Cilostazol, a selective type III phosphodiesterase inhibitor,
decreases triglyceride and increases HDL cholesterol levels by
increasing lipoprotein lipase activity in rats
Takeshi Tani
a, Kenji Uehara
b,*, Toshiki Sudo
b, Keiko Marukawa
b,
Yoshinobu Yasuda
a, Yukio Kimura
baTokushima Research Institute,Otsuka Pharmaceutical Co.Ltd.,463-10Kagasuno Kawauchi-cho,Tokushima,771-0192Japan bThrombosis and Vascular Research Laboratory,Department of Ad6anced Pharmacology,Otsuka Pharmaceutical Co.Ltd.,463-10Kagasuno,
Kawauchi-cho,Tokushima,771-0192Japan
Received 3 December 1998; received in revised form 10 September 1999; accepted 3 November 1999
Abstract
Cilostazol, a selective type III phosphodiesterase inhibitor, has antiplatelet and vasodilating effects. In this study, the effects of cilostazol on lipid metabolism and lipoprotein lipase (LPL) activity were studied in rats. Cilostazol was administered orally at doses of 30 or 100 mg/kg twice a day for 1 – 2 weeks to rats. Cilostazol decreased the serum triglyceride level in normolipidemic rats. The serum triglyceride level was reduced and HDL cholesterol level was increased by cilostazol in streptozotocin (STZ)-induced diabetic rats. The disappearance of exogenous triglyceride was accelerated by cilostazol in normolipidemic rats. Cilostazol increased post-heparin plasma LPL activity but had no effect on hepatic triglyceride lipase activity in STZ-induced diabetic rats. Cilostazol also increased LPL activity in the heart in STZ-induced diabetic rats. These findings suggest that an increase in LPL activity is responsible for the serum triglyceride lowering and HDL cholesterol elevating effects of cilostazol in rats. © 2000 Elsevier Science Ireland Ltd. All rights reserved.
Keywords:Cilostazol; Lipoprotein lipase; Triglyceride; HDL cholesterol
www.elsevier.com/locate/atherosclerosis
1. Introduction
Abnormal lipid metabolism has been extensively studied as a risk factor for atherosclerosis. Increasing evidence of an association between serum triglyceride levels and the incidence of coronary heart disease (CHD) has been obtained [1]. Chylomicrons and very low density lipoproteins, which are triglyceride-rich lipoproteins, are released from the intestine and liver into the circulation. These particles are metabolized to smaller lipoproteins, and in this process triglycerides are hydrolyzed by lipoprotein lipase (LPL). LPL activ-ity is also responsible for the production of high densactiv-ity lipoproteins (HDL), anti-atherogenic lipoproteins [2,3]. Decreased LPL activity results in lipid abnormalities
such as hypertriglyceridemia and hypo HDL choles-terolemia [4]. One of the major causes of hypertriglyce-ridemia with low HDL cholesterol level in diabetes is a decrease in LPL activity due to insulin deficiency [5]. Cyclic nucleotide phosphodiesterases (PDEs) regulate various physiological responses in many cells and tis-sues, including platelet aggregation [6,7], vascular relax-ation [8,9], and cardiac muscle contraction [10,11]. Since PDEs exist in multiple forms in cells and tissues and each form of PDE acts in a complex manner in these responses, selective PDE inhibitors are expected to be useful for the treatment of various diseases [12,13]. Cilostazol [14,15], one such drug, can be con-sidered the first representative of a new family of an-tithrombotic agents which act by selective inhibition of one of seven PDE families, the cGMP-inhibited cAMP PDEs (PDE type III) [16].
Since LPL activity has been reported to be affected by cAMP, we examined the effect of cilostazol on lipid
* Corresponding author. Tel.: +81-88-6652126, ext. 2986; fax: +81-88-6655662.
E-mail address:k –[email protected] (K. Uehara).
metabolism and LPL activity in normolipidemic and streptozotocin (STZ)-induced diabetic rats.
2. Materials and methods
2.1. Animal experiments
Seven- to nine-week-old Wistar rats (Japan Clea or Japan SLC) were used. Diabetes was induced in fasted rats by intravenous injection of 40 or 60 mg/kg of streptozotocin (STZ) (Wako, Osaka, Japan). Cilostazol was suspended in 2% arabic gum (Wako) – tap water and administered orally at 30 or 100 mg/kg twice a day for 1 – 2 weeks. Control rats were administered 5 ml/kg of 2% arabic gum orally twice a day for 1 – 2 weeks. Rats were fasted overnight, and 2 h after final adminis-tration of the drug or arabic gum all analytical mea-surements were performed. Blood samples were collected from the tail vein or vena cava, and the serum and plasma separated by centrifugation were used for measurement of lipid levels, half-life of exogenous triglyceride, and LPL activity, as described below.
In this study, the following effects of cilostazol on lipid metabolism in rats were examined: (1) effect of the drug on the serum lipid levels in normolipidemic rats and STZ-induced diabetic rats, (2) effect of the drug on the disappearance of exogenous triglyceride in nor-molipidemic rats, (3) effect of the drug on LPL activity in post-heparin plasma in STZ-induced diabetic rats, and (4) effect of the drug on the heart LPL activity in STZ-induced diabetic rats. Male and female normolipi-demic rats were used in (1), while in others male rats were used.
2.2. Analytical measurements
2.2.1. Lipid and glucose le6els
Serum triglyceride and cholesterol levels were assayed enzymatically using the Wako triglyceride G test (Wako) and the Wako cholesterol CII test (Wako). The HDL was separated by the phosphotungstic acid – mag-nesium precipitation method using an HDL-precipita-tion kit (Wako). Plasma glucose level was measured using the Wako glucose B test (Wako).
2.2.2. Disappearance of exogenous triglyceride
Triglyceride emulsion (20% Intralipid, Kbbi, Phar-macia AB, Uppsala, Sweden) was injected into the tail vein of rats at a dose of 1 ml/kg under ether anesthesia. Blood was collected before and 5, 10, 15 and 20 min after injection of triglyceride emulsion and the serum triglyceride level was measured. The half-life was calcu-lated by logarithmic regression.
2.2.3. LPL acti6ity in post-heparin plasma
For the measurement of LPL activity in post-heparin plasma, 200 U/kg of heparin (Novoheparin, Novoin-dustry, Bajsvaeld, Denmark) was injected into the femoral vein of rats under pentobarbital anesthesia. Ten minutes later, blood was collected with EDTA as an anticoagulant and the plasma was immediately sepa-rated by centrifugation. LPL activity in post-heparin plasma was assayed as described by Murase et al. [17]. Total lipase and LPL activity were measured using glycerol tri [1-14
C]oleate (Amersham, UK), and triolein (Sigma, St Louis, MO) as substrates. Hepatic triglyce-ride lipase (HTGL) activity in plasma was selectively blocked by treatment with the antiserum to rat hepatic triglyceride lipase. Plasma was incubated with substrate and rat serum as a source of coenzyme apoprotein CII, in a final volume of 1 ml at 37°C for 30 min. Enzymatic reaction was stopped by the addition of 3 ml of 0.6 N H2SO4– isopropanol (1:4, v/v). The free fatty acid
(FFA) released during incubation was extracted and the radioactivity of FFA was determined by the method of Schotz et al. [18] with minor modifications. Enzyme activity was expressed asmmol FFA hydrolyzed/ml per h. HTGL activity was calculated by subtracting LPL activity from total lipase activity.
2.2.4. Heart LPL acti6ity
The heart was dissected after rats were sacrificed by exsanguination under ether anesthesia, and homoge-nized in 200 mg/ml of 50 mM ammonium – HCl buffer (pH 8.5) containing 0.5 U/ml of heparin. The ho-mogenate was left to stand at 4°C for 1 h and cen-trifuged at 10 000 rev./min for 20 min. The lipolytic activity of the supernatant was assayed based on the method of post-heparin plasma LPL activity. Enzyme activity was expressed as nmol FFA hydrolyzed/min per g tissue.
2.3. Statistical analysis
Values are expressed as means9S.D. Statistical com-parisons were made between normal and diabetic groups using the unpaired Student’s t-test (two-tailed), and between control and cilostazol-treated groups using one-way analysis of variance, followed by the two-tailed Dunnett test. The level of significance was set at PB0.05.
3. Results
3.1. Serum lipid le6els in normolipidemic rats
Table 1
Effect of cilostazol on serum lipid levels in male normolipidemic ratsa
Group n Serum lipids (mg/dl)
Triglyceride Cholesterol HDL cholesterol
10
Control 83911 78912 5097
Cilostazol 10 71913* 6998 4596 (30 mg/kg)
10 69910* 72911 4799 Cilostazol
(100 mg/kg)
aValues are expressed as means9S.D.
* Significantly different from the respective values in control rats, PB0.05.
(Table 2). The change in triglyceride level in female rats (29% decrease) was greater than that in male rats (16% decrease). HDL cholesterol levels were increased by 20% in both the 30 and 100 mg/kg cilostazol-treated groups (PB0.01), and total cholesterol levels were increased by 16 and 19% at 30 and 100 mg/kg, respec-tively, (PB0.05 for 30 mg/kg and PB0.01 for 100 mg/kg).
3.2. Serum lipid le6els in STZ-induced diabetic rats
STZ-induced diabetic rats clearly exhibited hyper-glycemia and hypertriglyceridemia compared with nor-mal rats (Table 3). Cilostazol did not alter glucose levels but did decrease the triglyceride levels signifi-cantly at a dose of 100 mg/kg (PB0.05). Cilostazol increased HDL cholesterol level significantly and in a dose-dependent fashion (PB0.05 for 30 mg/kg and PB0.01 for 100 mg/kg), but did not affect the in-creased total cholesterol level.
3.3. Disappearance of exogenous triglyceride in normolipidemic rats
The half-life of exogenous triglyceride was 20.6 min in control rats (Fig. 1). Cilostazol significantly de-creased this half-life in a dose-dependent fashion by 30 and 37% (PB0.05 for 30 mg/kg and PB0.01 for 100 mg/kg).
3.4. LPL acti6ity in post-heparin plasma in STZ-induced diabetic rats
Cilostazol significantly increased LPL activity in post-heparin plasma at a dose of 100 mg/kg (PB0.01), and slightly increased LPL activity in post-heparin plasma at a dose of 30 mg/kg. Cilostazol did not significantly affect HTGL activity (Fig. 2).
Table 2
Effect of cilostazol on serum lipid levels in female normolipidemic ratsa
Serum lipids (mg/dl) n
Group
Cholesterol HDL Triglyceride
cholesterol
10 70923 5798
Control 3995
10
Cilostazol 49910* 6698* 4796** (30 mg/kg)
6898**
10 4795**
Cilostazol 50912* (100 mg/kg)
aValues are expressed as means9S.D.
* Significantly different from the respective values in control rats, PB0.05.
** Significantly different from the respective values in control rats, PB0.01.
Cilostazol had no effect on serum total cholesterol or HDL cholesterol levels in male rats. In female rats, cilostazol significantly reduced the serum triglyceride level at doses of 30 and 100 mg/kg (PB0.05, each case)
Table 3
Effect of cilostazol on serum lipid levels in STZ-induced diabetic ratsa
n Plasma glucose (mg/dl)
Group Serum lipids (mg/dl)
Triglyceride Cholesterol HDL cholesterol
8 10598***
Normal rats 101915*** 6195*** 4392***
Diabetic rats
9 582937
Control 3809132 133940 5599
6698*
Cilostazol (30 mg/kg) 9 564926 3779102 133938
Cilostazol (100 mg/kg) 9 584919 214982* 120930 7598**
aValues are expressed as means9S.D.
Fig. 1. Effect of cilostazol on the disappearance of exogenous triglyc-eride in normolipidemic rats. Values are expressed as means9S.D. (n=8 – 10). Significantly different from the respective values in con-trol rats: *PB0.05, **PB0.01.
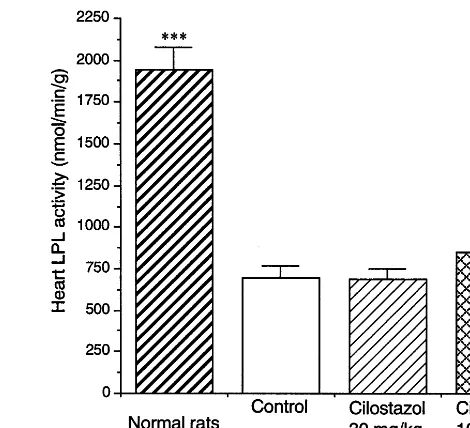
Fig. 3. Effect of cilostazol on the heart LPL activity in STZ-induced diabetic rats. Values are expressed as means9S.D. (n=8 – 9). Signifi-cantly different from the respective values in diabetic control rats: **PB0.01, ***PB0.001.
3.5. Heart LPL acti6ity in STZ-induced diabetic rats
Heart LPL activity was markedly decreased in STZ-induced diabetic rats compared with normal rats (PB
0.001) (Fig. 3). Cilostazol significantly increased heart LPL activity at a dose of 100 mg/kg (PB0.01), but at a dose of 30 mg/kg had no effect on this activity.
4. Discussion
Antiplatelet agents have been shown to be effective in preventing CHD and/or cerebral stroke as well as anti-hypertensive and lipid-lowering agents [19,20]. It is important that an agent selected for prevention of vascular disease has either no or a favorable side-effect profile for other risk factors. Many clinical studies of the effects of antihypertensive agents on lipid
metabolism have been carried out, and beta-blockers [21,22] and thiazide diuretics [23,24] have been shown to have adverse effects on lipid metabolism (i.e. induc-ing increases in triglyceride, increases in total choles-terol, and/or decreases in HDL cholesterol). However, the effects of antiplatelet agents on lipid metabolism have not been extensively studied. In this study, we showed that cilostazol significantly decreased serum triglyceride level and increased HDL cholesterol level in rats, and we also proposed a mechanism for the effects of this drug on lipid metabolism. In patients with intermittent claudication, cilostazol has been shown to improve walking distance with concomitant beneficial effects on plasma lipids (i.e. decreasing triglyceride and increasing HDL cholesterol) [25,26].
Serum triglyceride concentrations reflect the balance between triglyceride secretion into and triglyceride re-moval from the circulation. Maeda et al. previously studied the effect of cilostazol on triglyceride secretion from the liver using Triton WR-1339 in rats, and found that the rate of triglyceride secretion was decreased after cilostazol treatment [27]. However, plasma triglyc-eride levels were not significantly changed by cilostazol, and HDL cholesterol levels were not measured in that study. Therefore, it was not clear whether the alteration of plasma triglyceride level is caused by the reduced triglyceride secretion rate. In the present study, cilosta-zol significantly decreased serum triglyceride levels in male normolipidemic rats and concomitantly increased HDL cholesterol levels in female normolipidemic rats and male STZ-induced diabetic rats. It is known that a decrease in VLDL triglyceride itself increases HDL cholesterol by decreasing the triglyceride – cholesterol ester exchange activity of cholesterol ester transfer protein (CETP) in humans [28]. However, in rats lack-ing CETP [29], decrease in VLDL triglyceride secretion from the liver does not increase HDL cholesterol. Thus, a mechanism other than the effect of cilostazol on triglyceride secretion has to be considered for the serum triglyceride-decreasing and HDL cholesterol-elevating effects of the drug in rats. In this study, we focused on whether the drug enhances the triglyceride removal from the circulation. In an experiment using male nor-molipidemic rats, cilostazol significantly decreased the half-life of exogenous triglyceride after intravenous in-jection of Intralipid, suggesting that the drug increases triglyceride removal from the circulation. In addition, cilostazol significantly increased post-heparin plasma LPL activity in STZ-induced diabetic rats. Thus, the most likely mechanism by which cilostazol decreases serum triglyceride appeared to involve its effect on LPL, an enzyme playing a central role in the catabolism of triglyceride-rich lipoproteins.
Plasma HDL originates from three sources: the small intestine, liver, and LPL-mediated lipolysis of triglyce-ride-rich lipoproteins. Although the production of HDL by the liver and small intestine may be important in determining the plasma HDL cholesterol level, LPL also contributes to the determination of plasma HDL cholesterol levels. The increase in HDL cholesterol level induced by cilostazol might thus be the result of an increased degradation of triglyceride-rich lipoproteins by LPL.
Metabolism of cilostazol is slow in female rats, and the serum concentration of this drug is about 10-fold higher than that found in male rats [30]. Thus it ap-pears that the effect of the drug is more pronounced in female rats. In fact, although the serum triglyceride levels were decreased by 14% with a dose of 30 mg/kg of the drug in male normolipidemic rats, the level was decreased by 30% in female normolipidemic rats.
Cilostazol concomitantly increased serum HDL choles-terol levels in female rats, but affected only serum triglyceride levels in male rats. In female rats, cilostazol also increased the serum total cholesterol level, reflect-ing an increase in serum HDL cholesterol level, and resulting in a decrease in the ratio of non HDL choles-terol/HDL cholesterol. The same phenomenon, produc-ing increases in both HDL cholesterol and total cholesterol levels, was observed in a study of a recently-developed compound, NO-1886, which specifically in-creases LPL activity [31].
Insulin upregulates LPL activity [32], and therefore in diabetes, low LPL activity results in lipid abnormali-ties such as hypertriglyceridemia, particularly in insulin-dependent diabetes mellitus [33]. In fact, in STZ-induced diabetic rats, which is an insulin-depen-dent diabetic animal model, the serum triglyceride level was markedly increased and heart LPL activity was approximately 36% of that in normolipidemic rats. On the other hand, many investigators have shown that LPL activity is also affected by cAMP [34,35]. In studies using db-cAMP, which mimics cAMP, and agents which increase cAMP level such as IBMX, forskolin and cholera toxin, an increase in LPL activity and a comparable increase in LPL mRNA were ob-served in rat heart cells [36] and in a neonatal mouse hepatoma cell line [37]. Since type III PDE exists in heart, liver and adipocytes [38], cilostazol may be ex-pected to increase cAMP levels in such tissues. In this fashion, the drug affects tissue LPL activity. With regard to the effect of cilostazol, a decrease in serum triglyceride is more pronounced in male STZ-induced diabetic rats than that in male normolipidemic rats and an increase in HDL cholesterol is only observed in STZ-induced diabetic rats. The difference in the effect of cilostazol on serum lipid levels in male normolipi-demic and STZ-induced diabetic rats may be related to the LPL activity and insulin level in those rats. In STZ-induced diabetic rats, tissue cAMP levels may be a dominant regulator of LPL activity because insulin, a regulator of LPL activity, is depleted in these rats. Thus the potency of cilostazol on lipid metabolism may be more clearly detected in male STZ-induced diabetic rats than in male normolipidemic rats.
skeletal muscle needs to be examined. It may be impor-tant to clarify the effect of other type III PDE in-hibitors on LPL activity and LPL mRNA levels because the effect of cilostazol on LPL found in this study may be a common action of type III PDE inhibitors.
In summary, we found that cilostazol increased LPL activity, resulting in a decrease in serum triglyceride levels and an increase in HDL cholesterol levels. In addition to their antiplatelet and inotropic effects [39], in this study type III PDE inhibitors were shown to have other important effects. Compounds such as cilostazol may be useful to prevent vascular lesions by ameliorating both platelet hyperactivity and lipid abnormalities.
Acknowledgements
We thank Toshiro Murase, M.D. (Toranomon Hos-pital) for his gift of the anti-hepatic triglyceride lipase antibody.
References
[1] Austin MA. Plasma triglyceride and coronary heart disease. Arterioscler Thromb 1991;11:2 – 14.
[2] Taskinen M, Nikkila EA. High density lipoprotein subfractions in relation to lipoprotein lipase activity of tissues in man-evi-dence for reciprocal regulation of HDL2 and HDL3 levels by
lipoprotein lipase. Clin Chim Acta 1981;112:325 – 32.
[3] Patsch JR, Gotto AM Jr, Olivecrona T, Eisenberg S. Formation of high density lipoprotein2-like particles during lipolysis of very
low density lipoproteins in vitro. Proc Natl Acad Sci USA 1978;75:4519 – 23.
[4] Iglesias A, Contreras JA, Martinez-Pardo M, Entrala A, Herrera E, Lasuncion MA. Cholesterol ester transfer activity in lipo-protein lipase deficiency and other primary hypertriglyce-ridemias. Clin Chim Acta 1993;221:73 – 89.
[5] Haward BV. Lipoprotein metabolism in diabetes mellitus. J Lipid Res 1987;28:613 – 28.
[6] Hidaka H, Hayashi H, Kohri H, Kimura Y, Hosokawa T, Igawa T, Saitoh Y. Selective inhibitor of platelet cyclic adenosine monophosphate phosphodiesterase, cilostamide, inhibits platelet aggregation. J Pharmacol Exp Ther 1979;211:26 – 30.
[7] Simpson AWM, Reeves ML, Rink TJ. Effects of SK F94120, an inhibitor of cyclic nucleotide phosphodiesterase type III, on human platelets. Biochem Pharmacol 1988;37:2315 – 20. [8] Kauffman RF, Schenck KW, Utterback BG, Crowe VG, Cohen
ML. In vitro vascular relaxation by new inotropic agents: rela-tionship to phosphodiesterase inhibition and cyclic nucleotides. J Pharmacol Exp Ther 1987;242:864 – 72.
[9] Tanaka T, Ishikawa T, Hagiwara M, Onoda K, Itoh H, Hidaka H. Effects of cilostazol, a selective cAMP phosphodiesterase inhibitor on the contraction of vascular smooth muscle. Pharma-cology 1988;36:313 – 20.
[10] Farah AE, Alousi AA, Schwarz PP Jr. Positive inotropic agents. Annu Rev Pharmacol Toxicol 1984;24:275 – 328.
[11] Weishaar RE, Kobylarz-Singer DC, Steffen RP, Kaplan HR. Subclasses of cyclic AMP-specific phosphodiesterase in left ven-tricular muscle and their involvement in regulating myocardial contractility. Circ Res 1987;61:539 – 47.
[12] Pang SC. Cyclic AMP and cyclic GMP phosphodiesterases: target for drug development. Drug Dev Res 1988;12:85 – 92. [13] Weishaar RE, Michael HC, Bristol A. A new generation of
phosphodiesterase inhibitors: multiple molecular forms of phos-phodiesterase and the potential for drug selectivity. J Med Chem 1985;28:537 – 45.
[14] Kimura Y, Tani T, Kanbe T, Watanabe K. Effect of cilostazol on platelet aggregation and experimental thrombosis. Arzneim-Forsch Drug Res 1985;35:1144 – 9.
[15] Tani T, Sakurai K, Kimura Y, Ishikawa T, Hidaka H. Pharma-cological manipulation of tissue cyclic AMP by inhibitors: Ef-fects of phosphodiesterase inhibitors on the functions of platelets and vascular endothelial cells. Adv Second Messenger Phospho-protein Res 1992;25:215 – 27.
[16] Beavo JA, Conti M, Heaslip RJ. Multiple cyclic nucleotide phosphodiesterases. Mol Pharmacol 1994;46:399 – 405.
[17] Murase T, Yamada N, Ohsawa N, Kosaka K, Morita S, Yoshida S. Decline of postheparin plasma lipoprotein lipase in acromegalic patients. Metabolism 1980;29:666 – 72.
[18] Schotz MC, Garfinkel AS, Huebotter RJ, Stewart JE. A rapid assay for lipoprotein lipase. J Lipid Res 1970;11:68 – 9. [19] Gent M, Easton JD, Hachinski V, Panak E, Sicurella J, Blakely
JA. The Canadian American ticlopidine study (CATS) in throm-boembolic stroke, Lancet 1989; 1215 – 1220.
[20] Ridker PM, Manson JE, Buring JE, Goldhaber SZ, Hennekens CH. The effect of chronic platelet inhibition with low-dose aspirin on atherosclerotic progression and acute thrombosis: clinical evidence from the Physicians’ Health Study. Am Heart J 1991;122:1588 – 92.
[21] Tanaka N, Sakaguchi S, Oshige K, Niimura T, Kanehisa T. Effect of chronic administration of propranolol on lipoprotein composition. Metabolism 1976;25:1071 – 5.
[22] Day JL, Simpson N, Metcalfe J. Metabolic consequences of atenolol and propranolol in treatment of essential hypertension. Br Med J 1979;1:77 – 80.
[23] Grimm RH Jr, Leon AS, Hunninghake DB, Lenz K, Hannan P, Blackburn H. Effects of thiazide diuretics on plasma lipids and lipoproteins in mildly hypertensive patients. Ann Intern Med 1981;94:7 – 11.
[24] Helgeland A. The impact on serum lipids of combinations of diuretics anda-blockers and ofb-blockers alone. J Cardiovasc Pharmacol 1984;6(Suppl.):S474 – 6.
[25] Elam MB, Heckman J, Crouse JR, Hunninghake DB, Herd JA, Davidson M, Gordon IL, Bortey EB, Forbes WP. Effect of the novel antiplatelet agent cilostazol on plasma lipoproteins in patients with intermittent claudication. Arterioscler Thromb Vasc Biol 1998;18:1942 – 7.
[26] Dawson DL, Cutler BS, Meissner MH, Strandness DE Jr. Cilostazol has beneficial effects in treatment of intermittent claudication. Results from a multicenter, randomized, prospec-tive double-blind trial. Circulation 1998;98:678 – 86.
[27] Maeda E, Yoshino G, Nagata K, Matsuba K, Matsushita M, Sakurai K, Tani T, Kimura Y, Kazumi T. Effect of cilostazol on triglyceride metabolism in rats. Curr Ther Res 1993;54:420 – 4. [28] Barter P, Rye KA. Cholesterol ester transfer proteins: its role in
plasma lipid transport. Clin Exp Pharmacol Physiol 1994;21:663 – 72.
[29] Tall AR. Plasma lipid transfer proteins. J Lipid Res 1986;27:361 – 7.
[30] Akiyama H, Kudo S, Shimizu T. The absorption, distribution and excretion of a new antithrombotic and vasodilating agent, cilostazol, in rat, rabbit, dog and man. Arzneim-Forsch Drug Res 1985;35:1124 – 32.
in the coronary arteries of rats with experimental atherosclerosis. J Clin Invest 1993;92:411 – 7.
[32] Ong JM, Kirchgessner TG, Schotz MC, Kern PA. Insulin in-creases the synthetic rate and messenger RNA level of lipoprotein lipase in isolated rat adipocytes. J Biol Chem 1988;263:12933 – 8. [33] Nikkila EA. Plasma lipid and lipoprotein abnormalities in dia-betes. In: Jarrett RJ, editor. Diabetes and Heart Disease. Amster-dam: Elsevier, 1984:133 – 67.
[34] Friedman G, Chajek-Shaul T, Stein O, Nue L, Etienne J, Stein Y. b-adrenergic stimulation enhances translocation, processing and synthesis of lipoprotein lipase in rat heart cells. Biochim Biophys Acta 1986;877:112 – 20.
[35] Carneheim C, Nedergaard J, Cannon B. Cold-induced beta-adren-ergic recruitment of lipoprotein lipase in brown fat is due to increased transcription. Am J Physiol 1988;254(2 Pt. 1):E155 – 61.
[36] Freidman G, Reshef A, Ben-Naim M, Leitersdorf E, Stein O, Stein Y. Regulation of lipoprotein lipase by dibutyryl cAMP, cholera toxin, hepes and heparin in F1heart-cell cultures. Biochim Biophys
Acta 1992;1137:237 – 41.
[37] Peinado-Onsurbe J, Staels B, Deeb S, Auwerx J. Lipoprotein lipase expression in undifferentiated hepatoma cells is regulated by progesterone and protein kinase A. Biochemistry 1992;31:10121 – 8.
[38] Nicholson CD, Challiss RAJ, Shahid M. Differential modulation of tissue function and therapeutic potential of selective inhibitors of cyclic nucleotide phosphodiesterase isozymes. TiPS 1991;12:19 – 27.
[39] Thompson WJ. Cyclic nucleotide phosphodiesterases: pharmacol-ogy, biochemistry and function. Pharm Ther 1991;51:13 – 33.