 (E. coli disease resistance), HAL (halothane – meat quality, stress), IGF2 (carcase), MC4R (growth and fat), PRKAG3 (meat quality), RN (meat quality) (Walters, 2011).
2.3.5 Horse
The equine genome map was completed in the year 2007. Its applications are comparable to the development of antibiotics. Antibiotics benefited the health of everything; this is going to benefit the health of horses to the same extent in the next 50 years because of the things we can find out .More than 45 years ago, the introduction of antibiotics for use in animals greatly improved animal health and productivity. Today, genetic testing, gene therapy and the identification of genetic markers for certain diseases offer an even bigger opportunity for advancement in the equine industry (Steffanus, 2010).The Equine Genetic Diversity Consortium (EGDC) represents a collaborative, international community of equine researchers who are working to build a comprehensive understanding of genetic diversity among equine populations across the world and their work in future going to stimulate new studies into the origins of breeds and breed-defining traits and guide efforts to preserve genetic diversity (Petersen et al .,2011).
2.3.6 Poultry
Advances in DNA sequencing technology, the chicken genome project results, and a lot of hard work by researchers and geneticists are being combined by poultry primary breeders to change the way selection is done in pedigree flocks. Research projects involving pedigree lines from poultry primary breeders are demonstrating that genomic selection can increase the rate of progress in all traits, including economically important ones. Chickens have hundreds of thousands of SNPs in their genome. Phenotype information like growth rate, feed conversion in Broilers and egg production, egg quality and life span of the birds in layers are gathered from individuals within pedigree populations, and this is correlated with the individuals’ SNPs. With the use of statistics, an individual’s SNPs can be compared to information in the database and a genomic breeding value can be assigned to the animal. Analysis of an individual’s SNPs can allow selection to take place earlier in the life cycle of the animal, thus genomic selection can cut the generation time down, which speeds the rate of progress (Keefe, 2011).
2.3.7 Buffalo
Analyzer II with a paired end of 101 bp. A full run generated more than 230 million reads, which resulted in approximately 46 Gb of high quality sequences. In this pilot study, we tested 210 pairs of water buffalo sequences for their bovine orthologs using a cross species mega BLAST approach developed at NCBI. Among these 210 pairs of sequences, only 7 pairs (3.3%) hit absolutely nothing in the bovine genome. One hundred twenty pairs of sequences (57%) had sole unique hits with one or both ends. For the remaining 83 pairs that had multiple hits in the bovine genome, 31 unique hits can be identified manually based on the aligned length and sequence similarity to the bovine orthologs. The alignment size between both species ranged from 42 bp to 122 bp, but with 93% alignments having more than 85 bp in length. This pilot study provides initial evidence that de novo water buffalo genome sequences can be comparatively assembled based on the cattle genome assembly (Jiang et al ., 2011).
3. Conclusions
The genetic improvement of animals is a fundamental, incessant, and complex process. In recent years many methods have been developed and tested. The genetic polymorphism at the DNA sequence level has provided a large number of markers and revealed potential utility of application in animal breeding. The invention of polymerase chain reaction (PCR) in accordance with the constantly increasing accuracy in DNA sequencing methods also represents a milestone in this endeavor. Selection of markers for different applications are influenced by certain factors - the degree of polymorphism, the automation of the analysis, radioisotopes used, reproducibility of the technique, and the cost involved. Presently, the huge development of molecular markers by DNA sequencing will continue in the near future. It is expected that molecular markers will serve as an underlying tool to geneticists and breeders to create animals as desired and needed by the society.
4. References
Adam,D. (2002). Draft cow genome heads the field. Nature, Vol 417, pp 778.
Antin,P.B. and Konieczka,J.H., (2005).Genomic resources for chicken. Developmental Dynamics, Vol 232, pp 877–882.
Bentley, D.R., Balasubramanian, S., Swerdlow, H.P., Smith, G.P., Milton, J. and Brown, C.G., et al., (2008). Accurate whole human genome sequencing using reversible terminator chemistry. Nature, Vol 456, pp 53-59.
Blakesley,R.W., Hansen,N.F., Mullikin,J.C., Thomas,P.J., McDowell,J.C., Maskeri,B., Young,A.C., Benjamin,B., Brooks,S.Y., Coleman,B.I. et al. (2004) An intermediate grade of finished genomic sequence suitable for comparative analyses. Genome Research, Vol 14, pp 2235–2244.
Bleck, G.T., Conroy, J.C. and Wheeler, M.B., (1996). Polymorphisms in the bovine ß-casein 5’ flanking region, Journal of Dairy Science, Vol 79, pp 347–349.
Cockett,N.E., Shay,T.L. and Smit,M. (2001). Analysis of the sheep genome. Physiological. Genomics, Vol 7, pp 69–78.
Cole, J. B., VanRaden, P.M., O’Connell, J.R., Van Tassell, C.P., Sonstegard,T.S., Schnabel,R.D., Taylor,J.F. and Wiggans, G.R., (2009). Distribution and location of genetic effects for dairy traits. Journal of Dairy Science, Vol 92, pp 2931-2946.
Da, Y., (2003). Statistical analysis and experimental design for mapping genes of complex traits in domestic animals. Bioinformatics, Vol 30, No12, pp 1183–1192.
Dequeant,M.L. and Pourquie,O., (2005). Chicken genome: new tools and concepts. Developmental Dynamics, Vol 232, pp 883–886.
Eggen, A. (2008). Whole genome sequencing in livestock species: The end of the beginning? European Animal Disease Genomics Network of Excellence for Animal Health and Food Safety Animal Disease Genomics: Opportunities and Applications 10th - 11th June 2008 , Edinburgh, UK.
Fadiel, A., Anidi, I. and Eichenbaum, K. D.,(2005). Farm animal genomics and informatics: an update. Nucleic Acids Research, Vol 33, No 19, pp 6308–6318.
Fields, C., Adams, M. D., White, O., Venter, J. C. (1994). How many genes in the human genome. Nature Genetics,Vol 7, pp 345–346.
Harismendy, O., Ng, P.C., Strausberg, R.L., Wang, X., Stockwell, T.B., Beeson, K.Y., Schork N.J., Murray, S.S., Topol, E.J., Levy, S. and Frazer, K.A.,(2009). Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biology,Vol 10, pp 32.
Hocquette, J. F., Lehnert, S., Barendse, W., Cassar-Malek, I. and Picard, B.,(2007). Recent advances in cattle functional genomics and their application to beef quality. Animal, Vol 1, pp 159–173.
Jeffreys, A.J., Wilson V. and Thein S.L., (1985). Individual-specific fingerprint of human DNA. Nature,Vol 316, pp 76-79.
Jiang, Z. and Ott, T.L., (2010).Quantitative Genomics of Reproduction. Reproductive Genomics in Domestic Animals. Cockett, N.E., pp 1-7. Wiley and Blackwell publishers, July 2010. ISBN: 978-0-470-96182-7, Iowa USA.
Johnston, B. T. D. J. and Graser, H.U.,(2009). Integration of Dna markers into breedplan EBVs Proceedings of the Eighteenth Conference Matching Genetics and the Environment a New Look at and Old Topic. Proceedings of Association Advancement of Animal Breeding and. Genetics, 2009. Vol 18,pp 30-33.
Kambadur,R., Sharma, M., Smith, T.P.L. and Bass,j. J.(1997). Mutations in myostatin (GDF8) in Double-Muscled Belgian Blue and Piedmontese Cattle. Genome research, Vol 7, No1,pp 910-915.
Keefe, T. O., (2011). Poultry breeders see paradigm shift with genomic selection. More rapid improvement of economically important traits may be just around the corner for the egg layer and broiler industries. Egg industry, Vol 116, No 8, pp 8-11.
Kerstens, H.H.D., (2010). Bioinformatics approaches to detect genetic variation in whole genome sequencing data. PhD thesis submitted to Wageningen University.
Klymiuk,N. and Aigner,B., (2005). Reliable classification and recombination analysis of porcine endogenous retroviruses. Virus Genes, Vol 3, pp 357–362.
Koczan, D., Hobom, G.and Seyfert, H.M., (1991). Genomic organization of the bovine alpha-s1 casein gene. Nucleic Acid Research,Vol 19, pp 5591-5596.
Koopaei, H. K. and Koshkoiyeh, A. E. (2011).Application of genomic technologies to the improvement of meat quality in farm animals Biotechnology and Molecular Biology Review. Vol 6, No6, pp, 126-132.
Kumar,S. and Hedges,S.B., (1998).A molecular timescale for vertebrate evolution. Nature, Vol 392, pp 917–920.
Larkin,D.M., Everts-van der Wind,A., Rebeiz,M., Schweitzer,P.A., Bachman,S., Green,C., Wright,C.L., Campos,E.J., Benson,L.D., Edwards,J. et al. (2003). Acattle–human comparative map built with cattle BAC-ends and human genome sequence. Genome Research, Vol 13, pp 1966–1972.
Leif Andersson,(2001). Genetic dissection of phenotypic diversity in farm animals, Nature review, Vol 2, pp 130-138.
Li, H., Ruan, J., and Durbin, R., (2008). Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Research, Vol 18, pp 1851-1858. Li, R., Fan, W., Tian, G., Zhu, H., He, L., Cai, J., Huang, Q. and Cai, Q., et al .,(2009).The
sequence and de novo assembly of the giant panda genome. Nature, Vol463, 311-317.
Liu, x., Zhang, H., Li, H., Li, N., Zhang, Y., Zhang, Q., Wang, S., Wang, Q. and Wang, H., (2008). Fine mapping quantitative trait loci for body weight and abdominal fat traits: Effects of marker density and sample size. Poultry Science, Vol 87, pp 1314-1319.
Loftus, R.T., MacHugh, D. E., Bradley, D. G., Sharp, P. M. & Cunningham,P.,( 1994).Evidence for two independent domestications of cattle. Proceedings of National Academy of Sciences. USA,Vol 91, pp 2757–2761.
Manfredi, E., Ricordeau, G., Barbieri, M.E., Amigues, Y. and Bibe, B., (1995). Genotype caseines s1 et selection des boucs sur descendance dans les races Alpine et Saanen. Genetics Selection and Evolution, Vol 27, pp 451-458.
Montaldo, H.H. and Meza-Herrera, C.A., (1998). Use of molecular markers and major genes in the genetic improvement of livestock. Journal of Biotechnology. Vol 1, No 2, pp 1-7. Mouchel,N., Tebbutt,S.J., Broackes-Carter,F.C., Sahota,V., Summerfield,T., Gregory,D.J. and
Harris,A. (2001), The sheep genome contributes to localization of control elements in a human gene with complex regulatory mechanisms. Genomics, Vol 76, pp 9–13. Novaes, E., Drost, D.R, Farmerie, W.G., Pappas, G.J., Grattapaglia, D., Sederoff, R.R., and
Kirst, M.(2008). High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics,Vol 9, pp 312.
Petersen, J. L., Mickelson, J. R., Andersson, L. S., Bailey, E., Bannasch, D. L. and and Binns ,M. M. et al., (2011). The Equine Genetic Diversity Consortium: An International Collaboration To Describe Genetic Variation In Modern Horse Breeds. Plant & Animal Genomes XIX Conference, San Diego, CA January 15-19. P 617: Equine Perez-Enciso ,M. and Ferretti, L., (2010). Massive parallel sequencing in animal genetics:
wherefroms and Wheretos. Animal Genetics,Vol 41, pp 561–569.
Pushkarev, D., Neff, N.F. and Quake, S.R., (2009). Single-molecule sequencing of an individual human genome. Nature Biotechnology,Vol 27, 847-852.
Rempel, W.E., Lu, M., El-Kandelgy, S., Kennedy, C.F., Irvin, L.R., Mickelson, J.R. and Louis, C.F., (1993). Relative accuracy of the halothane challenge test and a molecular genetic test in detecting the gene for porcine stress syndrome. Journal of Animal Science, Vol 71, pp 1395-1399.
Rothschild, M. F. and Soller, M. (1997). Candidate gene analysis to detect genes controlling traits of economic importance in domestic livestock. Probe , Vol 8,pp 13–22.
Rothschild,M.F. (2003). From a sow’s ear to a silk purse: real progress in porcine genomics. Cytogenet. Genome Res., 102, 95–99.
Rothschild,M.F., (2004). Porcine genomics delivers new tools and results: this little piggy did more than just go to market. Genetic Research, Vol 83,pp 1–6.
Schild, T.A., Wagner, V. and Geldermann, H., (1994). Variants within the 5’ - flanking regions of bovine milk protein genes: kappa -casein gene. Theoretical and Applied Genetics, Vol 89,pp 116-120.
Shuster, D.E., Kehrli Jr, M.E., Ackermann, M.R. and Gilbert, R.O., (1992). Identification and prevalence of a genetic defect that causes leukocyte adhesion deficiency in Holstein cattle. Proceedings of National Academy of Sciences. USA, Vol 89, pp 9225-9229. Steffanus, D. (2010). Genetics: The new frontier .The equine genome map is as important to
the future of horses as the development of antibiotics was 45 years ago. America’s Horse Daily. March 11.
Tabor, H. K., Risch, N.J. and Myers, R.M., (2002). Candidate-gene approaches for studying complex genetic traits: Practical considerations. Nature Reviews Genetics, Vol 3: 391-397.
Walsh, B. (2000). Minireview: Quantitative genetics in the age of genomics. Theoretical. Population Biology, Vol 59, pp 175-184.
Walters, R., (2011). More commercial benefits on horizon as pig genome project nears completion. Information from pig genome already being used in the industry. Pig International ,Vol 41, No2: pp 16.
Wernersson,R., Schierup,M.H., Jorgensen,F.G., Gorodkin,J., Panitz,F., Staerfeldt,H.H., Christensen,O.F., Mailund,T., Hornshoj,H., Klein,A. et al., (2005). Pigs in sequence space: a 0.66X coverage pig genome survey based on shotgun sequencing. BMC Genomics,Vol 6,pp 70.
Wheeler, D.A., Srinivasan, M., Egholm, M., Shen. Y., Chen, L., McGuire, A., et al .,(2008). The complete genome of an individual by massively parallel DNA sequencing. Nature, Vol 452, pp 872-876.
Wiedmann, R.T., Smith, T.P.L., Nonneman, D.J., (2008). SNP discovery in swine by reduced representation and high throughput pyrosequencing. BMC Geneomics ,Vol 9,pp 81. Usai, M. G., Sechi, T., Salaris, S., Cubeddu, T., Roggio, T., Casu, S. and Carta, A.,(2010).
Analysis of a representative sample of Sarda breed artificial insemination rams with the OvineSNP50 BeadChip. Proceedings ICAR 37th Annual Meeting – Riga, Latvia (31 May - 4 June, 2010), pp 7-10.
VanRaden, P. M., Van Tassell, C.P., Wiggans,G.R., Sonstegard,T.S., Schnabel,R.D., Taylor, J.F. and Schenkel, F.,(2009). Reliability of genomic predictions for North American Holstein bulls. Journal of Dairy Science, Vol 92, pp 16-24.
VanRaden, P., Connell, J. R. O., Wiggans, G. R. and Weigel, K.A.,(2011). Genomic evaluations with many more genotypes. Genetics Selection Evolution,Vol 43, pp1-11. Van Tassell, C. P., Smith, T. P. L., Matukumalli, L. K., Taylor, J. F., Schnabel, R. D., C
Lawley, C.T., Haudenschild,C.D., Moore,S.S., Warren,W.C., and Sonstegard,T.S.,(2008). SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nature Methods, Vol 5, pp247–252. Yahyaoui, M. H., (2003). Genetic polymorphism in goat, Study of the kappa casein,beta
lactoglobulin, and stearoyl coenzyme A desaturase genes. Ph.D thesis submitted to Universidad Autónoma de Barcelona., Bellaterra, 2003.
Yan, H., Kinzler, K.W. and Volgelstein, B., (2000). Genetic testing, present and future. Science, Vol 289, 1890-1892.
Zhang, W. , Sayre, B. L., Xu, X., Tosser, G. , Li, J. and Wan, W., (2011). Goat genome sequencing and its annotation. Plant & Animal Genomes XIX Conference San Diego, CA January 15-19. W135 Cattle/sheep.
ISBN 978-953-51-0564-0 Hard cover, 174 pages
Publisher InTech
Published online 20, April, 2012
Published in print edition April, 2012
InTech Europe
University Campus STeP Ri Slavka Krautzeka 83/A 51000 Rijeka, Croatia Phone: +385 (51) 770 447 Fax: +385 (51) 686 166 www.intechopen.com
InTech China
Unit 405, Office Block, Hotel Equatorial Shanghai No.65, Yan An Road (West), Shanghai, 200040, China Phone: +86-21-62489820
Fax: +86-21-62489821
This book illustrates methods of DNA sequencing and its application in plant, animal and medical sciences. It has two distinct sections. The one includes 2 chapters devoted to the DNA sequencing methods and the second includes 6 chapters focusing on various applications of this technology. The content of the articles presented in the book is guided by the knowledge and experience of the contributing authors. This book is intended to serve as an important resource and review to the researchers in the field of DNA sequencing.
How to reference
In order to correctly reference this scholarly work, feel free to copy and paste the following: