STUDI PENENTUAN KETIDAKPASTIAN PENGUKURAN
(MEASUREMENT UNCERTAINTY) PADA MINYAK
SAWIT MENTAH (CPO) DENGAN BEBERAPA
METODEANALISIS KIMIA
SKRIPSI
ILMAN PANGKAL DOLOK HARAHAP
090802011
DEPARTEMEN KIMIA
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN
ALAM
UNIVERSITAS SUMATERA UTARA
MEDAN
STUDI PENENTUAN KETIDAKPASTIAN PENGUKURAN
(MEASUREMENT UNCERTAINTY) PADA MINYAK
SAWIT MENTAH (CPO) DENGAN BEBERAPA
METODE ANALISIS KIMIA
SKRIPSI
Diajukan untuk melengkapi tugas dan memenuhi syarat mencapai gelar sarjana sains
ILMAN PANGKAL DOLOK HARAHAP
090802011
DEPARTEMEN KIMIA
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM
UNIVERSITAS SUMATERA UTARA
MEDAN
PERSETUJUAN
Judul : Studi Penentuan Ketidakpastian Pengukuran (Measurement Uncertainty) Pada Minyak Sawit Mentah (CPO) Dengan Beberapa Metode Aalisis Kimia
Kategori : Skripsi
Nama : Ilman Pangkal Dolok Harahap Nomor Induk Mahasiswa : 090802011
Program Studi : Sarjana (S1) Kimia Departemen : Kimia
Fakultas : Matematika Dan Ilmu Pengetahuan Alam Universitas Sumatera Utara
Disetujui di
Medan, Januari 2014
Komisi Pembimbing :
Pembimbing 2 Pembimbing 1
Prof. Dr. Zul Alfian, M.Sc Drs. Chairuddin, M.Sc.
NIP. 195504051983031002 NIP. 195912311987011001
Diketahui/Disetujui oleh :
Departemen Kimia FMIPA USU Ketua,
PERNYATAAN
STUDI PENENTUAN KETIDAKPASTIAN PENGUKURAN
(MEASUREMENT UNCERTAINTY) PADA MINYAK SAWIT MENTAH (CPO) DENGAN BEBERAPA
METODE ANALISIS KIMIA
SKRIPSI
Saya mengakui bahwa skripsi ini adalah hasil kerja saya sendiri, kecuali beberapa kutipan dan ringkasan yang masing-masing disebutkan sumbernya.
Medan, Januari 2014
PENGHARGAAN
Bissmillahirramanirrahim,
Alhamdullilah, segala puji bagi Allah SWT karena berkat rahmat, nikmat, taufik, serta hidayah-Nya semata saya dapat menyelesaikan skripsi ini sebagai salah satu persyaratan untuk meraih gelar Sarjana Kimia pada Fakultas Matematika dan Ilmu Pengetahuan Alam Universitas Sumatera Utara. Serta shalawat dan salam saya hadiahkan pada Rasullulah, Muhammad SAW sosok yang sangat saya idolakan semoga kelak mendapat safaat beliau, Amin.
STUDI PENENTUAN KETIDAKPASTIAN PENGUKURAN (MEASUREMENT UNCERTAINTY) PADA MINYAK
SAWIT MENTAH (CPO) DENGAN BEBERAPA METODE ANALISIS KIMIA
ABSTRAK
Telah dilakukan studi penentuan ketidakpastian pengukuran (measurement uncertainty) pada minyak sawit mentah (CPO) dilakukan dengan penentuan asam lemak bebas metode titrasi netralisasi berdasarkan AOCS Ca5a-40, dan penentuan kadar ion besi Fe2+ dengan menggunakan metode spektrofotometri. Proses penentuan ketidakpastian pengukuran berdasarkan spesifikasi measurand, identifikasi sumber-sumber ketidakpastian, perhitungan ketidakpastian dengan menggunakan perhitungan ketidakpastian tipe A(menggunakan analisis statistik sederetan pengamatan) dan tipe B(menggunakan cara-cara lain selain analisis statistik sederetan pengamatan) kemudian mengubahnya menjadi deviasi standar, dan tahap terakhir yaitu perhitungan ketidakpastian standar gabungan dan ketidakpastian terekspansi. Ketidakpastian standar gabungan dihitung dengan menggunakan program komputer MS Exel 2007 berdasarkan pada metode perhitungan ketidakpastian Kragten spreadsheet. Dengan mengalikan ketidakpastian standar gabungan dengan faktor coverage 2, ketidakpastian terekspansi untuk penentuan asam lemak bebas metode titrasi netralisasi berdasarkan AOCS Ca5a-40 yang konsentrasinya 0,1612 mol/L dalam larutan sampel adalah ±0,0002 mol/L, sedangkan ketidakpastian terekspansi untuk penentuan kadar ion besi Fe2+ dengan menggunakan metode spektrofotometri uv-vis yang konsentrasinya 0,131 mg/L dalam larutan sampel adalah ±0,083 mg/L yang dihitung dengan menggunakan faktor coverage 2,14.
STUDY OF THE USE MEASUREMENT UNCERTAINTY ON CRUDE PALM OIL (CPO) WITH SOME
CHEMICAL ANALYSIS METHOD
ABSTRACT
DAFTAR ISI
2.3 Penentuan Ketidakpastian 5
2.3.1 Spesifikasi Measurand 5
2.6.1 Spektroskopi Ultraviolet dan Tampak (Visible) 13 3.2.1.7 Pembuatan Larutan Standar Fe2+ 10 mg/L 20 3.2.1.8 Pembuatan Larutan Seri Standar Fe2+ 0,2; 0,4;
0,6;
0,8 dan 1,0 mg/L 20
3.2.2 Prosedur Penentuan Fe2+ Dengan Metode
4.1.1.1.1 Perhitungan Ketidakpastian Tipe A 28 4.1.1.1.2 Perhitungan Ketidakpastian Tipe B 32 4.1.1.1.3 Perhitungan Ketidakpastian Standar
Gabungan 36 4.1.1.1.4 Perhitungan Ketidakpastian
Terekspansi 36 4.1.2 Perhitungan Ketidakpastian Untuk Analisis Penentuan
Asam Lemak Bebas Dengan Metode Titrasi Netralisasi 37 4.1.2.1 Perhitungan Komponen-komponen
Ketidakpastian Standar 37
4.1.2.1.1 Perhitungan Ketidakpastian Tipe A 37 4.1.2.1.2 Perhitungan Ketidakpastian Tipe B 38 4.1.2.1.3 Perhitungan Ketidakpastian Standar
Gabungan 39 4.1.2.1.4 Perhitungan Ketidakpastian
Terekspansi 40
4.2 Pembahasan 40
4.2.1 Perhitungan Ketidakpastian Untuk Analisis Penentuan
Asam Lemak Bebas Dengan Metode Titrasi Netralisasi 40 4.2.1.1 Sumber-sumber Ketidakpastian 40
BAB 5 Kesimpulan dan Saran 61
5.1 Kesimpulan 61
5.2 Saran 61
Daftar Pustaka 62
DAFTAR TABEL
Halaman
Tabel 4.1 Data absorbansi larutan standar Fe2+ 28
Tabel 4.2 Data perhitungan persamaan garis regresi 29
Tabel 4.3 ANAVA untuk uji linearitas 33
Tabel 4.4 Data ketidakpastian untuk elemen-elemen unsur
KHP(C8H5O4K) 44
DAFTAR GAMBAR
Halaman Gambar 2.1 Bagan alat spektrofotometri UV-VIS 14 Gambar 4.1 Grafik least square linear untuk analisis Fe2+ dengan metode
spektrofotometri 30 Gambar 4.2 Diagram Ishikawa sederhana untuk penentuan asam lemak bebas 42 Gambar 4.3 Detail diagram Ishikawa untuk penentuan asam lemak bebas 42 Gambar 4.4 Diagram Ishikawa untuk penentuan asam lemak bebas 42 Gambar 4.5 Metode perhitungan ketidakpastian Kragten Spreadsheet untuk
penentuan asam lemak bebas dengan metode titrasi netralisasi 48 Gambar 4.6 Histogram hu bungan ketidakpastian standar gabungan dengan
kontribusi masing-masing komponen pada penentuan asam lemak bebas dengan metode titrasi netralisasi 48 Gambar 4.7 Detail diagram ishikawa untuk analisis larutan Fe2+ dengan metode
spektrofotometri 50 Gambar 4.8 Diagram ishikawa untuk analisis larutan Fe2+ dengan metode
spektrofotometri 51 Gambar 4.9 Metode perhitungan ketidakpastian Krgaten Spreadsheet untuk
analisis larutan Fe2+ dengan metode spektrofotometri 59 Gambar 4.10 Histogram hubungan ketidakpastian standar gabungan dengan
DAFTAR LAMPIRAN
Halaman
Lampiran A. Data hasil pengukuran absorbansi dari larutan standar Fe2+ pada panjang gelombang maksimum maks) 510 nm. 64 Lampiran B. Data hasil pengukuran absorbansi dari larutan sampel pada
panjang gelombang maksimum maks) 510 nm. 65 Lampiran C. Hasil Perhitungan Ketidakpastian Untuk Analisis Dengan Metode
Titrasi Netralisasi dan Spektrofotometri. 65 Lampiran D. Daftar Ketidakpastian Untuk Analisis Fe2+ Dengan Metode
Spektrofotometri. 65 Lampiran E. Daftar Ketidakpastian Untuk Analisis Fe2+ Dengan Metode
Spektrofotometri. 66 Lampiran F. Daftar Simbol Parameter-Parameter Pada Perhitungan
STUDI PENENTUAN KETIDAKPASTIAN PENGUKURAN (MEASUREMENT UNCERTAINTY) PADA MINYAK
SAWIT MENTAH (CPO) DENGAN BEBERAPA METODE ANALISIS KIMIA
ABSTRAK
Telah dilakukan studi penentuan ketidakpastian pengukuran (measurement uncertainty) pada minyak sawit mentah (CPO) dilakukan dengan penentuan asam lemak bebas metode titrasi netralisasi berdasarkan AOCS Ca5a-40, dan penentuan kadar ion besi Fe2+ dengan menggunakan metode spektrofotometri. Proses penentuan ketidakpastian pengukuran berdasarkan spesifikasi measurand, identifikasi sumber-sumber ketidakpastian, perhitungan ketidakpastian dengan menggunakan perhitungan ketidakpastian tipe A(menggunakan analisis statistik sederetan pengamatan) dan tipe B(menggunakan cara-cara lain selain analisis statistik sederetan pengamatan) kemudian mengubahnya menjadi deviasi standar, dan tahap terakhir yaitu perhitungan ketidakpastian standar gabungan dan ketidakpastian terekspansi. Ketidakpastian standar gabungan dihitung dengan menggunakan program komputer MS Exel 2007 berdasarkan pada metode perhitungan ketidakpastian Kragten spreadsheet. Dengan mengalikan ketidakpastian standar gabungan dengan faktor coverage 2, ketidakpastian terekspansi untuk penentuan asam lemak bebas metode titrasi netralisasi berdasarkan AOCS Ca5a-40 yang konsentrasinya 0,1612 mol/L dalam larutan sampel adalah ±0,0002 mol/L, sedangkan ketidakpastian terekspansi untuk penentuan kadar ion besi Fe2+ dengan menggunakan metode spektrofotometri uv-vis yang konsentrasinya 0,131 mg/L dalam larutan sampel adalah ±0,083 mg/L yang dihitung dengan menggunakan faktor coverage 2,14.
STUDY OF THE USE MEASUREMENT UNCERTAINTY ON CRUDE PALM OIL (CPO) WITH SOME
CHEMICAL ANALYSIS METHOD
ABSTRACT
BAB 1 PENDAHULUAN
1.1.Latar Belakang
Analisis kuantitatif merupakan penentuan berapa zat tertentu ada di dalam suatu
sampel. Zat yang ditentukan, sering ditunjukkan sebagai zat yang diinginkan atau
analit, dapat terdiri dari sebagian kecil atau besar sampel yang dianalisis. Dalam
analisis kimia kuantitatif, banyak sekali dilakukan analisis dengan menggunakan
metode analisis kimia. (Underwood, A. L.1980). Hasil analisis yang diperoleh
diharapkan dapat digunakan untuk tujuan tertentu, misalnya pemeriksaan kualitas
air minum, analisis forensik bagian tubuh dalam beberapa kasus kriminal, dan
penentuan kualitas suatu produk industri yang akan diekspor ke luar negeri. Oleh
karena itu, penting untuk mengetahui kualitas dari hasil-hasil pengukuran dengan
mengutamakan jaminan kualitas (quality assurance) terhadap pengukuran-pengukuran yang dilakukan oleh laboratorium yang bersangkutan. Ini
dimaksudkan sebagai aturan bahwa suatu laboratorium tersebut mampu dan
memiliki data dengan kualitas yang diperlukan. Analisis kimia sangat
menekankan ketelitian dan keakuratan hasil-hasil analisis yang diperoleh dengan
menggunakan metode-metode standar. Untuk itu, penting untuk menyatakan
kualitas hasil-hasil pengukuran yang diperoleh sehingga dapat dilihat
kesesuaiannya dengan cara mencantumkan tingkat kepercayaan pengukuran.
Salah satu pengukuran yang bermanfaat diantaranya adalah ketidakpastian
pengukuran (measurement uncertainty).
Ketidakpastian pengukuran mulai dipublikasikan pada tahun 1993 oleh
(International Organization on Legal Metrology)dalam “Guide to the Expression of Uncertainty in Measurement” (GUM) yang menulis tentang peraturan umum dalam menghitung da mengekspresikan ketidakpastian pengukuran dalam
laporan-laporan analisis akan menjadi hal biasa di masa yang akan datang.
(Ellison et al. 1999)
Dalam penelitian ini, penulis tertarik untuk menghitung ketidakpastian
pengukuran pada minyak sawit mentah (CPO) dengan menggunakan
metode-metode analisis dengan titrasi netralisasi dan spektrofotometri UV-VIS.
1.2Permasalahan
1. Berapa besar interval ketidakpastian dari hasil analisis dengan penentuan
asam lemak bebas berdasarkan AOCS Ca5a-40.
2. Berapa besar interval ketidakpastian dari hasil analisis penentuan kadar
ion besi Fe2+ dengan menggunakan metode spektrofotometri UV-VIS.
1.3Pembatasan Masalah
Dalam penelitian ini permasalahan dibatasi pada :
1. Analisis dilakukan dengan penentuan asam lemak bebas metode titrasi
netralisasi berdasarkan AOCS Ca5a-40.
2. Analisis dilakukan dengan penentuan kadar ion besi Fe2+ dengan menggunakan metode spektrofotometri uv-vis.
3. Sampel yang digunakan adalah minyak sawit mentah (CPO).
1.4Tujuan Penelitian
1. Untuk menentukan nilai ketidakpastian pada hasil analisis asam lemak
bebas dengan metode titrasi netralisasi berdasarkan AOCS Ca5a-40.
2. Untuk menentukan nilai ketidakpastian pada analisis kadar ion besi Fe2+ dengan metode spektrofotometri UV-VIS.
1.5Manfaat Penelitian
Penelitian ini dapat digunakan sebagai informasi untuk menentukan
Ketidakpastian setiap hasil-hasil minyak sawit mentah (CPO) di laboratorium.
1.6Metodologi Penelitian
Penelitian ini merupakan eksperimen laboratorium. Sampel yang digunakan
adalah minyak sawit mentah dari salah satu pabrik di daerah Belawan. Pada
penentuan % FFA dengan metode titrimetri netralisasi, dimana minyak sawit
mentah ditambahkan alkohol netral dan indikator phenolftalein, kemudian dititrasi
dengan NaOH sampai diperoleh perubahan warna menjadi merah lembayung.
Pada penentuan konsentrasi Fe2+ dengan metode spektroskopi UV-VIS.
1.7Lokasi Penelitian
BAB 2
TINJAUAN PUSTAKA
2.1 Pengertian Ketidakpastian
Kata ketidakpastian berarti suatu keraguan, dan dengan demikian pengertian
ketidak pastian dalam arti yang luas adalah suatu pengukuran dimana validitas
dan ketepatan hasilnya masih diragukan. Berdasarkan “International Vocabulary Of Basic and General Terms in Metrology”, pengukuran didefinisikan sebagai sederetan operasi yang mempunyai objek untuk ditentukan nilai kuantitasnya.
(Choi et al. 2002)
Parameter yang diuji terdiri dari distribusi statistik hasil-hasil beberapa
pengukuran yang ditentukan sebagai deviasi standar. Dapat juga berupa
komponen-komponen lain, yang termasuk juga sebagai deviasi standar, namun
yang dihitung berupa distribusi eluang berdasarkan percobaan ataupun informasi
lainnya.
Dalam proses analisis kimia, measurand dapat berupa konsentrasi suatu
analit. Tetapi di dalam analisis kimia juga digunakan untuk menentukan
pengukuran kuantitas, meliputi warna, pH, dan lain-lain. Oleh karena itu
digunakanlah istilah umum yaitu measurand.
Ketidakpastian pengukuran berbeda dengan kesalahan. Kesalahan dapat
diartikan sebagai perbedaan antara hasil-hasil pengukuran yang diperoleh dengan
nilai measurand yang sebenarnya dan dapat juga sebagai acuan untuk suatu hasil pengukuran. Sebaliknya, ketidakpastian yang berupa jangkauan nilai, tidak dapat
2.2 Metode Validasi
Dalam praktiknya, metode validasi yaitu kesesuaian metode pengujian analisis
yang dilakukan. Studi ini menghasilkan data pada kinerja keseluruhan dan
pengaruh faktor-faktor individu yang diterapkan untuk estimasi ketidakpastian.
Metode validasi bergantung pada penentuan parameter kinerja metode
keseluruhan. Studi validasi untuk analisa kuantitatif, biasanya metode analisis ini
menentukan beberapa atau semua parameter berikut : akurasi, bias, linearitas,
batas deteksi, dan spsifikasi.
2.3 Penentuan Ketidakpastian
Estimasi ketidakpastian memiliki prinsip yang sederhana. Tahap-tahap
yang perlu dilakukan untuk menentukan ketidakpastian pengukuran adalah
sebagai berikut:
1. Spesifikasi measurand
2. Identifikasi sumber-sumber ketidakpastian
3. Perhitungan komponen-komponen ketidakpastian
4. Perhitungan ketidakpastian standar gabungan
2.3.1 Spesifikasi measurand
Pada tahap ini semua pernyataan yang sedang diukur dan rumus kuantitatif yang
menghubungkan measurand dengan parameter-parameter (kuantitatif measurand, konstan, kalibrasi standart dan sebagainya) harus ditulis dengan jelas. Dalam
menentukan measurand, perlu diketahui juga apakah tahap sampling yang dilakukan sesuai dengan standard operating procedure (SOP) atau dengan
2.3.2 Identifikasi sumber-sumber ketidakpastian
Kita harus memfokuskan pada suatu sumber ketidakpastian yang paling utama
untuk digunakan dalam penentuan ketidakpastian pengukuran. Sumber-sumber
yang mungkin dapat mengakibatkan timbulnya ketidakpastian pengukuran adalah
sampling, transfortasi, penyimpanan dan penanganan sampel, pembuatan sampel,
kondisi lingkungan dan pengukuran, orang yang melakukan uji, variasi dalam
prosedur uji, instrumentasi, standar kalibrasi atau bahan-bahan pembanding,
perangkat lunak, dan metode-metode pengukuran karena efek-efek
sistematik.(ILAC-G17,2002) .
2.3.2.1. Sampling
Dimana pengambilan sampel merupakan prosedur yang paling penting untuk
suatu analisa. Namun, bila pengambilan sampel secara acak ini merupakan
komponen ketidakpastian yang mempengaruhi nilai hasil akhir.
2.3.2.2. Penyimpanan dan penanganan sampel
Bila sampel yang diuji disimpan untuk waktu yang lama sebelum analisis, kondisi
penyimpanan dapat mempengaruhi hasil. Lama penyimpanan serta kondisi selama
penyimpanan ini dapat dianggap sebagai sumber ketidakpastian
2.3.2.3. Instrumentasi
Instrumentasi juga dapat termasuk sebagai sumber ketidakpastian, misalnya batas
akurasi untuk kesetimbangan analitis, pengotrol suhu yang dapat mempertahankan
2.3.2.4. Kemurnian Reagen
Walaupun molaritas larutan volumetrik tidak akan diketahui sebenarnya bahkan
jika larutan induk telah diuji, hal ini harus tetap dilakukan prosedur pengujian
ketidakpastian. Misalnya, zat organik, apabila tidak diketahui kemurniannya
mungkin di dalam zat organik tersebut masih terdapat garam-garam anorganik
yang merupakan sumber dari ketidakpastian.
2.3.2.5. Kondisi Pengukuran
Misalnya, pada suhu gelas ukur yang kita gunakan tidak sesuai dengan suhu yang
sudah dikalibrasi. Demikian pula, kelembaban mungkin penting dimana apabila
bahan sangat sensitif, maka ini dapat mempengaruhi nilai ketidakpastian.
2.3.2.6. Efek Sampel
Misalnya dari sampel yang berbentuk kompleks, dimana sampel ini akan
mempengaruhi respon dari instrument yang disebabkan oleh unsur-unsur yang
terdapat pada sampel tersebut.
2.3.2.7. Efek Komputasi
Pada efek ini yang dibahas adalah ketidakpastian yang disebabkan oleh kalibrasi,
misalnya pada saat menggunakan kalibrasi garis lurus pada saat respon
2.3.2.8. Pengujian Larutan Blanko
Pada suatu analisis sangat penting untuk menguji larutan blanko, yaitu untuk
menghindari ketidakpastian pada sampel. Hal ini sangat penting dalam analisa
yang dilakukan berulang-ulang.
2.3.2.9. Efek dari operator
Pada efek ini yang dapat menyebabkan nilai ketidakpastian itu adalah salah satu
kemungkinan seorang operator itu salah pada saat membaca meter atau skala
konsisten tinggi atau rendah.
2.3.2.10. Efek acak
Efek acak mempunyai kontribusi yang besar pada penentuan ketidakpastian.
Catatan ini harus dimasukkan dalam daftar sebagai hal yang biasa.
Untuk memudahkan analisis semua sumber-sumber ketidakpastian tersebut, dapat
digunakan gambar diagram Ishikawa yang menjelaskan hubungan sumber-sumber
ketidakpastian dengan parameter-parameter measurand. (Ellison et al. 1999)
2.3.3 Perhitungan Komponen-Komponen Ketidakpastian
Ada 2 cara yang digunakan untuk menghitung komponen-komponen
ketidakpastian, yaitu:
1. Menghitung sumber-sumber ketidakpastian yang muncul dan kemudian
2. Setelah penentuan secara langsung nilai gabungannya kemudian
mengubahnya menjadi ketidakpastian hasil dari beberapa atau
sumber-sumber tersebut dengan menggunakan data perlakuan metode.
Ada dua tipe perhitungan ketidakpastian, yaitu:
1. Perhitungan ketidakpastian melalui analisis statistik beberapa seri
pengamatan yang diistilahkan dengan perhitungan (ketidakpastian) tipe A.
2. Perhitungan ketidakpastian dengan menggunakan cara-cara lain selain
analisis statistik beberapa seri pengamatan yang diistilahkan dengan
perhitungan (ketidakpastian) tipe B.(Taylor,B.N and Kuyat,C.E.,1994)
2.3.4 Perhitungan Ketidakpastian Standar Gabungan
2.3.4.1 Ketidakpastian Standar
Ketidakpastian standar merupakan ketidakpastian tiap-tiap komponen yang
merupakan nilai dari hasil ketidakpastian suatu pengukuran yang berupa perkiraan
deviasi standar yang sama dengan variasi akar kuadrat yang positif.
Hasil dari perhitungan tipe A, yang biasanya berupa rata-rata deviasi
standar dapat digunakan langsung untuk mencari ketidakpastian standar
gabungan. Sebaliknya, hasil dari perhitungan tipe B harus diubah terlebih dahulu
menjadi ketidakpastian standar dengan menggunakan distribusi peluang.
(Vetter,T.M.,2001). Persamaan-persamaan yang digunakan untuk menghitung
ketidakpastian standar untuk model distribusi peluang, yaitu distribusi normal,
distribusi seragam atau segiempat, dan distribusi segitiga dapat dilihat dari
lampiran.
2.3.4.2Ketidakpastian Standar Gabungan
menggabungkan ketidakpastian-ketidakpastian individual dengan menggunakan
metode “akar penjumlahan kuadrat” biasa, ataupun metode-metode lain yang
sederajat yang telah diterbitkan dan dipublikasikan.
(Taylor,B.N and Kuyat,C.E.,1994)
Hubungan antara ketidakpastian standar gabungan uc (y) dari nilai y
dengan ketidakpastian dari parameter-parameter bebas x1 , x2 , …, xn adalah:
, , … , ∑ , ∑ , ,
(2.1)
Dimana y(x1, x2, …, xn) adalah fungsi beberapa x1, x2, …, xn, ci
merupakan koefisien sensitifitas yang dihitung sebagai , diferensial parsial dari y terhadap xi , dan melambangkan ketidakpastian dalam y yang muncul karena
ketidakpastian dalam xi.
Jika variabel-variabel tersebut tidak bebas, hubungannya menjadi sangat
rumit, yaitu:
, … ∑ , ∑, , ,
(2.2)
Dimana u(xi,xk) merupakan kovarians antara xi dan xk, ci dan ck merupakan
koefisien sensitivitas. Kovarians dihubungkan dengan koefisien penghubung rik
yaitu u(xi,xk)= u(xi) u(xk) rikdimana -1 ≤ rik ≤ 1.
Rumus-rumus penggabungan ketidakpastian tersebut dapat dibuat menjadi lebih
sederhana dengan menggunakan dua aturan penggabungan ketidakpastian standar,
yaitu:
1. Aturan Pertama
Jika rumus hanya mengandung operasi pengurangan atau penjumlahan,
⋯ (2.3)
2. Aturan Kedua
Jika rumus mengandung operasi perkalian atau pembagian, contohnya
y=(p x q x r…) atau y= p/(q x r x …), maka ketidakpastian gabungan uc(y)
adalah:
⋯
(2.4)
(Ellison et al. 1999)
2.3.4.3 Ketidakpastian Terekspansi
Ketidakpastian terekspansi merupakan suatu nilai dari kuantitas yang
menunjukkan interval di sekitar hasil suatu pengukuran yang diharapkan bias
memberikan fraksi distribusi yang besar dari nilai-nilai yang terdapat dalam
measurand. (ISO.Guide to the Expression of Uncertainty in Measurement,1999)
Ketidakpastian terekspansi pengukuran U, dihasilkan melalui perkalian ketidakpastian standar uc(y) dengan fakor coverage k,
U= k x uc (y) (2.5)
Jika distribusi normal (Gaussian) dapat digunakan pada penentuan measurand dan ketidakpastian standar mempunyai reliabilitas yang sesuai, maka digunakan faktor
coverage k = 2 dengan peluang coverage sekitar 95%. (EAL. Expression of the Uncertainty of Measurement in Calibration,1999)
Jika keadaan normalitas dan reliabilitas tersebut tidak terpenuhi, maka
kebebasannya, atau yang dikenal dengan derajat kebebasan efektif Veff dari uc(y),
yang ditentukan dengan menggunakan persamaan sebagai berikut:
∑
(2.6)
Dimana vi merupakan derajat kebebasan dari u(xi), dan
∑ (2.7)
Nilai Veff dapat digunakan untuk menentukan factor coverage efektif, kp, dengan
menggunakan table distribusi-t sesuai dengan tingkat kepercayaan yang
diinginkan. (Taylor,B.N and Kuyat,C.E.,1994)
2.4 Penulisan Ketidakpastian Pengukuran
Jika suatu penentuan ketidakpastian untuk hasil tunggal hendak dilakukan, dimana
hasil dari ketidakpastian tersebut harus dipublikasikan, adapun bentuk
penyampaiannya adalah:
([hasil] ± [ketidakpastian]) (satuan)
Jika sejumlah hasil disampaikan secara bersamaan, ketidakpastian yang
disampaikan adalah ketidakpastian yang dapat digunakan untuk semua hasil yang
dilaporkan. Nilai hasil ketidakpastian pengukuran harus ditulis dengan dua bentuk
yang nyata. Nilai numerik hasil pengukuran biasanya dibulatkan sampai bentuk
yang nyata yang paling kecil dari nilai ketidakpastian terekspansi. (EAL.
Expression of the Uncertainty of Measurement in Calibration,1999)
Untuk menyederhanakan perhitungan ketidakpastian pengukuran dapat digunakan
perangkat lunak (software). Dimana cara ini memberikan keuntungan dalam metode penurunan angka, dan hanya memerlukan pemahaman dalam
menggunakan rumus unuk menurunkan hasil akhir. (Ellison et al. 1999)
2.6Metode Analisis Kimia
2.6.1 Spektroskopi Ultraviolet dan Tampak (Visible)
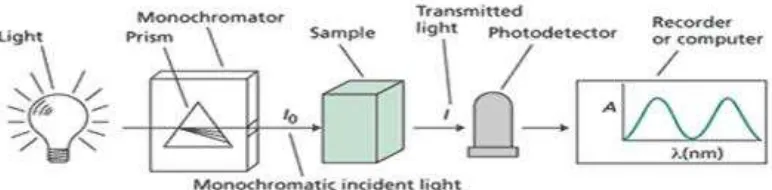
Spektrofotometer sesuai dengan namanya adalah alat yang terdiri dari
spektrometer dan fotometer. Spektrometer menghasilkan sinar dari spektrum
dengan panjang gelombang tertentu dan fotometer adalah alat pengukur intensitas
cahaya yang ditransmisikan atau yang diabsorpsi. Jadi spektrofotometer
digunakan untuk mengukur energi secara relatif jika energi tersebut
ditransmisikan, direfleksikan atau diemisikan sebagai fungsi dari panjang
gelombang. Sedangkan pada spektrofotometer, panjang gelombang yang
benar-benar terseleksi dapat cahaya seperti prisma.
Sumber yang biasa digunakan pada spektroskopi absorpsi adalah lampu
wolfram, lampu hidrogen atau deuterium digunakan untuk sumber pada daerah
UV. Kebaikan lampu wolfram adalah energi radiasi yang dibebaskan tidak
bervariasi pada berbagai panjang gelombang. Untuk memperoleh tegangan yang
stabil dapat digunakan transformator. Monokromator digunakan untuk
memperoleh sumber sinar yang monokromatis. Alatnya dapat berupa prisma
ataupun grating. Untuk mengarahkan sinar monokromatis yang diinginkan dari
hasil penguraian ini dapat digunakan celah.
Pada pengukuran daerah tampak kuvet kaca atau kuvet kaca corex dapat
digunakan, tetapi untuk pengukuran daerah UV kita harus menggunakan sel
kuarsa karena gelas tidak tembus cahaya pada daerah ini. Umumnya, tebal
kuvetnya adalah 10 mm, tetapi yang lebih kecil ataupun yang lebih besar dapat
digunakan. Peranan detektor penerima adalah memberikan respon terhadap
Gambar 2.1 : Bagan alat spektrofotometri UV-VIS
(www.google.com.spektrofotometri.valdisreinaldo)
Secara umum untuk mempelajari secara kuantitatif berkas radiasi yang
dikenakan cuplikan, maka caranya adalah dengan membandingkan intensitas sinar
mula-mula (Io) dengan sinar yang dilewatkan dari cuplikan (It). Ada tiga kemungkinan fenomena yang terjadi yaitu:
1. Io = It, artinya tidak ada sinar yang diserap atau semua ditransmisikan (dilewatkan).
2. It = 0, artinya semua sinar diserap.
3. It > Io, artinya sebagian sinar diserap dan sebagian lagi dilewatkan.
Kejadian 1 dan 2 tidak memberikan informasi, tetapi kejadian 3 akan
memberikan informasi sebagai dasar analisa baik kualitatif maupun kuantitatif.
Besarnya penurunan intensitas sinar (∆I = It – Io) tergantung jenis pengabsorpsi (dasar analisa kualitatif) dan tergantung dengan konsentrasi penyerap (dasar
analisa kuantitatif).
Metode spektrofotometri memerlukan dibuatnya suatu kurva standar
(disebut juga kurva referensi atau kalibrasi) untuk konstituen yang akan
ditentukan. Kuantitas-kuantitas yang sesuai dari konstituen itu diambil dan diolah
dengan cara yang sama seperti larutan contoh untuk pengembangan warna dan
pengukuran transmisi (ataupun absorbansi) pada panjang gelombang yang
optimal. Absorbansi (log I0/It) dialurkan terhadap konsentrasi sehingga diperoleh suatu alur garis lurus jika hukum Beer dipatuhi. Kurva itu kemudian digunakan
2.6.2 Titrasi Asidi-Alkali
Istilah analisis titrimetri mengacu pada analisis kimia kuantitatif yang dilakukan
dengan menetapkan volume suatu larutan yang konsentrasinya diketahui dengan
tetap, yang diperlukan untuk bereaksi secara kuantitatif dengan larutan dari zat
yang akan ditetapkan. Larutan dengan konsentrasi yang diketahui tepat itu,
disebut larutan standar. Bobot zat yang hendak ditetapkan, dihitung dari volume
larutan standar yang digunakan dan hukum-hukum stoikiometri yang diketahui.
Titrasi asam-basa merupakan cara yang cepat dan mudah untuk
menentukan jumlah senyawa-senyawa yang bersifat asam dan basa. Kebanyakan
asam dan basa organik dan anorganik dapat dititrasi dalam larutan berair, tetapi
sebagian senyawa itu, terutama senyawa organik tidak larut dalam air, karena itu
senyawa organik itu dapat ditentukan dengan cara titrasi asam-basa dalam pelaru
non-air.
Untuk menentukan basa digunakan larutan baku asam kuat (misalnya
HCl), sedangkan untuk menentukan asam digunakan larutan baku basa kuat
(misalnya NaOH). Titik akhir titrasi biasanya ditetapkan dengan bantuan peruahan
warna indikator asam-basa yang sesuai atau dengan bantuan peralatan (misalnya
potensiometer, spektrofotometer, dan konduktometer). (Khopkar, S.M.,2008)
Titrasi asam-basa dapat dianggap sebagai interaksi pasangan asam-basa
berpasangan menurut teori Bronsted-Lowry, yaitu:
Asam1 + Basa2 = Basa1 + Asam2
Bila titrasi dilakukan dalam pelarut air, maka perpindahan proton selalu
dinyatakan melalui molekul air. Akibatnya, persamaan umum untuk titrasi
asam-basa dalam pelarut air ditulis sebagai persamaan reaksi antara ion hidronium dan
ion hidroksida, yakni lawan reaksi autoprotolisis air:
Selama proses titrasi pH larutan berubah perlahan-lahan, tetapi di daerah
titik kesetaraan perubahan pH sangat besar. Untuk titrasi asam-basa biasanya
dibuat larutan-larutan asam atau basa dengan sekitar konsentrasi yang diinginkan
dan kemudian distandarisasi salah satu dari larutan dengan suatu standar primer.
Larutan yang dengan demikian telah distandarisasikan dapat dipakai sebagai suatu
larutan standar skunder untuk memperoleh normalitas larutan yang lainnya. Untuk
pekerjaan yang sangat teliti, sepatutnya kedua asam dan basa distandarisasikan
sendiri-sendiri terhadap standar primer. Satu dari standar primer yang secara luas
dipergunakan untuk larutan-larutan basa ialah senyawa kalium hydrogen ftalat,
BAB 3
METODE PENELITIAN
3.1 Alat dan Bahan 3.1.1 Alat
‐ Gelas Beaker 250 mL PYREX
‐ Gelas Erlenmeyer 250 mL PYREX
‐ Neraca Analitis Presisi ±0,001 g Mettler PM 400
‐ Gelas ukur 50 mL PYREX
‐ Gelas ukur 10 mL PYREX
‐ Buret 10 mL PYREX
‐ Labu Takar 1000 mL PYREX
‐ Labu Takar 100 mL PYREX
‐ Labu Takar 50 mL PYREX
‐ Spatula ‐ Pipet Tetes ‐ Karet Penghisap
‐ Pipet Volume 5 mL PYREX
‐ Pipet skala 5 mL PYREX
‐ Botol Reagen Coklat ‐ Cawan porselen
‐ Tanur Thermo Scientific
‐ Kuvet
‐ Statif dan Klem ‐ Botol Aquades
‐ Spektrofotometer Agilent Carry 40
3.1.2 Bahan
‐ Crude Palm Oil ‐ Aquadest
‐ Asam Asetat Glasial p.a. (E. Merck)
‐ HCl(p) p.a. (E. Merck)
‐ Hidroksilamin p.a. (E. Merck)
‐ 1,10-Fenantrolin p.a. (E. Merck)
‐ Fe(NH4OH)2S04 p.a (E. Merck)
‐ ZnO p.a. (E. Merk)
‐ Natrium Asetat p.a. (E. Merck)
3.2. Prosedur Penelitian 3.2.1 Penyediaan Reagent
a. Pembuatan Larutan Hidroksilamin 10 %
Sebanyak 10,00 gram NH2OH.HCl dilarutkan dengan aquades dan diencerkan dalam labu ukur 100 mL sampai garis tanda kemudian dihomogenkan.
b. Pembuatan Larutan Buffer Amonium Asetat
Sebanyak 25,00 gram CH3COONH4 dilarutkan dengan 15 mL aquades dan ditambahkan 70 mL CH3COOH glacial, kemudian diencerkan di dalam labu ukur 100 mL sampai garis tanda dan dihomogenkan
c. Pembuatan Larutan 1,10-Fenantrolin
Sebanyak 0,10 gram 1,10-fenantrolin monohidrat dilarutkan dalam 100 mL
akuades yang telah ditambahkan 2 tetes HCl(p)
d. Pembuatan Larutan NaOH 0,1 N
Sebanyak 4,00 gram NaOH dilarutkan dengan aquadest dan diencerkan dengan
akeadest dalam labu ukur 1 L sampai garis tanda kemudian dihomogenkan.
e. Pembuatan Larutan Induk Fe2+ 1000 mg/L dari Kristal Fe(NH4OH)2SO4
Dilarutkan sebanyak 3,9640 gram Kristal Fe(NH4OH)2SO4 dengan sedikit akuadest lalu dipindahkan secara kuantitatif ke dalam labu ukur 1 L. Selanjutnya
diencerkan dengan akuadet hingga tanda tera dan dihomogenkan.
f. Pembuatan Larutan Standar Fe2+ 100 mg/L
Sebanyak 10 mL larutan induk Fe2+ 1000 mg/L dimasukkan ke dalam labu ukur 100 mL, kemudian diencerkan dengan akuadest sampai garis tanda dan
dihomogenkan.
g. Pembuatan Larutan Standar Fe2+ 10 mg/L
Sebanyak 5 mL larutan induk Fe2+ 100 mg/L dimaasukkan ke dalam labu ukur 50 mL, kemudian diencerkan dengan akuadest sampai garis tanda dan
dihomogenkan.
h. Pembuatan Larutan seri standar Fe2+ 0,2; 0,4; 0,6; 0,8; 1,0 mg/L
Sebanyak 2, 4, 6, 8, dan 10 mL larutan standar 10 mg/L dimasukkan
masing-masing ke dalam labu ukur 100 mL, kemudian diencerkan dengan akuadest
3.2.2. Prosedur Penentuan Fe2+ dengan Metode Spektrofotometri
3.2.2.1Penentuan konsentrasi Fe2+ pada larutan standar
Sebanyak 50 mL larutan seri standar 0,2 mg/L ion Fe2+, dimasukkan ke dalam beaker glass, ditambah 1 mL HCl(p), ditambah 3 mL Hidroksilamin-HCl 5%,
kemudian diuapkan hingga volume awal, lalu didinginkan. Setelah
didinginkan ditambahkan Kristal CH3CCOONa sambil diaduk hingga pH=3, ditambah 10 mL buffer asetat, ditambah 2 mL larutan 1,10-fenantrolin 0,1 %,
kemudian dipindahkan ke dalam labu ukur 50 mL, diencerkan dengan aquadest
sampai garis tanda, lalu dihomogenkan dan didiamkan selama 15 menit, lalu
diukur absorbansinya pada λ = 510 nm. Dilakukan perlakuan yang sama untuk
0,4; 0,6; 0,8; dan 1,0 mg/L dan blanko.
3.2.2.2Penentuan konsentrasi Fe2+ pada sampel
Sebanyak 50 mL minyak sawit mentah (CPO), dimasukkan ke dalam cawan
porselen, ditambahkan 4,00 gram ZnO, selanjutnya dibakar sampai api pada saat
pembakaran padam. Setelah dibakar, diabukan di dalam tanur pada suhu 600 oC selama 2 jam. Dimasukkan ke dalam labu takar 50 mL, ditambah 1 mL HCl(p), ditambah 3 mL Hidroksilamin-HCl 5%, ditambah 10 mL buffer asetat, ditambah 2
mL larutan 1,10-fenantrolin 0,1 %, diencerkan dengan aquadest sampai garis
tanda, lalu dihomogenkan dan didiamkan selama 15 menit, lalu diukur
absorbansinya pada λ = 510 nm.
Konsentrasi larutan FeSO4 diperoleh dengan menggunakan rumus :
CFe2+ = Konsentrasi ion Fe2+ dalam larutan sampel [mg/L]
L = Faktor pengenceren
3.2.3 Prosedur Penentuan Asam Lemak Bebas dengan Metode Titrasi Netralisasi.
Penentuan Asam lemak bebas meliputi tahap-tahap berikut :
Sebanyak 7,05 gram minyak sawit mentah dimasukkan ke dalam Erlenmeyer,
ditambahkan 50 mL alkohol netral, ditambah 3 tetes indikator phenolftalein,
selanjutnya dititrasi dengan NaOH 0,1 N sampai terbentuk merah lembayung.
Konsentrasi NaOH diperoleh dengan menggunakan rumus:
. .
. . (2.9) Dimana :
CNaOH = Konsentrasi larutan NaOH [mol.1-1] FKHP = Berat molekul KHP [g.mol-1] MKHP = Berat KHP [g]
PKHP = Kemurnian KHP
VT = Volume rata-rata larutan NaOH [mL]
3.3. Prosedur Penentuan Ketidakpastian
3.3.1 Spesifikasi measurand
Spesifikasi measurand adalah penjelasan tentang prosedur pengukuran yang terdiri dari langkah-langkah pengukuran dan persamaan matematika yang
digunakan untuk menentukan measurand. Dalam hal ini, measurand adalah konsentrasi Fe2+.
Sumber-sumber ketidakpastian dapat diidentifikasi dari paremeter-parameter yang
terdapat pada persamaan matematika yang digunakan untuk menentukan
meaasurand, dimana semua parameter-parameter tersebut juga mempunyai
sumber-sumber ketidakpastian masing-masing.
3.3.3 Perhitungan Ketidakpastian
Karena hanya sedikit data perlakuan metode yang tersedia, perhitungan
ketidakpastian dilakukan dengan cara menghitung komponen-komponen
ketidakpastian secara terpisah. Berikut ini adalah metode yang digunakan untuk
memperoleh komponen-komponen ketidakpastian individual :
a. Perhitungan ketidakpastian tipe A : diperoleh dari repeatability eksperimen dan dihitung sebagai deviasi standar dari nilai yang diukur.
b. Perhitungan ketidakpastian tipe B, yang terdiri dari :
i) Perhitungan berdasarkan hasil-hasil atau data-data lain, seperti
toleransi peralatan gelas, kemurnian zat, drift dan pembulatan
angka neraca analitik, drift dan pembulatan angka absorbansi pada
spektrofotometer.
ii) Melalui prinsip-prinsip teoritis, seperti pengaruh suhu terhadap
volume yang dapat mempengaruhi hasil.
3.3.4 Perhitungan Ketidakpastian Gabungan
3.3.4.1 Ketidakpastian standar
Semua kontribusi ketidakpastian diubah menjadi ketidakpastian standar, yaitu
sebagai deviasi standar. Berikut ini adalah cara pengubahan kontribusi
ketidakpastian menjadi ketidakpastian standar, yaitu:
i) Jika ketidakpastian berasal dari perhitungan ketidakpastian tipe A,
ketidakpastian standarnya adalah deviasi standar mean yang
ii) Jika ketidakpastian berasal dari perhitungan ketidakpastian tipe B,
maka digunakan tiga distribusi peluang untuk mengubahnya menjadi
ketidakpastian standar, dengan kriteria sebagai berikut:
1. Jika batas-batas ± a diberikan dengan tingkat kepercayaannya, maka untuk menghitung deviasi standar, nilai a dibagi dengan titik persentasi distribusi normal yang sesuai untuk tingkat kepercayaan tersebut.
2. Jika batas-batas ± a diberikan tanpa tingkat kepercayaan dan cukup beralasan untuk menganggap batas-batas marginnya lebih disukai, maka
digunakan distribusi seragam dengan deviasi standar √ .
3. Jika batas-batas ± a diberikan tanpa tingkat kepercayaan, tetapi cukup beralasan untuk menganggap batas-batas marginnya tidak disukai,
melainkan titik tengahnya, maka digunakan distribusi segitiga, dengan
deviasi standar.
3.3.4.2 Ketidakpastian standar gabungan
Untuk menghitung ketidakpastian standar gabungan yaitu dihitung dengan
menggunakan metode perhitungan ketidakpastian Kragten spreadsheet dengan menggunakan software program MS Excel 2007 (Microsoft Inc.)
3.3.4.3Ketidakpastian terekspansi
Untuk menghitung ketidakpastian terekspansi yaitu dihitung dengan berdasarkan
persamaan (2.5) dimana faktor coverage-nya diperoleh dengan menggunakan table distribusi-t pada tingkat kepercayaan 95% dan derajat bebas aktif(Veff)
diperoleh dari persamaan (2.6).
3.4 Bagan Penelitian
3.4.1 Analisa Besi (Fe2+) dengan Metode Spektroskopi UV-Vis
Dimasukkan ke dalam labu erlenmeyer
Ditambahkan 1 mL HCl(p)
Ditambahkan 3 mL hidroksilamin-HCl 5 %
Diuapkan hingga ½ volume awal
Didinginkan
Ditambahkan Kristal CH3COONa sambil diaduk hingga berada pada kisaran pH 3
Ditambahkan 10 mL buffer asetat
Ditambahkan 2 mL larutan 1,10-fenantrolin 0,1 %
Dipindahkan secara kuantitatif ke dalam labu ukur 50 ml
Diencerkan dengan akuadest sampai garis tanda
Dihomogenkan dan didiamkan selama 15 menit
Diukur %T nya pada λ = 510 nm
Dilakukan prosedur yang sama untuk larutan seri standar
0,4; 0,6; 0,8; 1,0 mg/L dan blanko
Hasil
3.4.1.2 Penentuan Fe2+ pada Minyak Sawit Mentah (CPO)
3.4.2. Penentuan Asam Lemak Bebas (AOCS Ca5a-40)
Ditimbang 7,05 gram
Dimasukkan ke dalam erlenmeyer
Ditambahkan 50 mL alkohol netral
Ditambahkan 3 tetes indikator penolftalein
BAB 4
HASIL DAN PEMBAHASAN
4.1 Hasil Penelitian
Setelah dilakukan penelitian penentuan nilai ketidakpastian dengan menggunakan
metode analisis kimia, yaitu analisis dilakukan dengan penentuan asam lemak
bebas metode titrasi netralisasi berdasarkan AOCS Ca5a-40, analisis dilakukan
dengan penentuan kadar ion besi Fe2+ dengan menggunakan metode spektrofotometri uv-vis.
4.1.1 Perhitungan Ketidakpastian untuk Analisis Fe2+ dengan Metode Spektrofotometri
4.1.1.1Perhitungan komponen – komponen ketidakpastian standar
4.1.1.1.1 Perhitungan ketidakpastian tipe A
Pembuatan kurva kalibrasi larutan standar logam Fe2+ dilakukan dengan membuat
larutan standar dengan berbagai konsentrasi yaitu pada pengukuran 0,0; 0,2; 0,4;
0,6; 0,8; dan 1,0 mg/L, kemudian diukur absorbansinya dengan alat
spektrofotometri UV-VIS. Data absorbansi untuk larutan standar Fe2+ dapat dilihat pada tabel 4.1 di bawah ini.
Tabel 4.1. Data absorbansi larutan standar Fe2+
Konsentrasi (mg/L) Absorbansi
0,0 0,0003
0,2 0,0570
0,4 0,1170
0,6 0,1720
0,8 0,2250
1,0 0,2790
Hasil pengukuran absorbansi kemudian diplotkan terhadap konsentrasi sehingga
diperoleh kurva kalibrasi larutan standar logam Fe2+ pada gambar 4.1 berikut.
y = 0,2789x + 0,558
Gambar 4.1.Grafik Least Square Linear untuk Analisis Fe2+ dengan Metode
Spektrofotometri
Perhitungan persamaan garis regresi
Untuk menentukan persamaan garis regresi dari kurva kalibrasi dapat ditentukan
dengan menggunakan metode least square sebagai berikut :
Tabel 4.2. Tabel data perhitungan persamaan garis regresi
Persamaan garis regresi untuk kurva kalibrasi dapat diturunkan dari persamaan
garis :
Y = ax + b (4.1) dimana :
Selanjutnya harga slope dapat ditentukan dengan mengunakan metode least square sebagai berikut :
Harga intersep (b) diperoleh melalui substitusi (a) kepersamaan berikut :
Y = aX + b (4.3) b =Y - aX
b = 0,19525 – (0,2789)(0,5) b = 0,19525 – 0,13945 b = 0,0558
Sehingga diperoleh harga intersep (b) = 0,0558 Maka pesamaan garis regresi yang diperoleh adalah :
Y = 0,2789X + 0,0558 (4.4)
Koefisien Korelasi
Koefisien korelasi dapat ditentukan dengan menggunakan persamaan sebagai
r =
Koefisien korelasi untuk logam Besi Fe2+ adalah:
r =
Dengan menggunakan persamaan 4.4, dapat dihitung konsentrasi larutan sampel
(X0) yang diukur absorbansinya (Y0), yaitu :
Ketidakpastian u(x0,y) pada nilai x0 karena variasi dalam yi
Ketidakpastian konsentrasi Fe2+ dalam larutan sampel (X0) karena pengaruh
variasi absorbansi larutan kalibrasi dapat ditentukan dengan mensubstitusikan
nilai pada tabel 3.1. kedalam persamaan :
(4.6)
Karena dari Sx/y = dan diperoleh
Ketidakpastian (x0, xi) pada nilai x0 karena variasi dalam xi
Persamaan yang digunakan untuk menentukan ketidakpastian konsentrasi Fe2+ dalam larutan sampel yang diukur absorbansinya karena pengaruh variasi
konsentrasi larutan standar kalibrasi adalah sebagai berikut :
Hubungan Linear antara xi dan yi
Untuk menguji hubungan linear antara xi dan yi digunakan uji F dengan
persamaan yang digunakan untuk memperoleh nilai Fhitungsebagai berikut : F = MSLOF / MSPE
Tahap – tahap perhitungan nilai Fhitungadalah sebagai berikut :
Perhitungan penjumlahan kuadrat total (SST), dikoreksi untuk mean
Perhitungan penjumlahan kuadrat karena kesalahan murni (SSPE)
Perhitungan penjumlahan kuadrat karena regresi (SSREG)
Perhitungan penjumlahan kuadrat karena tidak linear (SSLOF) SSLOF = SST – SSREG - SSPE
Dalam hal ini, hipotesis nol adalah terdapat hubungan linear antara xi dan yi. Dan
Tabel 4.3. Tabel ANAVA untuk uji Linearitas
Rincian perhitungan ketidakpastiant ipe B yang berasal dari kontribusi konstan
nilai x dan y karena nilai–nilai ini diperoleh dari sederetan pengenceran larutan stok dan pengukuran absorbansi dengan menggunakan peralatan
spektrofotometer, adalah sebagai berikut :
Ketidakpastian standar konsentrasi ion Fe2+ dalam larutan stok
Ketidakpastian standar massa Kristal Fe(NH4)2(SO4)2.6H2O [u(mkristal)]
Mkristal = mkristal_rep + mkristal_drifth + mkristal_read
Ketidakpastian standar labu takar 100 mL [u(V100)]
V100 = V100_rep + V100_cal + V100_temp
Ketidakpastian standar konsentrasi ion Fe2+ dalam larutan stok
Ketidakpastian standar konsentrasi ion Fe2+ dalam larutan standar kerja
Ketidakpastian standar volume pipet volume 5 ml [u(V5)]
V5 = V5_rep + V5_cal + V5_temp V5_temp = V5_cal x x ∆t
Ketidakpastian standar volume pipet volume 500 ml [u(V500)]
V500 = V500_rep + V500_cal + V500_temp V500_temp = V500_cal x x ∆t
Ketidakpastian standar konsentrasi ion Fe2+ dalam larutan standar kalibrasi
Ketidakpastian standar volume larutan standar kerja yang diambil untuk membuat larutan standar kalibrasi [u(Vi_st)]
Vi_st = Vi_st_rep + Vi_st_cal + Vi_st_temp
Vi_st_temp = Vi_st_cal x x ∆t
Ketidakpastian volume labu takar 50 mL [u(Vi_50)]
Vi_50 = Vi_50_rep + Vi_50_cal + Vi_50_temp Vi_50_temp = Vi_50_cal x x ∆t
Ketidakpastian standar pengukuran absorbansi
Ketidakpastian standar pengukuran absorbansi larutan standar kalibrasi [u(Ai)]
Ai = Ai_rep + Ai_drifth+ Ai-round)
Ketidakpastian standar pengukuran absorbansi larutan sampel [u(Asampel)]
Asampel = Asampel_rep + Asampel_drifth+ Asampel-round)
Ketidakpastian standar faktor pengenceran
Ketidakpastian volume labu takar 50 mL [(V50)]
V50 = V50_rep + V50_cal + V50_temp V50_temp = V50_cal x x ∆t
Ketidakpastian volume larutan sampel yang diambil untuk analisis spektrofotometri [u(V40)]
Ketidakpastian standar faktor pengenceran [u(L)]
4.1.1.1.3 Perhitungan ketidakpastian standar gabungan
Persamaan yang digunakan untuk menentukan konsentrasi Fe2+ dalam larutan sampel adalah sebagai berikut :
x L x Sx0
Jika persamaan tersebut diadaptasikan dengan persamaan (2.4), maka
perhitungan ketidakpastian konsentrasi Fe2+ dalam larutan sampel adalah sebagai berikut :
4.1.1.1.4 Perhitungan ketidakpastian terekspansi
U(C
Fe(II)) = k
px u
c(C
Fe2+)
4.1.2 Perhitungan ketidakpastian untuk analisis penentuan asam lemak bebas dengan metode titrasi netralisasi
4.1.2.1Perhitungan komponen – komponen ketidakpastian standar
4.1.2.1.1 Perhitungan ketidakpastian tipe A
Ketidakpastian tipe A berasal dari pengolahan statistik data perlakuan metode
yang dihasilkan yaitu berupa repeatability hasil pengukuran. Repeatability hasil pengukuran dihitung dengan menggunakan persamaan deviasi standar berikut :
Berdasarkan perlakuan metode, pada lampiran 1 dilakukan dua kali
penentuan konsentrasi, yaitu konsentrasi larutan standar dan konsentrasi larutan
sampel. Oleh karena itu, standar deviasi masing-masing konsentrasi tersebut
digabung dengan menggunakan persamaan deviasi standar mean gabungan
berikut :
Karena deviasi standar ini bervariasi terhadap tingkat konsentrasi analit,
maka digunakan persamaan deviasi standar relatif berikut untuk menentukan
repeatability pengukurannya, yaitu :
4.1.2.1.2 Perhitungan ketidakpastian tipe B
Dalam hal ini, ketidakpastian tipe B berasal dari ketidakpastian yang muncul
dalam perlakuan metode. Rincian persamaan-persamaan yang digunakan adalah
sebagai berikut :
Ketidakpastian standar massa KHP [u(mKHP)]
Ketidakpastian standar kemurnian massa KHP [u(PKHP)]
Ketidakpastian standar berat molekul KHP [u(FKHP)]
4.1.2.1.3 Perhitungan ketidakpastian standar gabungan
Persamaan untuk menghitung konsentrasi larutan NaOH adalah sebagai berikut :
Jika persamaan tersebut diadaptasikan dengan perhitungan ketidakpastian
gabungan aturan 2, persamaan (2.4), maka perhitungan ketidakpastian konsentrasi
larutan NaOH adalah sebagai berikut :
4.1.2.1.4 Perhitungan ketidakpastian terekspansi
U(CNaOH) = kp x (CNaOH)
4.2 Pembahasan
4.2.1 Penentuan ketidakpastian analisis dilakukan dengan penentuan asam
lemak bebas metode titrasi netralisasi berdasarkan AOCS Ca5a-40 4.2.1.1 Sumber-sumber ketidakpastian
Analisis sumber-sumber ketidakpastian akan lebih mudah dipahami dengan cara
menggambarkan diagram Ishikawa, seperti yang tampak pada gambar 4.2 - 4.4.
Tahap pertama untuk menggambarkan diagram Ishikawa adalah dengan
menggambarkan empat parameter yang terdapat pada persamaan 2.9 sebagai
cabang utama (tampak pada gambar 4.2). Kemudian ketidakpastian
masing-masing parameter tersebut ditambahkan ke dalam diagram, seperti yang terlihat
pada gambar 4.3. Komponen ketidakpastian repeatability yang terdapat pada parameter massa dari titrimetri standar KHP dan volume titrasi dari NaOH.
4.2.1.2Perhitungan ketidakpastian standar
4.2.1.2.1 Perhitungan ketidakpastian tipe A
Repeatability hasil pengukuran (rep)
Berdasarkan hasil pengolahan data diperoleh deviasi standar relatif (RSD) sebesar
0,1 %. Nilai repeatability ini sudah termasuk nilai repeatability untuk seluruh sumber – sumber ketidakpastian, yaitu massa KHP, standar kemurnian massa
KHP, standar berat molekul, standar volume NaOH yang digunakan dalam titrasi.
P(KHP) m(KHP)
Gambar 4.2 : Diagram Ishikawa sederhana untuk penentuan asam lemak bebas
P(KHP) m(KHP)
Gambar 4.3 : Detail diagram Ishikawa untuk penentuan asam lemak bebas
P(KHP) m(KHP)
4.2.1.2.2 Perhitungan ketidakpastian tipe B
Ketidakpastian massa KHP
1. Repeatability
Log dari kualiti kontrol menunjukkan ketidakpastian standar dari 0,05 mg
beratnya sama pengecekan sampai menjadi 100 gram. Harga ini untuk
repeatability ditentukan oleh sepuluh pengukuran dari tare dan berat gross diikuti oleh perhitungan yang berbeda dari masing – masing pasangan pengukuran
tersebut dan evaluasi dari standar deviasinya berbeda. Kontribusi dari
repeatability ini hanya dilakukan penjumlahan satu kali karena standar deviasinya berbeda secara langsung yang ditentukan dari eksperimen yang diberikan.
2. Calibration / Linearity
Keterangan kalibrasi dari keseimbangan kuota sekitar ± 0,15 mg untuk linearitas
nilai ini memberikan perbedaan yang maksimum antara berat sebenarnya pada
data dan pembacaan skala. Pembuat keseimbangan merekomendasikan evaluasi
yang tidak pasti untuk menggunakan distribusi segiempat untuk mengubahnya
menjadi kontribusi linier menjadi standar yang tidak tentu. Keseimbangan
kontribusi linier menurut gambar 4.4. Kontribusi ini telah dihitung dua kali yang
pertama untuk tare dan yang satu lagi untuk berat gross.
Kombinasi dari dua kontribusi ini memberikan standar yang tidak pasti
P(KHP)
Penyediannya memberikan informasi yang tidak lebih jauh menghubungkan
ketidakpastian dalam daftar itulah sebabnya ketidakpastian ini memiliki distribusi
segiempat.
F(KHP)
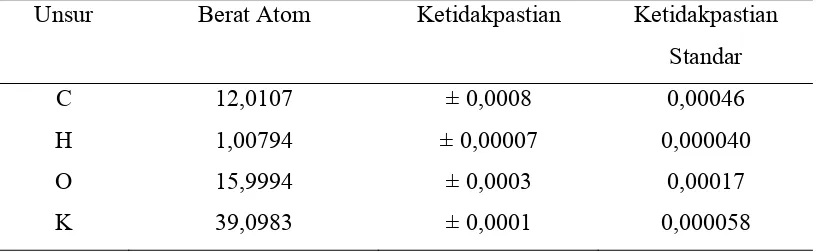
Dari tabel IUPAC(International Union Of Pure and Applied Chemistry) yang terakhir berat atom dan data yang tidak pasti untuk elemen – elemen unsur dari
KHP(C8H5O4K) adalah
Tabel 4.4 Data ketidakpastian untuk elemen-elemen unsur KHP(C8H5O4K)
Unsur Berat Atom Ketidakpastian Ketidakpastian
Standar
C 12,0107 ± 0,0008 0,00046
H 1,00794 ± 0,00007 0,000040
O 15,9994 ± 0,0003 0,00017
K 39,0983 ± 0,0001 0,000058
(Ellison et al. 1999)
Untuk masing – masing elemen, standar tidakpasti ditemukan dengan cara
mengolah kuota IUPAC yang tidak pasti sebagai bentuk ikatan dari distribusi
segiempat. Penyesuaian standar yang tidak pasti diperoleh dengan cara membagi
nilai tersebut dengan akar
Kontribusi dari elemen yang terpisah memberikan formula berat yang
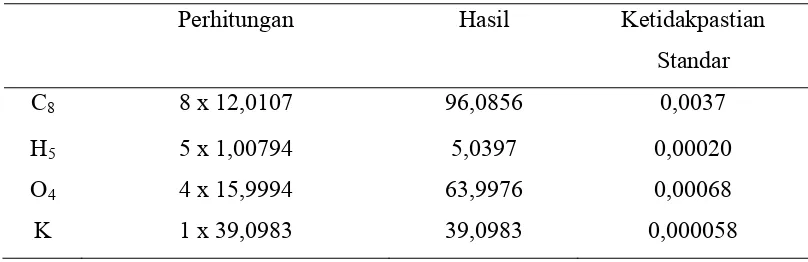
Tabel 4.5 Data ketidakpastian untuk KHP(C8H5O4K)
Perhitungan Hasil Ketidakpastian
Standar
C8 8 x 12,0107 96,0856 0,0037
H5 5 x 1,00794 5,0397 0,00020
O4 4 x 15,9994 63,9976 0,00068
K 1 x 39,0983 39,0983 0,000058
(Ellison et al. 1999) Ketidakpastian dari masing – masing nilai ini dihitung dengan melipat
gandakan standar tidak pasti dari tabel sebelumnya dari jumlah atom tersebut. Hal
itu memberikan berat formula untuk KHP. Karena pernyataan ini merupakan jumlah dari nilai yang bebas, standar tidak pasti u(FKHP) adalah akar kuadrat dari
jumlah kuadrat dari kontribusinya masing – masing.
V(T)
1. Repeatability dari volume yang digunakan
Kegunaan dari piston buret, tidak seperti tabung volumetrik, yang umumnya
tidak meliputi penggunaan secara lengkap dalam jumlahnya. Bila diperkirakan
variabel dari penggunaan volum tersebut akan dibutuhkan untuk sejumlah sasaran
akhir diantara penggunaannya. Untuk buret yang digunakan, beberapanya disusun
dengan menggunakan volume yang terbatas yang harus diperiksa dan dicatat;
sebagai contoh 0-10, 10-20, 5-15 dan seterusnya. Persamaannya harus diselidiki
secara repeatability dari perbedaan volume yang digunakan, seperti 5, 10, 15 mL dan seterusnya. Contohnya repeatability dari data yang sebelumnya menggunakan 19 mL yang disimpan, diberikan sampel standar deviasi dari 0,004 mL,
menggunakan pengukuran secara langsung sebagai standar yang tidak pasti.
Keterbatasan akurasi dari voilume yang digunakan ditunjukkan oleh
pembuatannya seperti yang digambarkan. Untuk 20 mL piston buret jumlah ini
khususnya sekitar 0,03 mL. Dianggap distribusi segitiga memberikan standar
tidak pasti dari 0,03 / akar 6 = 0,012 mL.
3. Temperature
Ketidakpastian dikarenakan kekurangan kontrol temperatur yang dikalkulasi
dengan cara yang sama seperti contoh yang sebelumnya, tetapi pada saat ini akan
diambil variasi temperatur yang mungkin dari 3oC (dengan keakuratan 95 %). Kemudian menggunakan koefisien dari perluasan volume untuk air yaitu 2.1.10-4 o
C-1.
4. Repeatability dari titik yang diperoleh
Repeatability dari titik akhir yang diperoleh diperiksa secara teliti selama metode yang sebenarnya. Kemudian memberikan kondisi standar tidak pasti sebesar 0,004
mL yang merupakan data yang cocok.
5. Penyimpangan dari titik akhir yang diperoleh
Titrasi ini ditunjukkan menurut lapisan Argon untuk meniadaakan beberapa
penyimpangan karena absorpsi dari CO2 dalam larutan titrasi. Pendekatan ini mengikuti petunjuk, yang lebih baik mencegah beberapa penyimpangan yang
terdiri dari kebenaran untuk senyawa tersebut. Tidak ada petunjuk lain dimana
titik akhir ditentukan dari bentuk kurva pH yang tidak cocok dengan titik
ekuivalen karena asam kuat dititrasi dengan basa kuat. Oleh karena itu, hal ini
pasti dapat diabaikan. VT ditemukan menjadi 18,64 mL dan dipakai untuk
mencampurkan 4 sisa dari kontribusi ketidakpastian u(VT) dari volume VT.
4.2.1.2.3 Perhitungan dari kombinasi standar ketidakpastian
Nilai dari parameter pada persamaan diatas, standar ketidakpastiannya dan standar
ketidakpastian relatipnya digabungkan di dalam lampirannnnn A2.1. Karena
kombinasi ketidakpastian dengan masing – masing komponen dari persamaan
yang beragam seperti diatas.
Lembaran yang menyebar standar yang digunakan untuk
menyederhanakan perhitungan diatas dari kombinasi standar tidak pasti. Secara
luas diperkenalkan ke dalam metode yang diberikan dalam lampiran E. Lembaran
yang menyebar diisi ke dalam nilai yang diperkirakan yang ditunjukkan ke dalam
nilai yang ditunjukkan ke dalam tabel.
Pada akhirnya lembaran yang menyebar tadi menguji kontribusi relatip
dari parameter yang berbeda. Bagian dari masing – masing kontribusi dapat
dengan mudah dilihat dengan menggunakan tampilan histogram dari standar
ketidakpastian relatip.
Kontribusi dari ketidakpastian pada volume titrasi VT menjadi lebih jauh.
Prosedur berat dan pemurnian dari titrasi standar menunjukkan urutan yang sama
dari jarak untuk standar ketidakpastian relatipnya, dimana ketidakpastian dalam
berat formula hampir mendekati dari jarak yang lebih kecil dari sebelumnya.
Perluasan ketidakpastuan u(CNaOH) diperoleh dengan mengalikan kombinasi
Gambar 4.5 : Metode perhitungan ketidakpastian Kragten Spreadsheet untuk
penentuan asam lemak bebas dengan metode titrasi netralisasi.
Gambar 4.6 : Histogram hubungan ketidakpastian standar gabungan dengan
kontribusi masing – masing komponen pada penentuan asam lemak
bebas dengan metode titrasi netralisasi.
Nilai parameter yang dibrikan dalam baris kedua dari C2-F2. Standar ketidakpastiannya
dimasukkan ke dalam baris dibawahnya(C3-F3). Nilai dari spreadsheet dari C2-F2 di
dalam kolom kedua dari B5-B8. C5 menunjukkan nilai dari mKHP dari C2 ditambahkan
4.2.2 Penentuan ketidakpastian dalam analisis Fe2+ dengan metode spektrofotometri
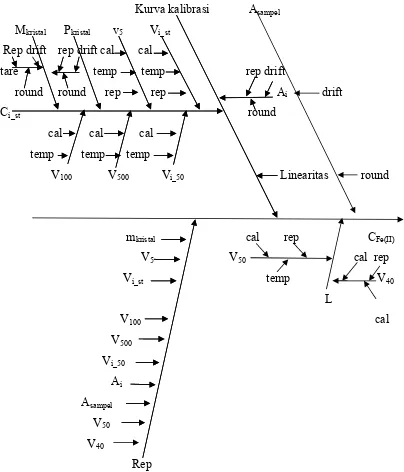
4.2.2.1 Sumber-sumber ketidakpastian
Sumber-sumber ketidakpastian yang terdapat pada analisis Fe2+ dengan metode spektrofotometri adalah:
a. Variasi random dalam pengukuran y, yang mempengaruhi respons larutan standar kalibrasi yi dan respon larutan yang diukur y0.
b. Variasi random yang berasal dari kesalahan dalam memperoleh nilai
larutan standar kalibrasi xi.
c. Hubungan linier antara xidan yi.
d. Nilai-nilai xi dan yi yang mengandung ketidakpastian konstan, yang
muncul karena nilai x dihasilkan daari sederetan pengenceran larutan stok, dan juga karena nilai y diperoleh dari pengukuran absorbansi dengan menggunakan spektrofotometer.
Keempat sumber-sumber ketidakpastian diatas akan lebih jelas dilihat
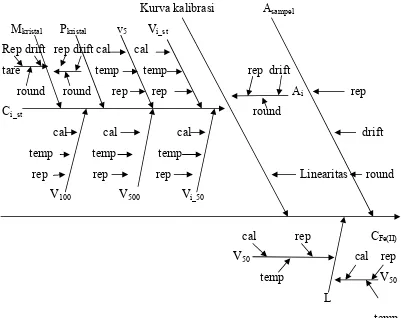
Kurva kalibrasi Asampel Mkristal Pkristal v5 Vi_st
Rep drift rep drift cal cal
tare temp temp rep drift
round round rep rep Ai rep Ci_st round
cal cal cal drift
temp temp temp
rep rep rep Linearitas round
V100 V500 Vi_50
cal rep CFe(II) V50 cal rep
temp V50 L
temp
Kurva kalibrasi Asampel Mkristal Pkristal v5 Vi_st
Rep drift rep drift cal cal
tare temp temp rep drift
round round rep rep Ai drift Ci_st round
cal cal cal
temp temp temp
V100 V500 Vi_50 Linearitas round
mkristal cal rep CFe(II) V5 V50 cal rep
Vi_st temp V40 L
V100 cal V500
Vi_50 Ai
Asampel V50 V40 Rep
Gambar 4.8 : Diagram Ishikawa untuk Analisis Larutan Fe2+ dengan Metode
4.2.2.2Perhitungan ketidakpastian standar 4.2.2.2.1 Perhitungan ketidakpastian tipe A
Repeatability (rep)
Dari hasil pengolahan data, diperoleh nilai Sx0 0,0039 mg/L. Nilai ini merupakan
nilai ketidakpastian dalam pengukuran y, yang mempengaruhi respon larutan standar kalibrasi yi dan respon larutan yang diukur y0 yang dilambangkan dengan u(x0, yi).
Nilai ketidakpastian yang berasal dari kesalahan dalam memperoleh nilai
larutan standar kalibrasi xi dilambangkan dengan u(x0,xi), biasanya sangat kecil
dibandingkan dengan ketidakpastian u(x0,yi) sehingga diabaikan.
Dari hasil pengolahan data pada tabel 3.3, tampak bahwa Fhitung lebih kecil
daripada Ftabel, sehingga hipotesis nol diterima dan gterdapat hubungan antara y
dan x. Hal ini berarti ketidakpastian karena hubungan linear x dan y tidak cukup besar untuk mempengaruhi ketidakpastian standar gabungan sehingga diabaikan.
Oleh karena komponen ketidakpastian u(x0,xi) dan hubungan linearitas
antara x dan y diabaikan, maka hanya kontribusi ketidakpastian Sx0 yang
digunakan dalam perhitungan ketidakpastian standar gabungan, dan nilai ini sudah
termasuk nilai komponen ketidakpastian repeatability untuk semua sumber ketidakpastian pada analis dengan metode spektrofotometri.
4.2.2.2.2 Perhitungan ketidakpastian tipe B
Nilai-nilai x dan y mengandung ketidakpastian konstan yang dikarenakan xi
dihasilkan dari sederetan pengenceran larutan stok dan juga karena yi dihasilkan
Ketidakpastian standar kemurnia kristal Fe(NH4)3(S04)2.6H2O [u(Pkristal)]
Dari hasil pengolahan data, diperoleh nilai u(Pkristal) 0,0058 gram. Nilai ini
diperoleh dari nilai kemurnian kristal yang diberikan oleh supplier, yaitu 100% ±
1%, yang diasumsikan sebagai distribusi seragam karena tidak ada informasi
mengenai tingkat kepercayaan yang diberikan.
Ketidakpastian standar massa kristal [u(mkristal)]
Dari hasil pengolahan data, diperoleh nilai u(mkristal) 0,00017 gram. Dimana
perkalian dua pada persamaan ketidakpastian standar gabungan
komponen-komponennya, dikarenakan penimbangan dilakukan dengan cara mengurangkan
massa kristal dan wadah (gross) dengan massa sisa sedikit kristal dan wadah (tare), yang mana keduannya mempunyai komponen-komponen ketidakpastian yang sama, yaitu drift dan pembulatan angka (round) pada neraca, seperti yang ditunjukkan pada gambar 4.12. Kareena tidak dilakukan penelitian khusus untuk
menentukan nilai ketidakpastian drift, nilai ini diperoleh dari katalog supplier
neraca yang digunakan. Sedangkan ketidakpastian round adalah setengah digit
terakhir yang mampu dibaca oleh neraca.
Ketidakpastian standar labu takar 100 mL [u(V100)]
Dari hasil pengolahan data, diperoleh u(V100) 0,13 mL. Nilai ini diperoleh dari
penggabungan dua komponen ketidakpastian labu takar 100 mL, yaitu
ketidakpastian kalibrasi dan perbedaan suhu kalibrasi dengan suhu laboratorium.
Nilai ktidakpastian kalibrasi diperoleh dari nilai yang tertera pada peralatan labu
takar 100 mL, yaitu (100 ± 0,08) mL. Karena tidak ada informasi lain tentang
tingkat kepercayaan, dan karena asumsi supplier mengkalibrasi peralatan ini
dengan ketelitian yang tinggi, maka ketidakpastian ini diasumsikan sebagai
mengalikan perbedaan suhu kalibrasi dengan siuhu laboratorium, yang bervariasi
±10oC (suhu kalibrasi adalah 20oC), dengan koefisien ekspansi volume air. Karena variasi ini tidak diamati secara khusus, maka diasumsikan sebagai
distribusi seragam.
Ketidakpastian standar labu takar 500 mL [u(V500)]
Dari hasil pengolahan data, diperoleh u(V500) 0,61 mL. Nilai ini diperoleh dari
penggabungan ketidakpastian kalibrasi dan perbedaan suhu kalibrasi dengan suhu
laboratorium. Nilai ketidakpastian kalibrasi diperoleh dari nilai toleransi yang
tertera pada peralatan labu takar 500 mL, yaitu (500 ± 0,15), yang dikalibrasi pada
suhu 20oC, yang diasumsikan sebagai distribusi segitiga. Sedangkan komponen ketidakpastian suhu diperoleh dengan mengalihkan perbedaan suhu kalibrasi
dengan suhu laboratorium dengan koefisien ekspansi volume air.
Ketidakpastian standar pipet volume 5 M [u(V5)]
Dari hasil pengolahan data, diperoleh u(V5) 0,0073 mL. Nilai ini diperoleh dari
penggabungan ketidakpastian kalibrasi dan perbedaan suhu kalibrasi dengan suhu
laboratorium. Nilai ketidakpastian kalibrasi diperoleh dari nilai toleransi yang
tertera pada peralatan pipet volume 5 mL, yaitu (5 ± 0,001)mL, yang dikalibrasi
pada suhu 20oC. Sedangkan komponen ketidakpastian suhu diperoleh dengan mengalikan perbedaan suhu kalibrasi dengan suhu laboratorium dengan koefisien