PERBANDINGAN POLA PITA AMPLIFIKASI DNA DAUN, BUNGA,
DAN BUAH KELAPA SAWIT NORMAL DAN ABNORMAL
ALFINIA AZIZAH
PROGRAM STUDI BIOKIMIA
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM
INSTITUT PERTANIAN BOGOR
BOGOR
2009
ABSTRAK
ALFINIA AZIZAH. Perbandingan Pola Pita Amplifikasi DNA Daun, Bunga, dan
Buah Kelapa Sawit Normal dan Abnormal. Dibimbing oleh EDY DJAUHARI P
dan DARMONO TANIWIRYONO.
Kelapa sawit dimanfaatkan sebagai bahan baku industri minyak goreng,
makanan, kosmetik, sabun, lilin, logam, serta biodiesel. Permintaan Crude Palm
Oil (CPO) yang semakin meningkat kurang bisa dipenuhi sehingga perbanyakan
bibit unggul kelapa sawit dilakukan dengan kultur jaringan. Namun teknik ini
menghasilkan tanaman kelapa sawit yang abnormal, sehingga diperlukan penanda
molekuler yang mampu mendeteksi abnormalitas secara dini. Penelitian bertujuan
mengetahui perbedaan genotip daun, bunga, dan buah dari tanaman kelapa sawit
normal dan abnormal, berdasarkan perbedaan pola pita amplifikasi DNA sampel
terhadap primer acak menggunakan teknik amplifikasi DNA. Hasil penelitian
menunjukkan perbedaan pita amplifikasi yang muncul pada sampel DNA bunga
abnormal dengan primer OPA 8, DNA buah normal dengan primer OPB 2, dan
DNA buah abnormal (primer OPA 18, OPA 20, OPB 2, dan OPB 9). Fragmen
DNA yang berbeda kemudian diisolasi dan disekuensing. Sekuen basa yang
berhasil dibaca hanya DNA buah abnormal dengan primer OPB 9. Analisis hasil
sekuen dilakukan dengan program Basic Local Alignment and Search Tool for
Nucleotida (BLAST) namun tidak menunjukkan homologi yang tinggi dengan
gen dalam bank gen. Hasil BLAST juga tidak menghasilkan urutan basa yang ada
pada suatu gen penyandi tertentu dalam tanaman kelapa sawit.
ABSTRACT
ALFINIA AZIZAH. Comparison of DNA Amplification Pattern from Leaves,
Flowers, and Fruits Normal and Abnormal Oil Palm. Under the direction of EDY
DJAUHARI P and DARMONO TANIWIRYONO.
Many of palm oil are used as industrial raw materials to produce oil, food,
cosmetics, soap, candles, metal industry, and biodiesel. Demand for Crude Palm
Oil (CPO) that has been increasing can not be fulfilled so tissue culture can use to
get oil palm prime seed. However, this technique can produce oil palm plants with
a high level of abnormalities, so it is necessary to use molecular marker that is
able to detect seed abnormalities earlier in tissue culture. The aim of this research
was to determine the genotype differences in leaf, flower, and fruit from both
normal and abnormal oil palm, based on differences in DNA amplification pattern
sample using DNA amplification methode. Results of this research indicate that
the DNA amplification pattern appears in the DNA abnormal flower sample with
the primary OPA 8, normal fruit DNA with the primary OPB 2, and fruit
abnormal DNA (with the primary OPA 18, OPA 20, OPB 2, and OPB 9).
Different DNA fragment from PCR results were isolated and sequenced. Base
sequence successfully be read is only abnormal fruit DNA with primer OPB 9.
Sequence analysis from the results were done by the Basic Local Alignment and
Search Tool for Nukleotida (BLAST) program but it does not indicate a high
homology with the genes in the bank gene. BLAST results also did not produce
base sequence from specific gene in the oil palm plant.
PERBANDINGAN POLA PITA AMPLIFIKASI DNA DAUN, BUNGA,
DAN BUAH KELAPA SAWIT NORMAL DAN ABNORMAL
ALFINIA AZIZAH
Skripsi
sebagai salah satu syarat untuk memperoleh gelar
Sarjana Sains pada
Program Studi Biokimia
PROGRAM STUDI BIOKIMIA
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM
INSTITUT PERTANIAN BOGOR
BOGOR
2009
Judul Skripsi : Perbandingan Pola Pita Amplifikasi DNA Daun, Bunga, dan
Buah Kelapa Sawit Normal dan Abnormal
Nama
: Alfinia Azizah
NIM
: G44104020
Disetujui
Drs. Edy Djauhari P, M.Si
Ketua
Anggota
Dr.Ir. H. Darmono Taniwiryono, M.Sc
Diketahui
Dekan Fakultas Matematika dan Ilmu Pengetahuan Alam
Dr. drh. Hasim, DEA
PRAKATA
Puji dan syukur penulis panjatkan kepada Allah SWT atas segala rahmat,
karunia, dan ridlo-Nya sehingga penulis dapat menyelesaikan penelitian dan
penulisan laporan akhir yang berjudul Perbedaan Pola Pita Amplifikasi DNA
Daun, Bunga, dan Buah Kelapa Sawit Normal dan Abnormal. Penelitian
dilakukan di Laboratorium Biologi Molekuler dan Rekayasa Genetika, Balai
Penelitian Bioteknologi Perkebunan Indonesia yang berlangsung dari bulan
Februari hingga Juli 2008. Penelitian ini dilakukan sebagai prasyarat untuk
memperoleh gelar Sarjana Sains pada Program Studi Biokimia, Fakultas
Matematika dan Ilmu Pengetahuan Alam.
Terima kasih penulis ucapkan kepada Drs. Edy Djauhari P, M.Si selaku
pembimbing utama dan Dr. Ir.H. Darmono Taniwiryono, M.Sc selaku
pembimbing anggota dan Kepala Balai Penelitian Bioteknologi Perkebunan
Indonesia atas segala saran dan kritiknya, serta kepada Dr. Asmini Budiani, M.Si
atas segala masukan dan nasehatnya. Penulis juga mengucapkan kepada seluruh
peneliti dan staf di Laboratorium Biologi Molekuler dan Rekayasa Genetika, Balai
Penelitian Bioteknologi Perkebunan Indonesia. Terakhir penulis ucapkan rasa
terima kasih yang tulus dan rasa hormat yang setinggi-tingginya kepada Ayah dan
Ibu atas doa dan segala dukungan yang telah diberikan kepada penulis. Semoga
hasil penelitian ini dapat bermanfaat bagi ilmu pengetahuan dan memberikan ilmu
bagi yang membutuhkan.
Bogor, Januari 2009
RIWAYAT HIDUP
Penulis dilahirkan di Sleman, Daerah Istimewa Yogyakarta pada tanggal
27 Desember 1985 dari ayahanda Arifin dan ibunda Nanik Ngafiatun. Penulis
merupakan anak pertama dari empat bersaudara.
Penulis lulus dari SMU PPMI Assalaam Surakarta pada tahun 2004.
Selanjutnya lolos seleksi masuk IPB melalui jalur Undangan Seleksi Masuk IPB
(USMI) dan diterima pada program studi Biokimia, Fakultas Matematika dan
Ilmu Pengetahuan Alam. Penulis melakukan Praktik Kerja Lapang di
Laboratorium Biologi Molekuler dan Rekayasa Genetika, Balai Penelitian
Bioteknologi Perkebunan Indonesia dari bulan Juli sampai Agustus 2007 dengan
judul Perbandingan Pola Pita DNA Kelapa Sawit Normal dan Abnormal dengan
Metode Amplifikasi DNA.
Selama mengikuti masa perkuliahan penulis mengikuti kegiatan dalam
kampus baik dalam lembaga kemahasiswaan dan berbagai kepanitiaan. Unit
kegiatan mahasiswa yang penulis ikuti adalah kegiatan seni sunda Gentra
Kaheman dan fotografer magang pada Koran Kampus. Lembaga kemahasiswaan
yang pernah diikuti adalah Community of Research and Education of
DAFTAR ISI
Halaman
DAFTAR GAMBAR ... viii
DAFTAR LAMPIRAN ... viii
PENDAHULUAN ... 1
TINJAUAN PUSTAKA
Tanaman Kelapa Sawit ... 1
Polymerase Chain Reaction (PCR) ... 2
Random Amplified Polymorphic DNA (RAPD) ... 3
Sekuensing ... 4
BAHAN DAN METODE
Alat dan Bahan ... 6
Metode ... 6
HASIL DAN PEMBAHASAN
Isolasi DNA dari Daun, Bunga, dan Buah Kelapa Sawit ... 8
Amplifikasi DNA Hasil Isolasi dengan Primer Acak ... 9
Analisis Hasil Urutan Basa Fragmen DNA ... 12
SIMPULAN DAN SARAN ... 13
DAFTAR PUSTAKA ... 13
DAFTAR GAMBAR
Halaman
1 Tanaman Kelapa Sawit ... 2
2 Skema proses PCR ... 3
3 Skema proses PCR-RAPD ... 4
4 Skema proses sekuensing dengan metode Sanger ... 5
5 Hasil elektroforesis larutan DNA sampel ... 9
6 Sampel DNA daun, bunga, dan buah normal dan abnormal dengan primer
OPA 8 dan OPA 9 ... 10
7 Sampel DNA daun, bunga, dan buah normal dan abnormal dengan primer
OPA 18 ... 10
8 Sampel DNA daun, bunga, dan buah normal dan abnormal dengan primer
OPA 19 dan OPA 20 ... 10
9 Sampel DNA daun, bunga, dan buah normal dan abnormal dengan primer
OPB 2 ... 11
10 Sampel DNA daun, bunga, dan buah normal dan abnormal dengan primer
OPB 9 ... 11
11 Analisis BLAST hasil sekuensing fragmen DNA buah abnormal dengan
primer OPB 9 ... 12
12 Urutan basa yang diperoleh dari hasil sekuensing fragmen DNA buah
abnormal dengan primer OPB 9 ... 13
DAFTAR LAMPIRAN
Halaman
1 Alur penelitian ... 16
2 Pembuatan larutan sediaan ... 17
3 Proses isolasi DNA daun, bunga, dan buah kelapa sawit ... 18
4 Perhitungan pengenceran DNA dengan konsentrasi 100 ng/µl ... 20
5 Tabel urutan basa primer OPA dan OPB ... 21
6 Gambar pola pita hasil amplifikasi DNA ... 22
7 Hasil analisis BLAST dari urutan basa bunga abnormal dengan primer
OPB 9 ... 24
16
Lampiran 1 Alur penelitian
Sampel DNA kelapa sawit (normal dan abnormal)
Proses PCR-RAPD
Isolasi fragmen hasil PCR-RAPD
Sequencing
Analisis hasil sequencing
Isolasi DNA sampel
17
Lampiran 2 Pembuatan larutan sediaan
Pembuatan bufer ekstraksi DNA. (50 ml)
Bufer ekstraksi DNA dibuat dengan mencampurkan :
CTAB 10%
10 ml
EDTA 0.5 M pH 8.0
2 ml
tris-HCl 1M pH 8.0
5 ml
NaCl 5 M
12.6 ml
dH2O steril
20.4 ml
Pembuatan Bufer TE (100 ml)
Bufer TE dibuat dengan melarutkan :
Tris-HCl 1 M pH 8.0
1 ml
EDTA 0.5 M pH 8.0
0.2 ml
dH
2O steril
98.8 ml
Pembuatan Bufer TBE 5× (500 ml)
Bufer TBE 5× dibuat dengan komposisi :
Tris-Base
27gram
Asam borat
13.75 gram
EDTA 0.5 M pH 8.0
10 ml
dan ditepatkan dengan ddH2O hingga 500 ml.
Saat pemakaian dalam pembuatan gel agarosa, bufer TBE 5× ini diencerkan
menjadi 0.5×.
18
Lampiran 3 Proses isolasi DNA daun, bunga, dan buah kelapa sawit
Sterilisasi alat dan bahan dengan otoklaf (121°C, 1 atm)
Masing-masing sampel ditimbang sebanyak 1 gram
Sampel digerus dengan mortar
Ditambah 0.1 g PVPP dan N
2cair sampai halus
Tabung sentrifus berisi 5 ml bufer ekstraksi dan ß-merkaptoetanol yang sudah
dipanaskan (65°C)
Dipanaskan pada 65°C selama 30 menit (tiap 5 menit dikocok)
Larutan didinginkan pada suhu kamar
Diekstraksi dengan larutan CI sebanyak 1 volume
Homogenasi dengan vorteks
Disentrifugasi pada ±13500 g, 10 menit, 4 °C
Supernatan dipindahkan ke tabung baru Diulang 2×
Diekstraksi dengan larutan CI sebanyak 1 volume
Ditambah isopropanol dingin 1 volume
Disimpan dalam lemari es 4°C, 30 menit
Disentrifugasi pada ±13500 g, 10 menit, 4 °C
Pelet DNA dikeringkan
Ditambah 1 ml bufer TE
Kocok hingga pelet DNA semua larut
Ditambah Na-asetat 1/10 volume dan etanol absolut 2.5 ml
Dibekukan selama 30 menit atau semalam
19
Lanjutan Lampiran 3
Disentrifugasi pada ±16000 g, 10 menit, 4 °C
Pelet DNA dicuci dengan etanol 70%
Pelet dikeringkan dengan sentrifugasi pada ±16000 g, 5 menit, 4 °C
Pelet dilarutkan dalam MW
20
Lampiran 4 Perhitungan pengenceran DNA dengan konsentrasi 100 ng/µl
V
1×M
1= V
2×M
2 → V1=
1 2 2M
M
×
V
Keterangan:
V
1= volume larutan DNA yang diambil untuk diencerkan
M1= konsentrasi larutan DNA sebelum diencerkan
V2= volume akhir pengenceran (50 μl)
M2= konsentrasi larutan DNA yang diinginkan (100 ng/μl)
Volume DNA yang diencerkan (µl)
Penambahan MW (µl)
V
1(DNA Daun N) =
6
.
09
820
100
50
=
×
43.91
V
1(DNA Daun Ab) =
5
.
78
865
100
50
=
×
44.22
V
1(DNA Bunga N) =
4
.
28
1180
100
50
=
×
45.72
V1 (DNA Bunga Ab) =
3
.
86
1295
100
50
×
=
46.14
V1 (DNA Buah N) =
6
.
25
800
100
50
×
=
43.75
V1 (DNA Buah Ab) =
5
.
58
895
100
50
×
=
21
Lampiran 5 Tabel urutan basa primer OPA dan OPB
No
Kode
5’ to 3’
1
OPA 6
GGT CCC TGAC
2
OPA 7
GAA ACG GGTG
3
OPA 8
GTG ACG TAGG
4
OPA 9
GGG TAA CGCC
5
OPA 10
GTG ATC GCAG
6
OPA 14
TCT GTG CTGG
7
OPA 15
TTC CGA ACCC
8
OPA 17
GAC CGC TTGT
9
OPA 18
AGG TGA CCGT
10
OPA 19
CAA ACG TCGG
11
OPA 20
GTT GCG ATCC
12
OPB 1
GTT TCG CTCC
13
OPB 2
TGA TCC CTGG
14
OPB 3
CAT CCC CCTG
15
OPB 7
GGT GAC GCAG
16
OPB 8
GTC ACA ACGG
17
OPB 9
TGG GGG ACTC
18
OPB 10
CTG CTG GGAC
19
OPB 17
AGG GAA CGAG
22
1 2 3 4 5 6 1 2 3 4 5 6 M
OPA 6 OPA 7
1 2 3 4 5 6 M 1 2 3 4 5 6
1 2 3 4 M 5 6
1 2 3 4 5 6 M
Lanjutan Lampiran 6
Keterangan gambar: (1) DN (2) Dab (3) BN (4) BAb (5) FN (6) FAb (M) Marker 1 kb +a. Sampel DNA daun, bunga, dan buah normal dan abnormal dengan primer
OPA 6 dan OPA 7.
c. Sampel DNA daun, bunga, dan buah
normal dan abnormal dengan primer
OPA 10.
d. Sampel DNA daun, bunga, dan buah
normal dan abnormal dengan primer
OPA 17.
b. Sampel DNA daun, bunga, dan buah normal dan abnormal dengan primer
OPA 14 dan OPA 15.
23
1 2 3 4 5 6 M 1 2 3 4 5 6 M
1 2 3 4 5 6 M M 1 2 3 4 5 6
1 2 3 4 5 6 1 2 3 4 5 6 M
OPB 8
OPB 17
Lampiran 7 Hasil analisis BLAST dari urutan basa bunga abnormal dengan primer
OPB 9
Keterangan gambar: (1) DN (2) Dab (3) BN (4) BAb (5) FN (6) FAb (M) Marker 1 kb +e. Sampel DNA daun, bunga, dan buah
normal dan abnormal dengan primer
OPB 1.
f. Sampel DNA daun, bunga, dan buah
normal dan abnormal dengan primer
OPB 3.
g. Sampel DNA daun, bunga, dan buah
normal dan abnormal dengan primer
OPB 7.
h. Sampel DNA daun, bunga, dan buah
normal dan abnormal dengan primer
OPB 10.
i. Sampel DNA daun, bunga, dan buah normal dan abnormal dengan primer
OPB 8 dan OPB 17.
24
Deskripsi Max score Total score E value Max identMus musculus strain 129S6/SvEvTac clone rp21-580p20 map 10, complete sequence
51.8 51.8 0.003 80% Mus musculus strain C57BL/6J clone rp23-358m4,
complete sequence
51.8 51.8 0.003 80% Mus musculus strain 129S6/SvEvTac chromosome 10 clone
rp21-411d9, complete sequence
51.8 51.8 0.003 80% Canis lupus familiaris clone RP81-386E20, complete
sequence
50.0 50.0 0.011 78% Mus musculus chromosome 6, clone RP23-402P24,
complete sequence
50.0 50.0 0.011 76% Mus musculus chromosome 5, clone RP24-67M11,
complete sequence
50.0 50.0 0.011 79% Mus musculus chromosome 5, clone RP23-445H19,
complete sequence
50.0 50.0 0.011 79% Drosophila melanogaster chromosome 3R, complete
sequence
48.2 48.2 0.039 79% Mus musculus BAC clone RP23-119P9 from chromosome
13, complete sequence
48.2 48.2 0.039 80% Drosophila melanogaster clone BACR03J17, complete
sequence
48.2 48.2 0.039 79% Drosophila melanogaster clone BACR27G04, complete
sequence
48.2 48.2 0.039 79% Mus musculus BAC clone RP24-422I4 from chromosome
13, complete sequence
48.2 48.2 0.039 80% Mustela putorius furo clone CH237-388F7, complete
sequence
46.4 46.4 0.14 76% Felis catus clone RP86-181L4, complete sequence 46.4 90.9 0.14 76% Mus musculus BAC clone RP23-473K23 from chromosome
13, complete sequence
46.4 46.4 0.14 80% Canis familiaris, clone XX-397C16, complete sequence 44.6 44.6 0.47 77% Canis Familiaris chromosome 12, clone XX-93M5,
complete sequence
44.6 44.6 0.47 79% Canis Familiaris chromosome 3, clone XX-500D6, complete
sequence
44.6 44.6 0.47 77% Canis Familiaris chromosome 14, clone XX-483F1,
complete sequence
44.6 44.6 0.47 76% Canis Familiaris chromosome 6, clone XX-463G11,
complete sequence
44.6 44.6 0.47 76% Canis Familiaris chromosome 3, clone XX-393O18,
complete sequence
44.6 44.6 0.47 77% Canis lupus familiaris clone RP81-196D8, complete
sequence
44.6 44.6 0.47 76% Canis Familiaris, clone XX-10C7, complete sequence 44.6 44.6 0.47 76% Mus musculus chromosome 3, clone RP23-296I12,
complete sequence
44.6 44.6 0.47 78% Human DNA sequence from clone RP11-92B10 on
chromosome X Contains the 5' end of a variant of the CUL4B gene for cullin 4B and the 5' end of a variant of the gene for MCT-1 protein (MCT-1), complete sequence
44.6 44.6 0.47 85%
Mus musculus chromosome 11, clone RP23-179C3, complete sequence 44.6 44.6 0.47 75%
Lanjutan Lampiran 7
Deskripsi Max score Total score E value Max ident25
Mus musculus BAC clone RP23-25I3 from chromosome 7, complete sequence
44.6 44.6 0.47 79% Mouse DNA sequence from clone RP23-285D3 on
chromosome 11 Contains part of a novel gene, a heat shock protein (Hsp) pseudogene and a CpG island, complete sequence
44.6 44.6 0.47 75%
Mouse DNA sequence from clone RP24-246F8 on chromosome 13, complete sequence
44.6 44.6 0.47 79% Mouse DNA sequence from clone RP24-120H1 on
chromosome 16, complete sequence
44.6 44.6 0.47 86% Mouse DNA sequence from clone RP23-406N5 on
chromosome 4 Contains a ribosomal protein S19 (Rps19) pseudogene, a novel gene, the gene for a DNA segment Chr 4 Wayne State University 114 expressed (D4Wsu114e), the Mfn2 gene for mitofusin 2, a ribosomal protein L35a (Rpl35a) pseudogene, the Plod1 gene for procollagen-lysine 2-oxoglutarate 5-dioxygenase 1, a novel gene
(2510039O18Rik) and two CpG islands, complete sequence
44.6 44.6 0.47 76%
Dictyostelium discoideum AX4 chromosome 3 DDB0232519.02, whole genome shotgun sequence
42.8 42.8 1.6 76% Canis familiaris chromosome 33, clone XX-221P23,
complete sequence
42.8 42.8 1.6 75% Monosiga brevicollis MX1 predicted protein
MONBRDRAFT_7962 mRNA, complete cds
42.8 42.8 1.6 90% Mouse DNA sequence from clone RP23-102L17 on
chromosome 16, complete sequence
42.8 42.8 1.6 82% Canis Familiaris chromosome 26, clone XX-195P12,
complete sequence
42.8 42.8 1.6 81% Canis Familiaris chromosome 25, clone XX-122B22,
complete sequence
42.8 42.8 1.6 77% Vitis vinifera, whole genome shotgun sequence, contig
VV78X193509.12, clone ENTAV 115
42.8 42.8 1.6 78% Canis Familiaris chromosome 26, clone XX-497F13,
complete sequence
42.8 42.8 1.6 83% Canis Familiaris chromosome 37, clone XX-341P11,
complete sequence
42.8 42.8 1.6 75% Canis Familiaris chromosome 20, clone XX-263M4,
complete sequence
42.8 42.8 1.6 80% Canis Familiaris chromosome 26, clone XX-320A4,
complete sequence
42.8 42.8 1.6 80% Canis Familiaris chromosome 33, clone XX-84F14,
complete sequence
42.8 42.8 1.6 75% Mus musculus chromosome 15, clone RP23-85P16,
complete sequence
42.8 42.8 1.6 85% Canis Familiaris, clone XX-25G10, complete sequence 42.8 42.8 1.6 76% Felis catus clone RP86-439N5, complete sequence 42.8 42.8 1.6 78% Mus musculus chromosome 15, clone RP23-338F23,
complete sequence
42.8 42.8 1.6 85% Mus musculus BAC clone RP24-241I3 from 15, complete
sequence
42.8 42.8 1.6 77% Zebrafish DNA sequence from clone DKEY-83M22 in
linkage group 9, complete sequence
42.8 42.8 1.6 87%
Lanjutan Lampiran 7
Deskripsi Max score Total score E value Max ident26
T28A8
Mouse DNA sequence from clone DN-316O2 on chromosome 3 Contains two ribosomal protein, large, P0 (Rplp0) pseudogene, complete sequence
42.8 42.8 1.6 72%
Mus musculus genomic DNA, chromosome 16q clone:RP21-567F3, complete sequences
42.8 42.8 1.6 82% Dictyostelium discoideum AX4 chromosome 2
DDB0232469.02, whole genome shotgun sequence
41.0 41.0 5.7 93% Dictyostelium discoideum AX4 chromosome 1
DDB0232442.02, whole genome shotgun sequence
41.0 41.0 5.7 74% Canis familiaris chromosome 26, clone XX-397I18,
complete sequence
41.0 41.0 5.7 73% Populus trichocarpa clone POP037-N11, complete sequence 41.0 41.0 5.7 80% Canis familiaris chromosome 14, clone XX-54L5, complete
sequence
41.0 41.0 5.7 74% Canis familiaris chromosome 2, clone XX-83E16, complete
sequence
41.0 41.0 5.7 76% Canis familiaris chromosome 26, clone XX-93E11,
complete sequence
41.0 41.0 5.7 73% Zebrafish DNA sequence from clone CH211-51E8 in
linkage group 21, complete sequence
41.0 41.0 5.7 82% Lodderomyces elongisporus NRRL YB-4239 chromo
domain protein 1 (LELG_01549) partial mRNA
41.0 41.0 5.7 81% Canis familiaris chromosome 31, clone XX-361C3,
complete sequence
41.0 41.0 5.7 72% Tetrahymena thermophila SB210 hypothetical protein
(TTHERM_00188900) partial mRNA
41.0 41.0 5.7 100% Canis Familiaris chromosome 10, clone XX-480A12,
complete sequence
41.0 41.0 5.7 75% Canis Familiaris, clone XX-284L8, complete sequence 41.0 41.0 5.7 80% Canis Familiaris chromosome 11, clone XX-311M9,
complete sequence
41.0 41.0 5.7 80% Vitis vinifera contig VV78X194357.5, whole genome
shotgun sequence
41.0 41.0 5.7 87% Canis Familiaris chromosome 10, clone XX-181J11,
complete sequence
41.0 41.0 5.7 75% Canis Familiaris chromosome 14, clone XX-498K4,
complete sequence
41.0 41.0 5.7 74% Canis Familiaris chromosome 16, clone XX-195I11,
complete sequence
41.0 41.0 5.7 75% Canis Familiaris chromosome 8, clone XX-494F18,
complete sequence
41.0 41.0 5.7 83% Canis Familiaris chromosome 2, clone XX-361N13,
complete sequence
41.0 41.0 5.7 75% Canis Familiaris chromosome X, clone XX-242J14,
complete sequence
41.0 41.0 5.7 75% Canis Familiaris chromosome 6, clone XX-346D24,
complete sequence
41.0 41.0 5.7 74% Zebrafish DNA sequence from clone RP71-84K24 in
linkage group 3, complete sequence
41.0 41.0 5.7 76% Canis Familiaris chromosome 8, clone XX-368A9,
complete sequence 41.0 41.0 5.7 82%
Lanjutan Lampiran 7
Deskripsi Max score Total score E value Max identCanis Familiaris chromosome 6, clone XX-163J21, complete sequence
27
Canis Familiaris chromosome 32, clone XX-3D7, complete sequence
41.0 41.0 5.7 78% Canis Familiaris chromosome 32, clone XX-98B12,
complete sequence
41.0 41.0 5.7 78% Xenopus (Silurana) tropicalis chromosome CH216-62P2,
complete sequence
41.0 41.0 5.7 92% Dictyostelium discoideum AX4 component of 9-1-1
complex (rad9) partial mRNA
41.0 41.0 5.7 93% Canis lupus familiaris agouti signaling protein, nonagouti
homolog (mouse) (ASIP), mRNA >gb|AY714374.1| Canis familiaris agouti protein mRNA, complete cds
41.0 41.0 5.7 76%
Mus musculus BAC clone RP24-178A16 from chromosome 16, complete sequence
41.0 41.0 5.7 78% Mus musculus BAC clone RP23-227O11 from chromosome
12, complete sequence
41.0 41.0 5.7 79% Canis lupus familiaris clone RP81-55A4, complete
sequence
41.0 41.0 5.7 74% Homo sapiens chromosome 5 clone CTD-2170G1,
complete sequence
41.0 41.0 5.7 92% Homo sapiens BAC clone RP11-436F23 from 4, complete
sequence
41.0 41.0 5.7 75% Felis sp. NG192 DNA, INT1095 locus sequence 41.0 41.0 5.7 82% Homo sapiens chromosome 5 clone RP11-164A5, complete
sequence
41.0 41.0 5.7 92% Homo sapiens chromosome 5 clone RP11-54A24, complete
sequence
41.0 41.0 5.7 92% Mouse DNA sequence from clone RP23-137B16 on
chromosome 11 Contains a novel gene, the 5' end of the Meis1 gene for myeloid ecotropic viral integration site 1 and two Cpg islands, complete sequence
41.0 41.0 5.7 76%
Dictyostelium discoideum chromosome 2 map 1685067-2090751 strain AX4, complete sequence
41.0 41.0 5.7 93% Mus musculus strain C57BL/6J chromosome 2 clone
rp23-60i12, complete sequence
41.0 41.0 5.7 78% Mouse DNA sequence from clone RP23-259G10 on
chromosome 9, complete sequence
41.0 41.0 5.7 79% Mouse DNA sequence from clone RP23-300E5 on
chromosome 2 Contains the Lrp2 gene for low density lipoprotein receptor-related protein 2 and a CpG island, complete sequence
41.0 41.0 5.7 78%
Mus musculus BAC clone RP23-387C9 from 12, complete sequence
41.0 41.0 5.7 92% Mouse DNA sequence from clone RP23-393E22 on
chromosome 16, complete sequence
41.0 41.0 5.7 78% Mus musculus BAC clone RP23-299A14 from chromosome
6, complete sequence
PENDAHULUAN
Kelapa sawit (Elaeis guineensis Jacq) merupakan salah satu dari beberapa tanaman palma penghasil minyak yang memiliki nilai ekonomi tinggi dan termasuk industri padat karya. Pengusahaan tanaman ini untuk produksi minyak memiliki beberapa keunggulan, antara lain biaya produksi yang relatif murah, hasil per hektar tinggi, umur produktif yang panjang, serta pemanfaatannya beraneka ragam (Lubis 1992).
Industri yang memanfaatkan kelapa sawit sebagai bahan baku produknya antara lain industri minyak goreng, makanan, kosmetik, sabun, lilin, industri logam, serta biodiesel. Biodiesel sawit adalah minyak atau solar yang dibuat dengan menggunakan bahan baku minyak mentah kelapa sawit atau Crude Palm
Oil (CPO). Pencampuran biodiesel dengan
minyak solar biasanya diberikan sistem penamaan tersendiri seperti B2, B3 atau B5 yang berarti campuran biodiesel dan minyak solar yang masing-masing mengandung 2%, 3%, dan 5% biodiesel (Boedoyo 2006). Sumber energi alternatif ini bersifat ramah lingkungan karena tidak mencemari udara, mudah terbiodegradasi, dan berasal dari bahan baku yang dapat diperbaharui (BPPP 2006). Permintaan CPO yang semakin meningkat kurang bisa terpenuhi karena lahan yang terbatas sehingga diperlukan terobosan-terobosan baru yang menuntut dihasilkannya bahan tanaman kelapa sawit yang unggul baik dalam hal produktivitasnya maupun kualitas minyaknya. Perbanyakan masal bibit unggul kelapa sawit secara cepat dapat dilakukan dengan teknik kultur jaringan, namun teknik ini dapat menghasilkan tanaman kelapa sawit dengan tingkat abnormalitas yang tinggi.
Keunggulan teknik kultur jaringan adalah mampu menghasilkan bibit dalam jumlah banyak dengan waktu relatif singkat, seragam, sifatnya identik dengan induknya, masa non produktif lebih singkat dan produktivitasnya lebih tinggi. Abnormalitas pembuahan pada tanaman kelapa sawit asal kultur jaringan dikenal dengan istilah mantled, yaitu mesokarp tidak berkembang. Abnormalitas bisa menyebabkan terjadinya bunga jantan steril (Corley 1986). Abnormalitas terjadi pada rata-rata 5-10 % dari populasi bibit asal kultur jaringan (Jaligot 2000), dan bersifat epigenetik (Tregear et al 2002). Marmey (1991) menyatakan bahwa kalus remah yang disebut sebagai kalus sekunder menyebabkan terjadinya kalus embrioid yang abnormal. Menurut Jones (1991) abnormalitas yang
terjadi pada klon kelapa sawit hasil kultur jaringan disebabkan terhambatnya ekspresi gen yang mengatur pembungaan, sebagai akibat penambahan zat pengatur tumbuh tertentu ke dalam media. Terjadinya tingkat abnormalitas yang tinggi menyebabkan rendahnya minat konsumen terhadap bibit asal kultur jaringan.
Berdasarkan fenomena ini diperlukan penanda molekuler yang mampu mendeteksi abnormalitas secara dini sehingga bibit hasil kultur jaringan kelapa sawit yang dipasarkan pada masyarakat adalah bibit normal yang terjamin kualitasnya. Penggunaan penanda molekuler berbasis DNA untuk mendeteksi abnormalitas pada fase dini dianggap tepat untuk tujuan ini, karena memiliki tingkat akurasi yang tinggi dan tidak terpengaruh oleh cuaca serta dapat melibatkan contoh dalam jumlah yang banyak.
Teknik RAPD (Random Amplified
Polymorphic DNA) merupakan salah satu dari
beberapa teknik pembuatan penanda berbasis DNA dengan melibatkan penggunaan mesin PCR (Polymerase Chain Reaction). Teknik PCR-RAPD dapat digunakan untuk mengidentifikasi perbedaan genotip normal dan abnormal, berdasarkan perbedaan pada pita DNA yang dapat teramplifikasi dengan random primer. Pita DNA yang berbeda akan dianalisis lebih lanjut untuk mengetahui perbedaan urutan basa DNA antara genotip normal dan abnormal.
Penelitian ini bertujuan untuk mengetahui perbedaan genotip daun, bunga, dan buah dari tanaman kelapa sawit normal dan abnormal berdasarkan perbedaan pola pita amplifikasi cetakan DNA terhadap primer acak menggunakan teknik PCR-RAPD. Hipotesis penelitian ini adalah diperoleh genotip daun, bunga, dan buah dari tanaman kelapa sawit normal dan abnormal, serta diperoleh pola pita amplifikasi DNA sampel menggunakan teknik PCR-RAPD. Hasil penelitian ini diharapkan dapat memberikan sumbangan ilmu pengetahuan berupa data ilmiah yang membuktikan terdapat perbedaan genotip kelapa sawit normal dan abnormal.
TINJAUAN PUSTAKA
Tanaman Kelapa Sawit
Kelapa sawit (Gambar 1) adalah tanaman perkebunan atau industri dari famili Palmae. Menurut penelitian, tanaman ini berasal dari Nigeria di Afrika Barat, kemudian menyebar ke Afrika, Amerika, Asia, dan Pasifik Selatan.
2
Benih kelapa sawit pertama kali ditanam di Indonesia tahun 1848 berasal dari Mauritius, Afrika. Perkebunan kelapa sawit di Indonesia dipelopori oleh Adrien Hallet berkebangsaan Belgia di daerah Tanahitam, Hulu Sumatera Utara pada tahun 1911 (Setyamidjadja 2006).
Kelapa sawit dapat tumbuh pada tanah yang gembur, pengairan dan drainasenya baik, kaya akan humus, serta tidak memiliki lapisan padas. Nilai pH tanah sebaiknya berkisar antara 5.5-7.0. Curah hujan yang diterima minimal 1000-1500 mm/tahun yang terbagi rata sepanjang tahun, serta memiliki suhu tumbuh optimal 26°C dan kelembaban udara rata-rata 75% (Setyamidjadja 2006).
Kelapa sawit secara sistematika (taksonomi) termasuk dalam Divisi Spermatophyta, Sub divisi Angiospermae, Kelas Dicotyledonae, Famili Palmaceae, Subfamili Cocoideae, Genus Elaeis, Spesies: (1) Elaeis guineensis Jacq (kelapa sawit Afrika) dan (2) Elaeis melanococca atau
Corozo oleifera (kelapa sawit Amerika Latin).
Varietas atau tipenya dapat digolongkan berdasarkan tebal tipisnya cangkang dan warna buah. Terdapat tiga varietas berdasarkan tebal tipisnya cangkang (endocarp) yaitu, Dura, Pisifera, dan Tenera. Sedangkan berdasarkan warna buah dikenal tiga varietas yaitu, Nigrescens, Virescens, dan
Albescens (Setyamidjadja 2006).
Kelapa sawit yang tumbuh normal pada tahun kedua telah menunjukkan pembungaan. Buah yang terbentuk belum dapat diolah karena ukurannya masih terlalu kecil. Tandan buah telah masak atau siap panen sekitar 5.5 bulan setelah terjadi penyerbukan. Memasuki umur sekitar 30 bulan, tanaman kelapa sawit terutama varietas Tenera (Dura×Pisifera) siap dipanen bila tandan buahnya sudah mencapai berat ±3 kg. Pemanenan kelapa sawit perlu memperhatikan beberapa ketentuan agar tandan buah yang dipanen sudah matang, sehingga dihasilkan kelapa sawit dengan mutu yang baik (Setyamidjadja 2006).
Buah kelapa sawit terdiri atas perikarp dan kernel. Perikarp tersusun atas eksokarp (kulit), mesokarp (daging buah), dan endokarp (cangkang). Kernel memiliki kulit, endosperm, dan embrio. Umur dan warna mesokarp digunakan sebagai indikator kandungan minyak. Kelapa sawit yang berumur tiga bulan mesokarpnya berwarna putih kehijauan yang menunjukkan bahwa buah terdiri dari serat, klorofil, dan belum terbentuk minyak. Perubahan warna daging buah menjadi kuning kehijauan setelah berumur lebih dari tiga bulan dan
menunjukkan bahwa minyak sudah terbentuk dengan terbentuknya karotein (Lubis 1992).
Kelapa sawit yang dihasilkan dari kultur jaringan mengalami 10-40% perubahan ke arah abnormalitas pada organ bunga dan buah sehingga dihasilkan kelapa sawit yang abnormal (Corley 1986). Hal yang sangat ekstrim dari abnormalitas adalah tidak terbentuknya buah karena tandan buah dipenuhi oleh bunga jantan atau buah bermantel berat yang menyebabkan hilangnya produksi (Mathius 2001). Beberapa pendapat mengenai penyebab terjadinya abnormalitas pada tanaman kelapa sawit hasil kultur jaringan, perubahan tersebut dapat bersifat genetik (Rao & Danough 1990), gangguan ekspresi gen diakibatkan fitohormon (Jones 1991), struktur kalus, lamanya subkultur dan umur kalus, tekanan seleksi yang dipakai, jenis eksplan yang digunakan, level ploidi sumber eksplan dan kecepatan proliferasi kalus (Skirvin et al 1984 dan Karp 1995).
Gambar 1 Tanaman Kelapa Sawit.
Polymerase Chain Reaction (PCR)
Teknik ini dikembangkan oleh Kary Mullis tahun 1984 berdasarkan penemuannya tentang adanya aktivitas biologi dari DNA polimerase pada suhu tinggi dalam bakteri yang hidup pada sumber air panas (thermophiles). Reaksi rantai polimerase merupakan metode untuk memperbanyak potongan DNA yang diinginkan dengan sensitifitas, selektifitas yang tinggi, dan terjadi pada waktu yang sangat cepat. Spesifitas reaksi ditentukan pada penggunaan dua primer oligonukleotida yang berhibridisasi menjadi rangkaian komplementer pada untai DNA yang berlawan dan mengapit rangkaian sasaran (Murray 2003).
Lima bahan baku yang diperlukan untuk melakukan PCR adalah sampel target, primer, enzim taq polimerase, nukleotida (dNTP), dan bufer polimerase. Sampel target merupakan DNA yang ingin diamplifikasi. Primer merupakan untai DNA pendek yang
3
menempel pada fragmen DNA target, serta sebagai tempat awal terjadinya replikasi. Enzim taq polimerase berfungsi untuk replikasi DNA. Larutan dNTP (mengandung dATP, dGTP, dCTP, dan dTTP) perlu ditambahkan agar DNA polimerase dapat membentuk kompleks rantai baru yang komplementer. Reaksi PCR membutuhkan suatu bufer yang mengandung MgCl2 karena
aktivitas enzim polimerase dipengaruhi oleh konsentrasi ion Mg2+. Ion Mg2+ akan menstimulasi aktivitas enzim secara maksimal pada konsentrasi 2 mM. Jika konsentrasinya lebih tinggi, maka dapat bersifat sebagai inhibitor (Sambrook 1989).
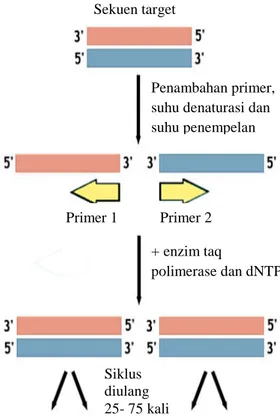
Proses PCR melibatkan serangkaian siklus temperatur berulang dan setiap siklus terdiri dari tiga tahap (Gambar 2). Pertama proses denaturasi dengan pemanasan (± 94 °C) untuk memisahkan untai ganda DNA menjadi untai tunggal. Kedua adalah penurunan suhu menjadi 50-70 °C untuk memungkinkan terjadinya penempelan (annealing) primer (oligonukleotida) dengan untai tunggal DNA sebagai cetakan. Suhu penempelan primer bergantung pada titik leleh (Tm) primer yang digunakan. Ketiga adalah ekstensi primer menjadi untai DNA baru dengan enzim DNA polimerase pada suhu optimumnya. Setiap satu siklus PCR akan menggandakan jumlah molekul cetakan DNA, sebab setiap utas baru yang disintesis akan berperan sebagai cetakan pada siklus selanjutnya. Penggandaan yang dihasilkan bersifat eksponensial sehingga dihasilkan produk PCR yang setara dengan 2n (n adalah jumlah siklus) (Sambrook 1989).
Random Amplified Polymorphic DNA
(RAPD)
Teknik PCR-RAPD merupakan salah satu teknik molekuler untuk mempelajari keanekaragaman genetika. Dasar analisis RAPD adalah menggunakan mesin PCR yang mampu mengamplifikasi sekuen DNA secara acak. Teknik ini melibatkan penempelan primer yang dirancang secara khusus sepuluh oligonukleotida pada cetakan DNA yang komplementer, selanjutnya akan dibentuk menjadi utas DNA baru. Proses selanjutnya sama dengan proses dasar PCR (Gambar 3). Jumlah produk amplifikasi PCR berhubungan langsung dengan jumlah dan orientasi sekuen yang komplementer terhadap primer di dalam genom tanaman (Azrai 2005).
Teknik RAPD hanya digunakan pada satu primer arbitrasi yang dapat menempel pada kedua utas DNA setelah didenaturasi pada
situs tertentu yang homolog dengan spesifitas penempelan yang tinggi. Potongan DNA yang teramplifikasi berdasarkan pilihan penempelan yang bersifat acak dan tidak harus berkaitan dengan gen tertentu. Penggunaan penanda RAPD relatif sederhana dan mudah dalam hal preparasi. Teknik RAPD memberikan hasil yang lebih cepat dibandingkan dengan teknik molekuler lainnya. Teknik ini juga mampu menghasilkan jumlah karakter yang relatif tidak terbatas, sehingga sangat membantu untuk keperluan analisis keanekaragaman organisme yang tidak diketahui latar belakang genomnya, baik organisme tingkat tinggi (eukariot) maupun organisme tingkat rendah (prokariot) (Bardakci 2001).
Keunggulan teknik ini antara lain kuantitas DNA yang dibutuhkan sedikit, hemat biaya, mudah dipelajari, dan primer yang diperlukan sudah banyak dikomersialisasikan sehingga mudah diperoleh. Tanaman tahunan menggunakan RAPD untuk meningkatkan efisiensi seleksi awal. Teknik RAPD telah banyak diaplikasikan dalam kegiatan pemuliaan tanaman, antara lain analisis keragaman genetik plasma nutfah tanaman (padi, kapas, dan jeruk mandarin), dan analisis populasi genetik tanaman (kakao dan kelapa) (Azrai 2005).
Gambar 2 Skema proses PCR. Penambahan primer, suhu denaturasi dan suhu penempelan Sekuen target Primer 1 Primer 2 + enzim taq polimerase dan dNTP Siklus diulang 25- 75 kali
4
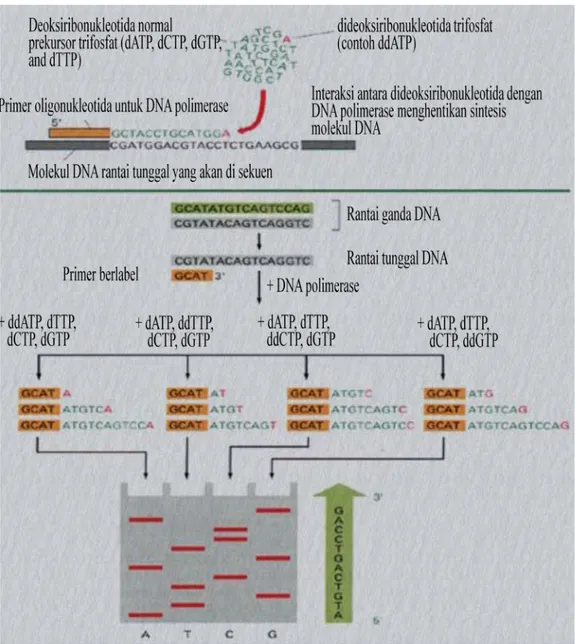
Sequencing
Sequencing adalah metode untuk
menentukan urutan basa-basa DNA dari sebuah potongan DNA. Tahun 1977, Frederick Sanger, Allan Maxam, dan Walter Gilbert merintis dikembangkannya metode
sequencing DNA yang kemudian dikenal
dengan metode sequencing Maxam dan Gilbert, yang melibatkan penggunaan suatu basa terdegradasi (Gilbert 2000). Metode
sequencing DNA yang umum digunakan saat
ini adalah metode Sanger yang bertumpu pada penggunaan analog dari rantai deoksinukleotida tripospat (dNTP) normal (Sanger 1980).
Analog-analog deoksinukleotida tripospat yang dimaksud adalah dideoksinukleotida tripospat (ddNTP). Analog-analog ini sama dengan deoksinukleotida tripospat normal, tapi tidak memiliki gugus hidroksil pada ujung 3’-nya. Ujung 5’ molekul-molekul ddNTP dapat bereaksi dengan ujung 3’ dNTP normal pada rantai DNA yang disintesis dengan bantuan enzim DNA polimerase yang juga bertindak sebagai pengendali reaksi, yaitu apabila sudah berikatan dengan satu
jenis ddNTP (ddATP, ddGTP, ddCTP, atau ddTTP) maka tidak ada nukleotida lain yang dapat ditambahkan karena ujung 3’nya tidak memiliki gugus hidroksil (Sanger 1980).
Komponen reaksi untuk tujuan sequencing terdiri dari potongan DNA yang akan disekuen, primer, campuran dNTP (dATP, dGTP, dCTP, dan dTTP, yang salah satunya diberi label radioaktif 32P) dengan konsentrasi normal, dan enzim polimerase. Campuran keempat bahan yang dilarutkan di dalam larutan penyangga tersebut kemudian dibagi rata ke dalam empat tabung. Masing-masing tabung ditambahkan ddNTP yang berbeda (ddATP, ddGTP, ddCTP, atau ddTTP) dengan konsentrasi rendah. Campuran diinkubasi pada suhu tinggi untuk memisahkan rantai ganda DNA, yang kemudian diikuti dengan perlakuan suhu rendah untuk memberikan peluang primer menempel pada DNA cetakan utas tunggal pada situs yang spesifik di ujung 5’. Enzim polimerase akan membantu sintesis rantai DNA melalui penambahan satu per satu basa yang jenisnya ditentukan oleh DNA cetakan, mulai dari ujung 3’ primer. Untuk memberikan ilustrasi kita ambil contoh pada tabung yang ditambah ddTTP. Adanya dTTP Gambar 3 Skema Proses PCR RAPD.
Suhu annealing Denaturasi DNA rantai ganda
Siklus pertama Siklus kedua
Penempelan primer Denaturasi Proses amplifikasi DNA Denaturasi DNA rantai ganda Penempelan primer Disintesis menjadi rantai DNA baru
5
pada konsentrasi normal reaksi akan terus berlanjut, tapi jika satu kali bereaksi dengan ddTTP maka reaksi akan berhenti. Setelah proses inkubasi berakhir terdapat hasil campuran reaksi dengan berbagai ukuran panjang rantai yang berakhir dengan molekul T pada ujung 3’ dan semuanya memiliki ujung 5’ yang sama (ujung 5’ dari primer), masing-masing dengan jumlah salinan yang banyak. Proses inkubasi yang sama juga dilakukan pada tiga dideoksinukleotida tripospat yang lain, masing-masing dengan memberi campuran reaksi ddCTP, ddATP, dan ddGTP yang dapat menghentikan reaksi pada posisi C, A, dan G secara berturut-turut (Sanger 1980).
Empat reaksi ini masing-masing kemudian dipisahkan menggunakan elektroforesis gel
akrilamid yang memiliki resolusi tinggi. Teknik ini memisahkan rantai berdasarkan ukuran, rantai yang berukuran lebih kecil akan bergerak lebih cepat sedangkan yang ukuran rantainya lebih besar akan bergerak lebih lambat. Metode ini dapat membandingkan secara akurat dan cepat, serta dapat membaca urutan basa sampai 300 nukleotida dari ujung 3’ primer. Empat lubang sisir pada gel akrilamid masing-masing akan diisi oleh hasil reaksi dengan dideoksinukleotida berbeda. Setiap sampel akan membentuk pola pita yang berbeda dan ukuran nukleotida yang lebih pendek ditunjukkan oleh pita yang terletak di bagian bawah gel. Urutan basa hasil sekuen kemudian dibaca dari dasar (ujung 5’) ke atas (ujung 3’) gel (Gilbert 2000). Skema proses
sequencing dapat dilihat pada Gambar 4.
6
BAHAN DAN METODE
Bahan dan Alat
Alat-alat yang digunakan adalah sentrifus
Beckman Coulter AllegraTM 64R Centrifuge,
Eppendorf sentrifuge 5417R, otoklaf, perangkat elektroforesis Toylab, pH meter
CyberScan 510, mesin PCR Biometra,
spektrofotometer UV-VIS Beckman
Coulter-DU 530, transilluminator Biorad, penangas es
dan air, pipet mikro, pipet Mohr, tabung Eppendorf, DNA speed vacum 110 savant, tabung sentrifus, neraca analitik Scaltec, alat yang mendokumentasikan hasil pengamatan elektroforesis dengan UV (UV T2201 Sigma dan Power Shot A640 Canon), skalpel, inkubator, cawan petri, mortar, vorteks, dan peralatan kaca laboratorium.
Bahan-bahan yang digunakan adalah ± 1 gram sampel kelapa sawit bagian daun (D), bunga (B), buah (F) yang normal (N) dan abnormal (Ab). Bahan yang digunakan untuk isolasi DNA yaitu larutan CTAB (cethyl
trimethyl ammonium bromide) 10% , Tris-HCl
1M pH 8.0, larutan EDTA (etilendiamin tetraasetat) 0.5 M pH 8.0, larutan NaCl 5 M , etanol 70% dingin, isopropanol dingin, etanol absolut dingin, β-merkaptoetanol 1%, larutan CI (kloroform:isoamilalkohol = 24:1), nitrogen cair, larutan Na-asetat 3 M pH 5.2,
polyvinyl-polypirollidone (PVPP), bufer TE
(tris-HCl dan EDTA), molecular grade water (MW), dan RNAse. Proses PCR-RAPD menggunakan kit merek Bioron yang terdiri dari complete buffer (mengandung MgCl2),
dNTP, dan enzim taq polimerase. Primer untuk PCR-RAPD adalah OPA dan OPB dari
Operon. Bahan untuk elektroforesis yaitu loading buffer (bromfenolblue 2.5%:sukrosa
40%), agarosa, EtBr 1% (w/v), bufer TBE, dan marker 1 kb (+) (M). Elusi fragmen DNA menggunakan kit AxyPrep DNA Gel
Extraction merek AXYGEN yang berisi bufer
DE-A, bufer DE-B, isopropanol, bufer W1,
bufer W2, dan larutan eluen. Proses
sequencing menggunakan kit sequencing.
Metode Isolasi DNA
Isolasi molekul DNA dilakukan dengan metode Castillo. Sebanyak satu gram sampel ditimbang dengan aluminium foil, kemudian direndam dalam N2 cair. Sampel digerus
sampai halus dengan mortar yang sudah didinginkan dengan N2 cair. Nitrogen cair
selalu ditambahkan hingga sampel selesai
digerus. Sebanyak ± 0.1 gram PVPP ditambahkan saat sampel digerus.
Serbuk halus yang dihasilkan dimasukkan dalam tabung sentrifus yang berisi 5 ml bufer ekstraksi dan 1% merkaptoetanol (50 μl) yang sudah dipanaskan. Campuran tersebut dikocok kemudian dipanaskan selama 30 menit pada suhu 65 °C sambil sesekali dikocok agar tidak mengendap kemudian didinginkan pada suhu kamar. Larutan CI ditambahkan sebanyak 1 volume ke dalam masing-masing tabung dan bobotnya diseimbangkan sebelum sentrifugasi. Campuran divorteks sampai homogen lalu disentrifugasi (±13500 g, 10 menit, 4 °C). Sebanyak 4 ml supernatan yang diperoleh dipindahkan ke tabung sentrifus baru dan ditambah larutan CI 1 volume. Campuran ini kemudian divorteks sampai homogen dan disentrifugasi seperti yang dilakukan sebelumnya. Supernatan yang diperoleh dipindahkan ke tabung baru sebanyak 3 ml dan ditambah larutan CI 1 volume. Campuran kemudian dihomogenasi dengan vorteks dan disentrifugasi seperti sebelumnya. Supernatan yang diperoleh dipindahkan ke tabung sentrifus baru sebanyak 1 ml dan ditambah isopropanol dingin 1 volume. Larutan dikocok perlahan kemudian disimpan dalam lemari es selama 30 menit. Larutan kemudian disentrifugasi kembali seperti sebelumnya dan supernatan yang diperoleh dibuang. Pelet yang dihasilkan kemudian dikeringkan.
Pelet DNA dilarutkan dalam 1 ml bufer TE, setelah itu ditambah CH3COONa dingin 3
M pH 5.2 sebanyak 1/10 volume (100 μl) dan 2.5 ml etanol absolut dingin secara berurutan kemudian disimpan dalam pendingin (-20 °C) selama 30 menit atau semalam. Suspensi disentrifugasi (±16000 g, 10 menit, 4 °C). Supernatannya dibuang dan pelet DNA yang diperoleh dicuci dengan etanol dingin 70% sebanyak 400 μl kemudian diendapkan dengan sentrifugasi (±16000 g, 5 menit, 4 °C). Pelet yang diperoleh dikeringkan dengan membalikkan tabung di atas tisu dan diangin-anginkan sebentar hingga bau etanol hilang. Pelet DNA dilarutkan dengan MW 100 μl. Jika pelet yang dihasilkan cukup banyak maka MW bisa ditambahkan lebih banyak. Setelah semua pelet DNA larut kemudian dipindahkan dalam tabung Eppendorf kecil dan disimpan dalam pendingin sebagai DNA stok.
Pemurnian DNA
Stok DNA dapat dimurnikan dengan ditambah RNAse. Volume RNAse yang ditambahkan 1/10 dari volume DNA.
7
Percobaan ini menggunakan 50 μl DNA ditambah 5 μl RNAse dan ditempatkan pada tabung Eppendorf baru dalam penangas es. Campuran larutan DNA dan RNAse dimasukkan dalam penangas air 37 °C selama 1 jam kemudian disimpan dalam pendingin sampai digunakan.
Uji Kualitatif dan Kualitas DNA
Tahapan ini dilakukan untuk mengetahui DNA yang berhasil diisolasi serta dapat dilihat kualitas DNA yang diperoleh. Gel agarosa 1% yang digunakan untuk elektroforesis dibuat dengan 0.3 g agarosa ditambah 30 ml TBE 0.5× untuk cetakan yang kecil atau 0.6 g agarosa dengan 60 ml TBE 0.5× untuk yang besar, kemudian dipanaskan hingga larut dan didinginkan pada suhu kamar sampai hangat. Selanjutnya larutan dituang ke cetakan gel elektroforesis yang sudah dipasangi sisir (cetakan sumur) dan ditunggu sampai gel padat. Gel yang sudah padat dipindahkan dalam bak elektroforesis yang berisi TBE 0.5×. Sampel yang akan dielektroforesis dicampur loading buffer dan MW dengan komposisi 2 μl DNA, 1 μl
loading buffer, dan 3 μl MW. Semuanya
dicampur dengan pipet mikro kemudian diinjeksikan ke dalam sumur pada gel agarosa. Alat elektroforesis dihubungkan pada power
supply yang dialiri arus listrik 50 Volt selama
±1 jam, kemudian gel direndam dalam larutan EtBr dengan konsentrasi 1% dan hasilnya diamati dengan bantuan lampu UV.
Elektroforesis gel agarosa juga digunakan untuk mengamati hasil PCR DNA. Perbandingan komposisi yang digunakan antara loading buffer dan sampel adalah 1:5. Marker yang digunakan memiliki ukuran 1 kb sebanyak 1 μl.
Uji Kuantitatif DNA
Metode spektrofotometer UV digunakan untuk uji kuantitatif DNA berdasarkan prinsip bahwa radiasi sinar ultraviolet akan diserap oleh nukleotida. Suspensi DNA hasil isolasi sebanyak 3 μl diencerkan menjadi 300 μl dengan MW. Larutan kemudian dituang dalam kuvet dan dibaca absorbansinya pada panjang gelombang (λ) 260 nm, 280 nm, dan 230 nm. Absorbansi dibaca pada tiga panjang gelombang untuk mengetahui adanya kontaminasi protein (280 nm) dan kontaminasi polisakarida dan senyawa fenol (230 nm). Kemurnian DNA ditentukan dengan nilai perbandingan A260/A280.
Penyerapan maksimal radiasi UV oleh DNA pada panjang gelombang 260 nm. Jika
absorbansi yang diperoleh 1.000, maka konsentrasi DNA setara dengan 50 μg/ml (Brown 2003). Nilai ini adalah faktor konversi, sehingga diperoleh rumus untuk menghitung konsentrasi DNA adalah:
DNA (μg/ml) = A260 × faktor konversi ×
faktor pengenceran
Proses PCR-RAPD
Senyawa DNA diamplifikasi dengan mesin PCR. Konsentrasi DNA yang digunakan untuk PCR disamakan menjadi 100 ng/μl. Sebanyak 1 μl larutan DNA (100 ng/μl) ditambahkan ke dalam campuran mix untuk PCR yang berisi campuran larutan 2.5 μl bufer yang mengandung MgCl2, 0.5 μl dNTP,
0.3 μl enzim taq polimerase DNA, 1.0 μl primer acak (OPA dan OPB), kemudian ditambah MW hingga volumenya menjadi 25 μl. Campuran tersebut kemudian dimasukkan ke dalam alat PCR dengan program 94 °C selama 3 menit 1 siklus, suhu 94 °C selama 1 menit untuk denaturasi, 36 °C selama 1 menit untuk penempelan primer, dan 72 °C selama 2 menit untuk pemanjangan rantai DNA baru (45 siklus), serta diakhiri dengan suhu 72 °C selama 4 menit untuk memastikan DNA yang diamplifikasi mengalami renaturasi sempurna. Hasil amplifikasi DNA dengan primer yang sama pada satu individu memiliki intensitas yang berbeda-beda. Penyebaran tempat penempelan primer pada DNA, jumlah fragmen yang teramplifikasi serta kemurnian dan konsentrasi cetakan DNA dalam reaksi akan mempengaruhi intensitas pita yang dihasilkan. Semakin banyak fragmen DNA yang teramplifikasi dengan primer maka intensitas pita akan semakin jelas. Kontaminasi oleh senyawa-senyawa fenolik dan polisakarida akan menghasilkan pita amplifikasi yang kabur atau kurang baik.
Tahap Pemurnian DNA Hasil PCR (Elusi)
Bagian gel yang menunjukkan fragmen DNA yang diinginkan dipotong dengan skapel, ditimbang, dan disimpan dalam tabung mikro berukuran 2 ml. PCR diulangi lagi dengan volume yang lebih besar. Fragmen DNA dimurnikan dari gel menggunakan kit
AxyPrep DNA Gel Extraction. Bobot gel
sebesar 100 mg setara dengan 100 μl volume bufer DE-A yang ditambahkan. Volume bufer DE-A yang ditambahkan sebanyak tiga kali bobot gel. Campuran tersebut kemudian dipanaskan suhu 75 °C hingga larut selama 6-8 menit dan dikocok setiap 2-3 menit. Bufer DE-B ditambahkan sebanyak setengah volume bufer DE-A. Jika fragmen DNA
8
berukuran kurang dari 400 bp maka harus ditambah isopropanol satu volume.
Larutan ditransfer ke kolom Axyprep yang ditempatkan dalam tabung mikro 2 ml kemudian disentrifugasi ±13500 g selama 2 menit pada suhu 25 °C. Filtrat yang diperoleh dibuang karena DNA dianggap menempel pada kolom. Bufer W1 ditambahkan sebanyak
500 μl kemudian disentrifugasi (±13500 g, 2 menit, 25 °C). Filtrat yang diperoleh dibuang kemudian ditambah 700 μl bufer W2 dan
disentrifugasi lagi (±13500 g, 2 menit, 25 °C). Proses ini diulang dua kali. Sentrifugasi dilakukan sekali lagi untuk membersihkan kolom dari larutan bufer. Kolom dipindahkan ke tabung mikro 1.5 ml kemudian ditambah 20-30 μl eluet tepat di tengah membran filter kolom dan biarkan selama ± 1 menit pada suhu ruang. Tabung yang berisi kolom kemudian disentrifugasi pada ±18500 g selama 2 menit dan hasilnya dicek dengan elektroforesis gel agarosa.
Pengurutan Basa Fragmen DNA
(Sequencing)
Fragmen DNA yang diduga berbeda hasil PCR yang telah dimurnikan kemudian diurutkan. Proses pengurutan dilakukan di Lembaga Biologi Molekular Eijkman Jakarta dengan menggunakan prinsip sequencing metode Sanger. Urutan basa DNA hasil pengurutan berupa elektroforegraf.
Urutan basa DNA hasil pengurutan dianalisis dengan program BLASTn (Basic
Local Alignment and Search Tool for Nucleotida) (www.ncbi.nlm.nih.gov) untuk
mengetahui kesesuaian urutan basa atau asam amino yang didapatkan dari suatu gen atau protein dengan urutan lain yang ada dalam bank data gen. Fragmen DNA hasil PCR yang diurutkan dianalisis dengan program BLASTn. Analisis dengan program ini dilakukan untuk mengetahui kesesuaian antara urutan basa yang didapatkan dengan urutan yang terdapat dalam bank data gen. Urutan yang didapatkan diharapkan memiliki tingkat homologi yang tinggi (e-value <10-4) dengan gen yang sesuai (Claverie 2003).
HASIL DAN PEMBAHASAN
Isolasi DNA dari Daun, Bunga, dan Buah Kelapa Sawit
Isolasi DNA dalam penelitian ini dilakukan menggunakan metode Orozco-Castillo yang sudah dimodifikasi. Metode ini banyak digunakan untuk isolasi DNA
tanaman karena mudah dan kemungkinan adanya enzim pendegradasi DNA lebih kecil dibanding dengan metode yang lain (Rogers & Bendich 1994). Isolasi dari organisme eukariot biasanya dilakukan melalui proses penghancuran sel (lysis), pemusnahan protein dan RNA, serta pemurnian DNA.
Metode isolasi diawali dengan penggerusan untuk membantu memecahkan dinding sel secara mekanik. Penambahan PVPP saat penggerusan dan merkaptoetanol dalam bufer ekstraksi bertujuan menghambat enzim polifenol oksidase yang dapat mendegradasi rantai DNA dan menyebabkan teroksidasinya senyawa fenol. Terjadinya oksidasi ditandai dengan terbentuknya warna coklat pada jaringan tanaman yang akan diisolasi. Penambahan nitrogen cair saat penggerusan juga dapat mencegah terjadinya oksidasi dan kerusakan DNA.
Campuran dalam bufer ekstraksi Castillo memiliki fungsi berbeda-beda. Suatu detergen kationik yang terdapat dalam bufer ekstraksi yaitu CTAB berfungsi membantu proses pemecahan membran sel. Bufer ekstraksi perlu dipanaskan terlebih dahulu pada suhu 65°C , karena CTAB yang berkemampuan melisis membran sel akan aktif pada kondisi panas (65°C). Larutan deterjen berfungsi menurunkan tegangan permukaan cairan dan melarutkan lipid sehingga membran sel mengalami degradasi, dan organel-organel di dalamnya dapat keluar dari sel. Isolasi DNA dari tanaman sering menghasilkan ekstrak yang banyak mengandung polisakarida. Penambahan CTAB yang bermuatan positif pada bufer ekstrak juga berfungsi untuk memisahkan polisakarida dari DNA dengan cara mengikat DNA yang bermuatan negatif. Penghancuran sel secara kimiawi dapat dilakukan dengan memanfaatkan senyawa kimia EDTA (etilendiamin tetraasetat). Fungsi EDTA adalah sebagai perusak sel dengan cara mengikat ion magnesium sebagai prekursor enzim sehingga enzim menjadi tidak aktif. Larutan NaCl sebagai larutan isotonik yang menjaga tekanan osmotik sel agar DNA tidak rusak. Larutan tris-HCl untuk memberikan kondisi pH yang optimum.
DNA yang tercampur dengan polisakarida, protein, dan pengotor lainnya perlu dibersihkan. Pembersihan DNA dilakukan dengan ekstraksi menggunakan larutan CI dan sentrifugasi. Larutan kloroform dapat menghilangkan kontaminasi akibat polisakarida sedangkan sentrifugasi akan memisahkan molekul-molekul berdasarkan bobot molekulnya. Larutan CI sebagai pelarut
9
organik dapat menghancurkan dan mengendapkan protein. Ekstraksi yang dilakukan berulang-ulang bertujuan agar DNA benar-benar terbebas dari pengotor. Ekstraksi dengan cara bertahap juga merupakan cara ekstraksi yang terbaik karena akan menghasilkan jumlah ekstrak yang lebih banyak dibandingkan ekstraksi secara langsung. Perbedaan kelarutan akan mengakibatkan organel-organel kecil dan pengotor terekstrak, sedangkan inti sel atau
nucleus diasumsikan tidak ikut terekstrak
sebab memiliki ukuran yang besar sehingga di dalam larutan hanya terdapat inti sel yang mengandung DNA.
Hasil larutan yang disentrifugasi akan menghasilkan campuran larutan yang terpisah menjadi tiga fase. Larutan CI memiliki densitas yang tinggi sehingga terletak di bagian bawah (fase organik) tabung sentrifus. Bagian tengah larutan terdapat protein yang dilarutkan oleh larutan CI. Larutan DNA yang terletak di bagian atas (fase air) dipipet dengan hati-hati agar bagian proteinnya tidak ikut terambil. Larutan DNA yang diambil dimasukkan dalam tabung sentrifus baru dan diekstraksi kembali dengan larutan CI.
Pemurnian DNA dilakukan dengan etanol dan isopropanol. Larutan-larutan tersebut dapat mengendapkan DNA sedangkan kontaminan yang lain tetap larut (Sambrook 1989). Penambahan Na-asetat berfungsi untuk membantu memekatkan dan mengendapkan DNA. Pencucian endapan DNA dengan etanol 70% bertujuan memisahkan senyawa-senyawa yang masih menempel pada DNA. DNA akan lebih stabil dalam bentuk larutan oleh karena itu DNA yang diperoleh dilarutkan dalam MW. Pemurnian DNA dari kontaminan RNA dilakukan menggunakan enzim RNAse yang berfungsi mendegradasi RNA sehingga dihasilkan DNA murni yang terbebas dari RNA dan siap digunakan sebagai cetakan DNA saat PCR.
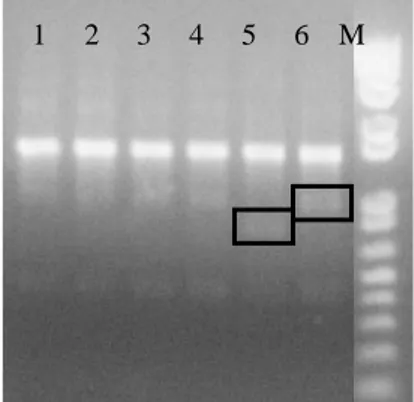
Konsentrasi DNA diukur dengan spektrofotometer pada panjang gelombang 260 nm. Ikatan rangkap terkonjugasi pada basa heterosiklik purin dan pirimidin menjadikan nukleosida, nukleotida, serta polinukleotida menyerap sinar UV (Murray 2003). Tabel 1 menunjukkan konsentrasi DNA hasil isolasi dan tingkat kemurniannya dari kontaminasi. Hasil elektroforesis DNA yang diisolasi (Gambar 5) menunjukkan bahwa konsentrasi DNA yang berhasil diisolasi tinggi, ditunjukkan oleh tebalnya pita DNA yang dihasilkan. Pita DNA hasil elektroforesis yang semakin tebal belum tentu
berbanding lurus dengan tingginya konsentrasi DNA. Kontaminasi senyawa polisakarida dan protein dapat mempengaruhi ketebalan pita, sehingga perlu dilakukan pengukuran kemurnian DNA terhadap senyawa kontaminan tersebut dengan spektrofotometer.Tingkat kemurnian DNA dapat dilihat dari perbandingan absorbansi panjang gelombang 260/280 nm (rasio F1) dan
260/230 nm (rasio F2). Rasio F1 menunjukkan
tingkat kemurnian terhadap kontaminan protein dan rasio F2 menunjukkan tingkat
kemurnian akibat kontaminan polisakarida. Nilai rasio yang baik berkisar pada 1.8-2.0 (Sambrook 1989).
Tabel 1 Data hasil spektrofotometer larutan DNA sampel Sampel* A260 [DNA] (µg/ml) 260/280 (rasio F1) 260/230 (rasio F2) DN 0.164 820 1.843 1.908 Dab 0.173 865 1.687 1.347 BN 0.236 1180 1.867 1.870 BAb 0.259 1295 1.847 1.828 FN 0.160 800 1.855 1.889 FAb 0.179 895 1.845 1.513 *Keterangan sampel; DN: daun normal, Dab: daun abnormal, BN: buah normal, BAb: buah abnormal, FN: buah normal, FAb: buah abnormal.
1 2 3 4 M 5 6
Gambar 5 Hasil elektroforesis larutan DNA sampel.
Amplifikasi DNA Hasil Isolasi dengan Primer Acak
Konsentrasi cetakan DNA yang akan diamplifikasi disamakan konsentrasinya menjadi 100 ng/μl. Tingginya konsentrasi cetakan DNA yang digunakan dalam PCR akan menghasilkan amplifikasi DNA yang kurang baik. Cetakan DNA yang terlalu banyak kemungkinan menyulitkan primer yang digunakan untuk menempel. Sebaliknya
Keterangan gambar: (1) DN (2) Dab (3) BN (4) BAb (5) FN (6) FAb (M) Marker 1 kb +
10
jika konsentrasi cetakan DNA terlalu rendah maka akan terjadi kompetisi tempat penempelan primer pada templat sehingga menyebabkan salah satu fragmen akan diamplifikasi dalam jumlah banyak sedangkan fragmen yang lain sedikit.
Amplifikasi cetakan DNA membutuhkan primer spesifik (sekuen oligonukelotida khusus) yang mengenali dan menempel pada situs tertentu pada cetakan DNA dengan sekuen nukleotida terkait. Primer biasanya terdiri dari 10-20 nukleotida dan dirancang berdasarkan daerah konservatif dalam genom tersebut. Semakin panjang ukuran primer, semakin spesifik daerah yang diamplifikasi. Jika suatu kelompok organisme memang berkerabat dekat, maka primer dapat digunakan untuk mengamplifikasi daerah tertentu yang sama dalam genom kelompok tersebut. Beberapa faktor yang berpengaruh seperti konsentrasi DNA contoh, ukuran panjang primer, komposisi basa primer, konsentrasi ion Mg, dan suhu hibridisasi primer harus dikontrol dengan hati-hati agar dapat diperoleh pita-pita DNA yang utuh dan baik (Suryanto 2003). Primer yang digunakan pada penelitian ini adalah primer acak OPA dan OPB yang memiliki panjang 10 basa yang urutannya berbeda-beda.
Hasil Amplifikasi DNA dengan Primer OPA
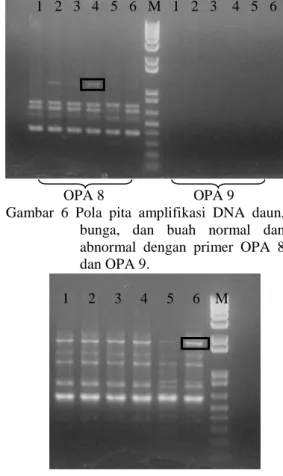
Pola pita amplifikasi DNA yang terbentuk antara sampel dengan primer OPA 6 dan OPA 7 hampir sama . Gambar 6 menunjukkan pola pita DNA yang teramplifikasi dengan primer OPA 8 dan OPA 9. Terdapat pola pita yang berbeda antara fragmen DNA bunga normal dan abnormal yang teramplifikasi dengan primer OPA 8. Terdapat pola pita pada bunga abnormal yang tidak terdapat pada bunga normal. Fragmen yang berbeda ini kemudian diisolasi dengan metode elusi menggunakan kit AxyPrep DNA Gel Extraction merek AXYGEN. Sequencing hasil elusi selanjutnya dilakukan untuk mengetahui perbedaan dengan yang normal. Fragmen cetakan DNA tidak menghasilkan pola pita amplifikasi DNA dengan primer OPA 9. Hal ini mungkin dapat disebabkan oleh primer yang belum larut sempurna, serta akibat adanya kontaminasi senyawa protein dan polisakarida dalam larutan DNA yang dapat mengganggu proses amplifikasi DNA. Pita amplifikasi yang dihasilkan cetakan DNA (daun, bunga, dan buah) normal dan abnormal dengan primer OPA 10 memiliki pola yang sama. Hal yang sama juga terjadi pada DNA sampel
(daun, bunga, dan buah) yang teramplifikasi dengan primer OPA 14, OPA 15, dan OPA 17. Hasil amplifikasi DNA cetakan buah abnormal dengan primer OPA 18 dan OPA 20 terdapat perbedaan, yaitu terdapat pola pita pada buah abnormal yang tidak terdapat pada buah normal (Gambar 7 dan Gambar 8).
Sequencing dilakukan pada fragmen yang
berbeda hasil elusi.
1 2 3 4 5 6 M 1 2 3 4 5 6
OPA 8 OPA 9
Gambar 6 Pola pita amplifikasi DNA daun, bunga, dan buah normal dan abnormal dengan primer OPA 8 dan OPA 9.
1 2 3 4 5 6 M
Gambar 7 Pola pita amplifikasi DNA daun, bunga, dan buah normal dan abnormal dengan primer OPA 18. 1 2 3 4 5 6 M 1 2 3 4 5 6
OPA 19 OPA 20 Gambar 8 Pola pita amplifikasi DNA daun,
bunga, dan buah normal dan abnormal dengan primer OPA 19 dan OPA 20.
11
Hasil Amplifikasi DNA dengan Primer OPB
Hasil amplifikasi cetakan DNA dengan primer OPB 1 hampir tidak terbentuk pita amplifikasi kecuali pada sampel DNA buah abnormal. Gambar 9 menunjukkan adanya pola pita yang diduga berbeda antara fragmen DNA buah normal dan abnormal yang diamplifikasi dengan primer OPB 2. Fragmen cetakan DNA normal dan abnormal dengan primer OPB 3 menghasilkan pola pita amplifikasi yang hampir sama, sama halnya dengan hasil amplifikasi menggunakan primer OPB 7, OPB 10, dan OPB 17. Gambar 10 menunjukkan terdapat pola pita yang diduga berbeda dihasilkan dari amplifikasi fragmen cetakan DNA buah normal dan abnormal dengan primer OPB 9.
Jumlah pita polimorfik hasil amplifikasi berbeda-beda. Semakin banyak pita polimorfik yang dihasilkan akan semakin mudah untuk mengamati adanya variasi. Adanya perbedaan pola pita yaitu berdasarkan jumlah dan ukuran pita menggambarkan adanya genom tanaman yang sangat kompleks (Grattapaglia dkk 1992). Hasil amplifikasi yang memperoleh jumlah pita yang tinggi serta banyaknya pita yang polimorfik dapat dijadikan sebagai kandidat penanda yang baik untuk sifat tertentu dan dapat diseleksi dengan uji selanjutnya.
Terdapat sampel yang tidak teramplfikasi dengan primer yang digunakan. Hasil amplifikasi yang kurang baik dapat disebabkan oleh ketidaksesuaian primer, efisiensi, dan optimasi proses PCR. Primer yang tidak spesifik atau sesuai dapat menyebabkan teramplifikasinya daerah lain dalam genom yang tidak dijadikan sasaran atau sebaliknya tidak ada daerah genom yang teramplifikasi. Optimasi PCR juga diperlukan untuk menghasilkan karakter yang diinginkan. Optimasi ini menyangkut suhu denaturasi dan annealing DNA dalam mesin PCR. Suhu denaturasi yang rendah dapat menyebabkan belum terbukanya DNA utas ganda sehingga tidak dimungkinkan terjadinya penempelan primer. Proses penempelan primer pada utas DNA yang sudah terbuka memerlukan suhu optimum, sebab suhu yang terlalu tinggi dapat menyebabkan amplifikasi tidak terjadi karena primer tidak menempel atau sebaliknya suhu yang terlalu rendah menyebabkan primer menempel pada sisi lain genom yang bukan sisi homolognya. Hal ini menyebabkan teramplifikasi banyak daerah tidak spesifik dalam genom tersebut. Suhu penempelan (annealing) ini ditentukan berdasarkan primer
yang digunakan yang dipengaruhi oleh panjang dan komposisi primer. Suhu penempelan ini sebaiknya sekitar 5°C di bawah suhu leleh. Suhu penempelan yang digunakan untuk primer OPA dan OPB adalah 36ºC (Toruan-Mathius N, Bangun SI, Maria-Bintang 2001). Secara umum suhu leleh (Tm) dihitung dengan rumus Tm = 4(G+C) + 2(A+T)°C (Rybicky 1996).
Pita-pita DNA (yang berbeda) yang muncul hanya pada tanaman normal atau abnormal dapat diisolasi dari gel agarosa. Sebelumnya perlu dilakukan PCR ulang dengan volume reaksi yang lebih besar (100 µl) dan dielektroforesis pada gel agarosa. Produk PCR kemudian dimurnikan dari gel agarosa menggunakan kit AxyPrep DNA Gel
Extraction merek AXYGEN. Hasil pemurnian
pita DNA dari gel di cek ulang pada gel agarosa 1% dan urutan basanya diketahui dengan sequencing. Urutan basa tersebut kemudian dianalisis dengan program BLAST untuk mengetahui tingkat kemiripannya dengan urutan basa dari beberapa gen yang sudah ada di bank data gen.
1 2 3 4 5 6 M
Gambar 9 Pola pita amplifikasi DNA daun, bunga, dan buah normal dan abnormal dengan primer OPB 2. 1 2 3 4 5 6 M
Gambar 10 Pola pita amplifikasi DNA daun, bunga, dan buah normal dan abnormal dengan primer OPB 9.
12
Analisis Hasil Urutan Basa Fragmen DNA Sequencing dilakukan satu arah pada 6
sampel DNA yang menghasilkan pola pita amplifikasi yang diduga berbeda, yaitu BAb (primer OPA 8), FN (primer OPB 2), dan FAb (primer OPA 18, OPA 20, OPB 9). Hasil
sequencing yang diperoleh tidak begitu bagus,
karena dari 6 sampel yang disekuen hanya sampel FAb dengan primer OPB 9 yang terbaca urutan basanya. Hal ini diduga disebabkan oleh pemurnian yang kurang baik, digunakannya produk PCR murni untuk
sequencing tanpa diklon terlebih dahulu, atau
proses sequencing yang kurang sempurna. Urutan basa yang diperoleh dari hasil
sequencing (Gambar 12) selanjutnya
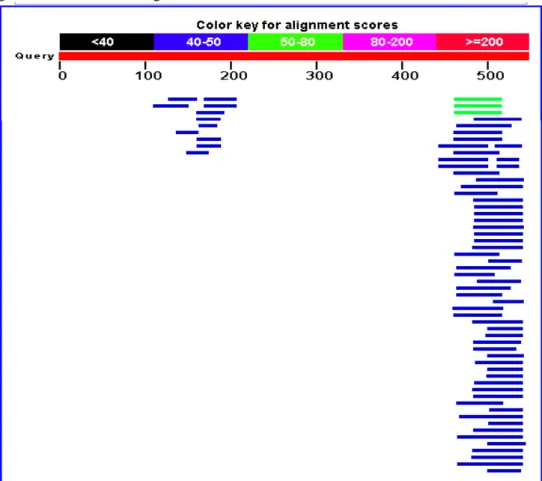
dianalisis dengan program BLAST dan hasilnya dapat dilihat pada grafik Gambar 11. Tampilan grafik menunjukkan fragmen DNA yang dianalisis memiliki tingkat homologi yang rendah dengan gen yang terdapat dalam bank gen ditunjukkan garis dalam grafik yang hampir semuanya berwarna biru. Hasil analisis juga tidak mendapatkan kesesuaian antara fragmen DNA dengan salah satu gen yang terdapat pada tanaman kelapa sawit yang sudah ada dalam bank gen. Garis
berwarna merah menunjukkan tingkat homologi yang sangat tinggi (≥ 200
nukleotida), garis merah muda menunjukkan tingkat homologi yang tinggi (80-200 nukleotida), garis hijau menunjukkan tingkat homologi sedang (50-80 nukleotida), garis biru menunjukkan tingkat homologi rendah (40-50 nukleoida), dan garis warna hitam menunjukkan tingkat homologi yang sangat rendah (< 40 nukleotida).
Hasil dari analisis yang kurang baik juga ditunjukkan dari nilai bit score yang rendah (kurang dari 150) dan e-value lebih dari 10-4 .
Bit score merupakan ukuran yang sangat
penting untuk penjajaran. Semakin tinggi bit
score maka tingkat homologi kedua sekuen
juga semakin tinggi. Sedangkan nilai e-value merupakan nilai dugaan yang memberikan ukuran statistik yang signifikan terhadap kedua sekuen. Nilai e-value yang semakin tinggi menunjukkan tingkat homologi antara kedua sekuen semakin rendah, dan jika nilai
e-value semakin rendah maka maka tingkat
homologi kedua sekuen semakin tinggi. Apabila nilai e-value 0 (nol) hal ini menunjukkan bahwa kedua sekuen tersebut identik (Claverie dan Notredame 2003).
Gambar 11 Analisis BLAST hasil sequencing fragmen DNA buah abnormal dengan primer OPB 9.

![Tabel 1 Data hasil spektrofotometer larutan DNA sampel Sampel* A 260 [DNA] (µg/ml) 260/280 (rasio F 1 ) 260/230 (rasio F2 ) DN 0.164 820 1.843 1.908 Dab 0.173 865 1.687 1.347 BN 0.236 1180](https://thumb-ap.123doks.com/thumbv2/123dok/4312567.2907893/31.892.478.774.440.610/tabel-data-hasil-spektrofotometer-larutan-sampel-sampel-rasio.webp)