ABSTRAK
Ilmu Bioinformatika meneliti tentang perubahan yang dialami oleh DNA, serta membantu memberikan tanda terhadap mutasi genetika yang terjadi. Untuk membandingkan sekuens DNA dan mencari tahu bagaimana kedua DNA memiliki kesamaan dapat digunakan algoritma-algoritma yang dapat mengolah pensejajaran DNA baik secara global maupun secara lokal. Pensejajaran secara global dilakukan dengan membandingkan semua karakter di dalam sekuens, sementara pensejajaran lokal hanya mengambil potongan dari sekuens dan membandingkannya. Pensejajaran global menjadi dasar penelitian laporan ini dengan menggunakan dua algoritma yaitu Needleman-Wunsch dan Lempel-Ziv. Kedua algoritma ini bekerja dengan membangun matriks skor dan mensejajarkan sekuens dari hasil matriks yang dibuat. Pengujian dilakukan dengan mengambil secara acak sekuens DNA dengan panjang kurang dari 1000 karakter hingga panjang melebihi 1000 karakter. Algoritma Needleman-Wunsch unggul dengan kecepatan proses hingga 1 miliseconds untuk dataset kurang dari 1000 karakter dan 42 miliseconds untuk dataset lebih dari 1000 karakter. Meskipun algoritma Lempel-Ziv memerlukan waktu untuk pembentukan frase, kecepatan algoritma Lempel-Ziv rupanya lebih cepat daripada algoritma Needleman-Wunch untuk kasus sekuens DNA yang memiliki frase sempurna.
ABSTRACT
Bioinformatics research is currently working on the changing of the DNA information, and marking the mutation for the DNA. For comparing DNA and finding out how two DNA can have similarities, bioinformatics using algorithms that works in global alignment and local alignment. The global alignment is comparing all the characters in sequence while the local only take a piece of characters from the alignment. This study proposes two algorithms for processing the DNA sequence in global alignment. The akgorithms are Needleman-Wunsch and Lempel-Ziv algorithms. These algorithms work with building a scoring matrix and create an alignment based on the matrix. The experiment is conducted by testing DNA sequences randomly with the length less than 1000 characters and more than 1000 characters. Needleman-Wunsch leading with processing speed up to 1 miliseconds for less than 1000 character dataset and 42 miliseconds for more than 1000 characters dataset, while Lempel-Ziv is leading the processing speed on specific case of perfect phrase in DNA sequence.
DAFTAR ISI
LEMBAR PENGESAHAN ... i
PERNYATAAN ORISINALISTAS LAPORAN PENELITIAN ... ii
PERNYATAAN PUBLIKASI LAPORAN PENELITIAN ... iii
PRAKATA ... iv
ABSTRAK ... vi
ABSTRACT ... vii
DAFTAR ISI ... viii
DAFTAR GAMBAR ... xi
DAFTAR TABEL ... xiii
DAFTAR NOTASI/ LAMBANG ... xiv
DAFTAR SINGKATAN ... xvi
DAFTAR ISTILAH ... xvii
BAB 1 PENDAHULUAN ... 1
1.1 Latar Belakang ... 1
1.2 Rumusan Masalah ... 3
1.3 Tujuan Pembahasan ... 3
1.4 Ruang Lingkup ... 3
1.5 Sumber Data ... 4
1.6 Sistematika Penyajian ... 4
BAB 2 KAJIAN TEORI ... 6
2.1 Bioinformatika ... 6
2.2 DNA, RNA, dan Protein ... 6
2.3 Rekayasa Perangkat Lunak ... 7
2.5 Algoritma Lempel-Ziv ... 9
2.6 Skor Dan Kecepatan Algoritma ... 10
BAB 3 ANALISIS DAN RANCANGAN SISTEM ... 11
3.1 Analisis Program ... 11
3.2 Needleman-Wunsch ... 11
3.2.1 Scoring Matrix ... 11
3.2.2 Traceback ... 14
3.2.3 Alignment ... 14
3.3 Lempel-Ziv ... 15
3.3.1 Lempel-Ziv Factorisation Phrase ... 15
3.4 Substitution Matrix ... 18
3.4.1 Gambaran Aplikasi... 18
3.5 Gambaran Keseluruhan ... 19
3.5.1 Persyaratan Antarmuka Eksternal ... 19
3.5.2 Persyaratan Antarmuka Perangkat Lunak ... 19
3.5.3 Persyaratan Antaruka Perangkat Keras ... 20
3.6 Pemodelan Perangkat Lunak ... 20
3.6.1 Use case Diagram ... 20
3.6.2 Package Dan Class Diagram ... 21
3.6.3 Skenario ... 22
3.6.3.1 Skenario Sistem: Input Sekuens ... 22
3.6.3.2 Skenario Sistem: Pilih Metode Pensejajaran ... 23
3.6.3.3 Skenario Sistem : Menentukan Nilai Pensejajaran ... 25
3.6.4 Activity Diagram ... 25
3.6.4.1 Activity Diagram: Input Sekuens ... 26
3.7 Rencana Pengujian ... 27
BAB 4 IMPLEMENTASI ... 28
4.1 Hasil Tampilan ... 28
4.2 Tampilan Awal Aplikasi ... 28
4.3 Tampilan Progress Dan Output ... 29
4.4 Implementasi Aplikasi ... 30
4.4.1 Scoring Matrix ... 30
4.4.2 Factor Sequence ... 34
4.4.3 Needleman-Wunsch ... 36
4.4.3.1 Compute Matrix ... 36
4.4.3.2 Pairwise Alignment Needleman-Wunsch ... 38
4.4.4 Lempel-Ziv ... 40
BAB 5 PENGUJIAN ... 42
5.1 Hasil Pengujian ... 42
5.2 Pengujian Sekuens Maksimal 1000 Karakter ... 42
5.3 Pengujian Sekuens Di Atas 1000 Karakter ... 47
5.4 Pengujian Spesifik Lempel-Ziv Factorisation Phrase Terhadap Nilai Gap 51 BAB 6 SIMPULAN DAN SARAN ... 53
6.1 Simpulan ... 53
6.2 Saran ... 54
DAFTAR PUSTAKA ... 55
DAFTAR GAMBAR
Gambar 2.1 Dogma sentral biologi molekuler ... 7
Gambar 3.1 Proses Awal Matriks Skor ... 12
Gambar 3.2 Proses Pengisian Tabel Matriks Skor ... 13
Gambar 3.3 Hasil Pengisian Matriks Skor ... 14
Gambar 3.4 Hasil Alignment Needleman-Wunsch ... 15
Gambar 3.5 Proses Pengisian Matriks Skor Lempel-Ziv ... 17
Gambar 3.6 Hasil Alignment Lempel-Ziv ... 17
Gambar 3.7 Contoh Substitution Matrix BLOSUM ... 18
Gambar 3.8 Use case Sistem Bioinformatika ... 20
Gambar 3.9 Package Bioinformatika ... 21
Gambar 3.10 Class Diagram Bioinformatika ... 22
Gambar 3.11 Input Sekuens ... 26
Gambar 3.12 Pilih metode pensejajaran... 26
Gambar 3.13 Menentukan nilai pensejajaran ... 27
Gambar 4.1 Tampilan Awal Aplikasi Bioinformatika ... 29
Gambar 4.2 Tampilan Progress ... 30
Gambar 4.3 Tampilan Output ... 30
Gambar 4.4 Kode Program Untuk Proses Scoring Matrix ... 33
Gambar 4.5 Contoh Proses Scoring Matrix ... 33
Gambar 4.6 Kode Program Untuk Factor Sequence ... 35
Gambar 4.7 Contoh Factor Sequence... 35
Gambar 4.8 Kode Program Untuk Compute Matrix ... 37
Gambar 4.9 Contoh Proses Compute Matrix ... 37
Gambar 4.10 Kode Program Untuk Pairwise Alignment Needleman-Wunsch .... 39
Gambar 4.11 Contoh Proses Sequence Alignment Needleman-Wunsch... 39
Gambar 4.12 Kode Program Untuk Pairwise Alignment Lempel-Ziv ... 41
Gambar 4.13 Contoh Pairwise Sequence Lempel-Ziv ... 41
Gambar 5.1 Grafik Waktu Untuk Pengujian Sekuens Maksimal 1000 Karakter . 44 Gambar 5.2 Grafik Panjang Sekuens Untuk Tiap Test Case ... 45
Gambar 5.4 Grafik Waktu Proses Terhadap Panjang Sekuens ... 46
Gambar 5.5 Grafik Waktu Proses Terhadap Test Case... 49
Gambar 5.6 Grafik Panjang Sekuens Terhadap Test Case ... 50
Gambar 5.7 Grafik Skor Terhadap Panjang Sekuens... 50
DAFTAR TABEL
Tabel 2.1 Jenis perangkat lunak dan contohnya... 7
Tabel 3.1 Lempel-Ziv Factorisation Phrase ... 16
Tabel 3.2 Skenario Input Sekuens ... 23
Tabel 3.3 Skenario Pilih Metode Pensejajaran ... 24
Tabel 3.4 Skenario Menentukan Nilai Pensejajaran ... 25
Tabel 5.1 Pengujian Sekuens Maksimal 1000 Karakter ... 42
Tabel 5.2 Pengujian Sekuens Di Atas 1000 Karakter ... 47
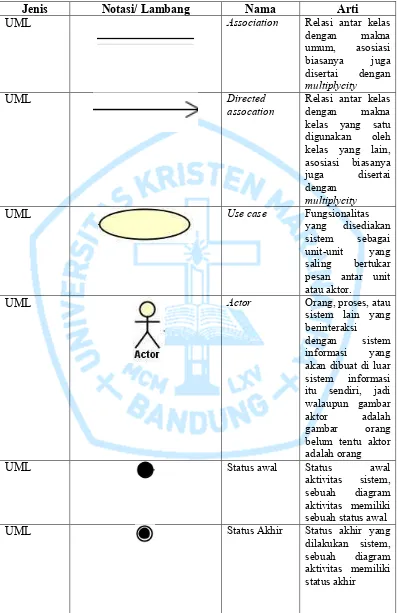
DAFTAR NOTASI/ LAMBANG
UML Use case Fungsionalitas
Jenis Notasi/ Lambang Nama Arti
UML Aktivitas Aktivitas yang
dilakukan sistem, biasanya di awali dengan kata kerja
UML Percabangan Asosiasi
percabangan di mana jika ada pilihan aktivitas lebih dari satu
Referensi:
DAFTAR SINGKATAN
DNA Deoxyribonucleic Acid
RNA Ribonucleic Acid
mRNA Messenger Ribonucleic Acid
BLAST Basic Local Alignment Search Tool
LASTZ Local Alignment Search Tool - Z
YASS Yet Another Similarity Searh
RPL Rekayasa Perangkat Lunak
DAFTAR ISTILAH
Sequence Sekuens atau kumpulan karakter DNA yang berupa urutan huruf-huruf.
Matrix Matriks, implementasi penyusunan karakter-karakter menjadi kotak yang tersusun sehingga dapat dihitung pensejajarannya.
Alignment Pensejajaran, teknik mencari kesejajaran dari sekuens DNA.
Phrase Frase
Data Compression Kompresi data menjadi lebih singkat/kecil.
BAB 1
PENDAHULUAN
1.1Latar Belakang
Kehidupan manusia merupakan rangkaian dari berbagai macam
penerimaan, pengolahan dan penyampaian informasi, baik itu cara bicara, pola
hidup, sejarah, dan semua yang berhubungan dengan kegiatan manusia.
Penyampaian informasi tidak hanya disampaikan di tahap komunikasi manusia
pada umumnya, misalnya berbicara atau menyampaikan sesuatu secara visual,
penyampaian informasi juga terjadi di tahap molekular sel, yaitu DNA. DNA adalah
struktur informasi unik terkecil yang dimiliki oleh setiap organisme dan DNA
berperan penting dalam seluruh kehidupan yang terjadi di muka bumi ini.
Ilmu yang mempelajari tentang DNA secara spesifik, salah satunya, adalah
mikrobiologi. Mikrobiologi sudah berkembang pesat dari abad ke abad dengan
meneliti bagaimana suatu informasi dari DNA tiap organisme dapat diwariskan atau
bahkan dimodifikasi. Seiring dengan perkembangan ilmu mikrobiologi, kehidupan
di dunia juga mengalami perkembangan dan perubahan yang kita sebut dengan
evolusi. Evolusi yang kita kenal berhubungan erat dengan perubahan penyampaian
informasi DNA oleh tiap organisme. Informasi DNA atau sequence dari DNA yang ada mengalami perubahan yang disebut mutasi [2].
Menurut Deepa Agashe dalam penelitiannya di jurnal biologi molekular dan
evolusi, mutasi atau perubahan informasi sel terjadi berulang kali pada banyak
varian gen [3]. Dengan semakin banyaknya perubahan informasi sel dari tiap
organisme, wujud atau cara bertahan hidup sebuah organisme semakin beragam.
Dalam hal ini, ilmu mikrobiologi mendapat tantangan dalam membandingkan
organisme satu dengan yang lainnya; apakah suatu organisme tersebut memiliki
kerabat atau hubungan erat dengan organisme yang memiliki wujud ataupun cara
bertahan hidup yang mirip.
Dengan berkembangnya data tentang informasi DNA yang dimiliki, ilmu
mikrobiologi saja tidak cukup dalam mengolah data dan melakukan perbandingan
2
sequence DNA yang ada, membandingkan, dan dapat mengambil kesimpulan dari hasil analisa tersebut.
Bioinformatika adalah aplikasi dalam bidang sains biologi yang berfokus
pada analisis data sekuens biologi. Bioinformatika cenderung digunakan dalam
komputasi mikrobiologi dan dengan adanya bioinformatika, perkembangan pesat
dari data sequence DNA yang muncul hampir tiap hari dapat diolah dan ditangani secara cepat. Bioinformatika mempunyai peran penting dalam pensejajaran atau
perbandingan sequence DNA. Dalam sequence DNA, bioinformatika menggunakan algoritma untuk mensejajarkan suatu DNA dan mencari kecocokan
dari DNA tersebut sehingga kemiripan dan korelasi antar DNA dari organisme
dapat dianalisis dengan baik.
Untuk melakukan pensejajaran sekuens DNA, program dari ilmu
bioinformatika menggunakan berbagai macam algoritma. Pensejajaran DNA
memiliki dua macam teknik, yaitu pensejajaran global ataupun lokal. Dalam
pensejajaran global, seluruh karakter DNA diproses sementara dalam pensejajaran
lokal hanya sebagian dari karakter DNA yang diproses [2]. Beberapa algoritma
yang digunakan dalam pensejajaran lokal antara lain adalah Smith-Waterman,
FASTA, BLAST, dan masih banyak algoritma yang sedang dikembangkan.
Sementara untuk pensejajaran global, algoritma Needleman-Wunsch masih kerap
digunakan dan juga dikembangkan agar lebih efisien [4]. Di sisi lain, pensejajaran
global mengalami banyak perkembangan dan banyak algoritma-algoritma baru
diciptakan dengan mengadaptasi dari beberapa algoritma dasar, salah satunya
adalah metode Lempel-Ziv, yaitu metode teknik data compression yang kemudian diadaptasi untuk pensejajaran DNA [5].
Kecepatan proses dan ketepatan pensejajaran, menjadi tolak ukur penting
terhadap pensejajaran yang baik oleh suatu algoritma. Untuk itu, mencari
keseimbangan yang optimal terhadap kecepatan dan ketepatan pensejajaran
menjadi dasar pembuatan aplikasi bioinformatika ini dengan
mengimplementasikan dan menganalisa kinerja dua algoritma pensejajaran global
yaitu Needleman-Wunsch dan Lempel-Ziv.
Dari studi literatur yang telah dilakukan kinerja kedua algoritma ini belum
3
meneliti lebih jauh mengenai fungsi, analisa, dan perbandingan dua algoritma ini,
khususnya mengenai kecepatan dan ketepatan serta implementasinya di dalam
sebuah program. Harapannya adalah tugas akhir ini dapat berkontribusi kepada
bidang bioinformatika, khususnya, pemahaman tentang kinerja dua algoritma
tersebut dan, umumnya, bidang ilmu informatika serta penerapannya dalam biologi
seluler.
1.2Rumusan Masalah
Dari latar belakang yang sudah disampaikan, maka dapat dirumuskan
beberapa masalah yang menjadi dasar pembuatan program bioinformatika, yaitu:
1. Bagaimana membuat implementasi Needleman-Wunsch dan Lempel-Ziv ke
dalam suatu program?
2. Bagaimana menentukan algoritma yang tepat untuk tiap kasus pensejajaran
DNA yang berbeda?
3. Bagaimana proses dan analisa dari kedua algoritma dapat membantu penelitian
biologi molekuler ?
1.3Tujuan Pembahasan
Tujuan pembahasan dari analisa algoritma bioinformatika, yaitu:
1. Mengembangkan aplikasi bioinformatika yang memiliki proses pengolahan
data sekuens dengan algoritma Needleman-Wunsch dan Lempel-Ziv.
2. Memberikan analisis dari proses kinerja tiap algoritma untuk menentukan
algoritma yang tepat di tiap kasus berbeda.
3. Memberikan analisis dari kedua algoritma dalam proses pensejajaran DNA
untuk membantu memberikan hasil yang cepat dan tepat dalam penelitian
biologi molekuler.
1.4Ruang Lingkup
Ruang lingkup untuk program ini meliputi 3 hal yaitu aplikasi, perangkat
lunak, dan perangkat keras.
4
a. Aplikasi ini akan memiliki dua metode pengolahan sekuens DNA
berdasarkan dua algoritma yaitu Needleman-Wunsch dan Lempel-Ziv.
b. Tiap metode pengolahan sekuens DNA akan dites dari maksimal 1000
karakter hingga di atas 1000 karakter.
2. Perangkat lunak yang digunakan:
a. Microsoft Office 2013
b. Netbeans
c. JDK 8.0.2
d. Windows 10 Pro
3. Perangkat keras yang digunakan untuk mendukung pembuatan aplikasi ini
adalah:
a. Prosesor Intel Celeron @ 1.50GHz, 64bit.
b. RAM 4GB
c. HDD 500GB
1.5Sumber Data
Sumber data untuk mendukung analisis dari program bioinformatika adalah
string barisan DNA yang didapat dari website National Center for Biotechnology Information (NCBI). String yang digunakan akan diambil secara acak sebanyak 100
buah sekuens DNA dengan panjang kurang dari 1000 karakter untuk 50 dataset
pertama dan panjang lebih dari 1000 karakter untuk 50 dataset kedua. Dataset ini digunakan untuk pengujian kecepatan dan skor dari hasil pensejajaran DNA
menggunakan algoritma Needleman-Wunsch dan Lempel-Ziv.
1.6Sistematika Penyajian
Sistematika penyajian untuk laporan analisa program bioinformatika
5
BAB 1 PENDAHULUAN
Bab ini menjelaskan dasar-dasar pembahasan mengenai DNA, proses
penyaluran informasi organisme, barisan DNA, dan peran bioinformatika dalam
pensejajaran barisan DNA.
BAB 2 KAJIAN TEORI
Berisi teori-teori yang menjadi dasar penyusunan laporan.
BAB 3 ANALISIS DAN PERANCANGAN
Berisi analisis dari tiap algoritma, serta perancangan program
bioinformatika yang memiliki empat algoritma pensejajaran barisan DNA.
BAB 4 IMPLEMENTASI
Berisi penerapan dari program terhadap barisan data DNA.
BAB 5 PENGUJIAN
Berisi pengujian dari tiap algoritma yang ada, serta pengujian keseluruhan
program bioinformatika.
BAB 6 SIMPULAN DAN SARAN
Berisi kesimpulan dari laporan, serta saran dari penulis untuk program yang
BAB 6
SIMPULAN DAN SARAN
6.1Simpulan
Simpulan yang dapat diambil dari hasil pembuatan aplikasi Bioinformatika ini adalah:
1. Aplikasi Bioinformatika dapat memproses alignment dari dua buah sekuens DNA yang dipilih dengan algoritma Needleman-Wunsch dan algoritma Lempel-Ziv.
2. Algoritma Needleman-Wunsch memiliki keunggulan dalam alignment sekuens dengan panjang karakter kurang dari 1000, Needleman-Wunsch memiliki waktu proses dengan nilai minimal 1 miliseconds dan nilai maksimal 75 miliseconds. sementara algoritma Lempel-Ziv memiliki waktu kinerja yang lebih lama dengan waktu proses minimal 25 miliseconds dan maksimal 960 miliseconds dalam penelitian untuk karakter dengan panjang maksimal 1000, namun Lempel-Ziv memiliki keunggulan dalam alignment untuk sekuens dengan faktorisasi sempurna. Faktorisasi sempurna dapat dilihat dari frase yang terurut secara bertahap dengan pengulangan dari tiap frase yang sudah tercatat di atasnya dengan tambahan satu karakter selanjutnya dari tiap frase. Needleman-Wunch dapat digunakan untuk pensejajaran sekuens pada umumnya namun Lempel-Ziv memiliki keunggulan dalam proses pengolahan dataset dengan faktorisasi sempurna.
3. Algoritma Needleman-Wunsch dan Lempel-Ziv dapat memberikan hasil alignment dengan visualisasi gap yang dapat dilihat jelas oleh
pengguna, sehingga hasil alignment tersebut dapat digunakan untuk penelitian dalam bidang biologi molekuler untuk mencari tanda mutasi
(mutation mark) dalam suatu kasus pensejajaran DNA.
4. Lempel-Ziv factorisation phrase memiliki keunggulan hanya dalam kasus sekuens dengan perfect factorisation phrase. Namun proses factorisation phrase tidak mengubah nilai gap terhadap hasil alignment
54
6.2Saran
Saran pengembangan aplikasi Bioinformatika yang dapat diberikan adalah sebagai berikut:
1. Memberikan batasan karakter maksimal untuk pengolahan sekuens yang tidak melebihi kapasitas memori komputer/laptop yang digunakan, karena untuk saat ini, proses alignment dengan karakter yang melebihi 8000 akan mengalami masalah kelebihan pemakaian memori.
2. Memberikan suggestion terhadap algoritma Lempel-Ziv yang memiliki keunggulan dalam pemrosesan data untuk sekuens yang memiliki faktorisasi sempurna. Suggestion dapat berupa disclaimer yang terdapat di aplikasi, sehingga pengguna mengerti bagaimana tiap algoritma menangani sekuens yang diberikan.
ANALISA PERBANDINGAN DAN
IMPLEMENTASI ALGORITMA DNA PAIRWISE
SEQUENCE ALIGNMENT
NEEDLEMAN-WUNSCH DAN LEMPEL-ZIV
TUGAS AKHIR
Diajukan untuk Memenuhi Persyaratan Akademik dalam
Menyelesaikan Pendidikan pada Program Studi
S1 Teknik Informatika Universitas Kristen Maranatha
Oleh
Mikhael Avner Malendes
1372042
PROGRAM STUDI S1 TEKNIK INFORMATIKA
FAKULTAS TEKNOLOGI INFORMASI
UNIVERSITAS KRISTEN MARANATHA
BANDUNG
PRAKATA
Segala puji syukur hanya kepada Tuhan Yesus Kristus atas segala berkat dan hikmat yang diberikan kepada penulis sehingga tugas akhir yang berjudul
“Analisa Perbandingan Dan Implementasi Algoritma DNA Pairwise Sequence
Alignment Needleman-Wunsch Dan Lempel-Ziv“ ini dapat diselesaikan. Penulis
sadar laporan ini masih jauh dari kesempurnaan, kekurangan, dan kesalahan yang terdapat di dalam laporan ini dikarenakan keterbatasan ilmu pengetahuan dan pengalaman penulis. Dengan demikian, penulis meminta kebijaksanaan dan pengertian dari para pembaca untuk memakluminya.
Pada kesempatan ini, penulis ingin mengucapkan terima kasih kepada pihak-pihak yang telah membantu dalam mengerjakan tugas akhir ini.
1. Kepada Bapak Dr. Hapnes Toba, M. Sc. selaku Dekan Fakultas Teknologi Informasi Universitas Kristen Maranatha Bandung.
2. Kepada Bapak Hendra Bunyamin, S.Si., M.T. selaku pembimbing yang telah banyak membantu penulis dalam penyelesaian tugas akhir ini.
3. Kepada Ibu Meliana Christianti J., S.Kom., M.T. selaku Koordinator Tugas Akhir S1 Teknik Informatika Universitas Kristen Maranatha Bandung.
4. Kepada dosen-dosen di Fakultas Teknologi Informasi yang telah memberikan masukan dan memberikan pedoman dalam menyelesaikan tugas akhir ini. 5. Kepada keluarga penulis papi Gerson Malendes, mami Elsye Malendes, kak
Meity dan kak Lian yang telah banyak membantu dan memberikan dukungan dalam doa dan finansial selama menyelesaikan tugas akhir ini.
6. Kepada rekan-rekan penulis Claudia, Silvi, Malvin, Imel, Stefi, Robby, Yoses, Nicky, Jordy, Kevin, Gio yang telah banyak memberikan dukungan dan motivasi sehingga penulis dapat menyelesaikan tugas akhir ini dengan sebaik-baiknya.
Akhir kata, penulis berharap semoga hasil tugas akhir ini, dapat memberikan sesuatu yang dapat berarti bagi semua pihak. Oleh karena itu, penulis akan menerima setiap kritik dan saran yang dapat membangun penulis agar dapat bekerja lebih baik lagi.
Bandung, 13 Desember 2016
DAFTAR PUSTAKA
[1] J. Simarmata, Rekayasa Perangkat Lunak, Yogyakarta: Penerbit ANDI, 2010.
[2] D. P. A. P. Ernawati, “Implementasi Algoritma Smith-Waterman Pada Local Alignment Dalam Pencarian Kesamaan Pensejajaran Barisan DNA (Studi
Kasus : DNA Tumor Wilms),” Jurnal Pseudocode, pp. 170-177, 2014.
[3] D. Agashe, “Large-Effect Beneficial Synonymous Mutations Mediate Rapid
and Parallel Adaptation in a Bacterium,” Molecular Biology and Evolution,
vol. 33, no. 6, pp. 1542-1553, 2016.
[4] W. H. B. R. Alex Aravind, “Pairwise sequence alignment algorithms : a
survey,” Association for Computing Machinery Journal, 2016.
[5] A. Chan, “Stanford Project,” 2004. [Online]. Available: biochem218.stanford.edu/Projects%202004.Chan.pdf.
[6] S. J. P. J. Z. Shanika Kuruppu, “Optimized Relative Lempel-Ziv Compression
of Genomes,” vol. XXXIV, pp. 1-8, 2011.
[7] Z. R. Yang, Machine Learning Approaches to Bioinformatics, Singapore: World Scientific Publishing Co. Pte. Ltd., 2010.
[8] W. S. Susan Elrod, Schaum's Genetika, Jakarta: Erlangga, 2007.
[9] V. Likic, “The Needleman-Wunsch Algorithm for Sequence Alignment,” dalam Bio21 Molecular Science and Biotechnology Institute, Melbourne, 2008.
[10] S. J. P. Dominik Kempa, “Lempel-Ziv Factorization: Simple, Fast, Practical,”
Algorithm Engineering and Experiments (ALENEX), pp. 103-112, 2013.
[11] J. C. G. C. N. L. J. L. J. F. D. B. S. C. L. Dandan Song, “Parameterized
BLOSUM Matrices for Protein Alignment,” Transactions on Computational Biology and Bioinformatics, vol. 12, no. 3, pp. 686-694, 2015.
[13] S. W. P. M. F. R. Muhamad Reza Firdaus Zen, “Penerapan Algoritma Needleman-Wunsch sebagai Salah Satu Implementasi Program Dinamis Pada Pensejajaran DNA dan Protein,” Jurnal STMIK, pp. 1-5, 2006.
[14] R. S. Harris, Improved Pairwise Alignment Of Genomic DNA, Pennsylvania: Pennylvania State University, 2007.
[15] A. A. B. R. Waqar Haque, “An Efficient Algorithm for Local Sequence