i SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Farmasi
Oleh: Ayesa Syenina NIM : 088114093
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
ii
Persetujuan Pembimbing
VALIDASI METODE KROMATOGRAFI CAIR KINERJA TINGGI (KCKT) FASE TERBALIK PADA PENETAPAN KADAR NIKOTIN
DALAM EKSTRAK ETANOLIK DAUN TEMBAKAU
Skripsi yang diajukan oleh:
Ayesa Syenina
NIM : 088114093
Telah disetujui oleh:
Pembimbing Utama
iv
Complaining does not work as a strategy
-The Last Lecture
Kupersembahkan untuk:
Orangtauku dan kakakku terkasih
vi
LEMBAR PERNYATAAN PERSETUJUAN PUBLIKASI KARYA ILMIAH UNTUK KEPENTINGAN AKADEMIS
Yang bertanda tangan di bawah ini, saya mahasiswa Universitas Sanata Dharma:
Nama : Ayesa Syenina
Nomor Mahasiswa : 088114093
Demi kepentingan ilmu pengetahuan, saya memberikan kepada Perpustakaan Universitas Sanata Dharma karya ilmiah saya yang berjudul:
VALIDASI METODE KROMATOGRAFI CAIR KINERJA TINGGI (KCKT) FASE TERBALIK PADA PENETAPAN KADAR NIKOTIN DALAM EKSTRAK ETANOLIK DAUN TEMBAKAU
Beserta perangkat yang diperlukan (bila ada). Dengan demikian saya memberikan kepada Perpustakaan Universitas Sanata Dharma hak untuk menyimpan, mengalihkan dalam bentuk media lain, mengelolanya dalam bentuk pangkalan data, mendistribusikan secara terbatas, dan mempublikasikannya di internet atau media lain untuk kepentingan akademis tanpa perlu meminta ijin dari saya ataupun memberi royalti kepada saya selama tetap mencantumkan nama saya sebagai penulis.
Demikian pernyataan ini yang saya buat dengan sebenarnya. Dibuat di Yogyakarta
Pada tanggal: 18 Agustus 2011 Yang menyatakan
vii
Kinerja Tinggi (KCKT) Fase Terbalik pada Penetapan Kadar Nikotin dalam Ekstrak Etanolik Daun Tembakau”. Skripsi ini disusun untuk memenuhi salah satu syarat memperoleh gelar Sarjana Farmasi (S.Farm) di Fakultas Farmasi Universitas Sanata Dharma.
Proses penyusunan skripsi ini takkan dapat terselesaikan tanpa adanya bantuan dari berbagai pihak, sehingga pada kesempatan ini penulis juga mengucapkan terima kasih kepada :
1. Ipang Djunarko, M.Sc., Apt. selaku Dekan Fakultas Farmasi Universitas Sanata Dharma.
2. Ibu Christine Patramurti, M.Si., Apt. selaku dosen pembimbing yang telah memberikan bimbingan, nasihat serta dukungan selama penelitian dan penyusunan skripsi.
3. Jeffry Julianus, M.Si., selaku dosen penguji yang telah memberikan kritik dan saran.
4. Yohanes Dwiatmaka, M.Si. selaku dosen penguji yang telah memberikan kritik dan saran.
5. Rini Dwi Astuti, M.Sc., Apt. selaku Kepala Laboratorium Farmasi Universitas Sanata Dharma yang telah memberikan ijin kepada penulis untuk melakukan penelitian di laboratorium.
6. Seluruh staf laboratorium kimia: Mas Bimo, Pak Parlan dan Mas Kunto atas bantuannya selama berlangsungnya penelitian.
7. Staf sekretariat Farmasi: Mas Dwi, Pak Mukimin, dan Mas Narto.
8. Keluarga tercinta, papa, mama, dan Agata yang telah mendukung dan memberikan semangat.
9. Teman satu kelompok skripsi Amel, Dina, Citra, Novi dan Helena.
viii
11.Teman-teman FST yang telah memberi semangat dan dukungan selama penelitian dan penyusunan skripsi.
12.Semua orang yang telah membantu dan mendukung penyusunan skripsi yang tidak dapat disebutkan penulis satu per satu.
Penulis menyadari bahwa karya ini masih belum sempurna, maka dari itu saran dan kritik sangat diharapkan.. Akhir kata penulis meminta maaf apabila ada kesalahan selama penulisan Tugas Akhir ini. Semoga ini dapat bermanfaat untuk semua.
Yogyakarta, 18 Agustus 2011
Ayesa Syenina
ix
HALAMAN PENGESAHAN ... iii
HALAMAN PERSEMBAHAN ... iv
PERNYATAAN KEASLIAN KARYA ... v
PERNYATAAN PERSETUJUAN PUBLIKASI ILMIAH ... vi
PRAKATA ... vii
C. Standarisasi Ekstrak ... 8
1. Aspek parameter spesifik. ... 8
2. Aspek parameter non spesifik. ... 9
x
1. Transisi sigma-sigma star (δ→δ*)... 11
2. Transisi n-sigma star (n→δ*) ... 11
3. Transisi n-phi star (n→π*) ... 11
4. Transisi phi-phi star (π→π*) ... 12
E. Kromatografi Cair Kinerja Tinggi (KCKT) ... 12
1. Definisi ... 12
2. Instrumentasi ... 14
3. Analisis Kualitatif dan Kuantitatif ... 21
F. Parameter Validasi Metode Analisis ... 22
1. Selektifitas atau spesifitas ... 23
2. Linearitas ... 24
3. Akurasi ... 24
4. Presisi ... 25
5. Sensitivitas ... 26
6. Rentang (Range) ... 27
7. Limit of detection (LOD) dan Limit of quantitation (LOQ) ... 27
G. Landasan Teori ... 27
H. Hipotesis ... 28
BAB III. METODOLOGI PENELITIAN ... 29
A. Jenis dan Rancangan Penelitian ... 29
B. Variabel Penelitian ... 29
C. Definisi Operasional... 30
D. Bahan Penelitian... 30
E. Alat Penelitian ... 31
F. Tata Cara Penelitian ... 31
1. Pembuatan Fase Gerak ... 31
2. Pembuatan Larutan Baku Nikotin ... 32
3. Penentuan Panjang Gelombang Pengamatan ... 32
4. Preparasi Sampel ... 32
5. Validasi Metode ... 33
xi
BAB IV. HASIL DAN PEMBAHASAN ... 37
A. Pembuatan Fase Gerak ... 37
B. Pembuatan Larutan Baku Nikotin ... 40
C. Penentuan Panjang Gelombang Pengamatan Nikotin ... 41
D. Preparasi Sampel ... 43
E. Pengamatan Waktu Retensi (tR) Nikotin ... 44
F. Pembuatan Kurva Baku Nikotin ... 48
G. Validasi Metode ... 50
BAB V. KESIMPULAN DAN SARAN ... 58
A. Kesimpulan ... 58
B. Saran ... 58
DAFTAR PUSTAKA ... 59
LAMPIRAN ... 62
xii
DAFTAR TABEL
Tabel I. Karakteristik fisika-kimia dari nikotin ...6
Tabel II. UV cutoff solvent yang digunakan sebagai fase gerak ... 17
Tabel III. Parameter validasi metode untuk tiap jenis prosedur uji ...22
Tabel IV. Rentang kesalahan yang diperbolehkan pada tiap konsentrasi analit... ...25
Tabel V. Kriteria penerimaan presisi berdasar kadar analit ...26
Tabel VI. Hasil persamaan regresi linear baku nikotin ...49
Tabel VII. Hasil perhitungan resolusi (Rs) sampel ...51
Tabel VIII. Hasil persen perolehan kembali (recovery) baku nikotin ...52
Tabel IX. Hasil penetapan recovery baku adisi... 55
xiii
Gambar 3. Senyawa hasil degradasi nikotin ... 6
Gambar 4. Tumbuhan Tembakau……….. 7
Gambar 5. Struktur anabasin, anatabin, dan nornikotin... 8
Gambar 6. Transisi elektronik diantara tingkatan energi dalam suatu molekul. ...11
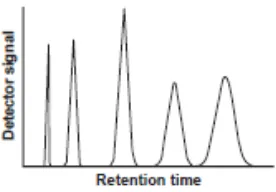
Gambar 7. Suatu kromatogram ...13
Gambar 8. Diagram sistem KCKT... 15
Gambar 9. Skema pompa piston resiprok tunggal ...18
Gambar 10. Skema pompa dual-piston dengan pompa paralel ...19
Gambar 11. Skema desain pompa dual-piston in-series ...19
Gambar 12. Skema loop injector ...20
Gambar 13. Reaksi silika dengan suatu organochlorosilane...21
Gambar 14. Protonasi cincin pirolidin pada nikotin dalam suasana asam ...38
Gambar 15. Gugus kromofor dan auksokrom pada nikotin ...41
Gambar 16. Spektra panjang gelombang maksimum nikotin ...42
Gambar 17. Reaksi penggaraman nikotin oleh asam klorida ... 44
xiv
Gambar 19. Gugus polar dan non polar pada nikotin ... 46
Gambar 20. Interaksi nikotin dengan fase diam oktadesilsilan (C18)... 46
Gambar 21. Interaksi nikotin dengan fase gerak buffer
asetat:metanol:asetonitril (40:54:6)... 47
Gambar 22. Kromatogram baku nikotin dan sampel ekstrak daun
tembakau... 48
Gambar 23. Kurva baku hubungan antara konsentrasi baku nikotin
dengan AUCnya... 50
Gambar 24. Kromatogram baku nikotin, sampel dan sampel adisi... 54
xv
Lampiran 3. Perhitungan pergeseran pKa nikotin dan pH buffer
berdasarkan Kazakevich dan LoBrutto (2007) ...67
Lampiran 4. Spektrum Nikotin ...68
Lampiran 5. Kromatogram seri baku nikotin ...69
Lampiran 6. Perolehan AUC seri baku nikotin... ...77
Lampiran 7. Persamaan dan gambar kurva baku nikotin ...78
Lampiran 8. Kromatogram dan perhitungan resolusi sampel ...79
Lampiran 9. Kromatogram baku nikotin untuk validasi metode ...81
Lampiran 10. Perolehan nilai AUC dan contoh perhitungan konsentrasi terukur baku nikotin ...89
Lampiran 11. Perhitungan persen perolehan kembali (recovery) dan koefisien variasi (KV)…………... 90
Lampiran 12. Kromatogram sampel dan sampel adisi………... 91
Lampiran 13. Perolehan nilai AUC sampel dan sampel adisi, contoh
xvi
dan KV baku nikotin adisi………... 96
Lampiran 14. Kromatogram blanko pelarut buffer
xvii
Nikotin merupakan suatu jenis alkaloid yang ditemukan dalam daun tembakau. Manfaat farmakologis dari nikotin belakangan ini telah banyak diteliti oleh sejumlah ilmuwan, dan beberapa diantaranya menemukan potensi nikotin dalam mengobati berbagai macam penyakit. Penetapan kadar nikotin di dalam ekstrak etanolik daun tembakau dapat dilakukan dengan menggunakan metode kromatografi cair kinerja tinggi (KCKT) fase terbalik. Untuk menjamin hasil yang terpercaya, maka perlu dilakukan validasi metode terlebih dahulu.
Penelitian ini mengikuti jenis dan rancangan penelitian observasional deskripif. Sistem kromatografi cair kinerja tinggi (KCKT) dalam penelitian ini menggunakan kolom fase diam okta desilsilan (C18) dan fase gerak buffer
asetat:metanol:asetonitril (40:54:6) dengan kecepatan alir 1,2 mL/menit. Detektor yang digunakan dalam penelitian ini adalah detektor UV pada panjang gelombang 260 nm.
Parameter validasi metode pada penelitian ini meliputi selektifitas, linearitas, akurasi, presisi dan rentang. Hasil penelitian menunjukkan selektifitas yang baik dengan nilai resolusi (Rs) sebesar 1,531 serta linearitas yang baik dengan koefisien korelasi (r) 0,9996 pada konsentrasi 0,01-0,09 ppm. Perolehan nilai rentang persen recovery dan KV untuk konsentrasi 0,01; 0,05 dan 0,09 ppm secara berurutan adalah 88,56-112,64% dengan KV 11,97%; 99,02-101,78% dengan KV 1,37% serta 100,34-101,62% dengan KV 0,63%, sedangkan nilai recovery adisi baku nikotin yang diperoleh adalah 82,7127-100,6181% dengan KV 9,767%. Metode KCKT fase terbalik ini memenuhi kriteria validasi metode pada rentang konsentrasi 0,05-0,09 ppm.
xviii
METHOD VALIDATION OF REVERSED PHASE HIGH PERFORMANCE LIQUID CHROMATOGRAPHY FOR THE
DETERMINATION OF NICOTINE IN TOBACCO LEAVES ETHANOLIC EXTRACT
ABSTRACT
Nicotine is an alkaloid found in tobacco leaves. Scientists have recently discovered that nicotine may have the pharmacological properties to be used in the treatment of various illnesses. The amount of nicotine in tobacco leaves ethanolic extract can be determined by using reversed phase high performance liquid chromatography (HPLC). To guarantee a reliable result, it is therefore necessary to validate the method.
This research is conducted with a descriptive observational plan and design. The HPLC system used for the quantitative analysis of nicotine consists of octadecylsilane (C18) as the stationary phase and a mixture of acetate buffer:
methanol: acetonitrile (40:54:6) as the mobile phase with a flow rate of 1.2 mL/minute. The detector used in this research is UV detector with the experimental wavelength of 260 nm.
The parameters of method validation used in this research are selectivity, linearity, accuracy, precision, and range. The results of this research indicates that this method is of good selectivity and linearity with a resolution (Rs) of 1.531 and a correlation coefficient (r) of 0.9996 at 0.05-0.09 ppm. The percentage of recovery and coefficient of variation obtained at 0.01; 0.05 and 0.09 ppm were 88.56-112.64% with a CV 11.97%; 99.022-101.778% with a CV 1.37% and 100.338-101.618% with a CV 0.63% respectively, as for the recovery for the standard addition was 82.7127-100.6181% with a CV of 9.767%. Therefore, this reversed phase HPLC method fulfills the criteria of method validation at a range of concentration 0.05-0.09 ppm.
1
A. Latar Belakang
Nikotin merupakan suatu jenis alkaloid alami yang terdapat dalam daun
dan batang tembakau, Nicotiana tabacum dan Nicotiana rustica, dalam kadar
0,5-8%. Sebagian besar masyarakat menganggap bahwa nikotin adalah zat yang
berbahaya dan merugikan kesehatan tubuh serta tidak memiliki manfaat. Hal ini
dikarenakan oleh sifatnya yang toksik dan adiktif (Landoni, 1991), namun
ternyata nikotin juga memiliki manfaat yang cukup besar dalam bidang kesehatan.
Beberapa ilmuwan menemukan bahwa nikotin merangsang pelepasan
neurotransmitter tertentu, diantaranya adalah serotonin, dopamine, dan
norepinephrine, dimana ketidakseimbangan ketiga neurotransmitter ini dapat
menyebabkan terjadinya depresi. Dengan demikian, nikotin berpotensi dalam
mengobati depresi, schizophrenia, penyakit Alzheimer dan penyakit Parkinson
(Anonim, 2006).
Besarnya potensi nikotin memungkinkan dikembangkannya suatu sediaan
farmasi menggunakan ekstrak daun tembakau dengan nikotin sebagai zat aktifnya.
Sebelum dapat digunakan dalam pembuatan sediaan farmasi, ekstrak daun
tembakau sangat penting untuk distandarisasi. Salah satu aspek standarisasi adalah
pemastian kadar senyawa aktif farmakologis melalui analisis kuantitatif metabolit
sekunder yang akan menjamin keseragaman khasiat. Melalui proses standarisasi
dapat digunakan sebagai acuan dalam formulasi sediaan farmasi ekstrak tembakau
(Saifudin dkk., 2011).
Dalam proses standarisasi, diperlukan suatu metode yang tervalidasi
dalam menetapkan kadar nikotin dalam ekstrak tembakau. Pemilihan metode
analisis kuantitatif yang memiliki spesifitas, linearitas, akurasi dan presisi yang
baik merupakan suatu aspek yang sangat penting agar diperoleh hasil yang
acceptable (dapat diterima) pada saat penetapan kadar. Pada penelitian ini peneliti
akan menggunakan metode kromatografi cair kinerja tinggi (KCKT) fase terbalik.
Dasar pemilihan metode kromatografi cair kinerja tinggi (KCKT) fase
terbalik pada penelitian ini adalah karena sampel yang digunakan, yaitu ekstrak
tembakau, tidak hanya mengandung nikotin, namun juga beberapa senyawa
lainnya, sehingga dibutuhkan suatu metode yang dapat memisahkan senyawa
multikomponen sekaligus mengkuantifikasinya. Berdasarkan hasil optimasi yang
telah dilakukan dalam rangkaian penelitian ini, sistem kromatografi cair kinerja
tinggi (KCKT) fase terbalik memberikan kondisi optimal menggunakan fase diam
kolom oktadesilsilan (C18) dan fase gerak buffer asetat:metanol:asetonitril
kinerja tinggi (KCKT) fase terbalik menggunakan fase diam kolom oktadesilsilan
(C18) dan fase gerak buffer asetat:metanol:asetonitril (40:54:6) dengan kecepatan
alir 1,2 mL/menit telah memenuhi parameter validasi: spesifitas, linearitas,
akurasi, presisi dan rentang ?
2. Keaslian karya
Penelitian mengenai nikotin yang telah dilakukan adalah penetapan kadar
nikotin dalam sampel biologis menggunakan metode kromatografi cair kinerja
tinggi (KCKT), kromatografi gas, spektrofotometri massa, dan kromatografi
cair-MS (LC-cair-MS) (Nakajima, Yamamoto, Kuroiwa, Yokoi, 2000); penetapan kadar
nikotin dalam macam-macam merk rokok (Alali, Massadeh, 2002); penetapan
kadar nikotin dalam tembakau dengan metode LC-MS-MS serta penetapan kadar
nikotin dalam sediaan farmasi dengan menggunakan metode kromatografi cair
kinerja tinggi (KCKT) (Vlase, Filip, Mîndruţău, and Leucuţa, 2005). Berdasarkan
studi pustaka di atas, maka belum pernah ada penelitian penetapan kadar nikotin
dalam ekstrak tembakau dengan menggunakan metode kromatografi cair kinerja
3. Manfaat penelitian
a. Manfaat metodologis. Hasil dari penelitian ini diharapkan dapat
memberikan alternatif metode penetapan kadar nikotin dalam ekstrak etanolik
daun tembakau, yaitu menggunakan metode kromatografi cair kinerja tinggi
(KCKT) fase terbalik dengan validitas yang baik.
b. Manfaat praktis. Diharapkan dengan penelitian ini metode
kromatografi cair kinerja tinggi (KCKT) fase terbalik dapat dijadikan metode
alternatif dalam penetapan kadar nikotin dalam ekstrak etanolik daun tembakau.
B. Tujuan Penelitian
Tujuan dari penelitian ini adalah menentukan apakah metode
kromatografi cair kinerja tinggi fase terbalik yang menggunakan fase diam kolom
oktadesilsilan (C18) dan fase gerak buffer asetat:metanol:asetonitril (40:54:6)
dengan kecepatan alir 1,2 mL/menit dalam penetapan kadar nikotin dalam ekstrak
etanolik daun tembakau telah memenuhi parameter validasi: spesifitas, linearitas,
akurasi, presisi dan rentang pada penetapan kadar nikotin dalam ekstrak etanolik
A. Nikotin
Nikotin adalah suatu jenis alkaloid yang paling banyak ditemukan dalam
daun tembakau Nicotiana tabacum dan Nicotiana rustica (Vlase dkk., 2005).
Gambar 1. Struktur molekul nikotin (Anonim, 1999).
Nikotin merupakan amin tersier yang tesusun atas cincin piridin dan
cincin pirolidin yang bersifat toksik dan karsinogenik (Alali dan Massadeh, 2002).
Nikotin dapat ditemukan dalam beberapa bentuk, yaitu sebagai bentuk
diprotonasi, monoprotonasi, dalam basa bebas. Saat berada pada bentuk
terprotonasi, atom nitrogen pada cincin piridin bersifat lebih asam dibandingkan
yang terdapat pada cincin pirolidin (Karbalaie, Ghotbi, Taghikhani dan Yamini,
2009).
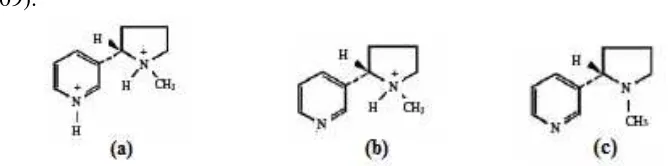
Gambar 2. Nikotin dalam bentuk yang berbeda-beda. a) diprotonasi, b) monoprotonasi, dan c) basa bebas (Karbalaie dkk., 2009).
Nikotin bila terpapar oleh udara dan cahaya dapat mengalami oksidasi
berwarna kecoklatan. Dalam suatu penelitian saat suatu larutan yang mengandung
nikotin dipaparkan pada udara terbentuk senyawa degradasi nikotin yaitu,
methylamine, myosmine dan nicotine-N-1-oxide, sedangankan saat larutan yang
mengandung campuran nikotin dan cotinine dibiarkan terpapar udara selama
beberapa minggu didapatkan jumlah cotinine bertambah (Crooks, 1999).
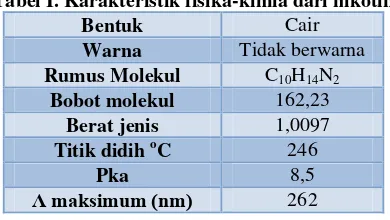
Karakteristik dari nikotin dapat dilihat pada Tabel I (Domino, 1999).
Gambar 3. Senyawa hasil degradasi nikotin. (a)1’S,2’S dan 1’R,2’S nicotine-N-1-oxide (b)
myosmine-N-oxide dan (c) cotinine (Crooks, 1999).
Tabel I. Karakteristik fisika-kimia dari nikotin
Bentuk Cair
Efek negatif nikotin telah lama diketahui masyarakat, namun dewasa ini
perkembangan medis telah ditemukan beberapa manfaat dari nikotin. Nikotin
dapat meningkatkan konsentrasi, proses belajar dan ingatan karena adanya
peningkatan asetilkolin (Karbalaie dkk., 2009). Selain itu, terdapat beberapa
manfaat nikotin yaitu untuk pengobatan: depresi/ kecemasan, dimana nikotin
bekerja pada bagian otak yang mempengaruhi perasaan, termasuk
neurotransmitter seperti serotonin dan dopamine, serta dalam pengobatan penyakit
bahwa nikotin telah mengurangi terjadinya tremor dan bahwa nikotin
kemungkinan berfungsi sebagai neuroprotector bagi pasien (LeChat, 2010).
B. Ekstrak Tembakau
Tembakau (Nicotiana tabacum L.) termasuk dalam famili Solanaceae
yang banyak ditemukan diberbagai daerah di Indonesia seperti di Provinsi
Sumatera Utara, Jawa Barat, Jawa Tengah, D.I. Yogyakarta, Jawa Timur, Bali,
NTB, Lampung dan Sulawesi Selatan. Terdapat dua jenis tembakau yang
dibedakan berdasarkan iklim yaitu tembakau musim kemarau/Voor-OOgst (VO)
dan tembakau musim penghujan/Na-Oogst (NO) (Anonim(a), 2011).
Ekstrak tumbuhan merupakan material yang diperoleh dengan cara
menyari bahan tumbuhan dengan pelarut tertentu. Sehingga, ekstrak tembakau
merupakan material yang berasal dari tumbuhan tembakau yang diperoleh dengan
cara menyari tumbuhan tembakau dengan pelarut yang sesuai (Saifudin, Rahayu
dan Teruna, 2011).
Terdapat beberapa jenis ekstrak yaitu: ekstrak cair, ekstrak kental, dan
ekstrak kering. Ekstrak cair jika hasil ekstraksi masih bisa dituang, biasanya kadar
air lebih dari 30%. Ekstrak kental jika memiliki kadar air antara 5-30%. Ekstrak
kering jika mengandung air kurang dari 5% (Saifudin dkk., 2011).
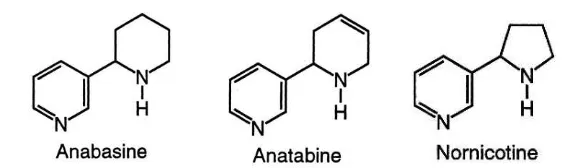
Dalam ekstrak daun tembakau, komponen yang paling banyak terkandung
di dalamnya adalah nikotin. Selain itu terdapat pula beberapa senyawa alkaloid
yang berada dalam jumlah yang lebih sedikit yaitu anabasin, anatabin, dan
nornikotin. Jumlah alkaloid-alkaloid ini bervariasi dalam tiap spesies Nicotiana
(Domino, 1999).
Gambar 5. Struktur anabasin, anatabin, dan nornikotin (Jacob, Hatsukami, Severson, Hall, Yu dan Benowitz, 2002).
C. Standarisasi Ekstrak
Standarisasi merupakan suatu rangkaian proses yang melibatkan
berbagai metode analisis fisik dan mikrobiologi berdasarkan kriteria umum
keamanan (toksikologi) terhadap suatu ekstrak alam (tumbuhan obat). Standarisasi
secara normatif ditujukan untuk memberikan efikasi yang terukur secara
farmakologis dan menjamin keamanan. Terdapat dua aspek dalam standarisasi
ekstrak yaitu aspek parameter spesifik dan non spesifik (Saifudin dkk., 2011).
1. Aspek parameter spesifik.
Aspek ini berfokus pada senyawa atau golongan senyawa dalam ekstrak
Berfokus pada aspek kimia, mikrobiologi dan fisis yang akan
mempengaruhi keamanan konsumen dan stabilitas (Saifudin dkk., 2011).
D. Spektrofotometer UV
Metode spektroskopik dalam analisis didasarkan pada pengukuran radiasi
elektromagnetik yang diserap oleh analit. Radiasi elektromagnetik didefinisikan
sebagai suatu bentuk energi yang ditransmisikan melalui ruang kosong pada
kecepatan yang tinggi dan tidak membutuhkan adanya media untuk transmisinya,
sehingga dapat melewati ruang yang vakum. Radiasi elektromagnetik ini berupa
aliran partikel-partikel kecil atau gelombang energi yang dinyatakan dengan foton
atau quanta. Dalam spektrofotometri UV radiasi, elektromagnetik yang dikenakan
pada analit adalah pada panjang gelombang antara 200-400 nm (Skoog, West,
Holler, 1994).
Energi dari foton bergantung pada frekuensi radiasi yang diberikan oleh :
(1)
Dimana h adalah konstanta Planck (6,63 x 10-34 J s), sehingga hubungan antara
energi radiasi dengan panjang gelombang dan angka gelombang adalah :
(2)
Penyerapan radiasi sinar ultraviolet oleh spesies atom atau molekul (M)
dapat dipertimbangkan sebagai proses dua langkah; yang pertama adalah
melibatkan eksitasi sebagaimana ditunjukkan oleh persamaan berikut :
M + hv → M* (3)
Hasil reaksi antara M dengan foton (hv) merupakan partikel yang tereksitasi
secara elektronik yang disimbolkan dengan M*. Keberadaan M* sangat singkat
dan umumnya diakhiri dengan sebuah relaksasi yang melibatkan konversi energi
eksitasi menjadi panas seperti pada persamaan di bawah :
M*→ M + panas (4)
Penyerapan sinar UV pada umumnya dihasilkan oleh eksitasi
elektron-elektron ikatan. Penyerapan radiasi ultraviolet dibatasi oleh sejumlah gugus
fungsional (yang disebut dengan kromofor) yang mengandung elektron valensi
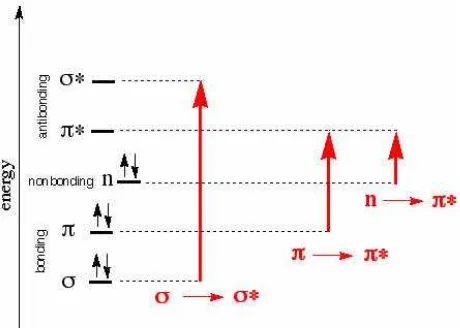
dengan tingkat energi eksitasi yang relatif rendah. Elektron yang terlibat pada
penyerapan radiasi ultraviolet ada tiga yaitu elektron sigma (δ); elektron phi (π);
dan non bonding electron (Gandjar dan Rohman, 2007).
Transisi-transisi elektronik yang terjadi diantara tingkat-tingkat energi di
dalam suatu molekul terdapat empat, yaitu transisi sigma-sigma star (δ→δ*);
Gambar 6. Transisi elektronik diantara tingkatan energi dalam suatu molekul
1. Transisi sigma-sigma star (δ→δ*)
Energi yang dibutuhkan untuk transisi ini besarnya sesuai dengan energi
sinar yang frekuensinya terletak diantara UV vakum (< 180 nm), sehingga jenis
transisi ini kurang bermanfaat untuk analisis dengan cara spektrofotometri UV
(Gandjar dan Rohman, 2007).
2. Transisi n-sigma star (n→δ*)
Jenis transisi ini terjadi pada senyawa organik jenuh yang mengandung
atom-atom yang memiliki elektron bukan ikatan (elektron n). Energi yang
diperlukan untuk transisi jenis ini lebih kecil dibanding transisi δ→δ*, sinar yang
diserap mempunyai panjang gelombang 150-200 nm (Gandjar dan Rohman,
2007).
3. Transisi n-phi star (n→π*)
Jenis transisi ini terjadi pada molekul organik yang mempunyai gugus
fungsional yang tidak jenuh sehingga ikatan rangkap dalam gugus tersebut
memberikan orbital phi yang diperlukan. Transisi ini paling cocok untuk analisis
gelombang ini secara teknis dapat diaplikasikan pada spektrofotometer (Gandjar
dan Rohman, 2007).
4. Transisi phi-phi star (π→π*)
Dalam kebanyakan transisi π→π*, molekul dalam keadaan dasar relatif
non polar, dan keadaan terkesitasinya lebih polar dibandingkan keadaan dasar
(Gandjar dan Rohman, 2007).
E. Kromatografi Cair Kinerja Tinggi (KCKT) 1. Definisi
Kromatografi cair kinerja tinggi (KCKT) atau biasa disebut juga dengan
HPLC (High Performance Liquid Chromatography) merupakan teknik pemisahan
yang diterima secara luas untuk analisis bahan obat, baik dalam bulk atau dalam
sediaan farmasetik, serta dalam cairan biologis. KCKT dapat digunakan baik
untuk analisis kualitatif maupun kuantitatif (Rohman, 2009).
KCKT ini tergolong kromatografi kolom, sebagaimana kromografi
kolom lainnya sampel yang melalui kolom akan mengalami pemisahan
senyawa-senyawa di dalamnya. Jika kekuatan interaksi masing-masing senyawa-senyawa
berbeda-beda maka senyawa-senyawa tersebut akan terpisah menjadi puncak-puncak
tersendiri. Progres dari pemisahan kromatografi ini akan dimonitor oleh suatu
detektor yang sesuai yang terletak pada ujung kolom. Hasil yang diperoleh akan
berbentuk suatu kromatogram yang terdiri atas puncak untuk masing-masing
Gambar 7. Suatu kromatogram
Pemisahan senyawa dalam KCKT diatur oleh distribusi senyawa dalam
fase gerak dan fase diam. Penggunaan kromatografi cair secara sukses terhadap
suatu masalah yang dihadapi membutuhkan penggabungan secara tepat dari
berbagai macam kondisi operasional seperti jenis kolom, fase gerak, panjang dan
diameter kolom, kecepatan alir fase gerak, suhu kolom, dan ukuran sampel
(Gandjar dan Rohman, 2007).
Teori mengenai KCKT dapat dibagi menjadi 2 aspek yaitu aspek kinetik
dan aspek termodinamik. Aspek kinetik dari KCKT bertanggung jawab atas
pelebaran kromatogram, sedangkan aspek termodinamik mempengaruhi waktu
retensi analit dalam kolom. Bila dilihat dari segi analisis, faktor kinetik
mempengaruhi lebar dari puncak kromatogram (efisiensi) dan faktor
termodinamik akan memperngaruhi letak puncak pada kromatogram
(selektivitas). Sedangkan dilihat dari segi praktis, efisiensi pemisahan pada KCKT
lebih berkaitan dengan optimasi instrumen, dimensi kolom dan geometri partikel.
Selektivitas lebih berkaitan dengan interaksi intermolekuler dan dipengaruhi oleh
tipe eluen, komposisi, temperatur dan variabel lainnya yang memungkinkan
Tipe KCKT yang paling banyak digunakan adalah kromatografi partisi,
dimana fase diamnya berupa cairan yang tidak bercampur (immiscible) dengan
cairan fase gerak. Bentuk awal dari kromatografi partisi yang digunakan adalah
kolom liquid-liquid, namun kolom tersebut sekarang telah digantikan dengan
kolom liquid-bonded-phase. Pada kromatografi liquid-liquid cairan ditahan pada
tempatnya dengan adsorpsi fisik, sedangkan pada kromatografi
liquid-bonded-phase fase diam cair ditahan dengan ikatan secara kimia sehingga menghasilkan
packing kolom yang jauh lebih stabil (Skoog dkk., 1998).
Model partisi dapat digunakan dalam menjelaskan mekanisme retensi.
Dalam model partisi, diumpamakan adanya dua fase yang berbeda (fase gerak dan
fase diam) dan terjadi ekuilibrium seketika dari analit yang terpartisi antara kedua
fase tersebut. Koefisien partisi dari analit dinyatakan sebagai :
(5)
dimana faktor retensi dari analit dinyatakan sebagai rasio jumlah analit pada fase
diam terhadap jumlah analit pada fase gerak (Kazakevich dan LoBrutto, 2007).
2. Instrumentasi
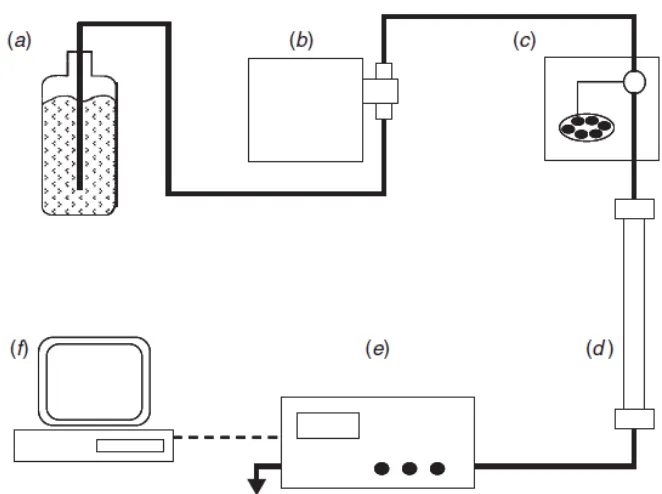
Instrumentasi KCKT pada dasarnya terdiri atas: wadah fase gerak,
pompa, alat untuk memasukkan sampel (tempat injeksi), kolom, detektor, wadah
penampung buangan fase gerak, dan suatu komputer atau integrator atau perekam
Gambar 8. Diagram sistem KCKT. (a) wadah fase gerak; (b) pompa; (c) autosampler atau injector; (d) kolom; (e) detector; (f) sistem pendataan (Snyder, Kirkland dan Dolan, 2010).
a. Wadah Fase gerak. Alat KCKT yang baru dilengkapi dengan satu atau
lebih wadah gelas, yang mengandung 500mL atau lebih fase gerak. Wadah fase
gerak harus bersih dan lembam (inert) (Gandjar dan Rohman, 2007). Degassing
(penghilangan gas) biasanya dilakukan terlebih dahulu pada fase gerak untuk
menghilangkan gas yang mungkin terdapat di dalamnya. Adanya gas dapat
menyebabkan flow rate yang tidak reprodusibel serta dapat mengganggu detektor
(Skoog, Holler dan Crouch, 1998).
Pada saat membuat pelarut untuk fase gerak, maka sangat dianjurkan
untuk menggunakan pelarut, buffer, dan reagen dengan kemurnian yang sangat
tinggi, dan lebih terpilih lagi jika pelarut-pelarut yang digunakan untuk KCKT
berderajat KCKT (HPLC grade). Adanya pengotor dalam reagen dapat
dapat terkumpul dalam kolom atau dalam tabung yang sempit, sehingga dapat
mengakibatkan suatu kekosongan pada kolom atau tabung tersebut. Oleh karena
itu, fase gerak harus disaring terlebih dahulu sebelum digunakan pada KCKT
(Gandjar dan Rohman, 2007).
b. Fase gerak pada KCKT. Fase gerak atau eluen biasanya terdiri atas
campuran pelarut yang dapat bercampur yang secara keseluruhan berperan dalam
daya elusi dan resolusi (Gandjar dan Rohman, 2007). Terdapat dua jenis elusi
yaitu elusi isokratik dimana komposisi dari fase gerak konstan selama proses
elusi, dan elusi gradient dimana komposisi fase gerak dapat diubah-ubah selama
proses elusi (Kar, 2005).
Fase gerak yang biasanya digunakan dalam KCKT fase terbalik adalah
campuran hidro organik. Senyawa organik yang umumnya digunakan adalah
metanol dan asetonitril atau campuran keduanya. Senyawa-senyawa lainnya yang
dapat digunakan dalam fase gerak untuk penyesuaian selektivitas adalah
tetrahidrofuran, IPA, dan DMSO (Kazakevich dan LoBrutto, 2007).
Konsentrasi dari larutan organik dalam fase gerak merupakan faktor
dominan yang mempengaruhi retensi analit dalam sistem KCKT. Pertimbangan
dalam memilih solven fase gerak meliputi kompatibilitas antar solven, kelarutan
sampel dalam eluen, polaritas, transmisi cahaya, viskositas, stabilitas dan pH.
Solven yang digunakan sebagai fase gerak harus dapat bercampur serta tidak
menimbulkan presipitasi saat dicampur. Sampel harus dapat terlarut dalam fase
gerak karena apabila tidak, maka dapat terjadi presipitasi di dalam kolom.
yang sering digunakan. Solven yang terlalu kental dapat menyebabkan bentuk
puncak yang melebar (Kazakevich dan LoBrutto, 2007).
Tabel II. UV cutoffsolvent yang digunakan sebagai fase gerak (Kazakevich dan LoBrutto, 2007).
Terkadang dalam fase gerak juga ditambahkan buffer. Buffer umumnya
digunakan dalam fase gerak untuk mengkontrol selektivitas dan resolusi analit,
saat pH dari fase gerak ≈ pKa analit (analit berada pada bentuk 50% terionisasi)
maka perubahan pada nilai pH akan memberikan perubahan waktu retensi dan
pemisahan yang maksimum. Namun hal ini hanya berlaku pada perubahan pH 1
unit dari nilai pKa analit, diluar rentang tersebut analit akan berada pada bentuk
terionisasi atau pada bentuk molekul (tidak terionisasi) dan waktu retensinya tidak
akan bergantung dengan adanya perubahan pH (Snyder dkk., 2010).
c. Pompa. Pompa yang digunakan dalam KCKT dalam pompa yang
memenuhi syarat wadah pelarut, yakni : pompa harus inert terhadap fase gerak.
dan mampu mengalirkan fase gerak dengan kecepatan alir 3 mL/menit (Rohman,
2009).
Pompa KCKT dapat diklasifikasikan berdasarkan rentang kecepatan
alir, mekanisme kerjanya atau berdasarkan metode pencampurannya. Pompa yang
biasa digunakan dalam analisis umumnya memilki rentang kecepatan alir
0,001-10 mL/menit. Kebanyakan pompa menggunakan mekanisme resiprok. Sedangkan
berdasarkan metode pencampurannya biasanya menggunakan kondisi
pencampuran tekanan rendah atau tekanan tinggi (Ahuja dan Dong, 2005).
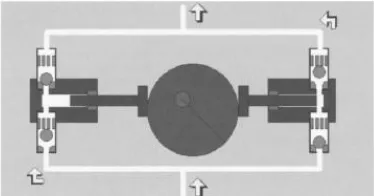
Gambar 9. Skema pompa piston resiprok tunggal
Kebanyakan pompa KCKT menggunakan desain piston resiprok seperti Gambar 9
diatas. Pada gambar dapat dilihat terdapat cam bermotor yang dapat menjalankan
piston secara depan ke belakang untuk mengalirkan solven melalui suatu vulva
inlet dan outlet. Sedangkan Gambar 10 merupakan pompa yang menggunakan
piston ganda dimana terdapat satu motor yang menjalankan dua piston pada
pompa yang berbeda. Hasil yang diperoleh pada pompa model ini lebih stabil
Gambar 10. Skema pompa dual-piston dengan pompa paralel.
Dalam perkembangannya, model pompa resiprok disempurnakan dengan
berbagai macam modifikasi seperti pada desain dual piston in-series yang kini
banyak digunakan, dimana pada model ini, dua piston dijalankan oleh motor yang
terpisah. Terdapat juga pre-piston yang disinkronisasi dengan piston sekunder
untuk memberikan aliran yang lebih halus dan komposisi yang lebih akurat.
Model pompa ini dapat dilihat pada gambar di bawah ini (Ahuja dan Dong 2005).
Gambar 11. Skema desain pompa dual-piston in-series
d. Tempat penyuntikan sampel. Sampel-sampel cair dan larutan
disuntikkan secara langsung ke dalam fase gerak yang mengalir di bawah tekanan
menuju kolom. (Gandjar dan Rohman, 2007).
Pada KCKT sampel-sampel cair dan larutan disuntikkan secara langsung
ke dalam fase gerak yang mengalir di bawah tekanan menuju kolom
menggunakan alat penyuntik yang terbuat dari tembaga tahan karat dan katup
loop ini tersedia untuk volume antara 0,5µL-2mL. Pada saat posisi mengisi, loop
untuk sampling terisolasi dari fase gerak dan terbuka terhadap atmosfer. Suatu
syringe digunakan untuk mengambil sampel dan memasukannya ke dalam loop.
Apabila terdapat sampel yang lebih maka akan dikeluarkan melalui pembuangan.
Setelah sampel terisi ke dalam loop, injektor akan menghadap posisi injek. Pada
posisi ini fase gerak diarahkan melalui loop untuk sampling dan sampel akan
dibawa melewati kolom (Harvey, 2000).
Gambar 12. Skema loop injector. (a) posisi mengisi, (b) posisi injek
e. Kolom. Kolom merupakan bagian KCKT yang terdapat fase diam di
dalamnya. Fase diam pada KCKT berupa lapisan film cair yang terikat pada basis
partikel silika. Tujuan terikatnya lapisan film ini adalah untuk mencegah
kemungkinan terjadinya kebocoran cairan fase diam dari dalam kolom. Lapisan
film cair ini terikat pada partikel silika melalui ikatan kovalen. Partikel silika
direaksikan dengan organochlorosilane Si(CH3)2RCl, dimana R merupakan suatu
alkil atau gugus alkil tersubstitusi. Kepolaran dari fase diam bergantung pada jenis
R, apabila R merupakan suatu gugus fungsi yang bersifat polar, maka fase diam
juga akan bersifat polar, sebaliknya fase diam akan bersifat non polar apabila R
Oktadesilsilan (ODS atau C18) yang merupakan fase diam dimana R pada
organochlorosilane berupa hidrokarbon n-octyldecyl paling banyak digunakan
karena mampu memisahkan senyawa-senyawa dengan kepolaran yang rendah,
sedang, maupun tinggi (Rohman, 2009).
f. Detektor. Suatu detektor harus memiliki karakteristik memiliki respon
terhadap solut yang cepat dan reprodusibel; sensitifitas tinggi; stabil; mempunyai
volume sel yang kecil sehingga mampu meminimalkan pelebaran pita; sinyal yang
dihasilkan berbanding lurus dengan konsentrasi solut; tidak peka terhadap
perubahan suhu dan kecepatan alir fase gerak (Rohman, 2009). Salah satu contoh
detektor yang sering digunakan adalah detektor UV-Vis. Detektor ini didasarkan
pada adanya peneyerapan radiasi ultraviolet (UV) dan sinar tampak (Vis) pada
kisaran panjang gelombang 190-800 nm oleh spesies solut yang mempunyai
struktur-struktur atau gugus-gugus kromoforik. Sel detektor umumnya berupa
tabung dengan diameter 1 mm dan panjang celah optiknya 10 mm, serta diatur
sedemikian rupa sehingga mampu menghilangkan pengaruh indeks bias yang
dapat mengubah absorbansi yang terukur (Kar, 2005).
3. Analisis Kualitatif dan Kuantitatif
Analisis kualitatif KCKT berupa pengamatan waktu retensi (tR) senyawa
secara berurutan dalam kondisi alat yang stabil dengan perbedaan waktu
pengoperasian anatar keduanya sekecil mungkin (Gandjar dan Rohman, 2007).
Untuk KCKT kuantifikasi dapat dilakukan dengan mengukur tinggi
puncak atau dengan luas puncak. Tinggi puncak diukur sebagai jarak dari garis
dasar ke puncak maksimum. Sedangkan luas puncak diukur sebagai hasil kali
tinggi puncak dan lebar pada setengah tinggi (W1/2) (Gandjar dan Rohman, 2007).
F. Parameter Validasi Metode Analisis
Validasi merupakan suatu proses dokumentasi atau membuktikan bahwa
metode analisis menghasilkan data analitik yang dapat diterima untuk tujuan
penggunaannya. Langkah awal dalam perkembangan suatu metode dan
validasinya adalah menentukan standar minimum yang merupakan spesifikasi dari
metode untuk tujuan yang ingin dicapai (Christian, 2004).
Parameter analitik yang diperlukan untuk validasi dapat bervariasi
bergantung pada tipe prosedur analitik seperti terlihat pada Tabel III di bawah ini :
Tabel III. Parameter validasi metode untuk tiap jenis prosedur uji (Harmita, 2004)
Parameter
bahan baku atau komponen sisa pada suatu sampel. Sedangkan kategori II adalah
metode analitik yang menentukan performa karakteristik (contoh: disolusi,
pelepasan obat) (Harmita, 2004).
Proses validasi biasanya meliputi pengujian parameter-parameter
selektivitas, linearitas, akurasi, presisi, sensitivitas, rentang, limit of detection
(LOD) dan limit of quantification (LOQ).
1. Selektifitas atau spesifitas
Selektifitas atau spesifitas merupakan kemampuan dari metode untuk
mendeteksi dan menganalisa analit dalam sebuah matriks tanpa gangguan dari
komponen lain yang berada dalam matriks tersebut (Ahuja dan Rasmussen, 2007).
Untuk deteksi yang spesifik, dimanfaatkan karakteristik unik dari analit, misalnya
spektrum analit (panjang gelombang UV yang spesifik, fluoresens), massa
molekul, fragmentasi molekul. Spesifitas juga dapat diperoleh melalui preparasi
sampel, contohnya dengan derivatisasi, ekstraksi, presipitasi, adsorpsi, dan lain
sebagainya (Ermer dan Miller, 2005).
Spesifitas dapat ditentukan melalui perhitungan resolusi dengan rumus :
Rs = (6)
Dimana:
t2 = waktu retensi puncak kedua t1 = waktu retensi puncak pertama
W0,5(1) = lebar puncak pertama pada setengah tinggi puncak
Nilai Rs harus mendekati atau lebih dari 1,5 karena akan memberikan
pemisahan puncak yang baik (base line resolution) (Snyder dkk., 2010).
2. Linearitas
Linearitas adalah kemampuan metode analisis yang memberikan respon
yang secara langsung atau dengan bantuan transformasi matematik yang baik,
proporsional terhadap konsentrasi analit dalam sampel (Harmita, 2004).
Linearitas dapat dilihat melalui kurva kalibrasi yang menunjukkan
hubungan antara respon dengan konsentrasi analit pada beberapa seri baku. Dari
kurva kalibrasi ini kemudian akan ditemukan regresi linearnya yang berupa
persamaan y=bx+a, dimana x=konsentrasi; y=respon, a=intersep y yang
sebenarnya dan b=slope yang sebenarnya. Tujuan dari dibuatnya regresi ini adalah
untuk menentukan estimasi terbaik untuk slope dan intersep y sehingga akan
mengurangi residual error, yaitu perbedaan nilai hasil percobaan dengan nilai
yang diprediksi melalui persamaan regresi linear (Harvey, 2000).
Sebagai parameter adanya hubungan linear digunakan koefisien korelasi
r pada analisis regresi linear. Hubungan linear yang ideal dicapai jika nilai b=0
dan r=+1 atau -1 tergantung arah garis (Harmita, 2004).
3. Akurasi
Akurasi sebuah metode analisis mencermikan kedekatan nilai atau harga
dari yang diperoleh saat penelitian dengan yang sebenarnya (true value). Akurasi
ditentukan dengan % recovery. Biasanya dilakukan terhadap minimal tiga
kimia) ditambahkan ke dalam campuran bahan pembawa sediaan farmasi
(plasebo) lalu campuran tersebut dianalisis dan hasilnya dibandingkan dengan
kadar analit yang ditambahkan (kadar yang sebenarnya). Dalam metode
panambahan baku, sampel dianalisis lalu sejumlah tertentu analit yang diperiksa
ditambahkan ke dalam sampel dicampur dan dianalisis lagi. Selisih kedua hasil
dibandingkan dengan kadar yang sebenarnya (hasil yang diharapkan). Dalam
kedua metode tersebut, persen peroleh kembali dinyatakan sebagai rasio antara
hasil yang diperoleh dengan hasil yang sebenarnya. Tabel IV menunjukkan
rentang kesalahan yang diperolehkan pada setiap konsentrasi analit pada matriks
(Harmita, 2004).
Tabel IV. Rentang kesalahan yang diperbolehkan pada tiap konsentrasi analit
Analit pada matrik sampel, % Rata-rata yang diperoleh, %
Presisi adalah ukuran yang menunjukkan derajat kesesuaian antara hasil
beberapa seri pengujian yang diperoleh dari sampel-sampel yang diambil dari
diterima berdasarkan kadar analit tertera dalam tabel di bawah ini (Huber, 2007):
Tabel V. Kriteria penerimaan presisi berdasar kadar analit
Analit (%) %RSD
Presisi diukur sebagai simpangan baku atau simpangan baku relatif
(koefisien variasi). Simpangan baku dalam presisi merupakan parameter yang
penting dalam mendeskripsikan lebarnya distribusi normal, misalnya derajat
persebaran data (Ermer dan Miller, 2005).
Presisi dapat dinyakatan sebagai keterulangan (repeatability) atau
keterulangan (reproducibility). Keterulangan adalah keseksamaan metode jika
dilakukan berulang kali oleh analis yang sama pada kondisi sama dan dalam
interval waktu yang pendek. Sedangkan ketertiruan adalah keseksamaan metode
jika dikerjakan pada kondisi yang berbeda. Kriteria seksama diberikan jika
metode memberikan simpangan baku relatif atau koefisien variasi 2% atau
kurang. Akan tetapi kriteria ini sangat fleksibel tergantung pada konsentrasi analit
yang diperiksa, jumlah sampel, dan kondisi laboratorium (Harmita, 2004).
5. Sensitivitas
Sensitivitas merupakan kemampuan suatu metode analisis untuk
membedakan dua konsentrasi yang berbeda dan ditentukan melalui slope dari
linearitas yang dapat diterima (Harmita, 2004).
7. Limit of detection (LOD) dan Limit of quantitation (LOQ)
LOD adalah jumlah terkecil analit dalam sampel yang dapat dideteksi
yang masih memberikan respon signifikan dibandingkan dengan blangko.
(Harmita, 2004). LOQ merupakan parameter pada analisis renik dan diartikan
sebagai kuantitas terkecil analit dalam sampel yang masih dapat memenuhi
criteria akurasi dan presisi (Harmita, 2004).
G. Landasan Teori
Nikotin merupakan suatu jenis alkaloid di dalam tumbuhan tembakau
(Nicotiana tabacum) yang memiliki cincin piridin dan pirolidin. Nikotin
berpotensi dalam mengobati penyakit seperti depresi dan penyakit Parkinson,
sehingga ekstrak tembakau yang mengandung nikotin dapat dikembangkan
menjadi suatu sediaan farmasi, oleh karena itu diperlukan standarisasi aspek
spesifik dari ekstrak daun tembakau untuk menjamin keseragaman kadar nikotin
di dalamnya.
Dalam proses standarisasi aspek spesifik ekstrak daun tembakau,
dibutuhkan suatu metode analisis yang memiliki sensitivitas dan selektivitas yang
tinggi untuk menetapkan kadar nikotin. Salah satu metode yang dapat digunakan
detektor UV. Nikotin memiliki kromofor yang dapat menyerap radiasi
elektromagnetik sehingga dapat ditetapkan kadarnya dengan detektor UV. Hasil
kerja yang maksimum dapat diperoleh pada kondisi optimum metode. Kondisi
optimum dari metode ini ditemukan pada penggunaan fase diam kolom
oktadesilsilan (C18), fase gerak buffer asetat, metanol, dan asetonitril dengan
perbandingan 40:54:6 dan laju alir 1,2 mL/menit.
Metode KCKT yang telah optimum harus divalidasi agar hasil yang
diperoleh dapat dipertanggungjawabkan serta memberikan jaminan bahwa metode
telah memenuhi persyaratan analisis. Parameter validasi metode meliputi akurasi,
yang ditentukan dengan persen perolehan kembali; presisi, yang dinyatakan dalam
Coefficient of Variation (CV); linearitas, yang ditentukan dengan nilai koefisien
korelasi (r) dan spesifitas, yang ditentukan dengan nilai resolusi.
H. Hipotesis
Metode kromatografi cair kinerja tinggi (KCKT) fase terbalik
menggunakan fase diam kolom oktadesilsilan (C18) dan fase gerak buffer
asetat:metanol:asetonitril (40:54:6) dengan kecepatan alir 1,2 mL/menit pada
penetapan kadar nikotin dalam ekstrak etanolik daun tembakau memenuhi
29
A. Jenis dan Rancangan Penelitian
Penelitian ini mengikuti jenis penelitian non eksperimental, karena tidak
dilakukan perlakuan atau manipulasi pada subjek uji yang digunakan dan
rancangan deskriptif karena hanya menggambarkan data yang diperoleh.
B. Variabel Penelitian
1. Variabel bebas dalam penelitian ini adalah sistem kromatografi cair kinerja
tinggi (KCKT) dengan fase diam C18 dan fase gerak buffer
asetat:metanol:asetonitril (40:54:6) pada kecepatan alir 1,2 mL/menit.
2. Variabel tergantung pada penelitian ini adalah parameter validitas yaitu
selektifitas, linearitas, akurasi, presisi dan rentang.
3. Variabel pengacau terkendali dalam penelitian ini adalah:
a. pH pelarut dan fase gerak yang dikendalikan dengan menggunakan buffer
pada pH 4.
b. Larutan baku yang bersifat mudah teroksidasi oleh udara dan cahaya
diatasi dengan menggunaan alumium foil untuk menutupi alat-alat gelas.
C. Definisi Operasional
1. Penelitian yang dilakukan termasuk dalam validasi metode kategori I, yaitu
metode yang digunakan untuk analisis kualitatif dan kuantitatif komponen
utama dalam suatu matriks. Validasi metode yang dilakukan meliputi
pengukuran terhadap parameter validasi yaitu selektifitas, linearitas, akurasi
dan presisi.
2. Sistem kromatografi cair kinerja tinggi (KCKT) yang digunakan dalam
penelitian ini menggunakan kolom fase diam okta desilsilan (C18) serta
komposisi fase gerak buffer asetat:metanol:asetonitril (40:54:6) dengan
kecepatan alir 1,2 mL/menit.
3. Kadar nikotin dinyatakan dalam satuan ppm (parts per million).
D. Bahan Penelitian
Bahan yang digunakan adalah baku nikotin (E.Merck), acetronitrile
(E.Merck), metanol (E.Merck), etanol (teknis), ammonium asetat (E.Merck),
natrium asetat (E.Merck), asam asetat glasial (E.Merck), asam klorida (teknis),
natrium hidroksida 5M (E.Merck), kloroform (E.Merck), aquades dan aquabides.
Semua bahan kimia yang digunakan memiliki grade pro analysis (p.a) kecuali
dinyatakan lain, sedangkan sampel yang diteliti berupa ekstrak etanolik daun
Shimadzu LC-10 AD No.C20293309457 J2) dengan sistem elusi gradien; detektor
UV-Vis (merek Shimadzu SPD 10 AV No.C20343502697 KG), kolom C-18
merek Bondapack C-18 dengan panjang kolom 25 cm No.P61271BO2,
sperangkat computer (merek Dell Vostro 220, printer merek HP D2566), alat
degassing ultrasonic (Retsch tipe T640 No.935922013), membran filter Whatman
ukuran pori 0,45 µm dan diameter 47 mm, neraca analitik merek Ohaus,
Millipore, mikropipet, indikator pH, seperangkat alat gelas.
F. Tata Cara Penelitian 1. Pembuatan Fase Gerak
a. Pembuatan buffer asetat pH 4. Ditimbang kurang lebih seksama
0,1683 g ammonium asetat p.a, 0,5599 g natrium asetat dan diambil 0,406 mL
asam asetat glasial. Ketiga zat tersebut dimasukkan ke dalam labu takar 250,0 mL
kemudian dilarutkan dengan aquabides hingga batas tanda. (Larutan buffer ini
harus selalu dibuat baru untuk mencegah tumbuhnya mikroorganisme).
b. Pembuatan fase gerak buffer asetat:metanol:asetonitril (40:54:6). Fase
gerak yang digunakan terdiri dari campuran buffer asetat, metanol, dan asetonitril
dengan perbandingan 40:54:6. Masing-masing larutan fase gerak disaring dengan
menggunakan kertas saring Whatman dengan bantuan pompa vakum. Selanjutnya
2. Pembuatan Larutan Baku Nikotin
a. Pembuatan larutan stok baku nikotin. Dibuat larutan stok baku nikotin
konsentrasi 2 ppm dengan cara mengambil sebanyak 10 µL baku nikotin,
dimasukkan ke dalam labu takar 5,0 mL, kemudian diencerkan dengan fase gerak
yang digunakan hingga tanda.
b. Pembuatan seri larutan baku nikotin. Dibuat seri larutan baku dengan
konsentrasi 0,01; 0,03; 0,05; 0,07 dan 0,09 ppm dengan cara mengambil sebanyak
25; 75; 125; 175 dan 225 µL dari larutan stok baku nikotin, dimasukkan labu takar
5,0 mL dan diencerkan dengan fase gerak yang digunakan hingga tanda.
3. Penentuan Panjang Gelombang Pengamatan
Dibuat seri larutan baku nikotin dengan konsentrasi 0,005; 0,007 dan
0,009 ppm. Masing-masing konsentrasi dibaca absorbansinya pada rentang
panjang gelombang 225-325 nm. Panjang gelombang pengamatan yang
digunakan adalah panjang gelombang yang memberikan serapan terbesar yang
sama pada 3 seri konsentrasi larutan baku nikotin.
4. Preparasi Sampel
Sebanyak 1,0 g ekstrak kental daun tembakau dilarutkan dalam 10 mL
asam klorida encer dengan bantuan ultrasonikator selama 30 menit hingga
semuanya larut. Selanjutnya larutan ditambah dengan 10 mL kloroform dan
diekstraksi selama 5 menit hingga terbentuk dua lapisan, lapisan kloroform
kemudian dibuang. Lapisan berair kemudian ditetesi dengan natrium hidroksida
4M hingga pH larutan mencapai 11-12, setelah itu ditambahkan 10 mL kloroform.
a. Penentuan resolusi sampel. Sebanyak 20 µL larutan fraksi kloroform
ekstrak tembakau yang telah disaring dengan millipore dan diawaudarakan selama
15 menit diinjeksikan pada sistem KCKT fase terbalik dengan fase diam C18 dan
fase gerak buffer asetat:metanol:asetonitril (40:54:6) pada kecepatan alir 1,2
mL/menit. Dilakukan repetisi tiga kali. Resolusi dihitung dengan memasukkan
selisih waktu retensi dan lebar setengah tinggi peak ke dalam rumus perhitungan
resolusi.
b. Pembuatan kurva baku dan penentuan linearitas. Dibuat seri larutan
baku dengan konsentrasi 0,01; 0,03; 0,05; 0,07 dan 0,09 ppm, masing-masing
larutan disaring dengan menggunakan millipore kemudian diawaudarakan 15
menit dan 20 µL dari masing-masing larutan diinjeksikan pada sistem KCKT fase
terbalik dengan fase diam C18 dan fase gerak buffer asetat:metanol:asetonitril
(40:54:6) pada kecepatan alir 1,2 mL/menit. Dari kromatogram akan diperoleh
luas area nikotin untuk masing-masing konsentrasi. Luas area ini kemudian
diplotkan terhadap konsentrasi nikotin untuk memperoleh regresi linear dengan
persamaan y = bx + a dan nilai koefisien korelasi (r) yang akan digunakan untuk
menentukan parameter validasi linearitas.
c. Penentuan persen perolehan kembali (recovery) dan penentuan
koefisien variasi baku nikotin. Sebanyak 20 µL larutan baku nikotin konsentrasi
selama 15 menit diinjeksikan pada sistem KCKT fase terbalik dengan fase diam
C18 dan fase gerak buffer asetat:metanol:asetonitril (40:54:6) pada kecepatan alir
1,2 mL/menit. Dilakukan replikasi sebanyak lima kali. Konsentrasi nikotin
diperoleh dengan cara memasukkan AUC yang diperoleh ke dalam persamaan
kurva baku. Kemudian dihitung persen recovery, Standard Deviation (SD) dan
Koefisien Variasi (KV).
d. Penentuan persen kembali (recovery) dan penentuan koefisien variasi
adisi baku nikotin dalam sampel. Dibuat dua macam larutan yaitu larutan sampel
dan larutan sampel adisi. Larutan sampel dibuat dengan cara mengambil 500 µL
ekstrak sampel ke dalam labu takar 5,0 mL dan diencerkan hingga tanda dengan
fase gerak yang digunakan. Larutan sampel adisi dibuat dengan cara mengambil
500 µL ekstrak tembakau dan 150 µL larutan stok baku nikotin ke dalam labu
takar 5,0 mL dan diencerkan dengan fase gerak yang digunakan hingga tanda.
Kedua larutan disaring dengan millipore dan diawaudarakan selama 15 menit,
kemudian diinjeksikan sebanyak 20 µL pada sistem KCKT fase terbalik dengan
fase diam C18 dan fase gerak buffer asetat:metanol:asetonitril (40:54:6) pada
kecepatan alir 1,2 mL/menit. Dilakukan replikasi sebanyak lima kali. Kadar baku
nikotin yang ditambahkan dalam sampel merupakan selisih nilai kadar sampel
yang dihasilkan oleh sampel isolat kloroform ekstrak daun tembakau. Menurut
Snyder dkk. (2010), syarat resolusi yang baik yaitu dimana senyawa analit
terpisah dari senyawa-senyawa yang lain adalah ≥ 1,5. Resolusi dihitung dengan
rumus :
Rs = (7)
Dimana : Rs = resolusi
t2= waktu retensi puncak kedua t1 = waktu retensi puncak pertama
W0,5(1) = lebar setengah tinggi puncak pertama
W0,5(2) = lebar setengah tinggi puncak kedua.
2. Linearitas dan Rentang
Linearitas ditentukan dengan nilai koefisien korelasi (r), yang diperoleh
dari AUC baku nikotin yang diplotkan terhadap konsentrasi baku. Nilai r yang
dipersyaratkan adalah ≥ 0,999. Sedangkan rentang diperoleh dari konsentrasi
terendah hingga tertinggi baku nikotin yang memberikan akurasi, presisi dan
linearitas yang baik.
3. Akurasi
Akurasi ditentukan dengan persen perolehan kembali (recovery), yang
dapat dihitung dengan rumus:
4. Presisi
Presisi dinyatakan dalam Koefisien Variasi (KV), yang dapat dihitung
dengan rumus :
KV =
(9)
37
Penelitian ini bertujuan untuk menentukan apakah penetapan kadar
nikotin dalam ekstrak daun tembakau dengan menggunakan metode kromatografi
cair kinerja tinggi fase terbalik dapat memenuhi persyaratan validasi yang berlaku.
Dasar pemilihan metode KCKT pada penelitian ini adalah karena analisis nikotin
dalam ekstrak daun tembakau termasuk analisis senyawa multikomponen.
Dikatakan demikian karena pada ekstrak daun tembakau tidak hanya terdapat
nikotin, melainkan senyawa-senyawa lainnya. Oleh karena itu dibutuhkan suatu
metode yang dapat memisahkan nikotin dari senyawa-senyawa lain dalam ekstrak
daun tembakau dan pada saat yang bersamaan dapat mengkuantifikasinya. KCKT
merupakan metode yang sesuai untuk kepentingan analisis ini karena KCKT dapat
memisahkan senyawa multikomponen sekaligus mengkuantifikasinya.
A. Pembuatan Fase Gerak
Pada penelitian ini, fase gerak yang digunakan berupa campuran buffer
asetat, metanol dan asetonitril dengan perbandingan 40:54:6. Berdasarkan
polaritas fase gerak dan fase diamnya, sistem kromatografi yang digunakan
merupakan sistem kromatografi fase terbalik, dimana fase gerak lebih polar
dibandingkan dengan fase diamnya, yaitu kolom oktadesilsilan (C18).
Metanol digunakan dalam fase gerak karena nikotin memiliki kelarutan
untuk menambah eluent strength dari fase gerak, dimana asetonitril memiliki
eluent strength yang lebih kuat dibandingkan metanol yaitu 3,1, sedangkan eluent
strength dari metanol adalah 1,0 (Sadek, 2002). Dengan bertambahnya eluent
strength dari fase gerak waktu retensi dari nikotin akan menjadi semakin singkat.
Fase gerak yang digunakan juga mengandung buffer. Buffer digunakan
dalam suatu sistem KCKT apabila analit merupakan senyawa yang mudah
terionisasi oleh pengaruh pH. Fungsi dari buffer adalah untuk mempertahankan
pH sistem, sehingga analit akan berada pada satu bentuk ionisasi.
Nikotin merupakan senyawa basa dengan pKa 8,5 yang mudah
terprotonasi dalam suasana asam dimana atom nitrogen pada cincin pirolidin
nikotin akan mengalami protonasi (Gambar 14), oleh sebab itu diperlukan buffer
untuk mengkontrol pH pada saat analisis. Pemilihan buffer dilakukan berdasarkan
pKa dari senyawa yang akan dianalisis (analitnya). pH dari buffer yang digunakan
± 2 unit dari pKa nikotin karena pada pH ± 2 pKa, nikotin akan berada dalam
bentuk terion (99% ionik) atau pada bentuk tidak terion (99% netral). Apabila pH
buffer sama dengan pKa nikotin, maka nikotin akan berada dalam bentuk 50%
terion dan 50% molekul, bila hal ini terjadi dapat menimbulkan masalah dimana
bentuk yang terion akan terelusi lebih dahulu sedangkan bentuk molekul akan
terelusi lebih lambat (Kazakevich dan LoBrutto, 2007).
dalam bentuk tak terion dengan pH buffer 2 unit di atas pKa nikotin, karena pada
pH > 7 dapat terjadi disolusi partikel-partikel silika pada kolom C18 yang
digunakan, terutama jika digunakan fase gerak yang kandungan airnya tinggi
(Kazakevich dan LoBrutto, 2007).
Berdasarkan perhitungan pergeseran pH oleh Kazakevich dan LoBrutto
(2007), pKa nikotin akan mengalami pergeseran menjadi 7,3 saat terlarut dalam
fase gerak yang mengandung 60% senyawa organik (54% metanol dan 6%
asetonitril), sedangkan pH dari buffer asam akan mengalami pergeseran sebanyak
1,2 unit ke atas, sehingga pH dari buffer harus ≤ 4,1 agar nikotin tetap berada
dalam bentuk terion, oleh sebab itu digunakan buffer asetat yang memiliki pKa
4,8 dengan rentang pH 3,8–4,8. Buffer asetat yang digunakan terdiri dari
ammonium asetat (0,349 mmol), natrium asetat (1,092 mmol) dan asam asetat
1,625 mL/L dengan pH 4.
Sebelum digunakan, masing-masing komponen fase gerak harus disaring
dengan menggunakan kertas Whatman. Tujuan dari penyaringan ini adalah untuk
menghilangkan adanya partikel-partikel asing dalam masing-masing larutan fase
gerak yang dapat menyumbat kolom dan akhirnya akan mengganggu analisis.
Terdapat dua macam kertas Whatman yang digunakan, yaitu kertas Whatman
organik yang digunakan untuk menyaring larutan organik (metanol, asetonitril)
Setelah semua larutan disaring, maka larutan diawaudarakan dengan
menggunakan ultrasonikator. Tujuannya adalah untuk menghilangkan
gelembung-gelembung udara yang terdapat pada larutan, adanya gelembung-gelembung udara dapat
mengganggu proses pemisahan sampel. Pencampuran masing-masing komponen
fase gerak dilakukan secara gradient dalam instrumen KCKT.
B. Pembuatan Larutan Baku Nikotin
Larutan baku nikotin dibuat dengan cara melarutkan sejumlah tertentu
baku nikotin dalam pelarut. Pelarut yang digunakan adalah sama dengan fase
gerak yaitu buffer asetat:metanol:asetonitril (40:54:6). Berdasarkan Snyder dkk.
(2010), untuk mencegah terjadinya perbedaan solvent strength antara pelarut dari
analit dan fase gerak maka lebih baik apabila pelarut yang digunakan sama
dengan fase gerak. Solvent strength ini merupakan salah satu faktor yang dapat
mempengaruhi retensi, sehingga dengan membuat pelarut sama dengan fase gerak
maka tidak akan terjadi perbedaan retensi antara pelarut analit dan fase gerak.
Dalam pembuatan larutan baku nikotin, dibuat larutan stok baku nikotin
dengan konsentrasi 2 ppm. Dari larutan stok ini kemudian dibuat 5 seri
Pada penelitian ini tidak terdapat senyawa lain selain nikotin yang dianalisis,
sehingga panjang gelombang pengamatan yang digunakan adalah panjang
gelombang dimana nikotin memberikan serapan maksimum (λ maksimum).
Penetapan panjang gelombang pengamatan nikotin ini dilakukan dengan
menggunakan spektrofotometer UV, secara teoritis nikotin memiliki serapan
maksimum pada panjang gelombang 262nm. Nikotin memiliki gugus kromofor
pada cincin piridinnya (Gambar 15) sehingga dapat memberikan serapan pada
daerah sinar ultraviolet.
Gambar 15. Gugus kromofor dan auksokrom pada nikotin
Pada penentuan panjang gelombang pengamatan digunakan tiga seri
konsentrasi nikotin dalam pelarut, dan terhadap masing-masing seri konsentrasi
dilakukan scanning panjang gelombang mulai dari 225 nm hingga 325 nm. Hasil
yang diperoleh berupa spektra panjang gelombang nikotin seperti yang terlihat
pada gambar dibawah ini:
Gambar 16. Spektra panjang gelombang maksimum nikotin pada tiga tingkat konsentrasi, 0,005ppm; 0,007ppm; dan 0,009ppm. Keterangan : A = konsentrasi 0,005ppm, absorbansi
0,205, λmaksimum 260nm; B = konsentrasi 0,007ppm, absorbansi 0,333, λmaksimum 260nm; C = konsentrasi 0,009ppm; absorbansi 0,374; λmaksimum 260nm
Gambar 16 di atas menunjukkan bentuk spektra nikotin, spektra ini
digunakan untuk analisis sekunder kualitatif, dimana spektra dari suatu senyawa
akan memiliki bentuk yang berbeda dengan senyawa yang lain sehingga dapat
digunakan untuk uji kualitatif. Dari Gambar 16 di atas terlihat bahwa pada tiga
konsentrasi yang berbeda bentuk spektra yang dihasilkan adalah sama, sehingga
disimpulkan bahwa spektra tersebut merupakan bentuk spektra dari nikotin. Dari
D. Preparasi Sampel
Sampel yang digunakan dalam penelitian ini adalah ekstrak etanolik
kental daun tembakau. Ekstrak daun tembakau ini mengandung bermacam-macam
senyawa lain selain nikotin, yaitu senyawa alkaloidal lainnya seperti anabasin,
anatabin dan nornikotin serta senyawa-senyawa non alkaloidal. Preparasi sampel
yang dilakukan bertujuan untuk mengekstraksi nikotin dari dalam sampel
sekaligus menghilangkan senyawa-senyawa non alkaloidal, khususnya yang
memiliki bobot molekul besar (contohnya tannin) yang dapat menyumbat kolom
C18 yang digunakan. Senyawa-senyawa alkaloidal akan ikut terekstraksi bersama
dengan nikotin, namun karena perbedaan struktur dan polaritasnya maka akan
terpisah saat dianalisis menggunakan KCKT.
Ekstrak kental daun tembakau dilarutkan dengan asam klorida encer
dengan bantuan ultrasonikator selama 30 menit. Nikotin yang tak terion dalam
sampel akan bereaksi dengan asam klorida membentuk nikotin hidroklorida yang
terlarut dalam fase asam klorida encer (Gambar 17), kemudian larutan ini
ditambahkan dengan kloroform sebanyak 10mL. Saat penambahan kloroform
nikotin hidroklorida akan tetap terlarut dalam fase asam klorida encer, sedangkan
senyawa-senyawa non alkaloidal akan terbawa dalam fase kloroform, sehingga
Gambar 17. Rekasi penggaraman nikotin oleh asam klorida
Fase air yang mengandung nikotin hidroklorida kemudian ditambah
dengan natrium hidroksida 4M hingga mencapai pH 11-12, tujuannya adalah agar
nikotin hidroklorida akan kembali menjadi nikotin yang tak terion (Gambar 18).
Oleh karena itu, dengan penambahan kloroform untuk kedua kalinya nikotin
hidroklorida yang telah kembali menjadi nikotin akan terlarut dalam fase
kloroform.
Gambar 18. Reaksi garam nikotin HCl dengan natrium hidroksida
Fase kloroform yang diperoleh kemudian diuapkan hingga tersisa residu
nikotin. Nikotin memiliki titik didih yang jauh lebih tinggi daripada kloroform,
yaitu 246oC sedangkan titik didih kloroform adalah 61,2oC, sehingga nikotin tidak
akan ikut menguap. Residu yang diperoleh kemudian dilarutkan dalam pelarut
sehingga siap untuk dianalisis.
E. Pengamatan Waktu Retensi (tR) Nikotin
Pengamatan waktu retensi (tR) merupakan parameter analisis kualitatif
dalam KCKT. Waktu retensi (tR) adalah waktu yang dibutuhkan oleh suatu analit