UNIVERSITAS INDONESIA
LAPORAN PRAKTEK KERJA PROFESI APOTEKER
DI PT. ASTRAZENECA INDONESIA – CIKARANG SITE
JL. TEKNO RAYA BLOK B1A - B1B, JABABEKA III
CIKARANG, BEKASI
PERIODE 7 JANUARI – 1 MARET 2013
LAPORAN PRAKTEK KERJA PROFESI APOTEKER
DITA ROSYITA DEWI, S.Farm
1206312984
ANGKATAN LXXVI
PROGRAM PROFESI APOTEKER
FAKULTAS FARMASI
DEPOK
JUNI 2013
UNIVERSITAS INDONESIA
LAPORAN PRAKTEK KERJA PROFESI APOTEKER
DI PT. ASTRAZENECA INDONESIA – CIKARANG SITE
JL. TEKNO RAYA BLOK B1A - B1B, JABABEKA III
CIKARANG, BEKASI
PERIODE 7 JANUARI – 1 MARET 2013
LAPORAN PRAKTEK KERJA PROFESI APOTEKER
Diajukan sebagai salah satu syarat untukmemperoleh gelar ApotekerDITA ROSYITA DEWI, S.Farm
1206312984
ANGKATAN LXXVI
PROGRAM PROFESI APOTEKER
FAKULTAS FARMASI
DEPOK
JUNI 2013
Puji dan syukur penulis panjatkan kepada Allah SWT atas segala rahmat dan karunia yang telah dilimpahkan sehingga penulisan laporan Praktek Kerja Profesi Apoteker (PKPA) di PT AstraZeneca Indonesia (PT. AZI) – Cikarang Site ini dapat terselesaikan. Shalawat dan salam senantiasa tercurah kepada Nabi Muhammad SAW beserta keluarga dan sahabatnya. Pada kesempatan ini, penulis ingin menyampaikan terima kasih dan rasa hormat kepada:
1. Bapak Drs. Rizman, M.S., Apt., selaku Plant Director PT. AZI – Cikarang Site atas izin dan kesempatan yang diberikan sehingga terlaksananya Praktek Kerja Profesi Apoteker di PT. AZI – Cikarang Site.
2. Ibu Dra. Sannaria Uliarta M.S., Apt., selaku Quality Assurance & Safety Health and Environment (QA & SHE) Manager PT. AZI – Cikarang Site, atas bimbingan, kesempatan, dan fasilitas yang telah diberikan untuk melaksanakan Praktek Kerja Profesi Apoteker di PT. AZI – Cikarang Site. 3. Ibu Haryanti Diah Astuti, S.Farm., Apt., selaku Quality Assurance
Supervisor, atas bimbingan dalam pengerjaan tugas umum dan khusus serta pembelajaran selama melaksanakan Praktek Kerja Profesi Apoteker di PT. AZI – Cikarang Site.
4. Ibu Prof. Dr. Yahdiana Harahap, M.Si., Apt., selaku Dekan Fakultas Farmasi UI yang telah memberi izin dan kesempatan untuk melakukan Praktek Kerja Profesi Apoteker.
5. Bapak Dr. Harmita, Apt., selaku Ketua Program Profesi Apoteker Fakultas Farmasi UI atas segala bantuan dan nasihatnya selama ini.
6. Bapak Dr. Iskandarsyah, M.S., Apt., selaku pembimbing di Program Profesi Apoteker Fakultas Farmasi UI yang telah memberikan arahan dan bimbingan pada penulis selama pelaksanaan dan penyusunan laporan PKPA di PT. AZI – Cikarang Site.
7. Seluruh karyawan di PT. AZI – Cikarang Site (QC, produksi, engineering, GA, warehouse, security) atas keramahannya dan kesediaannya dalam membantu dan memberikan informasi.
dan masukan selama ini.
9. Keluarga tercinta atas segenap kasih sayang, perhatian, dukungan serta motivasi yang diberikan.
10. Teman-teman farmasi angkatan 2008 dan Apoteker angkatan 76 yang sudah berjuang bersama selama 1 tahun ini.
11. Semua pihak yang tidak dapat disebutkan namanya satu persatu atas segala bantuan baik secara langsung maupun tidak langsung kepada penulis selama penulisan dan penyusunan laporan ini.
Penulis menyadari bahwa penulisan laporan ini masih jauh dari kesempurnaan. Oleh karena itu, penulis dengan senang hati menerima segala kritik dan saran demi perbaikan di masa yang akan datang. Akhir kata, penulis berharap semoga Tuhan Yang Maha Esa berkenan membalas segala kebaikan semua pihak yang telah banyak membantu. dan semoga laporan ini dapat memberi manfaat bagi pembacanya.
Penulis 2013
HALAMAN JUDUL ... ii
HALAMAN PENGESAHAN ... iii
KATA PENGANTAR ... iv
HALAMAN PENGESAHAN ... vi
DAFTAR ISI ... vii
DAFTAR GAMBAR ... ix DAFTAR TABEL ... x DAFTAR LAMPIRAN ... xi BAB 1. PENDAHULUAN ... 1 1.1. Latar Belakang ... 1 1.2. Tujuan ... 2
BAB 2. TINJAUAN UMUM ... 3
2.1. Industri Farmasi ... 3
2.2. Cara Pembuatan Obat yang Baik ... 6
2.2.1 Manajemen Mutu ... 7
2.2.2 Personalia ... 8
2.2.3 Bangunan dan Fasilitas ... 10
2.2.4 Peralatan ... 11
2.2.5 Sanitasi dan Higiene ... 12
2.2.6 Produksi ... 13
2.2.7 Pengawasan Mutu ... 14
2.2.8 Inspeksi Diri dan Audit Mutu ... 14
2.2.9 Penanganan Keluhan Terhadap Produk, Penarikan Kembali Produk, dan Produk Kembalian ... 15
2.2.10 Dokumentasi ... 15
2.2.11 Pembuatan dan Analisis Berdasarkan Kontrak ... 16
2.2.12 Kualifikasi dan Validasi ... 16
BAB 3. TINJAUAN KHUSUS PT ASTRAZENECA - CIKARANG SITE 18
3.1. Sejarah PT AstraZeneca Indonesia ... 18
3.2. Visi dan Misi PT AstraZeneca Indonesia ... 20
3.3. Struktur Organisasi Operasional ... 20
3.4. Lokasi dan Sarana Produksi ... 33
3.5. Bangunan dan Fasilitas serta Sarana Penunjang ... 34
3.6. Produk PT AZI - Cikarang Site ... 54
BAB 4. PEMBAHASAN ... 55
5.1. Manajemen Mutu ... 56
5.2. Personalia... 57
5.3. Bangunan dan Fasilitas ... 59
5.4. Peralatan ... 62
5.5. Sanitasi dan Higiene ... 62
5.9. Penanganan Keluhan Terhadap Produk, Penarikan Kembali
Produk dan Produk Kembalian ... 70
5.10. Dokumentasi ... 73
5.11. Pembuatan dan Analisis Berdasarkan Kontrak ... 74
5.12. Kualifikasi dan Validasi ... 75
BAB 6. KESIMPULAN DAN SARAN ... 78
6.1.Kesimpulan... 78
6.2. Saran ... 78
Gambar 3.1 Logo AstraZeneca ... 18 Gambar 3.2 Struktur organisasi sistem QA & SHE di PT. AZI – Cikarang Site 21 Gambar 3.3 PT. AZI - Cikarang Site... . 35
Tabel 3.1 Beberapa sertifikat GMP yang diperoleh PT. AstraZeneca
Indonesia ... 20 Tabel 3.2 Persyaratan aliran tekanan ... 50
Lampiran 1. Bagan struktur organisasi PT. AZI- Cikarang Site... 80 Lampiran 2. Produk PT. AstraZeneca Indonesia ... 81
1.1 Latar Belakang
Industri farmasi adalah badan usaha yang memiliki izin dari Menteri Kesehatan untuk melakukan kegiatan pembuatan obat atau bahan obat. Seperti yang telah diketahui, obat merupakan produk industri farmasi yang digunakan oleh masyarakat untuk mencegah, mengobati atau sebagai terapi pemeliharaan suatu penyakit. Oleh karena itu, pemerintah selalu berupaya untuk melindungi masyarakat dari beredarnya obat dengan kualitas rendah, tidak aman, dan membahayakan masyarakat. Salah satu upaya tersebut adalah dengan membuat regulasi atau kebijakan bagi industri farmasi dalam melakukan kegiatan pembuatan obat hingga obat tersebut siap didistribusikan ke masyarakat. Regulasi dibuat agar setiap industri farmasi selalu menghasilkan obat yang sesuai dengan tujuan penggunaannya, memenuhi persyaratan yang tercantum dalam dokumen izin edar (registrasi) dan tidak menimbulkan resiko yang membahayakan penggunanya.
Regulasi pemerintah yang ditujukan bagi industri farmasi dalam rangka menghindari beredarnya obat dengan kualitas, keamanan serta khasiat yang tidak diharapkan tertuang dalam Cara Pembuatan Obat yang Baik (CPOB). CPOB pertama kali diterbitkan pada tahun 1988, kemudian diikuti dengan penerbitan Petunjuk Operasional Penerapan CPOB pada tahun 1989 untuk memberikan penjelasan dalam penjabaran sehingga pedoman ini dapat diterapkan secara efektif di setiap industri farmasi. Sejauh ini, CPOB telah mengalami revisi sebanyak dua kali, yaitu pada tahun 2001 dan 2006. Pemerintah juga melakukan pembaharuan terhadap Penerapan Pedoman CPOB hingga tahun 2012 dalam rangka mengikuti perkembangan ilmu pengetahuan dan teknologi di bidang pembuatan obat dan bahan obat.
Salah satu aspek yang menentukan keberhasilan pelaksanaan CPOB di suatu industri farmasi adalah tersedianya sumber daya manusia yang memahami dan menjamin penerapan CPOB di setiap kegiatan perusahaan. Peran tersebut merupakan keahlian seorang apoteker karena profesi tersebutlah yang menguasai
konsep-konsep dan tujuan dari penerapan CPOB. Oleh karena itu,seorang apoteker di industri farmasi selain dituntut untuk selalu mengikuti perkembangan terkait obat juga dituntut untuk selalu memperbaharui pengetahuannya mengenai perkembangan regulasi pemerintah dalam mengatur CPOB dan mampu mengimplementasikannya dalam setiap kegiatan di perusahaan.
Praktek Kerja Profesi Apoteker (PKPA) merupakan salah satu sarana bagi calon apoteker untuk mendapatkan pengalaman praktis dan pemahaman yang lebih dalam mengenai tugas dan fungsi Apoteker di industri farmasi. Untuk merealisasikan tujuan tersebut, Program Pendidikan Apoteker Universitas Indonesia menjalin kerjasama dengan PT AstraZeneca Indonesia – Cikarang SIte untuk memberikan kesempatan kepada calon apoteker menyelenggarakan PKPA yang dilaksanakan mulai tanggal 7 Januari 2013 sampai dengan 1 Maret 2013.
1.2 Tujuan
Pelaksanaan Praktek Kerja Profesi Apoteker di industri farmasi bagi para calon apoteker bertujuan untuk:
a. Mengetahui penerapan CPOB di PT. AstraZeneca Indonesia – Cikarang Site.
b. Mengetahui serta memahami peran dan tanggung jawab apoteker di dalam industri farmasi.
2.1 Industri Farmasi (Menteri Kesehatan RI, 2010)
Berdasarkan Peraturan Menteri Kesehatan Republik Indonesia No. 1799/MENKES/PER/XII/2010, Industri Farmasi merupakan badan usaha yang memiliki izin dari Menteri Kesehatan untuk melakukan kegiatan pembuatan obat atau bahan obat. Obat dapat didefinisikan sebagai bahan atau paduan bahan, termasuk produk biologi yang digunakan untuk mempengaruhi atau menyelidiki sistem fisiologi atau keadaan patologi dalam rangka penetapan diagnosis, pencegahan, penyembuhan, pemulihan, peningkatan kesehatan dan kontrasepsi, untuk manusia. Bahan obat merupakan bahan baik yang berkhasiat maupun tidak berkhasiat yang digunakan dalam pembuatan obat dengan standar dan mutu sebagai bahan baku farmasi.
Pembuatan obat adalah seluruh tahapan kegiatan dalam menghasilkan obat yang meliputi pengadaan bahan awal dan bahan pengemas, produksi, pengemasan, pengawasan mutu, dan pemastian mutu sampai diperoleh obat untuk didistribusikan. Proses pembuatan obat dan/atau bahan obat hanya dapat dilakukan oleh industri farmasi dan Instalasi Farmasi Rumah Sakit untuk keperluan pelaksanaan pelayanan kesehatan di rumah sakit yang bersangkutan setelah memenuhi persyaratan CPOB yang dibuktikan dengan sertifikat CPOB. Industri farmasi dapat melakukan kegiatan proses pembuatan obat dan/atau bahan obat untuk semua tahapan dan/atau sebagian tahapan. Industri farmasi yang melakukan kegiatan proses pembuatan obat dan/atau bahan obat untuk sebagian tahapan harus berdasarkan penelitian dan pengembangan yang menyangkut produk.
Setiap pendirian industri farmasi wajib memperoleh izin dari Direktorat Jendral Bina Kefarmasian dan Alat Kesehatan yang merupakan bagian dari Kementrian Kesehatan dengan persyaratan sebagai berikut:
a) Berbadan usaha berupa perseroan terbatas
b) Memiliki rencana investasi dan kegiatan pembuatan obat c) Memiliki Nomor Pokok Wajib Pajak
d) Memiliki secara tetap paling sedikit 3 (tiga) orang Apoteker Warga Negara Indonesia masing-masing sebagai penanggung jawab pemastian mutu, produksi, dan pengawasan mutu; dan
e) Komisaris dan direksi tidak pernah terlibat, baik langsung atau tidak langsung dalam pelanggaran perundang-undangan di bidang kefarmasian.
Industri farmasi wajib memiliki Izin Usaha Industri Farmasi. Untuk memperoleh Izin Usaha Industri Farmasi, suatu perusahaan harus melewati tahap persetujuan prinsip yang diajukan secara tertulis kepada Direktur Jendral Bina Kefarmasian dan Alat Kesehatan dengan tembusan kepada Kepala Badan Pengawas Obat dan Makanan (Badan POM) dan Kepala Dinas Kesehatan Provinsi setempat. Sebelum pengajuan permohonan prinsip tersebut, industri farmasi wajib mengajukan persetujuan Rencana Induk Pembangunan (RIP) kepada Kepala Badan POM yang akan disetujui paling lama 14 hari setelah permohonan diajukan. Sementara itu, pemohon izin industri farmasi dengan status Penanaman Modal Asing (PMA) atau Penanaman Modal Dalam Negeri (PMDN) harus memiliki Surat Persetujuan Penanaman Modal dari instansi yang menyelenggarakan urusan penanaman modal sesuai peraturan perumdang-undangan. Persetujuan prinsip akan diberikan paling lama 14 hari kerja setelah pengajuan permohonan apabila disetujui oleh Direktur Jenderal Bina Kefarmasian dan Alat Kesehatan.
Persetujuan prinsip diberikan kepada industri farmasi untuk dapat langsung melakukan persiapan-persiapan, pembangunan, pengadaan, pemasangan instalasi peralatan, dan lain-lain yang diperlukan termasuk produksi percobaan dengan memperhatikan ketentuan perundang-undangan di bidang obat. Persetujuan prinsip tersebut berlaku selama jangka waktu 3 tahun dan dapat diperpanjang oleh Direktur Jenderal Bina Kefarmasian dan Alat Kesehatan untuk paling lama satu tahun. Selama melaksanakan pembangunan fisik, setiap enam bulan sekali perusahaan yang bersangkutan wajib menyampaikan informasi kemajuan pembangunan fisik kepada Direktur Jenderal Bina Kefarmasian dan Alat Kesehatan dengan tembusan kepada Kepala Badan POM dan Kepala Dinas Kesehatan Provinsi setempat. Persetujuan prinsip dapat batal apabila setelah
jangka waktu tiga tahun dan/atau setelah jangka waktu satu tahun perpanjangan, perusahaan yang bersangkutan belum menyelesaikan pembangunan fisik.
Pemohon yang telah selesai melaksanakan tahap persetujuan prinsip dapat mengajukan permohonan izin industri farmasi. Surat permohonan izin industri farmasi harus ditandatangani oleh direktur utama dan apoteker penanggung jawab pemastian mutu dengan kelengkapan sebagai berikut:
a) Fotokopi persetujuan prinsip Industri Farmasi
b) Surat Persetujuan Penanaman Modal untuk Industri Farmasi dalam rangka PMA atau PMDN
c) Daftar peralatan dan mesin-mesin yang digunakan d) Jumlah tenaga kerja dan kualifikasinya
e) Fotokopi sertifikat Upaya Pengelolaan Lingkungan dan Upaya Pemantauan Lingkungan / Analisa Mengenai Dampak Lingkungan
f) Rekomendasi pemenuhan persyaratan CPOB dari Kepala Badan POM g) Rekomendasi kelengkapan administratif dari Kepala Dinas Kesehatan
Kesehatan Provinsi
h) Daftar pustaka wajib seperti Farmakope Indonesia edisi terakhir
i) Surat pernyataan kesediaan bekerja penuh dari masing-masing apoteker penanggung jawab pengawasan mutu dan pemastian mutu
j) Fotokopi surat pengangkatan bagi masing-masing apoteker penanggung jawab produksi, pengawasan mutu, dan pemastian mutu dari pimpinan perusahaan
k) Fotokopi ijazah dan Surat Tanda Registrasi Apoteker (STRA) dari masing-masing apoteker penanggung jawab produksi, pengawasan mutu dan pemastian mutu
l) Surat penyataan komisaris dan direksi tidak pernah terlibat, baik langsung atau tidak langsung dalam pelanggaran perundang-undangan di bidang kefarmasian.
Setelah permohonan izin industri farmasi diajukan, Kepala Badan POM akan melakukan audit pemenuhan persyaratan CPOB dan Kepala Dinas Kesehatan Provinsi akan melakukan verifikasi kelengkapan persyaratan
administratif paling lama dalam waktu 20 hari kerja. Setelah dinyatakan memenuhi persyaratan CPOB oleh Kepala Badan POM dan memenuhi kelengkapan persyaratan administratif oleh Kepala Dinas Kesehatan Provinsi setempat, kedua lembaga tersebut akan mengeluarkan rekomendasi kepada Direktur Jenderal Bina Kefarmasian dan Alat Kesehatan paling lama 10 hari kerja. Direktur Jenderal Bina Kefarmasian dan Alat Kesehatan akan menerbitkan izin industri farmasi paling lama 10 hari kerja setelah menerima surat rekomendasi tersebut.
Izin usaha industri farmasi yang diberikan dapat berlaku untuk seterusnya selama perusahaan industri farmasi yang bersangkutan masih berproduksi dan tidak melanggar ketentuan perundang-undangan. Salah satu kelengkapan berkas permohonan izin usaha industri farmasi adalah surat rekomendasi pemenuhan persyaratan CPOB dari kepala Badan POM. Pemenuhan persyaratan CPOB dibuktikan dengan sertifikat CPOB yang berlaku selama 5 (lima) tahun sepanjang memenuhi persyaratan. Industri farmasi yang akan melakukan perubahan bermakna terhadap pemenuhan persyaratan CPOB, baik untuk perubahan kapasitas dan/atau fasilitas produksi wajib melapor dan mendapat persetujuan sesuai ketentuan peraturan perundang-undangan.
Kewajiban lain yang harus dilakukan oleh perusahaan yang telah memperoleh Izin Usaha Industri Farmasi yaitu membuat laporan secara elektronik terkait jumlah dan nilai produksinya sekali dalam enam bulan, sedangkan untuk laporan lengkap wajib disampaikan sekali dalam setahun yang disampaikan kepada Direktur Jenderal Bina Kefarmasian dan Alat Kesehatan dengan tembusan kepada Kepala Badan POM.
2.2 Cara Pembuatan Obat Yang Baik (CPOB)
Cara Pembuatan Obat yang Baik (CPOB) bertujuan untuk menjamin obat dibuat secara konsisten, memenuhi persyaratan yang ditetapkan dan sesuai dengan tujuan penggunaannya. CPOB mencakup seluruh aspek produksi dan pengendalian mutu. Pada pembuatan obat, pengendalian menyeluruh adalah sangat esensial untuk menjamin bahwa konsumen menerima obat yang bermutu tinggi. Pembuatan secara sembarangan tidak dibenarkan bagi produk yang
digunakan untuk menyelamatkan jiwa, atau memulihkan, atau memelihara kesehatan. Tidaklah cukup jika produk jadi hanya sekedar lulus dari serangkaian pengujian, tetapi yang lebih penting adalah bahwa mutu harus dibentuk ke dalam produk tersebut. Mutu obat tergantung pada bahan awal, bahan pengemas, proses produksi dan pengendalian mutu, bangunan, peralatan yang dipakai, dan personel yang terlibat. Pemastian mutu suatu obat tidak hanya mengandalkan pada pelaksanaan pengujian tertentu saja, namun obat hendaklah dibuat dalam kondisi yang dikendalikan dan dipantau secara cermat.
Aspek CPOB berdasarkan pedoman CPOB 2006 meliputi manajemen mutu; personalia; bangunan dan fasilitas; peralatan; sanitasi dan higiene; produksi; pengawasan mutu; inspeksi diri dan audit mutu; penanganan keluhan terhadap produk, penarikan kembali produk dan produk kembalian; dokumentasi; pembuatan dan analisis berdasarkan kontrak; serta kualifikasi dan validasi.
2.2.1 Manajemen Mutu (Badan Pengawas Obat dan Makanan RI, 2009)
Industri farmasi harus membuat obat sedemikian rupa agar sesuai dengan tujuan penggunaannya dan memenuhi persyaratan yang tercantum dalam dokumen izin edar (registrasi) serta tidak menimbulkan risiko yang membahayakan penggunanya karena tidak aman, mutu rendah, atau tidak efektif. Manajemen mutu bertanggung jawab untuk mencapai tujuan ini melalui suatu “Kebijakan Mutu”, yang memerlukan partisipasi dan komitmen dari semua jajaran di semua departemen di dalam perusahaan, para pemasok dan para distributor. Untuk mencapai tujuan mutu secara konsisten dan dapat diandalkan, diperlukan manajemen mutu yang didesain secara menyeluruh dan diterapkan secara benar. Unsur dasar manajemen mutu adalah suatu infrastruktur atau sistem mutu yang tepat mencakup struktur organisasi, prosedur, proses dan sumber daya. Tindakan yang sistematis diperlukan untuk mendapatkan kepastian dengan tingkat kepercayaan yang tinggi sehingga produk yang dihasilkan akan selalu memenuhi persyaratan yang telah ditetapkan.
Pemastian mutu adalah suatu konsep luas yang mencakup semua hal baik secara tersendiri maupun secara kolektif yang akan mempengaruhi mutu dari obat yang dihasilkan. Pemastian mutu adalah totalitas semua pengaturan yang dibuat
dengan tujuan untuk memastikan bahwa obat dihasilkan dengan mutu yang sesuai dengan tujuan pemakaiannya. Pengawasan mutu adalah bagian dari CPOB yang berhubungan dengan pengambilan sampel, spesifikasi dan pengujian, serta dengan organisasi, dokumentasi dan prosedur pelulusan yang memastikan bahwa pengujian yang diperlukan dan relevan telah dilakukan dan bahwa bahan yang belum diluluskan tidak digunakan serta produk yang belum diluluskan tidak dijual atau dipasok sebelum mutunya dinilai dan dinyatakan memenuhi syarat.
Setiap industri farmasi hendaklah mempunyai fungsi pengawasan mutu. Fungsi ini hendaklah independen dari bagian lain. Pengawasan mutu secara menyeluruh juga mempunyai tugas lain, antara lain menetapkan, memvalidasi dan menerapkan semua proses pengawasan mutu, mengevaluasi, mengawasi, dan menyimpan baku pembanding, memastikan kebenaran bahan dan produk, memastikan bahwa stabilitas dari zat aktif dan obat jadi dipantau, mengambil bagian dalam investigasi keluhan yang terkait dengan mutu produk, dan ikut mengambil bagian dalam pemantauan lingkungan. Personil pengawasan mutu hendaklah memiliki akses ke area produksi untuk melakukan pengambilan sampel dan investigasi sampel bila diperlukan.
Pengkajian mutu produk secara berkala hendaklah dilakukan terhadap semua obat terdaftar termasuk produk ekspor, dengan tujuan untuk membuktikan konsistensi proses, kesesuaian dari spesifikasi bahan awal, bahan pengemas, dan obat jadi untuk melihat tren dan mengidentifikasi perbaikan yang diperlukanuntuk produk dan proses.
Selain pengkajian mutu produk secara berkala, diperlukan juga manajemen risiko mutu berdasarkan pengetahuan secara ilmiah, pengalaman dengan proses dan pada akhirnya terkait dengan perlindungan pasien. Manajemen risiko mutu merupakan suatu proses sistematis untuk melakukan penilaian, pengendalian dan pengkajian risiko terhadap mutu suatu produk.
2.2.2 Personalia (Badan Pengawas Obat dan Makanan RI, 2006)
Sumber daya manusia sangat penting dalam pembentukan dan penerapan sistem pemastian mutu yang memuaskan dan pembuatan obat yang benar. Oleh sebab itu, industri farmasi bertanggung jawab untuk menyediakan personil yang
terkualifikasi dalam jumlah yang memadai untuk melaksanakan semua tugas. Tiap personil hendaknya memahami dan melaksanakan tugas dan tanggung jawab masing-masing. Seluruh personil hendaklah memahami prinsip CPOB dan memperoleh pelatihan awal dan berkesinambungan, termasuk instruksi mengenai higiene yang berkaitan dengan pekerjaan. Industri farmasi harus memiliki struktur organisasi. Tugas spesifik dan kewenangan dari personil pada posisi penanggungjawab hendaklah dicantumkan dalam uraian tugas tertulis. Tugas mereka boleh didelegasikan kepada wakil yang ditunjuk serta mempunyai tingkat kualifikasi yang memadai.
Personil kunci mencakup kepala bagian produksi, kepala bagian pengawasan mutu dan kepala bagian manajemen mutu (pemastian mutu). Posisi utama tersebut dijabat oleh personil purna waktu. Kepala bagian produksi dan kepala bagian manajemen mutu (pemastian mutu) / kepala bagian pengawasan mutu harus independen satu terhadap yang lain. Struktur organisasi perusahaan hendaklah sedemikian rupa sehingga ketiga bagian tersebut dipimpin oleh orang yang berlainan, yang tidak saling bertanggung jawab satu terhadap yang lain. Masing-masing hendaklah diberi wewenang penuh dan sarana pendukung yang diperlukan untuk dapat melaksanakan tugasnya secara efektif. Hendaklah personil tersebut tidak mempunyai kepentingan lain di luar organisasi yang dapat menghambat atau membatasi kewajibannya dalam melaksanakan tanggung jawab atau yang dapat menimbulkan konflik kepentingan pribadi atau finansial.
Kepala bagian produksi dan kepala bagian pengawasan mutu harus seorang apoteker yang cakap, terlatih, dan memiliki pengalaman praktis yang memadai di bidang industri farmasi dan keterampilan dalam kepemimpinan sehingga memungkinkan melaksanakan tugas secara profesional. Kepala bagian produksi hendaklah memiliki wewenang serta tanggung jawab penuh untuk mengelola produksi obat. Kepala bagian pengawasan mutu adalah satu-satunya yang memiliki wewenang untuk meluluskan bahan awal, produk antara, produk ruahan, dan obat jadi bila produk tersebut sesuai dengan spesifikasinya, atau menolaknya bila tidak cocok dengan spesifikasinya, atau bila tidak dibuat sesuai dengan prosedur yang disetujui dan kondisi yang ditentukan
Industri farmasi hendaklah memberikan pelatihan bagi seluruh personil karena tugasnya harus berada dalam area produksi, gudang penyimpanan atau
laboratorium (termasuk personil teknik, perawatan dan petugas kebersihan), dan bagi personil lain yang kegiatannya dapat berdampak pada mutu produk. Disamping pelatihan dasar dalam teori dan praktek CPOB, personil baru hendaklah mendapat pelatihan sesuai dengan tugas yang diberikan. Pelatihan berkesinambungan hendaklah juga diberikan, dan efektifitas penerapannya hendaklah dinilai secara berkala. Hendaklah tersedia program pelatihan yang disetujui kepala bagian masing-masing dan catatan pelatihan hendaklah disimpan. Setelah mengadakan pelatihan, prestasi karyawan dinilai untuk menentukan apakah mereka telah memiliki kualifikasi yang memadai untuk melaksanakan tugas yang diberikan kepadanya.
2.2.3 Bangunan dan Fasilitas
Bangunan dan fasilitas untuk pembuatan obat hendaklah memiliki desain, konstruksi dan letak yang memadai, serta disesuaikan kondisinya dan dirawat dengan baik untuk memudahkan pelaksanaan operasi yang benar. Tata letak dan desain ruangan harus dibuat sedemikian rupa untuk memperkecil risiko terjadinya kekeliruan, pencemaran silang dan kesalahan lain, dan memudahkan pembersihan, sanitasi, dan perawatan yang efektif untuk menghindari pencemaran silang, penumpukan debu atau kotoran, dan dampak lain yang dapat menurunkan mutu obat. Letak bangunan hendaklah sedemikian rupa untuk menghindari pencemaran dari lingkungan sekelilingnya, seperti pencemaran dari udara, tanah, dan air serta dari kegiatan industri lain yang berdekatan. Bangunan dan fasilitas hendaklah dikonstruksi, dilengkapi, dan dirawat dengan tepat agar memperoleh perlindungan maksimal dari pengaruh cuaca, banjir, rembesan dari tanah serta masuk dan bersarangnya serangga, burung, binatang pengerat, kutu, atau hewan lain. Bangunan dan fasilitas hendaklah dibersihkan dan bila perlu didesinfeksi sesuai prosedur tertulis yang rinci.
Adapun syarat-syarat bangunan dan fasilitas menurut CPOB adalah sebagai berikut:
a) Lokasi bangunan hendaklah sedemikian rupa untuk mencegah terjadinya pencemaran dari lingkungan sekelilingnya, seperti pencemaran dari udara, tanah dan air maupun dari kegiatan di dekatnya;
b) Bangunan dan fasilitas hendaklah dikonstruksi, dilengkapi dan dirawat dengan tepat agar memperoleh perlindungan maksimal dari pengaruh cuaca,
banjir, rembesan melalui tanah serta masuk dan bersarangnya binatang kecil, tikus, burung, serangga atau hewan lainnya;
c) Dalam menentukan rancang bangun dan tata letak hendaklah dipertimbangkan hal-hal sebagai berikut: kesesuaian dengan kegiatan lain, yang mungkin dilakukan dalam sarana yang sama atau dalam sarana yang berdampingan;
d) Tata letak ruang yang sedemikian rupa untuk memungkinkan kegiatan produksi dilaksanakan di daerah yang letaknya diatur secara logis dan berhubungan mengikuti urutan tahap produksi dan menurut kelas kebersihan yang disyaratkan; luasnya ruang kerja yang memungkinkan penempatan peralatan dan bahan secara teratur dan logis serta terlaksananya kegiatan, kelancaran arus kerja, komunikasi dan pengawasan yang efektif; pencegahan penggunaan kawasan industri sebagai lalu lintas umum;
e) Daerah pengolahan produk steril dipisahkan dari daerah produksi lain serta dirancang dan dibangun secara khusus;
f) Obat yang mengandung golongan penisilin dan sefalosporin diproduksi dalam suatu bangunan yang terpisah dilengkapi peralatan pengendali udara; g) Permukaan bagian dalam ruangan (dinding, lantai dan langit-langit)
hendaklah licin, bebas dari keretakan dan sambungan yang terbuka serta mudah dibersihkan dan bila perlu mudah didesinfeksi. lantai dan dinding di daerah pengolahan dibuat dari bahan kedap air, permukaannya rata dan memungkinkan pembersihan secara cepat dan efisien. sudut-sudut antara dinding, lantai dan langit-langit dalam daerah-daerah kritis hendaklah dibentuk lengkungan;
h) Saluran air limbah hendaklah cukup besar dan mempunyai bak kontrol serta ventilasi yang baik;
i) Bangunan memiliki penerangan yang efektif dan mempunyai ventilasi dengan fasilitas pengendali udara.
2.2.4 Peralatan
Peralatan untuk pembuatan obat hendaklah memiliki desain dan konstruksi yang tepat, ukuran yang memadai, serta ditempatkan dan dikualifikasi dengan
tepat, agar mutu obat terjamin sesuai desain serta seragam dari bets ke bets dan untuk memudahkan pembersihan serta perawatan. Peralatan hendaklah didesain dan dikonstruksikan sesuai dengan tujuannya. Permukaan peralatan yang bersentuhan dengan bahan awal, produk antara, atau produk jadi tidak boleh menimbulkan reaksi, adisi, atau absorbsi yang dapat mempengaruhi identitas, mutu atau kemurnian di luar batas yang ditentukan.
Peralatan sebaiknya dapat dibersihkan dengan mudah, baik bagian dalam maupun bagian luar, serta tidak boleh menimbulkan akibat yang merugikan terhadap produk. Pemasangan dan penempatan peralatan diatur sedemikian rupa sehingga proses produksi dapat berjalan secara efektif dan efisien. Peralatan hendaklah dirawat menurut jadwal yang tepat supaya tetap berfungsi dengan baik dan mencegah terjadinya pencemaran yang dapat mengubah identitas, mutu atau kemurnian produk. Peralatan yang rusak harus dikeluarkan dari area produksi dan pengawasan mutu, atau setidaknya diberi penandaan yang jelas.
2.2.5 Sanitasi dan Higiene
Tingkat sanitasi dan higienis yang tinggi hendaklah diterapkan pada setiap aspek pembuatan obat. Ruang lingkup sanitasi dan higienis meliputi personil, bangunan, peralatan dan perlengkapan, bahan produksi serta wadahnya, dan segala sesuatu yang dapat merupakan sumber kontaminasi produk. Sumber kontaminasi potensial hendaklah dihilangkan melalui suatu program sanitasi dan higienis yang menyeluruh dan terpadu, serta program tersebut senantiasa dievaluasi secara berkala untuk menjamin efektifitasnya.
Pembersihan mesin dapat mencegah adanya kontaminasi terhadap produk. Tiap kali sebelum dipakai, kebersihan peralatan diperiksa untuk memastikan bahwa semua produk atau bahan dari bets sebelumnya telah dihilangkan. Metode pembersihan dengan cara vakum atau cara basah lebih dianjurkan. Penggunaan udara bertekanan dan sikat sedapat mungkin dihindari karena dapat menambah risiko pencemaran produk. Pembersihan dan sanitasi peralatan serta wadah yang digunakan dalam pembuatan obat hendaklah tercakup dalam suatu prosedur tertulis yang cukup rinci. Penerapan higienis perorangan meliputi pemeriksaan kesehatan, menjaga kebersihan diri, memakai alat pelindung diri (APD) dengan
baik, menjaga kesehatan dan beberapa peraturan lain di area produksi. Semua personil hendaklah menjalani pemeriksaan kesehatan pada saat direkrut. Selain itu, hendaklah dilakukan juga pemeriksaan kesehatan kerja dan kesehatan personil secara berkala.
2.2.6 Produksi
Produksi obat hendaklah dilaksanakan dengan mengikuti prosedur yang telah ditetapkan dan memenuhi ketentuan CPOB yang menjamin senantiasa menghasilkan produk yang memenuhi persyaratan mutu serta memenuhi ketentuan izin pembuatan dan izin edar (registrasi). Produksi hendaklah dilakukan dan diawasi oleh personel yang kompeten. Penanganan bahan dan produk jadi, seperti penerimaan dan karantina, pengambilan sampel, penyimpanan, penandaan, penimbangan, pengolahan, pengemasan dan distribusi hendaklah dilakukan sesuai dengan prosedur atau instruksi tertulis dan bila perlu dicatat. Aspek produksi mencakup spesifikasi bahan awal; validasi proses (pembersihan, sterilisasi, dan lainnya); prosedur tetap; sistem penomoran bets/lot produk ruahan atau produk jadi; penimbangan dan penyerahan bahan baku obat; pengembalian bahan baku obat; pengolahan bahan baku menjadi produk obat jadi; monitoring; dan dokumentasi.
Penimbangan dan penyerahan bahan baku, bahan pengemas, produk antara dan produk ruahan dianggap suatu bagian dari siklus produksi dan memerlukan dokumentasi dan rekonsiliasi yang lengkap. Sebelum melakukan penimbangan dilakukan pemeriksaan kebenaran penandaan bahan baku termasuk label pelulusan. Kapasitas, ketepatan dan ketelitian alat timbangan dan alat ukur yang digunakan harus sesuai dengan jumlah bahan yang ditimbang. Semua prosedur produksi hendaknya divalidasi dengan tepat, sesuai dengan prosedur yang telah ditentukan dan catatan hasilnya hendaknya didokumentasikan. Perubahan yang penting dalam proses, baik itu penggantian alat maupun penggantian asal bahan baku, hendaknya dilakukan validasi ulang. Hal ini untuk menjamin bahwa perubahan tersebut akan tetap menghasilkan produk yang memenuhi spesifikasi yang telah ditentukan.
2.2.7 Pengawasan Mutu
Pengawasan mutu merupakan bagian yang esensial dari CPOB untuk memberikan kepastian bahwa produk secara konsisten mempunyai mutu yang sesuai dengan tujuan penggunaannya. Pengawasan mutu tidak terbatas pada kegiatan laboratorium, tapi juga harus terlibat dalam semua keputusan yang terkait dengan mutu produk. Pengawasan mutu hendaklah mencakup semua kegiatan analisis yang dilakukan di laboratorium, termasuk pengambilan sampel, pemeriksaan dan pengujian bahan awal, produk antara, produk ruahan dan produk jadi. Kegiatan ini mencakup juga uji stabilitas, program pemantauan lingkungan, pengujian yang dilakukan dalam rangka validasi, menyusun dan memperbaharui spesifikasi bahan dan produk serta metode pengujiannya.
2.2.8 Inspeksi Diri dan Audit Mutu
Tujuan inspeksi diri adalah untuk mengevaluasi apakah semua aspek produksi dan pengawasan mutu industri farmasi memenuhi ketentuan CPOB. Program inspeksi diri hendaklah dirancang untuk mendeteksi kelemahan dalam pelaksanaan CPOB dan untuk menetapkan tindakan perbaikan yang diperlukan. Inspeksi diri hendaklah dilakukan secara independen dan rinci oleh petugas yang kompeten dari perusahaan. Ada manfaatnya bila juga menggunakan auditor luar yang independen. Inspeksi diri dilakukan secara rutin dan pada situasi khusus, misalnya dalam hal penarikan obat jadi atau terjadi penolakan yang berulang.
Prosedur dan catatan inspeksi diri hendaklah didokumentasikan dan dibuat program tindak lanjut yang efektif. Penyelenggaraan audit mutu berguna sebagai pelengkap inspeksi diri. Audit mutu meliputi pemeriksaan dan penilaian semua atau sebagian dari sistem manajemen mutu dengan tujuan spesifik untuk meningkatkan mutu. Audit mutu umumnya dilaksanakan oleh spesialis dari luar atau independen atau tim yang dibentuk khusus untuk hal ini oleh manajemen perusahaan.
2.2.9 Penanganan Keluhan Terhadap Produk, Penarikan Kembali Produk, dan Produk Kembalian
Semua keluhan dan informasi lain yang berkaitan dengan kemungkinan terjadi kerusakan obat hendaklah dikaji dengan teliti sesuai dengan prosedur tertulis. Laporan dan keluhan mengenai produk dapat disebabkan oleh keluhan mengenai mutu yang berupa kerusakan fisik, kimiawi, atau biologis dari produk atau kemasannya. Keluhan lainnya adalah karena reaksi yang merugikan seperti alergi, toksisitas, reaksi fatal, dan reaksi medis lainnya, serta keluhan mengenai efek terapetik seperti produk tidak berkhasiat atau respon klinis yang rendah. Penarikan kembali produk adalah suatu proses penarikan kembali dari satu atau beberapa bets atau seluruh bets produk tertentu dari peredaran. Penarikan kembali produk dilakukan jika ditemukan produk yang cacat mutu atau jika ada laporan mengenai reaksi merugikan yang serius serta berisiko terhadap kesehatan. Penarikan kembali produk dapat berakibat penundaan atau penghentian pembuatan obat tersebut. Produk yang ditarik kembali hendaklah diberi identifikasi dan disimpan terpisah di area yang aman sementara menunggu keputusan terhadap produk tersebut.
Produk kembalian adalah obat jadi yang telah beredar, kemudian dikembalikan ke industri farmasi karena keluhan mengenai kerusakan, daluwarsa, atau alasan lain misalnya kondisi wadah atau kemasan yang menimbulkan keraguan akan identitas, mutu, jumlah, dan keamanan obat yang bersangkutan. Penanganan produk kembalian dan tindak lanjutnya hendaklah didokumentasikan dan dilaporkan. Bila produk harus dimusnahkan, dokumentasi hendaklah mencakup berita acara pemusnahan yang diberi tanggal dan ditandatangani oleh personel yang melaksanakan dan saksi.
2.2.10 Dokumentasi
Dokumentasi adalah bagian dari sistem informasi manajemen dan dokumentasi yang baik merupakan bagian yang esensial dari pemastian mutu. Dokumentasi yang jelas adalah fundamental untuk memastikan bahwa tiap personel menerima uraian tugas yang relevan secara jelas dan rinci sehingga memperkecil resiko terjadinya salah tafsir dan kekeliruan yang biasanya timbul
karena hanya mengandalkan komunikasi lisan. Keterbacaan dokumen sangat penting. Spesifikasi menguraikan secara rinci persyaratan yang harus dipenuhi produk atau bahan yang digunakan atau diperoleh selama pembuatan. Dokumen ini merupakan dasar untuk mengevaluasi mutu. Prosedur berisi cara untuk melaksanakan operasi tertentu, misalnya pembersihan, berpakaian, pengendalian lingkungan, pengambilan sampel, pengujian, dan pengoperasian peralatan. Dokumen hendaklah didesain, disiapkan, dikaji, dan didistribusikan dengan cermat. Dokumen hendaklah dikaji ulang secara berkala dan dijaga agar selalu up to date. Bila suatu dokumen direvisi, hendaklah dijalankan suatu sistem untuk menghindarkan penggunaan dokumen yang sudah tidak berlaku secara tidak sengaja.
2.2.11 Pembuatan dan Analisis Berdasarkan Kontrak
Pembuatan dan analisis berdasarkan kontrak harus dibuat secara benar, disetujui dan dikendalikan untuk menghindari kesalahpahaman yang dapat menyebabkan produk atau pekerjaan dengan mutu yang tidak memuaskan. Kontrak tertulis antara Pemberi Kontrak dan Penerima Kontrak harus dibuat secara jelas menentukan tanggung jawab dan kewajiban masing-masing pihak. Kontrak harus menyatakan secara jelas prosedur pelulusan tiap bets produk untuk diedarkan yang menjadi tanggung jawab penuh kepala bagian Manajemen Mutu (Pemastian Mutu).
2.2.12 Kualifikasi dan Validasi
CPOB menguraikan prinsip kualifikasi dan validasi yang dilakukan di industri farmasi. CPOB mensyaratkan industri farmasi untuk mengidentifikasi validasi yang perlu dilakukan sebagai bukti pengendalian terhadap aspek kritis dari kegiatan yang dilakukan. Perubahan signifikan terhadap fasilitas, peralatan dan proses yang dapat mempengaruhi mutu produk hendaklah divalidasi. Pendekatan dengan kajian risiko hendaklah digunakan untuk menentukan ruang lingkup dan cakupan validasi. Seluruh kegiatan validasi harus direncanakan terlebih dahulu. Unsur utama program validasi dirinci dengan jelas dan didokumentasikan dalam Rencana Induk Validasi (Validation Master Plan).
Protokol validasi tertulis hendaklah merinci kualifikasi dan validasi yang akan dilakukan. Hendaklah dibuat laporan yang mengacu pada protokol kualifikasi/validasi yang memuat ringkasan hasil yang diperoleh, tanggapan terhadap penyimpangan yang terjadi, kesimpulan dan rekomendasi perbaikan. Setelah kualifikasi selesai dilakukan, maka diberikan persetujuan tertulis untuk dapat melakukan tahap kualifikasi dan validasi selanjutnya.
PT. ASTRAZENECA INDONESIA – CIKARANG SITE
3.1 Sejarah PT. AstraZeneca Indonesia
AstraZeneca merupakan perusahaan biofarmasi global yang memproduksi dan mengembangkan obat resep untuk beberapa penyakit dunia yang paling serius, yaitu kanker, kardiovaskular, gastrointestinal, infeksi saluran pernapasan dan inflamasi (AstraZeneca, 2013). Berdirinya AstraZeneca berawal dari pendirian Astra AB di Soldeertatje, Stochlom bagian selatan, Swedia pada tahun 1913 dan Imperial Chemical Industries Ltd. (ICI) pada tahun 1926 di Inggris. Kedua perusahaan tersebut terus berkembang dan memperluas cabangnya di berbagai negara. Perluasan cabang ICI di Indonesia dimulai pada tahun 1970 dengan membentuk PT. ICI Farmasi Indonesia dan dua tahun kemudian dibangun pabrik Pandaan di daerah Pandaan – Pasuruan,Jawa Timur. Sementara itu, perluasan cabang Astra AB di Indonesia dimulai pada tahun 1987 dengan membentuk Divisi Astra Indonesia yang berada dibawah PT. Merck Indonesia.
Pada tahun 1993, kegiatan bahan kimia ICI memisahkan diri dari bisnis biosciences (termasuk obat-obatan) dengan membentuk Zeneca Pharma di Inggris. Setahun kemudian, Zeneca Pharma memperluas jaringannya ke Indonesia sehingga terbentuk Zeneca Pharma Indonesia.
AstraZeneca sendiri dibentuk pada tanggal 6 April 1999 melalui penggabungan antara Astra AB dari Swedia dan Zeneca Group PLC dari Inggris. Penggabungan tersebut bertujuan untuk meningkatkan pertumbuhan perusahaan jangka panjang dan meningkatkan nilai pemegang saham.
AstraZeneca telah mendistribusikan produknya ke lebih dari 100 negara di seluruh dunia. Perusahaan global ini memiliki karyawan berjumlah 57.200 orang di Dunia yang tersebar 46% di wilayah Eropa, 31% di Amerika dan 23 % di Asia-Pasifik. AstraZeneca terbagi menjadi beberapa bagian yang dibedakan berdasarkan perbedaan tanggung jawab, yaitu bagian Global Operations, R&D dan Finance. Global Operation yang terdiri dari 10.000 orang karyawan pada 19 site di 15 negara. Bagian ini terdiri dari 4 wilayah yang bertanggung jawab terhadap pembuatan dan suplai produk. Sementara itu, R&D (Research & Development) merupakan bagian yang bertanggung jawab mengembangkan dan menginovasikan obat dan produk biologi yang terletak pada 4 site AstraZeneca di 4 negara. Terakhir, bagian Finance bertanggung jawab membuat strategi manajemen keuangan AstraZeneca.
Terbentuknya AstraZeneca pada tahun 1999 berpengaruh terhadap perubahan PT. Zeneca Pharma Indonesia menjadi PT. AstraZeneca Indonesia. Pada tahun 2007, Pabrik Pandaan ditutup yang kemudian dibuatlah pabrik baru PT. AstraZeneca Indonesia di Cikarang, Bekasi. Pabrik PT. AstraZeneca Indonesia Cikarang-Site (PT. AZI-Cikarang Site) mulai didirikan pada tahun 2009 dan telah terkualifikasi pada bulan Mei 2010. Peresmian PT. AZI Cikarang-Site ini dilakukan pada tanggal 05 Oktober 2010 oleh Endang Rahayu Sedyaningsih selaku Menteri Kesehatan RI dan mulai produksi pada bulan November 2010.
PT. AZI dibangun di atas tanah seluas 5889 m2 dengan luas bangunan sebesar 2754 m2 yang terdiri dari ruang produksi, gudang, kantor, loker, laboratorium, ruang teknisi dan ruang utilitas. Ruangan tersebut dibangun untuk memfasilitasi kegiatan pengemasan (fase I), baik primer maupun sekunder, yang digunakan untuk mengemas sediaan padat untuk penggunaan oral dan memfasiitasi kegiatan pembuatan obat (fase II).
PT. AZI merupakan anggota AstraZeneca dengan pemegang saham utama Astrazeneca Holding B.V. Holland. PT. AZI telah mendapat lisensi untuk mengemas sediaan padat oral dari BPOM, Australia Therapeutic Good Administration dan Taiwan FDA (Tabel 3.1). Untuk fase I, PT. AZI hanya melakukan pengemasan sediaan padat dan repacking (mengemas ulang) produk impor AstraZenca sesuai ketentuan BPOM. PT. AZI juga menjalin kontrak
pembuatan sediaan padat dengan PT.Boehringer Ingelham Indonesia untuk di suplai ke pasar Indonesia (Astuti, 2012).
Tabel 3.1 Beberapa sertifikat GMP yang diperoleh PT. AstraZeneca Indonesia Cakupan
Inspeksi
Negara Tanggal Hasil Inspeksi
GMP Pre Audit BPOM 10 – 11 Juni 2010 Serifikat GMP pada
Januari 2011
GMP Audit BPOM 5 – 6 Agustus 2010 Serifikat GMP pada
Januari 2011
GMP Taiwan FDA 15 – 16 November 2010 Serifikat GMP pada
Juni 2011
GMP Australia TGA 23 – 25 November 2010 Serifikat GMP pada
Februari 2011
3.2 Visi dan Misi PT. AstraZeneca (Astuti, 2012) 3.2.1 Visi PT. AstraZeneca
Visi PT. AstraZeneca adalah menjadi partner yang terpercaya dan beretika bagi tenaga ahli kesehatan untuk meningkatkan kesehatan pasien dan menyediakan akses yang lebih luas untuk obat yang inovatif.
3.2.2 Misi PT AstraZeneca
Misi PT. AstraZeneca adalah untuk membuat perbedaan yang berarti bagi kesehatan pasien melalui obat yang hebat yang membawa keuntungan untuk pasien.
3.3 Struktur Organisasi Operasional (Astuti, 2012)
PT. AstraZeneca Indonesia - Cikarang Site dipimpin oleh seorang Direktur (Site Director) yang bertanggung jawab kepada RCP supply wilayah Asia Pasifik dan Jepang. Site Director membawahi tiga manager dan satu orang akuntan, yaitu:
a. Quality Assurance and Safety,Health, Enviroment (QA & SHE) Manager b. Supply Chain Manager
d. Plant Accountant
QA & SHE Manager selain bertanggung jawab pada site director PT. AZI – Cikarang Site juga bertanggung jawab langsung pada Region Quality Director dan membawahi QC Supervisor serta QA & SHE Supervisor. Sementara itu, Supply Chain Manager membawahi dua bagian yakni Warehouse Supervisor dan Supply Chain Supervisor. Plant Manager membawahi empat sub-departemen yakni Engineering Assc Manager, Production Supervisor, Assc Purchasing Manager dan General Affair & Personnel (GA & P) Supervisor. Bagan struktur organisasi PT. AZI- Cikarang Site secara lebih jelas dapat dilihat pada Lampiran 1.
3.3.1 Departemen Pemastian Mutu (QA & SHE Departement)
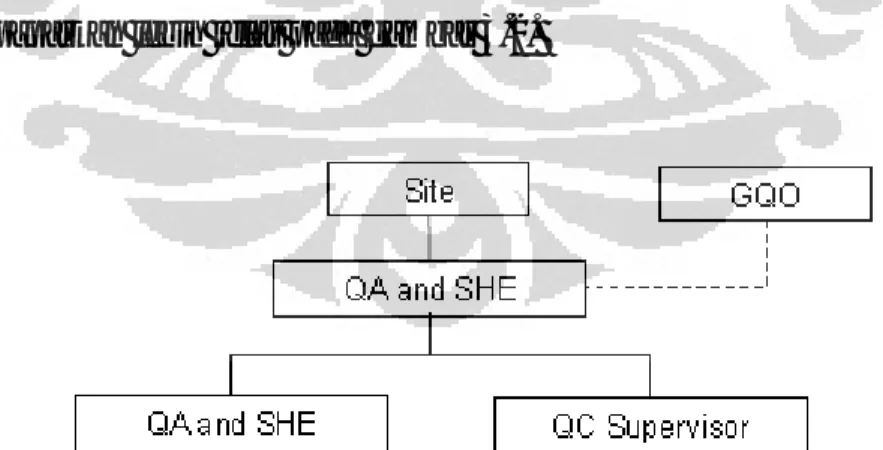
Departemen pemastian mutu atau Quality Assurance & Safety, Health, Environment Departement (QA & SHE Departement) bertanggung jawab terhadap aktivitas pemastian dan pengawasan mutu untuk memastikan bahwa produk yang dihasilkan sesuai dengan spesifikasi yang diinginkan, GMP dan syarat dari AstraZeneca internal. Departemen QA & SHE bertanggung jawab langsung kepada Site Director PT. AZI- Cikarang Site dan memiliki hubungan tidak langsung degan Global Quality Operation. Struktur organisasi sistem QA & SHE dipaparkan lebih jelas pada gambar 3.2.
Gambar 3.2 Struktur organisasi sistem QA & SHE di PT. AZI – Cikarang Site
[Sumber: Astuti, 2012]
Departemen QA & SHE dipimpin oleh seorang manager yang membawahi satu orang supervisor QA & SHE dan satu orang supervisor Quality Control (QC)
seperti yang dipaparkan dalam Gambar 3.2 di atas, dimana ketiganya merupakan seorang apoteker. Manager QA & SHEmemiliki tanggung jawab sebagai berikut:
a. Menyediakan dan mensuplai produk kualitas tinggi secara konsisten
b. Merencanakan, mengimplementasikan dan meninjau ulang Quality Management System yang mengacu pada standar AstraZeneca dan regualasi lokal
c. Menjalankan dan mengimplementasikan program dan strategi Global and Regional Quality pada level lokal
d. Merencanakan, mengimplementasikan, monitoring dan meninjau kembali SHE Management System yang mengacu pada standar AstraZeneca dan regulasi lokal untuk mencapai objektif lokal dan global SHE
e. Mengelola pihak kontraktor untuk mendukung bisnis AstraZeneca berdasarkan perspektif kualitas
f. Bertanggung jawab terhadap pelatihan GMP dan SHE di PT. AZI – Cikarang Site
g. Mengimplementasikan dan mengatur External Supplier Management Processes untuk kontraktor lokal (dapat pula bertanggung jawab mendukung di negara Asia Pasifik lainnya)
h. Berpartisipasi aktif dalam Regional External Supply Network
3.3.1.1 Sub-Departemen QA & SHE
Sub-Departemen QA & SHE dipimpin oleh seorang supervisor yang membawahi satu orang staff. Bagian ini memiliki tanggung jawab sebagai berikut:
a. Menyediakan dan mensuplai barang berkualitas tinggi secara konsisten b. Mengimplementasikan, memonitoring dan memperbarui Quality
Management System yang mengacu pada standar AstraZeneca dan regulasi lokal
c. Mengimplementasikan, memonitoring dan memperbarui SHE Management System yang mengacu pada standar AstraZeneca dan regulasi lokal untuk mencapai tujuan perusahaan
d. Memimpin pelatihan GMP dan SHE
f. Meninjau ulang AstraZeneca Quality Manual secara teratur
g. Mengeluarkan surat izin kerja dan mengimplementasikan sistem keamanan dalam bekerja
h. Berperan menyediakan pertolongan pertama pada kecelakaan kerja i. Bertindak sebagai sekretaris SHE dalam kepanitiaan SHE
j. Mendukung aktifitas kualifikasi
k. Menyiapkan protokol dan laporan validasi
l. Mengatur dan mengontrol prosedur Change Control, deviasi dan OOS m. Menangani keluhan
n. Menyiapkan kualifikasi personil untuk pekerja
o. Meninjau ulang batch record dan laporan quality conformity sebelum disetujui oleh pihak yang terkualifikasi
p. Memonitoring dan menginvestigasi Corrective Action and Preventive Action (CAPA) untuk audit eksternal
q. Menyiapkan dan melakukan audit internal
r. Menyiapkan Validation Master Plan dan Validation Master report s. Menyiapkan Product Review bersama Supervisor QC
t. Menyiapkan laporan SHE
u. Mengesahkan material yang datang dan produk jadi bila tidak ada deviasi v. Memberi nomor batch dan tanggal kadaluarsa produk jadi sesuai PON w. Mengatur distribusi SOP kepada departemen yang relevan
x. Menyiapkan sertifikat analisis produk jadi
y. Memonitoring dan mengevaluasi efektivitas pelatihan z. Menyimpan database pelatihan pekerja
3.3.1.2 Sub-Departemen Pengawasan Mutu (Quality Control)
Sub-Departemen Quality Control (QC) merupakan bagian dari Departemen QA & SHE yang terdiri dari satu orang supervisor dan tiga orang analis. Secara garis besar, QC bertanggung jawab terhadap pengujian kimia, fisika serta mikrobiologi komponen pengemasan dan produk untuk menjamin bahwa semua komponen tersebut sesuai dengan spesifikasi yang ditetapkan. Supervisor bagian ini memilki tanggung jawab sebagai berikut:
a. Menyediakan dan mensuplai produk dengan kualitas tinggi secara konsisten
b. Memimpin aktivitas QC di laboratorium QC termasuk inspeksi kualitas c. Memastikan terdapat sistem untuksampling yang sesuai, penanganan dan
penyimpanan sampel standar (reference sample)
d. Memastikan terdapat sistem yang diterapkan pada laboratorium QC termasuk instruksi pengoperasian alat
e. Memimpin sistem dokumentasi berdasarkan prosedur yang valid
f. Menyiapkan spesifikasi bahan pengemas, produk ruahan, dan produk jadi g. Menyiapkan lembar analisis untuk seluruh material yang diuji dan
memastikan bahwa analisis dilakukan sesuai prosedur
h. Meninjau ulang semua hasil analisis yang sebelumnya telah disahkan oleh QA
i. Melaporkan semua deviasi dan OOS yang terjadi serta menginvestigasi penyebabnya
j. Membuat proposal Change Control untuk setiap perubahan yang berkaitan dengan aktivitas QC
k. Memelihara stok dan reagen yang ada, media mikrobiologi, dan reference standard
l. Menyiapkan jadwal untuk analis QC
m. Mengecek semua log book yang terkait dengan aktivitas QC secara rutin n. Mengelola sampel pertinggal berdasarkan prosedur yang telah disetujui o. Memastikan monitoring sistem potable water dan udara bertekanan
dikakukan sesuai jadwal dan prosedur p. Mendukung validasi/kualifikasi proses
q. Memastikan terdapat prosedur untuk menganalisis material yang tidak diinventaris, misalnya minyak, dll.
Analis QC memiliki tanggung jawab yang dilimpahkan dalam rangka membantu memenuhi tanggung jawab Supervisor QC. Tanggung jawab analis QC diantaranya adalah melakukan sampling sesuai prosedur, melakukan inspeksi kualitas dari barang datang dan selama proses produksi, melakukan uji fungsi
terhadap peralatan QC sesuai jadwal dan prosedur masing-masing alat, melakukan validasi/verifikasi metode, melaporkan hasil deviasi atau OOS yang terjadi selama aktivitas, mengisi dokumen analisis berdasarkan hasil analisis yang diperoleh, menempelkan status material (release/reject) pada wadah material dan melakukan inspeksi pada produk jadi selama proses produksi.
Hasil pengujian yang tidak memenuhi persyaratan disebut sebagai Out of Specification (OOS) atau Hasil di luar Spesifikasi. Apabila terjadi OOS, analis wajib melaporkan hal tersebut untuk dilakukan investigasi, dilakukan perbaikan, dan didokumentasikan. OOS terbagi menjadi beberapa kategori, diantaranya disebabkan oleh kesalahan pada laboratorium, presisi metode yang buruk, kesalahan operator saat proses produksi, dan proses atau kemampuan produksi yang buruk.
3.3.2 Departemen Supply Chain
Dapartemen ini dikepalai seorang manager yang membawahi dua bagian, yaitu warehouse (gudang) dan supply chain. Kepala departemen ini bertanggung jawab memastikan suplai produk ke Pasar Indonesia dan Internasional berjalan dengan baik, yaitu dengan minimum inventaris dan maksimum efisiensi serta menyanggupi permintaan pasar. Kepala departemen supply chain juga bertanggung jawab mengembangkan kapasitas, kinerja, produktivitas gudang serta memastikan semua peralatan, fasilitas dan sistem gudang memenuhi persyaratan GMP, SHE dan persyaratan lain termasuk mengimplementasikan CAPA (Corrective and Preventive Action).
3.3.2.1 Sub-Departemen Gudang (Warehouse)
Gudang dikepalai oleh seorang supervisor dan dua orang operator. Supervisor bagian ini bertanggung jawab menerima seluruh bahan baku dan bahan pengemas, mendistribusikan material ke ruang produksi serta produk jadi ke distributor, menerapkan standar GMP pada sistem gudang, mengontrol dan menginventaris bahan baku, bahan pengemas, bulk dan produk jadi. Warehouse juga bertanggung jawab mengimplementasikan SHE AstraZeneca dan persyaratan Upaya Kesehatan Kerja (UKK) dari DEPNAKER.
Operator gudang bertanggung jawab untuk mengoperasikan aktivitas pergudangan dasar, seperti menerima barang datang dengan baik, mengeluarkan barang dari gudang dengan baik, menyimpan barang dalam gudang dengan baik, melakukan aktivitas gudang lainnya sesuai SOP AZI Cikarang. Operator gudang juga bertanggung jawab melakukan administrasi stok gudang, yaitu meng-input semua stok dan transaksi non-stok yang berkaitan dengan gudang dalam SAP untuk AZI Cikarang dan gudang di Pulogadung.
3.3.2.2 Sub-Departemen Supply Chain
Supevisor bagian ini bertanggung jawab melakukan kontak lokal untuk proyek transfer produk, merencanakan inventaris produksi dan material, mengembangkan kapasitas, kinerja dan produktivitas dalam mencapai permintaan produksi secara efektif dan efisien. Sub-Departemen Supply Chain juga bertanggung jawab menjaga kinerja supply chain and logistic mencapai atau diatas target yang diberikan serta mengimplementasikan SHE AstraZeneca dan persyaratan Upaya Kesehatan Kerja (UKK) dari DEPNAKER
3.3.3 Departemen Plant
Departemen Plant dikepalai oleh seorang Plant Manager yang bertanggung jawab mengembangkan kapasitas, kinerja dan produktivitas kegiatan produksi yang efektif dan efisien dalam memenuhi permintaan produksi serta memastikan peralatan dan fasilitas produksi memenuhi persyaratan GMP dan mengimplementasikan CAPA. Bebeapa tanggung jawab lain diantaranya adalah memelihara kebersihan ruang dan peralatan produksi, melakukan pelatihan SHE di ruang produksi serta memastikan semua GMP yang berkaitan dengan peralatan telah terkualifikasi dan terkalibrasi secara rutin sesuai prosedur yang ditetapkan.
Plant manager membawahi empat sub-departemen, yaitu Sub-Departemen Produksi, Teknik (Engineering), General Affair & Personnel (GA&P) serta Sub-Departemen Pembelian (Purchasing).
Sub-Departemen Produksi dikepalai oleh seorang supervisor yang memimpin satu orang koordinator pengemasan (packing coordinator) secara langsung. Packing coordinator akan membawahi dua orang operator dan lima orang packer. Sub-Departemen Produksi bertanggug jawab terhadap pengemasan produk obat, menyelesaikan proses produksi tepat waktu, validasi proses pengemasan, produk dan peralatan, memenuhi persyaratan GMP, mengimplementasikan SHE dan UKK dari DEPNAKER serta membuat laporan terkait produksi yang diserahkan kepada DepKes RI. Untuk memenuhi tanggung jawab tersebut, para packer diberi beberapa tanggung jawab yang terdiri dari:
a. Mengimplementasikan pedoman-pedoman CPOB (Cara Pembuatan Obat yang Baik).
b. Membantu melakukan persiapan yang perlu bagi pengemasan obat sesuai jalur dimana dia ditugaskan, antara lain: pembersihan, pelabelan, melakukan pemeriksaan kebenaran bahan-bahan komponen yang dikemas. c. Melakukan pekerjaan pengemasan di jalur yang telah diterapkan dan
melakukan pencatatan dengan benar dan lengkap serta dokumen pengemasan bets yang bersangkutan serta dokumen lain yang di atur dalam prosedur tetap.
d. Secara bersinambungan meningkatkan kemampuan diri dalam penguasaan mesin yang menjadi tanggung jawabnya.
e. Memperhatikan dan melapor atau member usulan terhadap segala kejadian yang memerlukan perbaikan saran kerja, mesin dan peralatan.
f. Melakukan pengemasan sesuai dengan prosedur pengemasan primer maupun sekunder dan mendokumentasikannya.
g. Melaksanakan In Process Control (IPC) untuk menjamin proses pengemasan tetap memenuhi standar yang ditetapkan selama pengemasan h. Membantu QC, dalam melakukan pengambilan sampel jika terjadi defect
pada penampilan fisik tablet.
Proses produksi yang dilakukan di PT. AZI – Cikarang Site saat ini hanya mencakup pengemasan primer dan sekunder sediaan solid oral non-steril (tablet). Hal tersebut sesuai dengan rancangan yang dibuat bahwa tahap awal produksi
yang dilakukan PT. AZI – Cikarang Site hanya meliputi proses pengemasan obat saja atau yang dikenal sebagai fase I. Namun demikian, beberapa ruangan di ruang produksi telah disiapkan untuk beranjak ke fase II, yaitu pembuatan obat dari mulai formulasi hingga pengemasan obat.
Pengemasan obat yang dilakukan dibagi menjadi tiga jenis. Pengemasan jenis pertama yaitu pengemasan primer dan sekunder obat yang diimpor dari Site AstraZeneca di negara lain. Pengemasan jenis ini hanya dilakukan untuk obat Inderal (propranolol hidroklorida). Pengemasan jenis kedua adalah repacking atau pengemasan ulang produk yang diimpor dari beberapa negara ke dalam kemasan baru yang berbeda dari kemasan sebelumnya, dimana pada kemasan baru ini telah didesain sedemikian rupa sehingga memenuhi spesifikasi dan standar lokal pada negara yang dituju tempat nantinya produk tersebut akan dijual. Pada kemasan yang baru ini juga dicantumkan batch number, expired date dan HET dari masing-masing produk. Pengemasan jenis terakhir adalah pengemasan sekunder obat yang diperoleh dari PT. Boehringer Ingelham Indonesia berdasarkan kontrak kerjasama (toll manufacturing) untuk didistribusikan di pasar lokal Indonesia. Daftar produk AstraZeneca secara lengkap dapat dilihat pada Lampiran 2.
3.3.3.2 Sub-Departemen Teknik (Engineering)
Sub-Departemen Teknik dikepalai oleh Engineering Asc. Manager dan membawahi seorang engineering administrator, pemimpin teknisi dan seorang anggota teknisi. Engineering asc. Manager bertanggung jawab memastikan jadwal perawatan engineering berjalan tepat waktu, menyediakan peralatan teknisi guna meminimalisir risiko rusaknya peralatan, memastikan bangunan, fasilitas dan peralatan telah memenuhi persyaratan GMP, memelihara fasilitas dan peralatan produksi, memastikan semua peralatan terkait GMP telah terkualifikasi dan terkalibrasi secara rutin sesuai prosedur
Engineering administrator memiliki tanggung jawab sebagai berikut: a. Melakukan administrasi dan kontrol dokumen di dalam area teknik b. Memperbarui status Work Order (WO) dan menyelesaikan laporan WO c. Memperbarui status Preventive Maintenance dan menyiapkan jadwal
d. Memperbarui status, menyiapkan jadwal dan memonitoring kemajuan proses kalibrasi
e. Melakukan administrasi pembelian untuk Engineering termasuk dalam sistem SAP
f. Mendukung proyek kualifikasi
g. Mengimplementasikan sistem pengelolaan dokumen dalam aktivitas sehari-hari
h. Mengimplementasikan sistem pengelolaan QA & SHE dalam aktivitas sehari-hari
i. Berpartisipasi dalam pelatihan GMP dan SHE j. Menjaga kebersihan ruang kontrol
Engineering Leader memiliki tanggung jawab sebagai berikut:
a. Melakukan, mengontrol dan mengevaluasi hasil preventive maintenance, kalibrasi dan status Job Order (JO).
b. Membuat rencana minggu proyek yang berkaitan dengan preventive maintenance, kalibrasi dan JO.
c. Memonitor proyek yang berkaitan dengan tugas dari vendor.
d. Melakukan pemeliharaan dan menjamin ketersediaan Consumable Part. e. Melakukan Follow-up CAPA yang terkait dengan dugaan risiko, audit
internal/eksternal, dll.
f. Mengimplementasikan sistem pengelolaan dokumen dalam aktivitas sehari-hari.
g. Mengimplementasikan sistem pengelolaan QA & SHE dalam aktivitas sehari-hari.
h. Berpartisipasi dalam training GMP dan SHE. i. Menjaga kebersihan ruang kontrol.
Sementara itu, tanggung jawab anggota teknisi meliputi: a. Melakukan monitoring pada sistem BAS
b. Melakukan monitoring tekanan, suhu yang dihasilkan dari filter-filter AHU dan ruang sampling
c. Melakukan program Preventive Maintenance sesuai jadwal
d. Melakukan pemeliharaan terhadap level klorin pada pompa air domestik (domestic water pump)/memonitor sistem air
e. Melakukan Job Order sesuai jadwal f. Melakukan kalibrasi sesuai persyaratan g. Mendukung proyek kualifikasi
h. Mengimplementasikan sistem pengelolaan dokumen dalam aktivitas sehari-hari
i. Mengimplementasikan sistem pengelolaan QA & SHE dalam aktivitas sehari-hari
j. Berpartisipasi dalam pelatihan GMP dan SHE k. Menjaga kebersihan ruang kontrol
Secara keseluruhan, bagian engineering bertanggung jawab dalam pengoperasian dan perawatan alat-alat penunjang fasilitas produksi, seperti AHU (Air Handling Unit), pendingin (chiller), kompresor (compressor), keran untuk kebakaran (fire hydrant) dan juga genset. AHU digunakan untuk mengatur dan menjaga tekanan, suhu dan kelembapan yang dibutuhkan di ruang produksi dan berbagai area lain dalam pabrik. Chiller berfungsi sebagai sumber air dingin yang nantinya akan diolah oleh AHU menjadi udara dingin. Udara bertekanan pada pabrik dihasilkan oleh kompresor bebas minyak. Sementara itu, Fire hydrant disediakan dalam setiap ruangan sebagai atisipasi apabila terjadi kebakaran. Fasilitas penunjang terakhir adalah genset yang berfungsi untuk menghasilkan arus listrik saat listrik mati sehingga diharapkan kebutuhan pabrik akan listrik dapat selalu tersedia.
Seperti yang telah dijelaskan sebelumnya, Sub-Departemen Teknik juga bertanggung jawab terhadap perawatan fasilitas penunjang. Perawatan tersebut meliputi breakdown maintenance dan preventive maintenance. Breakdown maintenance dilakukan ketika mesin/peralatan dalam kondisi rusak sementara preventive maintenance dilakukan sebagai tindakan pencegahan agar mesin/peralatan tidak rusak. Preventive maintenance biasanya dilakukan satu kali dalam sebulan atau sesuai jadwal yang ditentukan. Beberapa peralatan juga ada
yang dilakukan kalibrasi sesuai jadwal kalibrasi yag telah ditetapkan sedangkan kualifikasi dilakukan pada saat mesin pertama kali akan dioperasikan. Kualifikasi dilanjutkan dengan rekualifikasi setiap 3 tahun sekali.
Breakdown maintenance dilakukan berdasarkan Work Order (WO). Lembaran WO dapat diambil dan diisi oleh semua personil dari departemen manapun di dinding koridor lantai 2 apabila terdapat mesin/peralatan yang rusak atau tidak berfungsi dengan baik. Selanjutnya WO diserahkan ke bagian Teknik untuk selanjutnya dilakukan perbaikan terhadap kerusakan yang terjadi.
3.3.3.3 Sub-Departemen General Affair & Personnel (GA&P)
Bagian ini dikepalai oleh seorang supervisor yang membawahi satu orang karyawan GA & P yang bertanggung jawab melakukan perjanjian terhadap hal-hal yang berhubungan dengan tiket, akomodasi, logistik, vendor, pembayaran, perizinan, penyediaan makan (catering), serta mengurus faktur (invoice), inventaris dan barang non inventaris.
Bagian ini juga berhubungan langsung dengan kantor pusat di Jakarta. Secara umum, Sub-Departemen GA&P bertanggung jawab menyediakan pelayanan umum pada managemen fasilitas dan administrasi personil sesuai dengan standar PT.AZI – Cikarang Site. Pelayanan umum pada managemen fasilitas termasuk bertanggung jawab terhadap kebersihan PT.AZI – Cikarang Site. Oleh karena itu, supervisor GA&A juga membawahi tujuh personil dari PT. ISS Indonesia. Keenam orang dari personil tersebut bertanggung jawab terhadap kebersihan pabrik sementara satu personil bertanggung jawab terhadap pengendalian hama dan binatang pengerat pada PT.AZI – Cikarang Site. Personil-personil dari pihak PT.ISS tersebut bertanggung jawab pada Supervisor GA&A serta pimpinannya di PT.ISS Indonesia. Selain itu Sub-Departemen GA & P juga membawahi bagian security yang bertugas menjaga keamanan pabrik selama 24 jam.
3.3.3.4 Sub-Departemen Pembelian (Purchasing)
Sub-Departemen Pembelian dipimpin oleh seorang Asc.Purchasing Manager yang secara umum memiliki tanggung jawab menyusun dan mengelola
sistem procurement di PT. AZI – Cikarang Site. Sistem procurement merupakan sebuah sistem pembelian barang yang memiliki alur tersendiri dari mulai penawaran barang dari vendor hingga pembayaran tagihan barang yang dibeli oleh PT. AZI – Cikarang Site. Alur tersebut dimulai dari proses Quotation yaitu penawaran harga dari beberapa vendor. Kemudian User (kepala bagian departemen) akan menyiapkan Purchasing Requestation (permintaan internal) berdasarkan kebutuhan masing-masing bagian user yang diserahkan kepada Sub-Departemen Pembelian. Bagian Pembelian akan menyiapkan Purchase Order (PO) atau permintaan eksternal yang selanjutnya akan diserahkan kepada vendor. Vendor akan mengirimkan barang sesuai dengan PO dan menyerahkan Good Receive Note beserta Invoice (tagihan), surat jalan dan salinan PO. Sub-Departemen Pembelian akan melakukan validasi dokumen tersebut. Selanjutnya, apabila dokumen yang dicek sesuai dengan jumlah barang yang diterima, Sub-Departemen Pembelian akan menyerahkan dokumen-dokumen tervalidasi tersebut kepada bagian finance agar segera melakukan pembayaran pada vendor terkait.
Selain itu, tanggung jawab lain kepala bagian pembelian adalah melakukan penghematan biaya rumah tangga perusahaan sesuai target tahunan yang direncanakan, melakukan bidding (perbandingan minimal tiga penawaran dari tiga vendor) untuk menentukan vendor mana yang memiliki risiko terkecil terhadap kualitas produk dengan harga seminimum mungkin serta berperan dalam registrasi vendor ke dalam sistem komputasi perusahaan.
3.3.4 Plant Accountant
Plant Accountant berperan dalam pengadaan kerja sama dengan area bisnis yang relevan untuk memastikan ketepatan waktu dalam pengiriman; memastikan laporan manajemen dan anggaran sesuai dengan pedoman dan kebutuhan manajemen; bekerjasama dengan plant management, bagian produksi, QA, GA&P untuk menyokong anggaran dan analisis laporan bulanan, mengelola kerjasama kontrak antara Indonesia dan UK; serta terlibat dalam kontrol pembangunan, proses, pelaporan manajemen yang sedang berlangsung, transaksi keuangan dan masalah pajak.