LAPORAN PRAKTIKUM BIOFARMASI-FARMAKOKINETIKA FARMAKOKINETIKA SEDIAAN INTRAVENA
(MONO KOMPARTEMEN DAN MULTI KOMPARTEMEN)
Disusun Oleh:
Isman Maulia Reza A. (10060314140) Andri Nopriansyah (10060314141) Reza Nurwahyuni (10060314143) Sri Novi Mutmainah (10060314144) Adela Nursya’bani (10060314145) Humairani Rahman (10060314146) Safira Salsabila A. (10060314147) Shift/Kelompok : D-2 (12.30-16.30) Hari/TanggalPraktikum :Selasa, 28 November 2017 Hari/tanggal Penyerahan laporan :Selasa, 05 Desember 2017 Nama Asisten : Wulan Putri Saraswati, S.Farm
LABORATORIUM FARMASI TERPADU UNIT E PROGRAM STUDI FARMASI
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM UNIVERSITAS ISLAM BANDUNG
FARMAKOKINETIKA SEDIAAN INTRAVENA (MONO DAN MULTI KOMPARTEMEN)
I. Tujuan Percobaan
- Dapat memahami dan menganalisa : definisi dan perhitungan parameter-parameter farmakokinetika
- Dapat membandingkan antara model satu dan dua kompartemen sediaan intravena
II. Prinsip Percobaan
Prinsip dari percobaan ini adalah untuk untuk menentukan parameter farmakokinetik dan menentukan pemodelan farmakokinetik suatu obat dengan menggunakan data yang diberikan.
III. Teori Dasar
Farmakokinetik didefinisikan sebagai perubahan-perubahan kuantitatif dan tergantung kepada waktu dari konsentrasi obat dalam plasma dan jumlah total obat di dalam tubuh yang terjadi setelah pemberian obat dengan cara yang bermacam-macam (dua cara pemberian yang paling biasa adalah infusintravena dan regimen oral dengan dosis interval yang tetap, misalnya suatu tablet setiap 4 jam. (Mycek, 2004).
Fase farmakokinetik berkaitan dengan masuknya zat aktif ke dalam tubuh. Pemasukan in vivo tersebut secara keseluruhan merupakan fenomena fisiko – kimia yang terpadu di dalam organ penerima obat. Fase farmakokinetik ini merupakan salah satu unsur penting yang menentukan profil keadaan zat aktif pada tingkat biofase dan yang selanjutnya menetukan aktivitas terapetik obat (Devissaguet, Aiache, 1993).
farmakokinetika mempelajari segala sesuatu tindakan yang dilakukan tubuh terhadap obat (Tan. H.T, 2002).
Faktor fisiopatologik yang berpengaruh pada fase farmakokinetik dan farmakodinamik suatu obat di dalam tubuh (Devissaguet, Aiache, 1993).
Keturunan biofarmasi sekarang sudah dapat dikontrol, demikian juga absorbsi obat sudah dapat dipertimbangkan dengan seksama faktor – faktor yang mempengaruhinya. Kecepatan eliminai obat dari tubuh sangat ditentukan oleh parameter farmakokinetik obat tersebut. Dalam mengatur kecepatan pelepasan obat, diharapkan kita akan dapat suatu blood level yang terkontrol (Syukri, 2002).
Persamaan kinetika obat dalam darah pada pemberian bolus intravena dengan satu dosis D yang mengikuti model satu kompartemen diberikan dengan persamaan :
Cp = C0 . e-k.t
Dimana Cp adalah kadar obat dalam waktu tertentu, C0 adalah kadar obat
pada waktu 0, k atau ke adalah konstanta kecepatan eliminasi obat. Dengan menggunakan kadar obat pada berbagai waktu, harga C0 dan k dapat dihitung
dengan cara regresi linier setelah persamaan ditransformasikan ke dalam nilai logaritmik :
LnCp = LnC0 – k.t
Setelah ditentukan nilai C0 dan k, berbagai parameter farmakokinetik
obat yang berkaitan dengan cara pemberian obat secara bolus intravena dapat dihitung, seperti :
· volume distribusi (Vd): volume dalam tubuh di mana obat terlarut, · klirens (cl),
· waktu paruh eliminasi (t ½)
· Luas di bawah kurva dalam plasma (AUC) · Bioavalaibilitas (ketersediaan hayati)
Vd = DosisCp
Cl = Vd.k
t ½ = 0,693K
Absorbsi adalah transfer suatu obat dari tempat pemberian ke dalam aliran darah. Kecepatan dan efisiensi absorbsi tergantung pada cara pemberian. Untuk intra vena, absorbsi sempurna yaitu dosis total obat seluruhnya mencapai sirkulasi sistemik. Pemberian obat dengan sirkulasi lain hanya bisa menghasilkan absorbsi yang parsial dan karena itu merendahkan ketersediaan hayati. Tergantung pada sifat-sifat kimianya, obat-obat bisa diabsorbsi dari saluran cerna secara difusi pasif atau transpor aktif (Mycek, 2004).
1. Eliminasi melalui urin oleh filtrasi glomerulus 2. Metabolisme, biasanya oleh hati
3. Ambilan oleh hati dan selanjutnya dieliminasi memalui empedu.
Volume distribusi yang nyata adalah hitungan nilai yang menggambarkan sifat distribusi obat. Vd adalah volume yang dibutuhkan untuk
membuat dosis yang diberikan jika dosis itu didistribusikan dengan merata pada konsentrasi yang diukur di dalam plasma (Olson, 2002).
Kurva kadar plasma – waktu dihasilkan dengan mengukur konsentrasi obat dalam cuplikan plasma yang diambil pada berbagai jarak waktu setelah pemeberian suatu produk obat. Konsentrasi obat dalam tiap cuplikan plasma digambar pada koodinat kertas grafik rektangular terhadap waktu pengambilan cuplikan plasma. Selama obat mencapai sirkulasi umum sistemik, konsentrasi obat dalam plasma akan naik sampai maksimum. Pada umumnya absorbsi suatu obat terjadi lebih cepat dari pada eliminasi. Selama obat diabsorbsi ke dalam sirkulasi sistemik, obat didistribusikan ke semua jaringan dalam tubuh dan juga secara serentak dieliminasi. Eliminasi suatu obat dapat tercapai melalui ekskresi atau biotransformasi atau kombinasi dari keduanya (Shargel, 2012).
Dengan infus intravena yang kontinue, kecepatan obat masuk ke dalam tubuh adalah tetap. Dalam kebanyakan kasus eliminasi obat adalah first order artinya suatu fraksi obat yang tetap dibersihkan persatuan waktu. Oleh karena itu, kecepatan keluarnya obat dari tubuh meningkatkan secara proporsional bila konsentrasi plasma meningkat dan pada setiap saat selalu proporsional terhadap konsentrasi obat dalam plasma (Mycek, 2004).
Dengan model farmakokinetik yang kompleks dapat digunakan program computer untuk menghitung semua parameter menjadi jumlah titik data, seharusnya selalu melebihi jumlah parameter dalam model. Model farmakokinetik berguna untuk :
1. Memperkirakan kadar obat dalam plasma, jaringan dan urin pada berbagai pengaturan dosis.
2. Memperkirakan model kemungkinan akumulasi obat dan atau metabolit-metabolit.
3. Menghitung pengaturan dosis optimum untuk tiap penderita secara individual 4. Menghubungkan konsentrasi obat dengan aktifitas farmakologik atau
toksikologik.
5. Menilai perbedaan laju atau tingkat availabilitas antara formulasi (bioekivalen). 6. Menggambarkan perubahan faal atau penyakit yang mempengaruhi absorpsi atau
eliminasi obat.
7. Menjelaskan interaksi obat. (Syukri, 2002)
Jenis – jenis model farmakokinetik tubuh manusia. Model 1 kompartemen. Menurut model ini, tubuh dianggap sebagai 1 kompartemen tempat obat menyebar dengan seketika dan merata ke selruh cairan dan jaringan tubuh. Model ini terlalu sederhana sehingga untuk kebanyakan obat kurang tepat (Ganiswarna, 1995) .
Model 2 kompartemen. Tubuh dianggap terdiri atas kompartemen sentral dan kompartemen perifer. Kompartemen sentral terdiri dari darah dan berbagai jaringan yang banyak dialiri darah seperti jantung, paru, hati, ginjal dan kelenjar – kelenjar endokrin. Kompartemen perifer adalah berbagai jaringan yang kurang dialiri darah misalnya otot, kulit, dan jaringan lemak. Model 2 kompartemen ini pada prinsipnya sama dengan model kompartemen 1, bedanya hanya dalam proses distribusi karena adanya kompartemen perifer, eliminasi tetap dari kompartemen sentral. Model ini ternyata cocok untuk banyak obat (Ganiswarna, 2005).
Model 3 kompartemen, Kompartemen perifer dibagi atas kompartemen perifer yang dangkal dan kompartemen perifer yang dalam. Model mana yang cocok untuk suatu obat dan dapat diperkirakan dari profil kurva kadar obat dalam plasma terhadap waktu (Ganiswarna, 2005).
Respon biologis terhadap suatu obat, merupakan suatu hasil interaksi antara obat dengan molekul-molekul yang penting secara fungsional dalam sistem hidup atau reseptor. Respon disebabkan oleh suatu perubahan dalam suatu proses biologis yang ada sebelum pemberian obat. Besar respon berhubungan dengan konsentrasi obat yang dicapai pada tempat obat tersebut bekerja. Konsentrasi ini tergantung pada banyaknya dosis obat yang diberikan, besarnya absorbsi dan distribusi ke tempat tersebut dan laju serta besarnya obat yang dieliminasikan di dalam atau dari tubuh (Ansel, 1989).
IV. Prosedur Percobaan
V. Hasil Pengamatan
5.1 Pengamatan Mono Kompartemen
Dilakukan Persamaan Regresi antara ln Cp dan waktu ditiap pengujian obat dan ditentukan nilai R serta model kompartemen (Mono atau Multi)
Mono Kompartemen Multi Kompartemen
Didapatkan hasil persamaan regresi : y = bx + a
Ln Ct = Ke.Ln Cp0
Nilai R = Mendekati 1
Didapatkan hasil persamaan regresi : y = bx + a
Ln Ct = Ke.Ln Cp0
Nilai R = Tidak mendekati 1
Kemudian ditentukan parameter Farmkokinetika meliputi nilai : Ke, t½, lnCo,Dosis,Vd ,Cl dan AUC
Dibuat kurva kalibrasi Post
Distribusi dengan
diregresikan antara waktu dengan LnCp0 dimulai dari
waktu 36 jam.
Dihitung nilai Ln C’ dan C’ (C extrapolasi) dengan menggunakan persamaan regresi dari Post Distribusi (Eleminasi). Nilai x digantikan dengan nilai waktu 6,12 dan 18. Setelah itu Ln C’ diubah menjadi C’.
5.1.1. Tabel Data Mono Kompartemen
Time (menit) Cp (ng/mL) Ln Cp (ng/mL)
30 699 6,5497
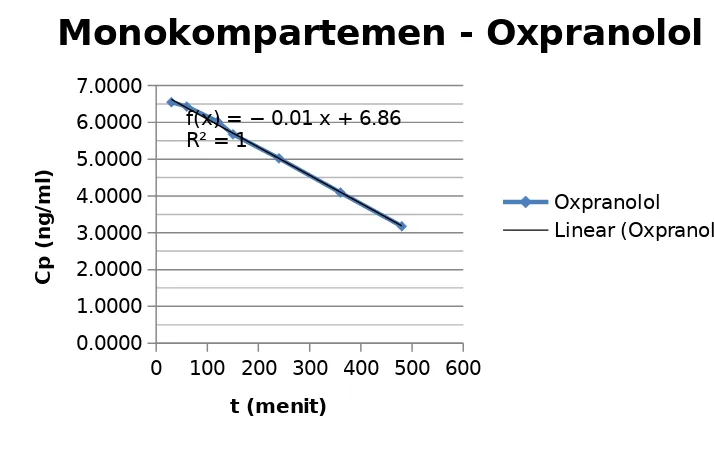
5.1.2. Kurva kalibrasi Mono Kompartemen Oxpranolol
0 100 200 300 400 500 600
5.1.3. Tabel Parameter farmakokinetik dan Hasil
Parameter Farmakokinetik Hasil
t1/2(menit) 91,184211
Ln Cpo (ng/mL) 6,8564
Co(ng/mL) 949,94112
Dosis (ng) 160000000
Vd (mL) 168431,49
Cl (mL/menit) 1280,0793
AUCo~(ng/mL.Menit 124992,25
5.2. Perhitungan Mono Kompartemen 5.2.1. Regresi Mono Kompartemen
Dilakukan Regresi Linear antara lnCp dengan t didapatkan persamaan : y = bx + a y = -0,0076x + 6,8564
r = 0,9984
bila dimasukan kedalam persamaan farmkokinetika : Ct = Cp0. e-Kt Ln Cp = Ln C0 . e-k.t
Ln Cp = 6,8564 – 0,0076x Cp = C0 . e-k.t
Cp = anti Ln C0 . e-k.t
Cp = 949,9411 ng/mL
Didapatkan nilai : Ln Cp0= 6,8564 ng/ml dan Ke = 0,0076 /menit
5.2.1. Parameter Farmakokinetika
t ½ (Waktu Paruh) = ln 2Ke=0,00760,693❑ ❑menit
=91,1842menit
Cp0= anti Ln 6,8564 ng/ml = 949,9411ng/ml
Dosis = 160 mg 160 x 106= 160x106nanogram
Vd (Volume Distribusi ) =DosisCp0 = 160000000ng
949,9411ng/ml=168431,4954ml Cl (Klirens) = Vd x Ke=168431,4954ml x0,0076menit =1280,0793ml/menit
AUCO~=
Dosis
Ke x Vd=0,0076160000000nanogram menit x168431,4954ml
6 360 5,8861 2,0944 8,1206 351,879
4 5,8633 12 70 4,2485 1,8052 6,0812 63,9188 4,1576 18 15 2,7081 1,516 4,5540 10,4460 2,3462
0 10 20 30 40 50 60 70 80 90 100
5.3.3. Tabel Distribusi dan Eliminasi
Parameter Farmakokinetik Hasil
5.4. Perhitungan Multi Kompartemen
5.4.1. Persamaan regresi Multi Kompartemen Methotrexate antara waktu dengan Ln Cp dimulai dari 6-90 jam:
y = -0,0865x + 4,9496
R² = 0,9457 Tidak mendekati 1 (MultiKompartemen)
5.4.2. Persamaan regresi untuk eliminasi antara waktu dengan LnCp dimulai dari waktu 36-90 jam:
y = -0,0482x + 2,3836
R² = 0,9775 Post Distribusi atau Fase Eleminasi 5.4.3. LnC’ atau C ektrapolasi
Didapat dari persamaan y = -0,0482x + 2,3836
dimana nilai x sebagai nilai waktu 6,12 dan 18 sehingga didapatkan nilai LnC’ dan C’ persamaan sebagai berikut :
lnC’ = -0,0482 (6) + 2,3836 = 2,0944 C’ = AntiLn 2,0944 = 8,1206 lnC’ = -0,0482 (12) + 2,3836 = 1,8052 C’ = AntiLn 1,8052 = 6,0812 lnC’ = -0,0482 (18) + 2,3836 = 1,516 C’ = AntiLn 1,516 = 4,5540
5.4.4. Cr dan Ln Cr (C Residual)
Cresidual = C Distribusi / C Observasi – Cp Extrapolasi (C’)
Cr(t=6) = 360 μg/mL – 8,1206 = 351,8794 μg/mL Ln Cr =5,8652
Cr(t=12) = 70 μg/mL – 6,0812 = 63,9188 μg/mL Ln Cr = 4,1576
Cr(t=18) = 15 μg/mL – 4,5540 = 10,4460 μg/mL Ln Cr = 2,3462
5.4.5. Persamaan regresi untuk distribusi antara waktu dengan Ln Cr
dimulai dari waktu ke 6-18 jam:
y = -0,2931x + 7,6394 R2 = 0,9997 Fase Distribusi
5.4.6. Fase Distribusi dan Fase Eliminasi a. Fase Distribusi ( α )
y = -0,2931x + 7,6394
α = b = 0,2931
Ln A = a = 7,6394
A = antiLn A = 2078,4963 b. Fase Eliminasi ( β )
y = -0,0482x + 2,3836 R² = 0,9775
β = b = 0,0482
Ln B = a = 2,3836
B = antiLn B = 10,8438
c. Dosis
400 mg/kgBB X 25 kgBB pasien = 1000 x 103=10000000μg
5.4.7. Parameter Farmakokinetika K (/jam) =
αβ (A + B) (Aβ+Bα) =
0,2931.0,0482(2078,4963+10,8439) (2078,4963.0,0482+10,8439.0,2931)=
0,0141(2089,3402) (103,3619) =¿ ¿103,361929,5169 =0,2856/jam
K12 (/jam) =
AB(β−α)2 (A+B) (Aβ+Bα)=
2078,4963x10,8439(0,0482−0,2931)2
(2078,4963+10,8439) (2078,4963x0,0482+10,8439x0,2931) ¿ 22539,0060(0,0599)
(2089,3402x103,3618)
¿215970,49991350,0864 =0,0063/jam
K21 (/jam) ¿ Aβ+A+BαB =(2078,4963x0,0482+10,8439x0,2931) (2078,4963+10,8439)
¿2089,3402103,3618 =0,0495/jam
t ½ eliminasi (Jam) = 0,693β =0,04820,693 =14,3776 jam
0,2856/jamx4786,1999ml=
10x106µg menggunakan aplikasi software microsoft excel untuk melilhat farmakokonetika sediaan intravena seperti zat aktif oxpranolol dan metrotreksat termasuk dalam mono kompartemen atau multi kompartemen. Tujuan dari penyederhaan farmakokinetika diantaranya untuk menggabrkan suatu sistem biologis yang kompleks berkaitan dengan pergerakan obat dalam tubuh dimana sebagian besar model farmakokinetika menganggap bahwa konsentrasi obat dalam plasma mencerminkan konsentrasi obat dalam tubuh secara global atau keseleruhan (Shargel., et al, 2012 : 11).
Manfaat dari penyederhanaan farmakokinetika tersebut untuk memprediksi kadar obat dalam plasma, jaringan dan urine pada berbagai pengaturan dosis dimana pengaturan dosis yang berbeda walau bentuk sediaan sama kemungkinana kadar obat akan berbeda pula. Menghubungkan kosentrasi yang ada dengan efek farmakologi atau toksikologi dimana obat tersebut harus dapat memberikan efek terapi yang diharapkan dan jangan sampai mencapai konsentrasi toksik atau racun. Berkaitan dengan menghitung pengaturan dosis optimum secara individual karena kondisi fisiologi individula setiap ras asia arau ras eropa atau afrika atau amerika akan menunjukan kondisi yang berbeda maka pengaturan dosis optimum akan berbeda pula (Shargel., et al, 2012 : 10-11).
model satu kompartemen atau multi kompartemen yang sangat tergantung pada proses yang dialami zat aktif selama dalam tubuh, jika obat tersebut ketika dimasukan kedalam tubuh langsung tersebar, seketika juga mengalami eliminasi dan dari hasil perhitungan mendapatkan kurva yang linier maka obat tersebut mengikuti model kompartemen 1. Dimana jika obat tersebut ketika dimasukan kedalam tubuh obat tersebut akan masuk kedalam kompartemen sentral setelah itu masuk kekompartemen perifer lalu kembali lagi kekompartemen sentral lalu mengalami eliminasi, dimana suatu zat tersebut tidak lain dan tidak bukan bahwa zat tersebut mengalami distribusi lalu mengalami eliminasi dapat dilihat dari grafik yang tidak linier.
Monokompartemen
Pada model satu kompartemen tubuh dianggap sebagai satu kesatuan. Jadi obat masuk dan secara cepat terdistribusi ke semua bagian lalu obat juga dapat keluar dari tubuh karena merupakan kompartemen terbuka. jika tubuh diasumsikan sebagai satu kompartemen, tidak berarti bahwakadar obat sama di dalam setiap jaringan atau organ, namun asumsi yang berlaku pada model tersebut ialah bahwa perubahan kadar obat di dalam darah mencerminkan perubahan kadar obat di jaringan. laju eliminasi (metabolism danekskresi) obat dari tubuh setiap saat sebanding dengan jumlah atau kadar obatyang tersisa di dalam tubuh pada saat itu
pada praktikum kali ini dilakukan simulasi in vitro model kompartemen satu terbuka dengan reaksi orde kesatu. Simulasi dilakukan baik dalam rute intravaskuler maupun rute ekstravaskuler. Rute intravaskuler dimodelkan untuk obat-obat intra vena dan rute ekstravaskuler dimodelkan untuk obat obat yang biasanya melalui fase absorpsi, seperti obat oral.
dengan protein plasma dan sebagian dalam bentuk bebas. Obat bebas selanjutnya didistribusikan sampai ditempat kerjanya dan menimbulkan efek. Kemudian dengan atau tanpa biotransformasi obat diekskresikan dari dalam tubuh melalui organ-organ ekskresi,terutama ginjal. Seluruh proses yang meliputi absorpsi, distribusi, metabolisme dan ekskresi disebut proses farmakokinetik dan proses ini berjalan serentak. Perbedaan jalur pemberian obat menyebabkan ketersediaan obat dalam aliran tubuh berbeda pula. Intravascular memiliki bioavailibilitas yang lebih tinggi (100%) karena obat langsung didistribusikan ke sistemik. Sedangan pada ekstravaskular, bioavailibilitasnya lebih rendah dibanding intravaskular. hal ini dikarenakan obat mengalami proses absorpsi terlebi dahulu.
0 100 200 300 400 500 600
Gambar diatas diumpamakan obat disuntikkan secara langsung ke dalam kompartemen ini (injeksi intravena) dan obat langsung masuk ke sistemik, tidak melalui proses absorpsi dulu mendistribusikan ke seluruh kompartemen. Konsentrasi obat pada waktu nol (Co) dapat dihitung dengan cara besarnya dosis obat (D) dibagi dengan besarnya volume distribusi.
keCepatan absorbs (ka") menggambarkankecepatan absorbsi, yaitu masuknya obat ke dalam sirkulasi sistemik dari absorbsinya (saluran cerna pada pemberian oral, jaringan otot pada pemberianintramuskular). Parameter inilah yang membedakan antara ekstravaskular dengan intravaskular.
Proses distribusi diilustrasikan oleh larutan dalam gelas beker. Parameter farmakokinetika yang digunakan yaitu volume distribusi (vd) merupakan volume hipotesis cairan tubuh yang akan diperlukan untuk melarutkan jumlah total obat pada konsentrasi yang sama seperti yang ditemukan dalam darah. Digunakan satu wadah sebagai ilustrasi model kompartemen satu terbuka. Model ini menganggap bahwa berbagai perubahan kadar obat dalam plasma mencerminkan perubahan yang sebanding dengan kadar obat dalam jaringan.
Klirens merupakan parameter farmakokinetika yang menggambarkan eliminasi obat yang merupakan jumlah volume cairan yang mengandung obat yang dibersihkan dari kompartemen tubuh setiap waktu tertentu. Secara umum eliminasi obat terjadi pada ginjal dan hati yang sering dikenal dengan istilah klirens total yang merupakan jumlah dari klirens ginjal (renalis) dan hati (hepatik)
Perbedaan model satu kompartemen dan 2 kompartemen yaitu Pada model 1 kompartemen, obat menganggap tubuh seperti 1 ruang yang sama dimana obat secara cepat terdistribusi ke semua jaringan, sehingga obat langsung di distibusikan tanpa proses abssorpsi. Sedangkan pada model 2 kompartemen, obat menganggap tubuh seperti 2 bagian:
Kompartemen sentral: organ2 dimana perfusi darahnya cepat (misalnya hati, ginjal)
Kompartemen perifer: organ2 dimana perfusi darahnya lambat (misalnya otot, lemak)
Sehingga obat masuk mengalami fase absorpsi, sehingga distribusi obat kedalam darah lambat.
Manfaat pemodelan farmakokinetik
menghitung pengaturan dosis optimum untuk tiap penderita secara individu,
memperkirakan kemungkinan akumulasi obat dan /atau metabolit-metabolit,
menghitung konsentrasi obat dengan aktivitas farmakologik atau toksikologik,
menilai perbedaan laju atau tingkat ketersediaan farmasetika dan hayati antar formulasi,
menggambarkan perubahan faal atau penyakit yang mempengaruhi absorpsi, distribusi, atau eliminasi obat,
menjelaskan interaksi obat.
Multi Kompartemen
Pada praktikum biofarmasi-farmakokinetika kali ini melakukan percobaan farmakokinetika sediaan intravena. Praktikum kali ini bertujuan agar dapat memahami dan menganalisa : definisi dan perhitungan parameter-parameter farmakokinetika dan dapat membandingkan antara model satu dan dua kompartemen sediaan intravena.
Penelitian farmakokinetik suatu zat aktif merupakan penelitian identifikasi dan penetapan konsentrasi obat dalam tubuh sebagai fungsi waktu sehingga dapat menggambarkan model parmakokinetik yang khas. Farmakokinetik adalah ilmu yang mempelajari kinetik zat aktif dalam tubuh (in vivo) dimulai dari absorpsi, distribusi, metabolisme, dan ekskresi. Obat yang masuk ke dalam tubuh akan mengikuti suatu model farmakokinetik yang khas. Model tersebut dapat berupa model satu kompartemen atau multi kompartemen yang sangat tergantung pada proses yang dialami zat aktif selama dalam tubuh (Shargel, 2005).
Penetapan kompartemen farmakokinetik dari obat pada setiap tahap perlu ditetapkan secara kuantitatif dan dijelaskan dengan bantuan parameter farmakokinetik. Parameter farmakokinetik ditentukan dengan perhitungan matematika dari data kinetika obat di dalam plasma atau di dalam urin yang diperoleh setelah pemberian obat melalui berbagai rutepemberian, baik secara intravaskular atau ekstravaskular. Parameter farmakokinetik dapat digunakan sebagai klasifikasi farmakokinetik dari obat-obatan yang digunakan dimana akhirnya akan berguna dalam penggunaannya dalam terapi pengobatan (Shargel, 2005).
jaringan-jaringan dengan perfusi tinggi. Kompartemen sentral secara cepat terdifusi oleh obat. Sedangkan kompartemen perifer yang berisi jaringan-jaringan yang berkesetimbangan lebih lambat dengan obat. Model ini menganggap bahwa obat dieleminasi dari kompartemen sentral. Pada multi kompartemen obat tidak langsung tereleminasi. Akan tetapi ada fase terdistribusi dan tereleminasi. (Shargel, 2005).
Setelah diketahui pemodelannya maka dilakukan perhitungan parameter-parameter farmakokinetika untuk multi kompartemen yang meliputi : Ln C’, C’, Cr’, Ln Cr yang diambil data dari waktu 6,12, 18 untuk menentukan nilai dari distribusi obat dari methotrexate. Kemudian K (/jam), K12 (/jam), K21 (/jam), t1/2
eleminasi (/jam), Vp (mL), AUC0~ (mg/mL.jam), Cl (ml/jam), t1/2 distribusi (/jam).
Dari hasil data pengamatan pemodelan farmakokinetika yang dilakukan dari methotreaxeta dengan dosis 400 mg/kg, diperoleh waktu paruh eleminasi obat yaitu 14,3776/jam dengan laju eleminasinya 0,2856/jam. Waktu paruh terminal yang dilaporkan untuk methotreaxeta kira-kira 3-10 jam untuk pasien yang menerima pengobatan psoriasis, atau rheumatoid arthritis atau terapi antineoplastik dosis rendah (kurangnya 30 mg). Bagi pasien yang menerima methotreaxeta dpsis tinggi, waktu paruh adalah 8-15 jam. (Roxane Labolatories. lnc, 2007)
menyerupai konsentrasi jaringan percobaan. kalaupun kompartemen-kompartemen jaringan bersifat hipotetik. Kadar dalam jaringanteoritik masih merupakan suatu informasi yang berharga untuk para dokter. Konsentrasi dalam jaringan teoritik dan konsentrasi dalam darah, merupakan cara yang teliti untuk menghitung jumlah keseluruhan obat yang tertinggal dalam tubuh pada setiap waktu. (Shargel, 2005).
Parameter farmakokinetika model multi kompartemen
Kompartemen jaringan merupakan konsentrasi obat rata-rata dalam suatu kelompok jaringan dan bukan merupakan konsetrasi obat yang sebenarnya dalam tiap jaringan anatomik.
Volume kompartemen sentral berguna untuk menggambarkan perubahan kosentrasi obat, oleh karena itu kompartemen sentral umumnya merupakan kompartemen yang diambil sebagai kompartemen cuplikan
a. Volume distribusi
Volume distribusi merupakan suatu parameter yang berguna dengan mengaitkan konsentrasi plasma dengan jumlah obat dalam tubuh. Untuk suatu obat yang dianga mengikuti model multi kompartemen, ada beberapa volume distribusi yang dapat diperhitungkan.
Volume distribusi dalam keadaan tunak
Obat masuk ke dalam kompartemen jaringan dari kompartemen sentral adalah sama dengan laju obat yang keluar dari kompartemen jaringan ke dalam kmpartemen sentral. Laju pemindahan obat ini dinyatakan sebagai : Dt K21 = Dp K12
Jumlah total obat dalam tubuh pada keadaan tunak adalah sama dengan jumlah obatdalam kompartemen jaringan. Dt dan jumlah obat dalam kompartemen sentral.
Volume distribusi yang dieksplorasikan
b. Klirens/bersihan (Cl)
menunjukkan berapa banyak urin yang dikeluarkan per waktu / kemampuan mengeliminasi (satuannya: volume/waktu). (Shargel, 2005). c. T1/2 Eleminasi
Jika terjadi gangguan pada ginjal yang menyebabkan clearance terganggu maka waktu paruh juga terpengaruh
Jika Clearance naik maka t1/2 turun -> karena obat cepet dieksresi Jika Clearance turun maka t1/2 naik -> karena obat lama dieksresi (Shargel, 2005).
d. Tetapan laju eliminasi
Dalam model kompartemen dua (pemberian IV) tetapan laju eliminasi K menyatakan eliminasi obat dari kompartemen sentral, sedangkan b menyatakan eliminsai obat dari seluruh tubuh setelah obat yang berdifusi mengalami kestimbangan. Oleh karena itu b berguna dalam perhitungan t1/2 dengan pengaturan dosis.
(Shargel, 2005).
1) Pemodelan farmakokinetik adalah untuk mengukur absorbsi, distribusi, metabolisme dan ekskresi obat di dalam tubuh yang diberikan secara intravena
2) Perbedaan satu kompartemen dan dua atau multi kompartemen yaitu fase absorpsi. Distribusi satu kompartemen lebih cepat dibandingkan dengan dua kompartemen
3) Oxpranolol mengikuti model kompartemen satu terbuka karena terbentuk kurva yang linier r = 0.9984
4) Methothrexate mengikuti model kompartemen dua terbuka karena kurva tidak menunjukkan linier serta adanya fase distribusi dan eliminasi.
Ansel.,Howard., C. 2004. Pengantar Bentuk Sediaan Farmasi. UI Press. Jakarta.
Devissaguet., Aiache. 1982. Farmaseutika 2 Biofarmasi Edisi ke-2. Tehnique et Documentation 11 Rue Lavoiser . Air langga University Press
Ganiswarna., 2005. Farmakologi Dan Terapi. Bagian Farmakologi Fakultas Kedokteran. Universitas Indonesia. Jakarta.
Michael., J., Neal. 2006. At a Glance Farmakologi Medis Edisi ke Lima. Penerbit Erlangga PT Gelora Aksara Pratama. Jakarta.
Mycek., 2004. Farmakologi Ulasan Bergambar. Widya Medika. Jakarta.
Olson James. 2004. Belajar Mudah Farmakologi. Penerbit Buku Kedokteran EGC. Jakarta. Hal 5
Shargel, Leon. 2012. Biofarmasetika Dan Farmakokinetika Terapan. Air Langga University. Jakarta.
Syukri.,Y. 2002. Biofarmasetika. UI Press. Yogyakarta.
Tan., H., Tjay dan Kirana Rahardja. 2002. Obat – Obat Penting. Elex Media Komputindo. Jakarta.