Protein evolution drives the evolution of the genetic code
and vice versa
Miguel A. Jime´nez-Montan˜o
a,b,*
aInno6ationskolleg Theoretische Biologie,Humboldt-Uni6ersita¨t zu Berlin,In6alidenstrasse43,D-10115Berlin,Germany bDepartamento de Fı´sica y Matema´ticas,Uni6ersidad de las Ame´ricas/Puebla,Sta.Catarina Ma´rtir,72820Puebla,Mexico
Received 30 March 1999; received in revised form 9 July 1999; accepted 12 July 1999
Abstract
A model for the developmental pathway of the genetic code, grounded on group theory and the thermodynamics of codon – anticodon interaction is presented. At variance with previous models, it takes into account not only the optimization with respect to amino acid attributes but, also physicochemical constraints and initial conditions. A ‘simple-first’ rule is introduced after ranking the amino acids with respect to two current measures of chemical complexity. It is shown that a primeval code of only seven amino acids is enough to build functional proteins. It is assumed that these proteins drive the further expansion of the code. The proposed primeval code is compared with surrogate codes randomly generated and with another proposal for primeval code found in the literature. The departures from the ‘universal’ code, observed in many organisms and cellular compartments, fit naturally in the proposed evolutionary scheme. A strong correlation is found between, on one side, the two classes of aminoacyl-tRNA synthetases, and on the other, the amino acids grouped by end-atom-type and by codon type. An inverse of Davydov’s rules, to associate the amino acid end atoms (O/N and non-O/non-N) of 18 amino acids with codons containing a weak base (A/U), extended to the 20 amino acids, is derived. © 1999 Elsevier Science Ireland Ltd. All rights reserved.
Keywords: Protein evolution; Developmental pathway; Primeval code; Codon reassignments; Codon – anticodon interaction; Aminoacyl-tRNA synthetases
www.elsevier.com/locate/biosystems
‘‘It is not the number of available signals but rather their distinguishability that matters in communication’’ Schumacher (1991).
1. Introduction
The problem of the origin and evolution of protein synthesis constitutes one of the major transitions in evolution, which is far from being solved at the present time. In the sensibly and acute statement of Smith and Szathma´ry (1995), ‘‘The origin of the code is perhaps the most
* Tel.: +52-28-29-2676; fax:+52-28-29-2045.
E-mail address: [email protected] (M.A. Jime´nez-Mon-tan˜o)
perplexing problem in evolutionary biology. The existing translational machinery is at the same time so complex, so universal, and so essential that it is hard to see how it could have come into existence, or how life could have existed without it’’.
Several authors have approached this problem by building possible scenarios in which the genetic code could have originated. See the book by Smith and Szathma´ry (1995) and the fast growing literature on the RNA world (Gesteland and Atkins, 1993). Most of these models are con-cerned with the catalytic properties of ribozymes. These are RNA molecules, assumed to play the role of enzymes in a primordial self-replicating system from which, it is presumed, the modern translational machinery originated. Another ap-proach is the search for ancestors of transfer-RNA (Eigen et al., 1989; Rodin et al., 1996).
The regularities of the codon catalogue were recognized from the very beginning (Sonneborn, 1965; Epstein, 1966; Goldberg and Wittes, 1966; Woese, 1967; Alff-Steinberg, 1969). However, the use of a not completely appropriate mathematical framework for their description had been the main drawback to obtain an understanding of the code’s possible evolution (Karasev and Sorokin, 1997). The customary expression ‘organization of the code’ really refers to two different problems, (i) the distribution of redundancy in the code, i.e. the ‘block structure’ of synonymous codons and the positions of the three stop codons (Gold-man, 1993), and (ii) the amino acid assign-ments. The answer to the first question is indepen-dent of the answer to the second one. It depends on the codon – anticodon interaction energy (Jime´nez-Montan˜o, 1994; Jime´nez-Montan˜o et al. 1995)1.
The standard approach to these problems em-ploys a three-dimensional sequence space of codons, equipped with a Hamming distance (the number of positions where the nucleotides differ in a codon pair). Because, implicitly or explicitly, it is assumed that ‘minimum change’ is synony-mous of single-nucleotide-change (Goldberg and Wittes, 1966). Further, in a recent paper (Xia and Li, 1998) the authors say that ‘‘it has long been proposed that the genetic code might have been arranged in such a way as to reduce the effect of non-synonymous mutations involving single nu-cleotide changes’’. This approach is inexact, be-cause not all single nucleotide changes are equivalent, mutational transitions and transver-sions (complementary and non-complementary) are not equal, and also because the three positions in a codon have different thermodynamic stability and mutation frequency.
An appropriate mathematical framework to represent the code, as a six-dimensional Boolean hypercube, was first proposed by Jime´nez-Mon-tan˜o and De la Mora-Basan˜ez (1992). This result was obtained building on pioneering algebraic approaches by Danckwerts and Neubert (1975), Bertman and Jungck (1979), Swannson (1984). The related early work of Rumer (1968) was unknown to the authors. More complete presenta-tions of the formalism appeared in later publica-tions (Jime´nez-Montan˜o et al., 1995, 1996). Independently, Klump (1993), Karasev and Sorokin (1997) made similar proposals, which il-luminate different aspects of the problem. A com-parison of the three geometrical representations of the code will be discussed in a forthcoming publi-cation (Jime´nez-Montan˜o and Klump, in prepara-tion). In the present contribution I further develop this approach, grounded on thermody-namics and group theory, and propose a model for the evolution of the code.
Besides the precursor – product relationships be-tween amino acids (or nucleotides) in biosynthetic pathways (Wong, 1975, 1976; Dillon, 1978; Tay-lor and Coates, 1989; Jime´nez-Sa´nchez, 1995), the only evidence we have from the time the code originated are ‘molecular fossils’. Among these, is the structure of the ‘universal’ code itself (Woese, 1965, 1967; Taylor and Coates, 1989;
Jime´nez-1It is important to notice, that in the present contribution
Montan˜o, 1994). Now supplemented with well-documented deviations (Jukes, 1990; Wolsten-holme, 1992), which are far from random (Schultz and Yarus, 1996). Therefore, any theory to infer prior codes should be consistent with this extant evidence. As we shall see, our model satisfies this requirement. Moreover, by its very nature, our phenomenological approach has the advantage of circumventing the difficulties that have plagued the proposals on the origin of the genetic code (Cedegren and Miramontes, 1997).
As Eigen (1971) first pointed out, the origin of the code should have been the result of a highly non-linear selection process. Therefore, it was strongly dependent on initial conditions, but ini-tial conditions cannot be derived from dynamics of any kind. It is an essential characteristic of the evolutionary process to involve a certain degree of contingency, at one or more points. However, in
our model the initial conditions are not assumed on the basis of the abundance of pre-biotically synthesized amino acids, nor on precursor – product relations in the biosynthetic pathways of pyrimidines (Jime´nez-Sa´nchez, 1995). This is so, because I do not appeal to prebiotic scenarios. From the point of view developed here, the ques-tion of the initial condiques-tions is not the quesques-tion of which ingredients appeared first, but the question of which were the first amino acids to be incorpo-rated into a primordial code. I assume as others did before, that ‘simple’ amino acids were intro-duced first. To have a comparison criterion, the amino acids are ranked according to two current measures to estimate chemical complexity, (i) the shortest-description of structural formulas (Pa-pentin, 1982), and (ii) the size/complexity score (Dufton, 1997), see Table 1.
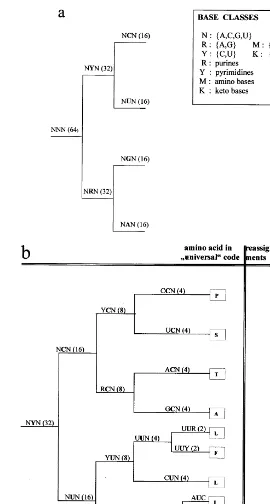
The structure of the code suggests that it evolved following a minimum change coding pathway, The development of an already-working system should happen by changes in its least significant features, without disturbing the major lines of the system (Swannson, 1984). Apart from this general assumption, the only additional as-sumption I make is that the codon – anticodon Gibbs free-energy of interaction induces a partial order (see Eq. (1) below) in the set of codon classes. This partial order defines a ‘time arrow’, in the sense that specific classes (e.g. NRN, CGY, etc.) correspond to the various stages of the pro-gressive differentiation of the code. These codon categories define amino acid groups, the amino acids belonging to a given category are the leaves from the node of that category in the develop-mental tree (Fig. 1). This approach gives a formal expression to pioneering ideas of Woese (1965, 1973) and coworkers (Woese et al., 1966), that envisioned a gradual development from a ‘sim-plest’ code. According to these authors, the first code was so imprecise because the ancestors of transfer RNAs were only able to recognize classes of similar codons (an extreme form of wobble) and classes of similar amino acids. As aptly sum-marized by Haig and Hurst (1991), ‘‘in this view, the modern version of the code evolved through a gradual increase in the discrimination of tRNA
Table 1
Ranking of amino acids (in isolation) and in side-chains of proteins, according to two measures to estimate chemical complexity
Shortest descrip- Size/complexity Shortest
descrip-tion of amino tion of side-chaina score of amino acidb
Fig. 1. (Continued)
for specific amino acids and specific codons within these ancestral sets’’.
By comparing a primeval code, coding only for seven amino acids, with the five letter alphabet employed by Riddle et al. (1997) to build a func-tional protein, it is shown that our proposed primeval code is enough to enable a primitive cell to produce functional proteins. These early proteins, in turn, drive the further evolution of the code. Similar simplified alphabets are obtained from randomly generated codes, under different optimization criteria, only after imposing the ini-tial conditions assumed in the model. However, a diverse outcome is attained with the primeval code proposed by Jime´nez-Sa´nchez (1995), that assumes radically different initial conditions.
It is shown that the developmental pathway of the genetic code is compatible with proposals about the co-evolution of the code and amino acid synthetic pathways (Wong, 1975; Dillon, 1978; Taylor and Coates, 1989), without invoking
non-testable assumptions about the temporal ap-pearance of nucleotides or amino acids. The ob-served departures from the ‘universal’ code, found in many organisms and cellular compartments (Jukes, 1990; Wolstenholme, 1992), fit naturally in the proposed evolutionary scheme. They can be explained in a way consistent with the ‘‘ambigu-ous intermediate’’ theory of Schultz and Yarus (1996).
In conclusion, the proposed model provides a simple and coherent scheme for the development of a coding system. It differs from previous mod-els in that it emphasizes the importance of physic-ochemical constraints and initial conditions, to delimit the possible developmental pathways.
2. The model
The model starts from the following established fact, the genetic code is the biochemical system for gene expression. Therefore, the genetic code is both, a physico-chemical and a communication system. On the physical side, molecular recogni-tion depends on complementary molecular sur-faces (by means of weak interactions); on the informational side, a prerequisite to define a code is the concept of distinguishability. Both aspects of the code are equally important to understand its structure and evolution.
As it was mentioned above, in previous publica-tions we have shown that the structure of the code (the relationship between codons) may be repre-sented as a six-dimensional Boolean hypercube (Jime´nez-Montan˜o et al., 1995, 1996). Accord-ingly, each base is determined by two independent dichotomic variables, chemical type (purine – pyrimidine) and H-bonding (weak – strong). Each codon corresponds to a node in the cube, and it is next to six nodes representing codons differing in a single property. Therefore, the hypercube simul-taneously represents the whole set of codons (and the corresponding amino acids and termination signal) and keeps track of which codons are one-bit neighbors of each other. See Figs. 2 – 4 in (Jime´nez-Montan˜o et al., 1996).
The distribution of redundancy depends on the local symmetries of different codons, with respect to the H-bonding categorization of the bases (Jime´nez-Montan˜o et al., 1996; Zhang, 1997). For example, the code separates into two almost iden-tical codes, with 32 codons each, according to Hydrogen-bonding of the third base, NNW and NNS (where W: (A, U) are weak and S: (C, G) strong bases). The symmetry is complete if al-lowance is made for two codon reassignments, AUA: (I)\(M); UGA (STOP)\(W), both of
which have been observed in mitochondria. These symmetries, in turn, have their physical origin in the codon – anticodon Gibbs free-energy of inter-action. Therefore, the two aspects of the code converge: it is the physical indistinguishability of some codon – anticodon interaction energies that makes the codons synonymous, and the code degenerate and redundant. This conclusion is sup-ported by thermodynamical measurements made by Klump and Maeder (1991). The thermody-namic approach to explain the origin of the distri-bution of the redundancy in the genetic code has the advantage of being independent of micro-scopic assumptions.
The explanations based on wobble rules (Crick, 1966) imply the existence of modified bases, which, in turn, require the existence of specific enzymes. Modified bases have an important effect on the codon – anticodon interaction, for example, pseudouridine has a very strong stabilizing effect on double-stranded, base pairing interactions when the modification is located within a base-paired region (Davies et al., 1998). However, all fine-tuning effects most probably are later refine-ments of the translation apparatus and, for this reason, they presumably did not play any role in primordial codes. The discovery of ‘four-way’ wobble (Jukes, 1990) in mitochondria led to revi-sion of the wobble rules (Heckman et al., 1980).
This conjecture was investigated experimentally by Langerkvist’s group (Langerkvist, 1978; Sa-muelsson et al., 1980), that postulated a ‘‘two out of three reading’’. Under the conditions of in vitro protein synthesis, a codon can be read by recogni-tion of only its first two nucleotides, the third position of the codon being disregarded. These authors proved their hypothesis only for codons of the SSN class. Jime´nez-Montan˜o et al. (1996) suggested a generalization of the hypothesis, based on the group-theoretical analysis of codon doublets made by Danckwerts and Neubert (1975). The main result was a classification of the codons of ‘mixed type’ class (WSN and SWN), with respect to the sets M1 and M2 of four-fold
and less than four-fold degenerate doublets, re-spectively. It was shown in that paper that the third base degeneracy of a codon does not depend on the exact base at the first position, but only of its H-bond character. Also Hasegawa and Miyata (1980) underlined the importance of the codon – anticodon interaction energy to understand the pattern of degeneracy. These authors noticed a strong correlation between codon composition and molecular weight of the coded amino acid. The further correlation, between molecular weight and the size/complexity score employed here (Table 1), has been fully discussed by Dufton (1997). Thus, our results extend their previous finding.
As already mentioned, the structure of the code suggests that it evolved following a course of minimal differentiation to diversify objects. In the context of the formalism we are employing, this means by changing a single distinctive feature of the codon at each time. From this assumption a dynamical evolutionary pattern of the code emerges naturally, envisioned as a refinement process.
3. The group-theoretical foundation
Nieselt-Struwe and Wills (1997) have discussed the intrinsic physical constraints that must be satisfied before a molecular genetic coding can evolve at all. As in their work, in the present model the selection process, which led to the
evolution of a coding system, has an underlying group-theoretical structure. However, our more specific model differs from their framework, which is too general. It also differs from the one proposed by Hornos and Hornos (1993), that employed a Lie group decomposition of the ‘uni-versal’ code to elucidate its structure. In this last paper, not only was no account given for the code’s observed deviations, but ‘‘the decomposi-tion was based on the properties of amino acids and did not reflect any obvious process of tempo-ral refinement in the differentiated recognition of amino acids and codons’’ (Nieselt-Struwe and Wills, 1997).
Instead, the mathematical background behind the present model consists of cartesian products of the Klein 4-group, which is the relevant group to describe the symmetries of nucleic acids (Zhang, 1997) and that underlies the hypercube structure (Jime´nez-Montan˜o and Klump, in preparation2). As shown by Danckwerts and
Neu-bert (1975), the transformations among the four bases obey a Klein 4-group. This result was ex-tended to B1B2 base doublets, by Bertman and
Jungck (1979), and to B2B3 base doublets by
Jime´nez-Montan˜o et al. (1995). However, the ex-tension to whole codons apparently fails because, in the universal code there are three amino acids with six codons each (S, L, R), a single amino acid with three codons (I), and two amino acids with one codon each. This is an insurmountable difficulty under the assumption of an immutable code. However, the situation changes if one as-sumes that the code evolves. As will be discussed below, in relation to the code’s deviations, the local symmetry of the codons may change. Under these circumstances, the above exceptions can be explained. The amino acids with six codons each are doubly assigned: the ‘normal’ assignment of four codons and a second assignment of two ‘extra’ codons (Taylor and Coates, 1989). The cases with one and three codons are the result of a single process in which the reassignment of a
2In a forthcoming paper, a detailed model of the structure
codon, belonging to an amino acid with two codons (B1B2R or B1B2Y), produces the 1-3
de-generacy (Fig. 1).
4. The informational problem
One of the main features of the present model is to give a rigorous characterization of appropri-ately differentiated codons, through the introduc-tion of codon classes with different local symmetries (Fig. 1). From this characterization, we propose a mechanism for the progressive dif-ferentiation of the coding assignments, starting from a point in which rudimentary translation machinery had a relative small threshold of trans-lational accuracy.
The four bases occurring in RNA, adenine (A), cytosine (C), guanine (G), and uracyl (U) (or thymine (T) in DNA) defines a four-letter alpha-bet X: {A, C, G, U (T)}. They may be categorized according to (i) chemical type C: {R, Y}, where
R: (A, G) are purines and Y: (C, U) are pyrimidi-nes, and according to (ii) H-bonding, H: {W, S}, where W: (A, U) are weak and S: (C, G) strong bases. The third possible partition into amino M:{A, C}, keto {U, G} classes is not independent from the former ones (Fig.1 in Jime´nez-Montan˜o et al., 1996), and is irrelevant for the description of the codon – anticodon mapping, but is impor-tant to distinguish between the two classes of aminoacyl tRNA synthetases (see Results).
Denoting the chemical type by C
i and the
H-bond category of the base Bi, by Hi at position i
of a codon (i=1, 2, 3), our basic assumption is that:
The codon – anticodon Gibbs free-energy of in-teraction obeys a hierarchical order, symbolically represented as
C2\H
2\C1\H1\C3\H3 (1)
This means, that the most important characteris-tic determining the codon – ancharacteris-ticodon interaction is the chemical type of the base in the second position. The next most important characteristic is whether there is a weak or strong base in this position, then the chemical type of the first base
and so on (Jime´nez-Montan˜o et al., 1996). Em-ploying the categorizations of the bases defined above, corresponding categorizations may be defined for codons, e.g. NSN, RGY, UNW, etc. where N=(A, C, G, U). With the help of these syntactic categories the codons can be partitioned into classes (Jime´nez-Montan˜o, 1994). The classes, displayed in Fig. 1, rigorously characterize ‘‘appropriately differentiated codons’’ (Nieselt-Struwe and Wills, 1997). They specify the differ-ent stages in the evolution of the code, and the possible amino acid groups at each stage. To derive them I make the following considerations. The expansion of the amino acid alphabet, following a minimum change coding principle (Swannson, 1984), is equivalent to fulfilling the following requirements, (a) at any stage in the development of the code the codon classes will differ, at most, in a single feature and, (b) these changes must obey a partial order, defined by a set of inequalities like Eq. (1) above. This means that the possible extensions of the vocabulary, by the incorporation of new amino acids, are ar-ranged in a subset relationship. According to the subset principle (Berwick, 1986), distinctive fea-tures are not determined independently of one another. Rather, some distinctive features can be fixed after other features are set. The system will make the smallest differentiation (distinction) consistent with its current state of development, characterized by the codon – anticodon recogni-tion fidelity. Since the capacity to distinguish dif-ferent codons depends on the stage of development of the system (i.e. fidelity increases in time), a minimum change is a relative concept. From the structure of the ‘universal’ code, we now that, at the present time, a minimum change corresponds to a change in H3, for example,
distinguish them. However, at the following stage, the threshold energy for minimal differentiation decreases (because of better enzymes). Therefore, the previous change is not the smallest any more. The H-bonding of the second base (H2) becomes
the new differentiation threshold, and so on. The key idea is that the inequalities Eq. (1) determine a definite order in which new features are used, to form new codon classes, which codify for the recently introduced amino acids. The new classes are formed by splitting of existing classes, with the split based on the order given by the basic as-sumption. In this way, a huge number of develop-mental pathways are eliminated because of the order in which a small number of parameters are set.
A split must be triggered by a detectable physico – chemical difference between at least one of the members of an existing codon class and the rest of the members. This event could be, e.g. the change of the middle base from a purine to a pyrimidine. In order to guarantee incremental evolution, the next available unused distinctive feature must be used as a point of refinement, i.e. the splitting occurs at the leading edge of the directed graph (Fig. 1) by successive refinement of existing classes. This is a powerful constraint for the possible evolution of a coding system. Sup-pose that this constraint did not exist, then a non-gradual evolution of the code would imply that codons differing in two or more features would be assigned to different amino acids. It would then be possible to form a new class parti-tion based on distinguishing both features without first using one of them to form a codon class. Therefore, there would be no class that would correctly accommodate a codon that has one value of the first feature and the contrary value of the second one. One way to remedy this problem would be to allow the system to go back and rebuild the classes that have already been formed, but this would violate the developmental order, with the corresponding perturbations at the protein level. Therefore, as a result of following a minimum change-coding path, at each stage the narrowest code is formed, consistent with the capability of the system to distinguish among different codon classes. If two codons cannot be
distinguished they must codify for the same amino acid, remaining synonymous3.
5. Results
5.1. De6elopmental pathway for the genetic code
According to the model, it is necessary to con-sider three different aspects to specify a possible developmental pathway for the genetic code:
1. Physico-chemical constraints.
2. Initial conditions, that require that simple amino acids enter the code before more com-plex ones.
3. An optimization principle to insure ‘small’ changes in protein structure when new amino acids are introduced.
Taking into account only the third requirement leads to ‘impossible’ pathways, in the sense that they may violate physico-chemical facts or the assumed initial conditions. Goldman (1993) gives examples showing that the natural code is far from optimal and easily improved, with respect to the optimization criterion proposed in (Haig and Hurst, 1991). He remarked that ‘‘the assignment of amino acids to synonymous codon sets, and the very existence of the observed synonymous codon sets, are being constrained by some as-yet-unmodeled factors’’. Further, he noticed that these factors might have significant bearing on the comment Haig and Hurst (1991) made that ‘‘the translation apparatus would be expected to evolve
3It is interesting to notice that the formal procedure
an inverse relationship between the frequency and severity of an error’’. These ‘as-yet-unmodeled factors’ are the physical constraints that we have taken into account in our model. The same concern has been expressed by Amirnovin (1997), with respect to the theories that take into account only biosynthetic relations between amino acids. He points out that, ‘‘the code has two main character-istics: first, it is degenerate with respect to the amino acids (i.e., there is more than one codon for all but two amino acids) but at the same time, a difference in degeneracy exists such that the number of codons per amino acid varies from one to six. Second, some of the biosynthetically related amino acids have closely related codons. The first characteristic of the code has been left mostly unexplained while the second has been discussed by Wong (1981)’’. He compared the codon correlations between codons of biosynthetically related amino acids in the uni-versal genetic code with those in randomly gener-ated codes. As a result, he found several randomly generated codes that have many more correlations than that found in the universal code.
These unexpected results, of finding ‘superior’ codes, in agreement with requirements (i) and (iii) above, do not imply that these codes developed in agreement with requirement (ii). As explicitly stated by Haig and Hurst (1991), their method of gener-ating variant codes is not meant to mimic the evolutionary process, but to test their null hypoth-esis. They remark that, ‘‘Our null model places strong constraints on the structure of the variant codes. All codes have the same level of degeneracy and the same probability of synonymous substitu-tions as the natural code. Therefore, our results detect error-minimizing features of the code that are additional to third (and second) base redun-dancy’’. Freeland and Hurst (1998) extended the mentioned work, lifting the second constraint by weighting transition errors differently from transversion errors and weighting each base differ-ently. However, in this later work no requirement on initial conditions is also imposed.
Next, I will discuss the results that follow from the developmental pathway of the genetic code obtained from the model. Then, I compare this pathway with the developmental pathways of two randomly generated codes. The first one was
ob-tained by maximizing the number of correlations between codons of biosynthetically related amino acids (Amirnovin, 1997), and the second one, reported by Goldman (1993), by optimizing polar requirement (Woese et al., 1966).
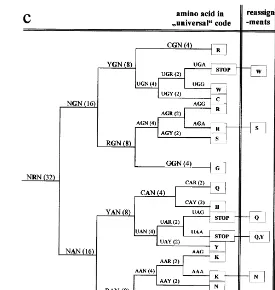
The developmental pathway of the genetic code, derived from the model, is displayed in Fig. 1. The first ramification (Fig. 1a) separates the codons of the Pyrimidine Branch (NYN) from those of the Purine Branch (NRN). From the first column of Fig. 1b it is clear that in the universal code the codons of the class NYN codify exclusively for nonpolar (hydrophobic) amino acids. This was first noticed by Klump (1993), in connection with his hypercube representation of the Genetic code. While the codons of the NRN class (Fig. 1c) codify polar (hydrophilic) amino acids, with the exception of tyr (Y) and trp (W). These two amino acids are among the five more complex amino acids, actually the most complex, according to Papentin’s struc-tural complexity measure (Table 1). Therefore, they are assumed to enter the code only in the last stage. Although, the first observations are well known, they acquire a new meaning in the present context: the differentiation of a single feature of the middle base of a codon implies the polar/nonpolar distinc-tion of amino acids. This is an important property to consider for understanding the evolution of the genetic code.
Recent results suggest the independence of protein folding kinetics of the details of the se-quence (Riddle et al., 1997). This finding confirms theoretical views, which hold that it is possible to construct globular proteins by specifying the se-quence only with respect to a hydrophobic/
hydrophilic alphabet (Chan and Dill, 1998). Fur-thermore, Hetch and co-workers (Kamtekar et al., 1993) have shown that four-helix bundles can be designed by using the same binary alphabet, with the appropriate sequence periodicity of polar and nonpolar residues. This periodicity is the major determinant of secondary structure in self-assem-bling oligomeric peptides (Xiong et al., 1995).
Surrogate codes can be constructed assuming different null hypotheses (see Amirnovin (1997) for three methods to generate surrogates). If the ‘block-structure’ is not assumed, there are more than 1065 possible codes (Goldman, 1993).
huge number to ‘only’ 21!=5×1019 possible
codes. The most conservative code found by Goldman (1993), without assuming the distribu-tion of degeneracy present in the universal code, clearly does not have the polar/nonpolar distinc-tion property (See Fig. 2 of Goldman’s paper). But even the more restricted, randomly generated codes, which do have the block-structure (see the two last columns of Fig. 1), do not share the polar/nonpolar distinction property.
The second stage in the development of the code is characterized by four codon classes, con-sisting of 16 codons each. According to both complexity measures (Table 1), Ala and Gly are the simplest amino acids, followed by Asp, Pro or Val, Leu. All their codons are of the SNN class. G, A, V and D belong to the GNN subclass and P, L to the CNN subclass. These codon classes are located in contiguous planes in the hypercube (See Figs. 2 – 4 in Jime´nez-Montan˜o et al., 1996). I assume that the first plane (GNN) is the primor-dial plane because it contains A and G. This implies that the first amino acids incorporated to the genetic code were, G, A, V and D. This result is in agreement proposals made by others, follow-ing very different criteria (Eigen and Schuster, 1979; Taylor and Coates, 1989 and references therein). However, it has only a partial overlap with the Klump (1993) result, that assumes that the primordial amino acids were G, A, P and R (see also Hartman, 1995). The last two belong to the CNN plane.
The hypercube representation of the genetic code proposed by Klump differs from the one assumed in our model, because he employed only the R/Y categorization of the bases to define the codon classes. To explore the relations between codons he introduces a scale to rank the codons according to their Gibbs energy of codon – anti-codon interaction (in kcal per mol triplet at 25°C). Then he defines a ‘mutational pathway’ (along the shallowest slope up the codon – anticodon interac-tion Gibbs energy levels) from the least stable codon – anticodon pair to the most stable one. See Fig. 1 in Klump (1993). He says, ‘‘as it turns out, this pass will always follow the same route. It starts with exchanging the nucleotide in the third position as the first step, which, with only two
exceptions (Stop/Trp and Ile/Met), preserves the amino acid encoded, it continues with exchanging the nucleotide in the first position (5%) as the second step, and only at the last step is the nucleotide in the middle position exchanged’’. Therefore his developmental pathway satisfies the inequalities:
C2\C1\C3 (2)
These are consistent with the inequalities we assumed in our model (inequalities Eq. (1)). How-ever, the inequalities involving the S/W catego-rization are not considered in Klump’s model. The assumed primordial amino acids have codons at the end of the mutational pathways, hence they have the highest codon – anticodon Gibbs energy and, for this reason, the more stable pairing. This point will be further discussed later on.
The chemical type of the first base,C1, dictates
the next developmental stage.The system has now
the capacity to make an R/Y differentiation of the first base. As can be seen from Fig. 1, all the amino acids introduced in the previous stage have codons in the class RNN. Therefore, the new amino acids should have codons of the class YNN. Following our ‘simple-first’ rule implies the introduction of one element of each of the follow-ing groups, (P, S) (L) (STOP, R) and (STOP). Therefore, we assume that besides A, G, V, D and the STOP signal, R, L, and P or S entered the amino acid alphabet. Of course, arginine (R) does not fit the rule (See Table 1). However, it is well known that arginine is an unusual amino acid (Taylor and Coates, 1989). It has been called an ‘‘early intruder’’ by Jukes (1973), who proposed that it replaced the simpler amino acid ornithine. Taylor and Coates (1989) made a similar pro-posal, suggesting that the probable explanation of this anomaly is that the codons AGN belonged to an amino acid no longer a member of the coded set, such as norvaline, norleucine,a-aminobutyric acid or ornithine. An even simpler candidate, b
amino alanine, as well as guanidioacetic acid have been proposed by Hartman (1995), in the place of arginine.
(Table 1). Essentially because each one measures different molecular aspects. However, taking into account Klump’s results above, it is proposed that the amino acids assigned at this stage were: A, G, V, D, P, L and ‘R’, where ‘R’ stands for the simpler basic amino acid which, it is assumed, was later replaced by arginine. The corresponding primeval code is displayed, in the conventional way, in Fig. 2. It leads to the important prediction that functional proteins can be coded with this limited amino acid alphabet.
In a recent paper, Riddle et al. (1997) asked the following question, ‘‘what is the minimum num-ber of amino acids that would have been needed to encode complex protein folds similar to those found in nature today? They found experimen-tally that is possible to build most of a src SH3 domain with a five-letter amino acid alphabet but not with a three-letter alphabet. SH3 is a 57-residue domain that has a complex b-barrel-like structure wherein residues spread throughout the sequence come together to create the binding site for a proline-rich peptide. They showed that the SH3 domain can be largely encoded by the fol-lowing amino acid alphabet, (A, G, I, E, K), which may be compared with our proposed primeval alphabet, (A, G, V, L, D, ‘R’, P). The differences between the two alphabets correspond to closely related amino acids. They can be ac-counted by the empirically conserved substitution groups found by Wu and Brutlag (1996). The
most prominent one of which is (I, V), found in 10 192 positions in the BLOCKS database (Henikoff and Henikoff, 1991), (D, E) was found in 5980, (K, R) in 6453 and (I, L, V) in 5328 positions, in the same database. They also occur in the theoretical groupings based on amino acid physical properties or structural comparisons pro-posed by many authors (e.g. Jime´nez-Montan˜o, 1984; Taylor, 1986; Bordo and Argos, 1991). As a matter of fact, these substitutions occur in the phylogenetic variation of src SH3 domains (Table 1 in Riddle et al., 1997).
Similar results are obtained from the randomly generated codes (Fig. 1), after imposing the initial condition (ii). For example, at the same level of resolution, from the first random code (Amirnovin, 1997), a corresponding primeval code could be (G, Q, S, C, P, L, D). However, a very different outcome is obtained with the prime-val code proposed by Jime´nez-Sa´nchez (1995), that assumes radically different initial conditions. According to table 2 from his paper, the corre-sponding amino acid alphabet is (K, N, Y, M, I, L, F). It is difficult to see how functional proteins could be constructed mainly with bulky, hydro-phobic, amino acids without incorporating G or A; and D or E (Riddle et al., 1997). Besides, it includes some of the most complex amino acids (Table 1).
In the next step, when the H-bond character of the first base (H1) enters the game, the codons
become specified by the two first bases. Thus, all the codons are of the form B1B2N. A primeval
code of this type, with the third base, N, having no specificity, was proposed long ago by Jukes (1965). He remarked that this code would have a maximum of 15 amino acids and four stop codons. In the following, for the amino acids with six codons (S, L, R), I adopt the convention of Taylor and Coates (1989) to call the additional pair of codons ‘extras’. The eight amino acids which are four-fold degenerate in the universal code are (G, A, P, S, V, T, L, R), counting the ‘extras’ separately. They are ranked among the first ten according to, at least, one complexity measure (Table 1), with the mentioned exception of arginine. It is not possible to know for sure
which of the two amino acids that share codons with common doublets entered at this stage. How-ever, from the ranking in Table 1, one possibility would be the following group, (I, C, Q, K, S, D). Thus, there are, altogether, 13 amino acids, {G (4), A (4), P (4), S (8), V (4), T (4), L (8), R (4), I (4), C (4), Q (4) K (4), D (4)} and STOP (4), where the numbers in parenthesis indicate the number of codons. In other variants there could be N (4) instead of K (4), or R (8) S (4) instead of R (4) S (8), etc.
These speculations are of little value, because (N, K) as other amino acids that share codon doublets, are degenerate with respect to the Gibbs free energy of codon – anticodon interaction (Klump and Maeder, 1991). On the contrary, the four-fold degenerate amino acids have doublets with the following composition, (G+G)=3/4, (A+U)=1/4. Therefore, they form the more sta-ble codon – anticodon pairs. Long ago, Goldberg and Wittes (1966) pointed out that triplets with a high GC content have greater pairing specificity and greater resistance to mutation. In their own words ‘‘the correlations described suggest that protective mechanisms may act at two levels, (i) the nucleotide level, where a high GC content may reduce the rate of mutation errors in protein synthesis, and (ii) the organizational level where the effects of a base change are minimized by degeneracy and by connectedness of codons for similar amino acids. The best-protected amino acids would be those with maximum GC content and degeneracy’’. Six of the seven amino acids in the proposed primeval code are among the best-protected amino acids.
In the following stage, the distinction of the chemical type (C3) leads to the R/Y degeneracy of
the third base. At this point there are already the 20 amino acids, {G (4), A (4), P (4), S (6), V (4), T (4), L (6), F (2), R (6), I (2), M (2), C (2), W (2), Q (2), H (2), K (2), N (2), D (2), E (2), Y (2), STOP (2)}. Where the ‘extra’ codons of serine (S), leucine (L) and arginine (R) are assigned accord-ing to the universal code. However, it is only in the final period when a distinction in the H-bond character of the third base (H3) becomes possible.
This last step breaks the R/Y symmetry of Trp (W), Ile (I) and Met (M) codons, leading to the
present assignments, W (1), I (3), M (1); and STOP (3).
5.2. De6iations from the uni6ersal code
Schultz and Yarus (1996) have underlined that the codon reassignments found in various ganelles, several species of ciliates and other or-ganisms, are ‘very nonrandom’. According to Table 1 of their paper, in 14 instances involving six codons to be reassigned, reassignment appar-ently proceed by single nucleotide changes. That is, by one or two bit changes. As can be seen from Fig. 1, they are connected with features in the right hand side of the set of inequalities Eq. (1). Therefore, they are associated with low values of the codon – anticodon interaction energy, and the later stages in the evolution of the code. This is in agreement with the view that these are relatively ‘‘modern changes’’ (Andersson and Kurland, 1995; Schultz and Yarus, 1996). Some reassign-ments restore the local symmetry of a codon, e.g. the assignment of the UGA (STOP) codon to tryptophan (W), restores the symmetry of the codon class UGR, which is broken in the univer-sal code. According to the results obtained by Inagaki et al. (1998), the ancestral mitochondrion was bearing the universal genetic code and subse-quently reassigned the codon UGA to Trp, inde-pendently, in various lineages. From the point of view developed in this contribution, this means a return to a less differentiated code. This interpre-tation agrees with a reductive mode of genomic evolution (Andersson and Kurland, 1995). On the contrary, the reassignment of AAA (K) to N breaks the symmetry of the codon class AAR, producing a 1-3 degeneracy. This case is com-pletely equivalent to the case of AUR, which in the universal code is split in AUA (I) and AUG (M). The reassignment of AUA in mitochondria brings back the symmetry. These variations clearly show that changes in the ‘least significant feature’ of a codon do occur, without a major disruption of the cell functioning.
5.3. Relationship between codon classes and aminoacyl tRNA synthetases
Fig. 3. ‘Alignment’ of amino recognized by class I (II) aminoa-cyl tRNA synthetase, with two amino acid groupings: (1) by codon type, with respect to K/M categorization; (2) by end atom type (Davydov, 1998). S’ and R’ stand for the ‘extra’ serine and arginine codons, respectively.
early stages of the development of the code. This assumption is supported by two independent observations:
1. The result shown in Fig. 3, of a remarkable correlation between the amino acids coded by NMN codons and the amino acids recognized by Class II aaRSs. Correspondingly, between those coded by NKN codons and the amino acids recognized by the synthetases of Class I. Already Wentzel (1995) pointed out a correla-tion between the middle base of a codon and aaRS class, but he did not notice the relation to the M/K categorization of the bases. 2. The implications of the tRNA ‘‘gene
recruit-ment’’ model (Saks et al., 1997)4
for the evolu-tion of the code. According to this model, a single anticodon mutation may affect both mappings realized by the genetic code. An interesting example of the connection be-tween the two mappings realized by tRNA adap-tor molecules, when there exists an anticodon identifier, is E. coli tRNAIle. This tRNA, specific
for the codon AUA, has the modified anticodon LAU, where L is lysidine. This is a modification of cytosine (C), whose 2-keto group is replaced by amino acid lysine (K). The L in this context pairs with A rather than G, a unique case of base modification altering base pairing specificity. The replacement of this L with unmodified C yields a tRNA that recognizes the codon AUG (M). As noticed by Voet and Voet (1995), ‘‘Surprisingly, treated in our model, I will recall some well know
facts about transfer-RNA molecules in order to have a proper perspective of its relation with the underlying microscopic picture. In extant organ-isms, the physical carriers of the genetic code are, of course, tRNAs adaptor molecules. They may be considered of as comprised of two informa-tional domains, the acceptor- TCC minihelix en-coding the operational mapping for amino acids (Schimmel et al., 1993) and the anticodon-con-taining domain with the three nucleotides of the codon – anticodon mapping. A model to relate the evolution of these two parts of the genetic code, assuming a tRNA ancestor, has been proposed by Rodin et al. (1996). The connection between amino acids and specific triplets is accomplished through the aminoacylation reactions catalyzed by aminoacyl tRNA synthetases (see, e.g. Voet and Voet, 1995). These enzymes select both an amino acid and a tRNA. As is well known, these enzymes come in two groups, called Class I and Class II aaRSs (Eriani et al., 1990). To each class correspond exactly ten amino acids.
The structure of the hypercube, which depends on the R/Y and W/S categorizations of the bases, was derived from the codon – anticodon mapping. Therefore, I assume that the only unused feature of the bases, the M/K categorization, was used to distinguish the two classes of aminoacyl syn-thetases. Since the most important base of a codon (anticodon) is the second base, it was natu-ral to assume that this base was also responsible for the differentiation of the synthetases in the
4Notwithstanding the existence of an operational mapping
however, this altered tRNAIle is also a much
better substrate for MetRS than is for IleRS. Thus, both the codon and the amino acid spe-cificity of this tRNA are changed by a single posttranscriptional modification.’’
In this paper I have emphasized the codon – anticodon mapping, i.e. the assignment of codon classes to amino acids. Recently, Davydov (1998) has proposed a set of rules to associate the amino acid end atoms (O/N and non-O/ non-N) of 18 amino acids with codons containing weak bases (A/U). These rules correctly predict all the codons in which the third base is non-re-dundant, that is, all the codons with doublets B1B2 in the set M2=(AU, UU, UA, AA, GA,
CA, UG, AG), plus AC, GU and CU. The amino acids that, according Davydov’s rules, al-low the correct association of codons are, (I, M, L, F, Y, K, N, D, E, Q, H, C, W, T, V, R’, S’). R’, S’ correspond to the ‘extra’ codons of R and S, respectively. Davydov’s rules give incorrect re-sults for the codons of R, S and A. Glycine and proline were not considered in the analysis per-formed by Davydov, for reasons explained in his article.
We have found an inverse of Davydov’s rules: 1. Codons of the form NAN+WCN have
amino acids with O/N end atoms.
2. Codons of the form NUN+WGN have amino acids with non-O/non-N end atoms. The exceptions being R and S.
3. Codons of the form SCN+SGN code for (P, A, R, G). These amino acids either were left out by Davydov or did not obey his rules.
Thus, it is seen that the amino acids beyond Davydov’s rules do not form a random set. All of them have codons of the class SSN, which are the most stable codons (Klump, 1993). With the exception of arginine (once more), the other three have no proper side-chain (see Davydov’s paper for further details). In Fig. 3 the amino acid recognized by the two classes of aminoacyl-tRNA synthetases are ‘aligned’ with two amino acid groupings defined by, (i) end atom type and (ii) codon type. It is clear from this figure, that the three ways of grouping amino acids are strongly correlated.
6. Discussion
The model presented above is aimed to under-stand not just what now is, but the ways what now is might plausibly be expected to have arisen (Kauffman, 1993). It is based on two assump-tions, the first is simply the hypothesis of a grad-ual change of the whole translation machinery. It is difficult to imagine how changes in this vital part of the cell could have been possible other-wise. The second follows from physical con-straints, which are consistent with measurements of codon – anticodon Gibbs free-energy of interac-tion (Klump and Maeder, 1991). Together with the assumed initial conditions, they lead to a coherent picture of the code’s evolution, consis-tent with extant evidence. Thus, we claim, the whole developmental process is imprinted in the ‘universal’ code not only the amino acid synthetic pathways, as assumed by Wong (1975), Wong (1976).
It is a current belief that, in present-day cells, the entire process of translation in the ribosome is optimized towards maximal efficiency. That means towards the optimization of the average translation rate of any one messenger being de-coded, with the highest fidelity. Thus, it is com-monly accepted that speed and accuracy are the two basic parameters of the dynamics of the ribosomal machinery (Chavancy and Garel, 1981 and references therein). It is important to notice that in this process the proper tRNA is selected only through codon – anticodon interactions, the aminoacyl group does not take part (Voet and Voet, 1995). It is natural to assume that this optimization dominated the different stages of the code’s evolution, specified in our model, from proto-cells to modern cells.
is equivalent to choosing the most accurate pair-ing, especially with respect to the third (wobble) position. The most accurate pairing is equivalent to the longest residence time for the codon – anti-codon reading. This inevitably leads to the slowest transcription rate’’, and further, ‘‘...there are vari-ations of this strategy, but the take-home message seems to be, eukaryotes go for precision. Their prime aim is to keep the large genome as intact as possible’’.
As noticed by Woese (1965), it is clear that the early cell followed the same evolutionary strategy. However, to picture later developments, following an optimization principle, it would be necessary to consider both speed and accuracy. Translation error minimization alone is not enough.
7. Conclusions
As in the paper written by Taylor and Coates (1989) 10 years ago, others have seen many of the patterns described by our model. But apart from the novelty of the approach, ‘‘it is the synthesis that adds up to the most interesting and, to us, most profound new insights’’. It contributes to explain how both the physicochemical properties of amino acids, and the biosynthetic pathways, are reflected in the genetic code. It emphasizes the importance of physicochemical constraints, im-posed by the codon – anticodon interaction, and of initial conditions to delimit the possible develop-mental pathways. And, finally, it shows how physical determinism, random variation and selec-tion are not contradictory concepts but, necessary ingredients to understand the evolution of the genetic code.
Acknowledgements
The author thanks Werner Ebeling and Hanspeter Herzel for many fruitful discussions and encouraging comments, Thomas Pohl for making the figures, and Maria E. Castellanos – Gallegos for suggestions to improve the presenta-tion. He also thanks the Innovationskolleg Theoretische Biology Berlin for the hospitality
during a sabbatical year and CONACYT and SNI, Mexico, for partial support.
References
Alff-Steinberg, C., 1969. The genetic code and error transmis-sion. Proc. Natl. Acad. Sci. U.S.A. 64, 584 – 591. Amirnovin, R., 1997. An analysis of the metabolic theory of
the origin of the genetic code. J. Mol. Evol. 44, 473 – 476. Andersson, S.G.E., Kurland, C.G., 1995. Genomic evolution drives the evolution of the translation system. Biochem. Cell. Biol. 73, 775 – 787.
Bertman, M.O., Jungck, J.R., 1979. Group graph of the genetic code. J. Heredity 70, 379 – 384.
Berwick, R., 1986. Learning from positive-only examples: The subset principle and three cases studies. In: Michalski, R., Carbonell, J., Mitchel, T. (Eds.), Machine learning, an artificial intelligence approach, vol. II. Morgan Kaufmann, California.
Bordo, D., Argos, P., 1991. Suggestions for ‘safe’ residue substitutions in site-directed mutagenesis. J. Mol. Biol. 217, 721 – 729.
Cedegren, R., Miramontes, P., 1997. Reply to ‘‘The origin of the genetic code’’. Trends Biochem. Sci. 22, 50.
Chan, H.S., Dill, K.A., 1998. Protein folding in the landscape perspective: chevron plots and non-Arrhenius kinetics. Proteins Struct. Funct. Genet. 30, 2 – 33.
Chavancy, G., Garel, J.P., 1981. Does quantitative tRNA adaptation to codon content in mRNA optimize the ribo-somal translation efficiency? Proposal for a translation system model. Biochemie 63, 187 – 195.
Crick, F.H.C., 1966. Codon anticodon pairing: The wobble hypothesis. J. Mol. Biol. 19, 548 – 555.
Danckwerts, H.J., Neubert, D., 1975. Symmetries of genetic code-doublets. J. Mol. Evol. 5, 327 – 332.
Davies, R.D., Veltri, C.A., Nielsen, L., 1998. An RNA model system for investigation of pseudouridine stabilization of codon anticodon interaction in tRNALys, tRNAHis and
tRNATyr. J. Biomol. Struct. Dyn. 15, 1121 – 1132.
Davydov, O.V., 1998. Amino acid contribution to the genetic code structure: end-atom chemical rules of doublet compo-sition. J. Theor. Biol. 193, 679 – 690.
Dillon, L., 1978. Chapter 6. In: The genetic mechanism and the origin of life. Plenum Press, New York.
Dufton, M.J., 1997. Genetic code synonym quotas and amino acid complexity: cutting the cost of proteins? J. Theor. Biol. 187, 165 – 173.
Eigen, M., 1971. Self-organization of matter and the evolution of biological macromolecules. Naturwiss. 58, 465 – 532. Eigen, M., Schuster, P., 1979. The hypercycle: a principle of
natural self-organization. Springer – Verlag, Heidelberg. Eigen, M., Lindemann, B.F., Tieze, M., Winkler-Oswatitsch,
Epstein, C., 1966. Role of the amino-acid ‘code’ and of selection for conformation in the evolution of proteins. Nature 210, 25 – 28.
Eriani, G., Delarue, M., Poch, O., Gangloff, J., Moras, D., 1990. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature 347, 203 – 206.
Freeland, S.J, Hurst, D.H., 1998. The genetic code is one in a million. J. Mol. Evol. 47, 238 – 248.
Gesteland, Atkins, 1993. The RNA World. Cold Spring Har-bor Lab. Press, Plainview, NY.
Goldberg, A.L., Wittes, R.E., 1966. Genetic code: aspects of organization. Science 153, 420 – 424.
Goldman, N., 1993. Further results on error minimization in the genetic code. J. Mol. Evol. 37, 662 – 664.
Haig, D., Hurst, L.D., 1991. A quantitative measure of error minimization in the genetic code. J. Mol. Evol. 33, 412 – 417.
Hasegawa, M., Miyata, T., 1980. On the antisymmetry of the amino acid code table. Orig. Life 10, 265 – 270.
Hartman, H., 1995. Speculations on the origin of the genetic code. J. Mol. Evol. 40, 541 – 544.
Heckman, J.E., Sarnoff, J., Alzner-de Weerd, B., Yin, S., Raj Bhandary, U.L., 1980. Novel features in the genetic code and codon reading patterns in Neurospora crassa mito-chondria based on sequences of six mitomito-chondrial tRNAs. Proc. Natl. Acad. Sci. U.S.A. 77, 3159 – 3163.
Henikoff, S., Henikoff, J.G., 1991. Automated assembly of protein blocks for data base searching. Nucleic Acids Res. 19, 6565 – 6572.
Hornos, J.E.M., Hornos, Y.M.M., 1993. Algebraic model for the evolution of the genetic code. Phys. Rev. Lett. 71, 4401 – 4404.
Inagaki, Y., Ehara, M., Watanabe, K.I., Hayashi-Ishimaru, Y., Ohama, T., 1998. Directionally evolving genetic code: The UGA codon from stop to tryptophan in mitochondria. J. Mol. Evol. 47, 378 – 384.
Jime´nez-Montan˜o, M.A., 1984. On the syntactic structure of protein sequences, and concept of grammar complexity. Bull. Math. Biol. 46, 641 – 660.
Jime´nez-Montan˜o, M.A., 1994. On the syntactic structure and redundancy distribution of the genetic code. BioSystems 32, 11 – 23.
Jime´nez-Montan˜o, M.A., de la Mora-Basa´n˜ez, R., Po¨schel, T., 1995. On the hypercube structure of the genetic code. In: Lim, H., Cantor, C.R. (Eds.), Bioinformatics and Genome Research. World Scientific Publishing Co. Ltd, Singapore. Jime´nez-Montan˜o, M.A., de la Mora-Basan˜ez, R., Po¨schel, T., 1996. The hypercube structure of the genetic code explains conservative and non-conservative aminoacid substitutions in vivo and in vitro. BioSystems 39, 117 – 125.
Jimenez-Montan˜o, M.A., De la Mora-Basan˜ez, R., 1992. The genetic code as a six-dimensional boolean hypercube. In: Abstracts of Proc. Soc. Math. Biol. Annual Meeting, July 23 – 26. U.C., Berkeley, CA.
Jime´nez-Sa´nchez, A., 1995. On the origin and evolution of the genetic code. J. Mol. Biol. 41, 712 – 716.
Jukes, T.H., 1965. Coding triplets and their possible evolution-ary implications. Biochem. Biophys. Res. Commun. 19, 391 – 396.
Jukes, T.H., 1973. Arginine as an evolutionary intruder into protein synthesis. Biochem. Biophys. Res. Comm. 53, 709 – 714.
Jukes, T.H., 1990. Genetic code 1990 Outlook. Experientia 46, 1149 – 1157.
Kamtekar, S., Schiffer, J.M., Xiong, H., Babik, J.M., Hecht, M.H., 1993. Protein design by binary patterning of polar and nonpolar amino acid sequences. Science 262, 1680 – 1685.
Karasev, V.A., Sorokin, S.G., 1997. Topological structure of the genetic code. Russ. J. Genet. 33, 622 – 628.
Kauffman, S.A., 1993. The origins of order. Oxford University Press, New York, p. 367.
Klump, H.H., 1993. The physical bases of the genetic code: the choice between speed and precision. Archv. Biochem. Bio-phys. 301, 207 – 209.
Klump, H.H., Maeder, D.L., 1991. The thermodynamic basis of the genetic code. Pure Appl. Chem. 63, 1357 – 1366. Kurland, C.G., Rigler, R., Ehrenberg, M., Blomberg, C., 1975.
Allosteric mechanism for codon-dependent tRNA selection on ribosomes. Proc. Natl. Acad. Sci. U.S.A. 72, 4248 – 4251.
Langerkvist, U., 1978. Two out of three: an alternative method for codon reading. Proc. Natl. Acad. Sci. U.S.A. 75, 1759 – 1762.
Nieselt-Struwe, K., Wills, P.R., 1997. The emergence of ge-netic coding in physical systems. J. Theor. Biol. 187, 1 – 14. Papentin, F., 1982. On order and complexity. II. Application to chemical and biochemical structures. J. Theor. Biol. 95, 225 – 245.
Riddle, D.S., Santiago, J.V., Bray – Hall, S.T., Doshi, N., Grantcharova, V.P., Yi, Q., Baker, D., 1997. Functional rapidly folding proteins from simplified amino acid se-quences. Nat. Struct. Biol. 4, 805 – 809.
Rodin, S., Rodin, A., Ohno, S., 1996. The presence of codon – anticodon pairs in the acceptor stem of tRNAs. Proc. Natl. Acad. Sci. U.S.A. 93, 4537 – 4542.
Rumer, Yu. B., 1968. Systematization of codons in the genetic code. Dokl. Akad. Nauk. SSSR 183, 225 – 226.
Saks, M.E., Sampson, J.R., Abelson, J., 1997. Evolution of a transfer RNA gene through a point mutation in the anti-codon. Science 279, 1665 – 1670.
Samuelsson, T., Elias, P., Lustig, F., Axberg, T., Fo¨lsch, G., Akesson, B., Lgerkvist, U., 1980. Aberrations of the classic codon reading scheme during protein synthesis in vitro. J. Biol. Chem. 255, 4583 – 4588.
Schimmel, P., Giege´, R., Moras, D., Yokoyama, S., 1993. An operational RNA code for amino acids and possible rela-tionship to genetic code. Proc. Natl. Acad. Sci. U.S.A. 90, 8763 – 9768.
Schultz, D.W, Yarus, M., 1996. On malleability in the genetic code. J. Mol. Evol. 42, 597 – 601.
the Physics of Information. Santa Fe Institute Studies in the Sciences of Complexity, vol. VIII. Addison – Wesley, Reading, MA, pp. 29 – 37.
Smith, J.M., Szathma´ry, E., 1995. The major transitions in evolution. Oxford University Press, London, p. 81. Sonneborn, T.M., 1965. Degeneracy of the genetic code,
ex-tent, nature, and genetic implications. In: Bryson, V., Vogel, H.J. (Eds.), Evolving genes and proteins. Academic Press, New York, pp. 377 – 397.
Swannson, R., 1984. A unifying concept for the amino acid code. Bull. Math. Biol. 46, 187 – 203.
Taylor, W.R., 1986. The classification of amino acid conserva-tion. J. Theor. Biol. 119, 205 – 218.
Taylor, F.J.R., Coates, D., 1989. The code within the codons. BioSystems 22, 177 – 187.
Voet, D., Voet, J.G., 1995. Biochemistry, 2nd edition. Wiley, New York, pp. 971 – 981.
Wentzel, R., 1995. Evolution of the aminoacyl-tRNA syn-thetases and the origin of the genetic code. J. Mol. Evol. 40, 545 – 550.
Woese, C.R., 1965. On the evolution of the genetic code. Proc. Natl. Acad. Sci. U.S.A. 54, 1546 – 1552.
Woese, C.R., 1967. The genetic code. The molecular bases for genetic expression. Harper and Row, New York. Woese, C.R., 1973. Evolution of the genetic code. Naturwiss.
60, 447 – 459.
Woese, C.R., Dugre, D.H., Saxinger, W.C., Dugre, S.A., 1966. The molecular basis of the genetic code. Proc. Natl. Acad. Sci. U.S.A. 55, 966 – 974.
Wolstenholme, D.R., 1992. Animal mitochondria DNA: struc-ture and evolution. Int. Rev. Cytol. 141, 173 – 216. Wong, J.T.F., 1975. A co-evolution theory of the genetic code.
Proc. Natl. Acad. Sci. U.S.A. 72, 1909 – 1912.
Wong, J.T.F., 1976. The evolution of a universal genetic code. Proc. Natl. Acad. Sci. U.S.A. 73, 2336 – 2340.
Wong, J.T.F., 1981. Coevolution of the genetic code and amino acid biosynthesis. Trends Biochem. Sci. 6, 33 – 35. Wu, T.D., Brutlag, D.L., 1996. Discovering empirically
con-served amino acid substitution groups in databases of protein families. In: States, D.J., Awarwal, P., Gaaster-land, T., Hunter, L., Smith, R. (Eds.) Proceedings of the IV International conference on intelligent systems for molecular biology, AAAI Press, California, pp. 230 – 240. Xia, X., Li, W.H, 1998. What amino acid properties affect
protein evolution? J. Mol. Evol. 47, 557 – 564.
Xiong, H., Buckwalter, B.L., Shieh, H.M., Hecht, M.H., 1995. Peridocity of polar and nonpolar amino acids is the major determinant of secondary structure in self-assembling oligomeric peptides. Proc. Natl. Acad. Sci. U.S.A. 92, 6349 – 6353.
Zhang, C.T., 1997. A symmetrical theory of DNA sequences and its applications. J. Theor. Biol. 187, 297 – 306.
.