BAB II
TINJAUAN PUSTAKA
A.
Cedera Kepala
1.
Neuropatologi
Cedera kepala dapat ditandai dengan
coup
dan
contra coup
serta dengan
shearing
dan
tearing
akson di otak akibat akselerasi rotasional dari
kepala.Diagnosis umum sehubungan cedera kepala meliputi fraktur tengkorak,
contusion, confusion
, laserasi, dan lesi fokal (Silver et al., 2005). Cedera sekunder
yang meliputi perdarahan lebih lanjut, deformitas mekanis, dan peningkatan
tekananan intrakranial dapat terjadi sebagai komplikasi sebagai cedera awal
(Bigler & Maxwel., 2011). Saat ini telah terbukti bahwa penderita cedera kepala
mengalami kehilangan sebagian volume otak sampai setidaknya satu tahun setelah
kecelakaan (Bendlin et al., 2008; Sidaros et al., 2009; Trivedi et al., 2007).
Pemahaman patologi seluler pada cedera kepala merupakan hal penting
dalam pengembangan terapi baru cedera kepala.Perubahan neurokimiawi otak
sesudah cedera dapat
berupa agen “neuroprotektif” dan
“autodestruksi”.Peningkatan
acetylcholine
terjadi segera setelah cedera (Donat et
al., 2008; Lyeth & Hayes, 1992).Peningkatan epinefrin dan norepinefrin pada
serum terjadi seiringan dengan tingkat keparahan cedera.Pada daerah sekitar
tempat cedera, ditemui peningkatan serotonin dan dopamin (Kobori et al.,
2011).Namun sampai sekarang bagaimana peranan agen neurokimiawi ini
minggu setelah cedera terjadi peningkatan faktor neurotropic, seperti
nerve
growth factor (NGF)
dan
fibroblast growth factor (FGF).
Faktor-faktor
neurotropik ini diduga akan membantu pemulihan (Ziebell & Morganti –
Kossmann, 2010).
Autofagi merupakan proses dimana otak membuang jaringan yang mati
atau rusak agar sel sehat dapat berfungsi dengan lebih efektif. Proses ini juga
diduga akan membantu pemulihan (Clark et al., 2008). Meskipun demikian,
proses ini dapat menyebabkan nekrosis dan apoptosis yang akan memicu
degenerasi Wallerian dalam jumlah yang tidak diketahui (Zhou et al., 2012).
Degenerasi Wallerian merupakan perluasan dari cedera kepala. Cedera yang
awalnya terjadi hanya pada akson akan meluas sampai ke badan sel (Kelley et al.,
2006). Proses ini sepertinya memegang peranan, baik dalam perubahan
white
matter
yang banyak dijumpai pada cedera kepala, maupun dalam degenerasi dan
reorganisasi. Proses perbaikan dan degenerasi yang berlangsung sekaligus ini
menyebabkan
microenvironment
otak yang sangat beragam.
2. Degenerasi Axon Post Trauma
Cedera kepala akan menghasilkan cedera akson, terutama akibat regangan
(Meythaler et al., 2001). Berbeda dengan cedera fokal, cedera akson pada cedera
kepala bersifat lebih difus.Transeksi komplit jarang terjadi, tetapi regangan
menyebabkan kerusakan struktur akson, yang dapat menyebabkan disfungsi sel
Permeabilitas membran akson akan segera terganggu, bahkan setelah
cedera kepala sedang dan ini akan disertai dengan pemadatan lokal dari
neurofilamen (Pettus et al., 1994). Pada model kucing percobaan, gangguan
permeabilitas lokai ini akan muncul dalam lima menit setelah trauma dan
pemadatan ini bertahan setidaknya selama enam jam setelah kejadian (Pettus &
Povlishock, 1996). Model trauma lain yang sering digunakan adalah regangan
pada neuron
in vitro
. Setelah regangan, terjadi distorsi berbentuk undulasi di
sepanjang akson yang terjadi akibat kerusakan mikrotubulus. Gangguan
mikrotubulus ini akan menyebabkan gangguan transport akson dan akumulasi
protein (Tang-Schomer et al., 2010).
3. Mekanisme Degenerasi Akson
a. Konsep Mekanis
Seperti apoptosis, kebanyakan bentuk degenerasi akson merupakan proses
self-destructing
seluler yang aktif dan melibatkan kaskade tertentu dengan
keterlibatan banyak faktor (Raff et al., 2002). Meskipun demikian, pada dasarnya,
apoptosis dan degenerasi akson melibatkan proses biokimia yang berbeda dan
dapat terjadi secara terpisah (Whitmore et al., 2003). Beberapa bentuk degenerasi
akson telah dijelaskan berdasarkan lokasinya pada akson dan waktu
terjadinya.Penelitian yang paling banyak dilakukan adalah mengenai degenerasi
sesudah trauma, baik degenerasi akut pada tempat trauma maupun degenerasi
b. Degenerasi Akson Akut
Istilah degenerasi akson akut mengacu pada disintegrasi akut akson dalam
beberapa jam setelah trauma susunan saraf pusat. Dalam 10-30 menit setelah
trauma, akson akan relatif stabil dengan gambaran makroskopis yang normal.
Meskipun demikian, pada tingkat molekuler, suatu sinyal sudah diaktivasi dengan
hasil akhir berupa fragmentasi akson. Proses ini diawali dengan influks kalsium
ke dalam akson dalam waktu cepat yang menyebabkan peningkatan konsentrasi
kalsium yang transien dalam 40 detik setelah cedera. Pemberian calcium channel
inhibitor pada saat ini akan menghambat peningkatan kalsium aksonal dan
degenerasi akut (Knoferle et al., 2010). Influks kalsium menyebabkan aktivasi
calpain yang mencapai nilai maksimal dalam 30 menit setelah cedera.
Perubahan pertama struktur mikroskopis dapat dinilai dalam 30 menit
pertama setelah cedera.Perubahan ini meliputi kondensasi dan perubahan
alignment
neurofilamen yang diikuti fragmentasi mikrotubulus (Knoferle et al.,
2010). Pada kelainan SSP lain, pemadatan neurofilamen fokal dan proteolisis
mikrotubulus terbukti berhubungan dengan aktivasi calpain (Veerana et al., 2004).
Karena itu, aktivasi calpain pada saat awal kemungkinan besar juga berperan
dalam degenerasi akson akut. Kerusakan sitoskeleton akan menyebabkan
gangguan transport akson.
Gambaran degenerasi akson akut lain adalah aktivasi autofagi lokal.
jam pertama setelah cedera. Inhibisi autofagi dengan obat-obatan seharusnya
mengurangi degenerasi akut, namun tidak seperti setelah pemberian calcium
channel blocker, pengurangan degenerasi tidak ditemui. Ini kemungkinan
menggambarkan bahwa autofagi merupakan proses lanjutan influks kalsium
(Knoferle et al., 2010).
Meskipun pemberian obat-obatan seharusnya akan mengurangi degenerasi
akson akut, efek jangka panjang dari terapi ini masih belum jelas. Karena
degenerasi akut hanya terjadi pada sekitar 400 μm akson di sekitar tempat
cedera,
keuntungan menyelamatkan bagian ini tidak bermakna.
c. Degenerasi Wallerian
Degenerasi Wallerian secara sederhana didefenisikan sebagai degenerasi
akson yang terjadi distal dari tempat cedera. Setelah trauma, bentuk bagian akson
yang tidak terkena degenerasi akut akan tetap normal dalam 24 sampai 72 jam
pertama. Kemudian, bagian distal akson akan menjalani fragmentasi progresif
yang menyerupai fragmentasi pada degenerasi akson akut (Kerschensteiner et al.,
2005) yang pada akhirnya menyebabkan
removal
seluruh bagian distal akson.
Degenerasi Wallerian berlangsung dengan kecepatan mulai 0,4 mm/jam pada
penelitian
in vitro
(Sievers et al., 2003) sampai 24 mm/jam pada saraf sciatic tikus
(Beirowski et al., 2005).Pada susunan saraf tepi, arah degenerasi Wallerian pada
akson tampaknya bergantung jenis lesi. Transeksi komplit saraf menyebabkan
al., 2005). Meskipun makrofag dan glia sepertinya memegang peranan penting,
mekanisme degenerasi Wallerian tampaknya bersifat intrinsik (MacDonald et al.,
2006).
Mekanisme molekuler yang mendasari degenerasi Wallerian belum
dipahami sepenuhnya.Kemajuan pesat terjadi dengan ditemukannya tikus mutan
WldS (Lunn et al., 1989). Pada tikus ini, potongan akson distal dari lesi bertahan
sepuluh kali lebih lama dibandingkan akson pada binatang
wild-type
, dan survival
badan sel tidak terganggu (Adalbert et al., 2006).
Protein mutan WldS merupakan produk gen yang terdiri dari potongan
faktor poliubiquiti UFD2a/UBE4b dan
nicotinamide monocleotide
adenytransferase -1
(NMNAT1; Mack et al., 2001).NMNAT1 adalah protein
kunci jaras
nicotinamide-adenine dinucleotide
+(NAD
+) pada mamalia.UBE4b
adalah ubiquitin ligase tipe E4 yang dapat menambahkan rantai multiubiquitin
pada jaras ubiquitin/proteasome (Hatakeyama et al., 2001).Tempat molekuler
WldS yang penting adalah tempat pengingkatan ATP dan tempat pengikatan
NMN
+Protein WldS berada terutama pada nukleus, meskipun juga dideteksi pada
aksoplasma dan organel aksoplasma (Yahata et al., 2009).Ekspresi NMNAT1
pada NMNAT1 dan tempat pengikatan
valocin-containing protein
(VCP)
pada UBE4b. NMNAT1 dan UBE4b yang fungsional diperlukan untuk aktivitas
neuroproteksi dari WldS. Ini didukung oleh fakta bahwa ekspresi NMNAT1 saja
tidak cukup untuk mencegah degenerasi akson pada neuron mammalia meskipun
penurunan aktivitas NMNAT1 pada tikus WldS transgrenik menyebaban
lokal pada kompartemen akson menyebabkan efek protektif yang menyerupai
tikus transgenik WldS (Babetto et al., 2010).Data ini menunjukkan bahwa
aktivitas protektif SldS dimediasi oleh transport protein terus-menerus pada
akson. Sesuai dengan penjelasan di atas, isoform NMNAT lain meningkatkan
survival akson lokal. NMNAT2 terus menerus ditranspor dari badan sel menuju
akson dengan waktu
turnover
yang singkat, sekitar 4 jam.
Down regulation
dari
NMNAT2 atau inhibisi transportnya menuju akson menyebabkan degenerasi
akson. Sebaliknya, overekspresi akan menghambat degenerasi (Gilley &
Coleman, 2010). Efek yang sama juga terlihat pada overekspresi isoform
mitokondria NMNAT3 (Yahata et al., 2009). Target isoform NMAT untuk
meningkatkan survival akson masih belum jelas. Seluruh NMNAT mempunyai
domain katalis sintesis NAD
+(Berger et al., 2005), meskipun data yang
mendukung peranan NAD
+dalam mempertahankan survival akson tidak
konsisten.Pemberian NAD
+konsentrasi tinggi pada ekstraseluler menyebabkan
perlindungan akson yang cedera.Sebaliknya, berbagai usaha untuk meningkatkan
konsentrasi NAD
+intrasel tidak memberikan efek pada degenerasi akson (Sasaki
et al., 2009).
d. Mekanisme Molekuler Degenerasi Akson
Degenerasi akson akut, degenerasi fokal akson, dan degenerasi Wallerian
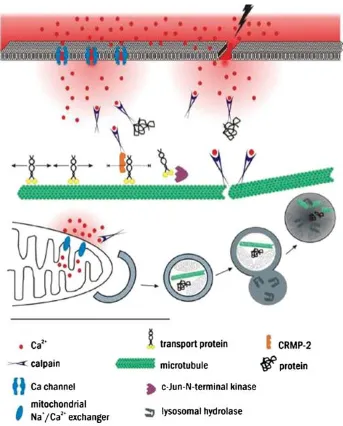
Gambar 1 Beberapa mekanisme molekul yang terlibat dalam degenerasi akson. (Lingor et al, 2012)
e. Kalsium
Beberapa proses yang menyebabkan peningkatan konsentrasi kalsium
akson pada berbagai lesi antara lain : (1) influks kalsium dari ruang ekstrasel
channel dari rongga ekstrasel, dan (3) lepasan kalsium dari depot kalsium intrasel
(Stirling & Stys, 2010).
Cedera mekanis akson menyebabkan kerusakan kontinuitas membrane dan
influks kalsum ekstrasel ke dalam sitoplasma.Sesuai percobaan Ziv dan Spira
(1995) pada akson in vitro, gelombang kalsium diinduksi oleh transeksi satu
cabang dendrit yang menyebar dengan cepat (dalam satuan detik) sampai
percabangan dendrit. Kalsium intra akson akan menurun dengan cepat beberapa
menit kemudian menuju tingkat tertentu setelah ujung akson yang cedera ditutup.
Meskipun demikian, kadar kalsium intrasel bervariasi, mulai >1 mM di dekat lesi
sampai beberapa ratus mikromolar distal dari lesi (Ziv & Spira, 1995). Pada
penelitian in vitro, diketahui bahwa diperlukan kadar kalsium ekstra akson > 200
μM untuk m
enyebabkan peningkatan kalsium intra akson setelah
axotomy
.
Kalsium juga terbukti memasuki akson melalui
L-type calcium channel,
bukan
N
type
(George et al., 1995).
Ruang ekstra akson bukanlah satu-satunya sumber kalsium.Depot kalsium
intrasel juga memberikan kontribusi yang cukup besar pada peningkatan kalsium
sitoplasimik.Misalnya, pada kerusakan iskemia di akson pada kolum dorsalis,
kalsium dilepaskan melalui reseptor
ryanodine
oleh reticulum endoplasma
(Ouardouz et al., 2003) atau mitokondria (Nikolaeva et al., 2005).
Pada akson dengan myelin, reseptor
ryanodine
dapat diaktivasi melalui
L-type calcium channel
. Baik reseptor
ryanodine
dan
L-type calcium channel
berada
pada axolemma.Namun, penghambatan pelepasan kalsium intra akson, misalnya
menggambarkan kemungkinan sumber kalsium lain atau adanya suatu mekanisme
lain yang tidak tergantung kalsium (Ouardouz et al., 2003). Pada proses iskemia,
kebanyakan lepasan kalsium intraaksonal dimediasi oleh pompa Na
+/Ca
2+yang
dibuktikan pada percobaan dengan CGP37157, suatu penghambat pompa
Na
+/Ca
2+Selain itu, kalsium intrasel yang berlebihan pada neuron dapat berasal dari
pompa pada membrane sel, seperti plasma-membrane calcium ATPase isoform 2.
Penurunan kadar ponpa ini akan memperbaiki patologi akson pada hewan
percobaan.
pada mitokondria (Nikolaeva et al., 2005).
f. Kejadian Lanjutan Setelah Influks Kalsium
Influks kalsium diikuti aktivasi
calcium-dependent protease
, seperti
calpains, yang akan membelah dan mendegradasi protein sitoplasma. Peningkatan
aktivitas calpains sudah terbukti pada diffuse axonal injury setelah trauma kepala,
stroke, cedera spinal cord, dan kelainan neurodegenerative (Vosler et al., 2008).
Calpains akan mendegradasi sejumlah target, seperti protein sitoskeleton, enzim,
reseptor,
channel,
dan faktor transkripsi melalui proses proteolisis (Saatman et al.,
2010). Calpains juga akan mendegradasi substrat yang penting untuk stabilitas
akson. Ini akan diikuti oleh pemecahan
collapsing response mediator protein-2
(CRMP-2) secara proteolotik (Touma et al., 2007). Karena CRMP-2 berikatan
dengan kinesin-1 dan berpartisipasi dalam transport akson, transport akson pada
Selain calpain, ada beberapa enzim yang tergantung kalsium yang
berpartisipasi dalam degenerasi akson. Cedera akson transien akibat regangan
akan menyebabkan lepasan kalsium yang pertama kali terjadi dari depot intrasel.
Ini akan diikuti dengan peningkatan kadar kalsium intrasel dalam 48 jam. Dalam
proses ini, penghambatan calcineurin, suatu
calcium-dependent phosphatase
akan
menghalangi degenerasi akson sekunder (Staal et al., 2010). Calpain dan
calcineurin hanyalah dua contoh dari protein tergantung kalsium yang terlibat
dalam proses degenerasi lebih lanjut.
g. Cedera Mitokondria
Cedera mitokondria memegang kunci penting dalam lokalisasi gangguan
kontinuitas akson. Pada model cedera kepala dengan
diffuse axonal injury
, influks
kalsium diduga dimediasi melalui pori-pori axolemma. Namun, kerusakan akson
dan aktivasi calpain tidak terjadi secara simultan sekaligus pada seluruh
akson.Kerusakan terjadi pada titik-titik fokal disertai akumulasi mitokondria
(Kilinc et al., 2009). Akumulasi fokal mitokondria mungkin akan menyebabkan
gangguan sitoskeleton fokal dan penumpukan substrat. Pada FAD, mitokondria
dirusak oleh oksigen dan nitrogen reaktif yang kemungkinan besar berasal dari
makrofag. Proses ini sendiri akan memacu degenerasi akson lebih lanjut (Nikic et
al., 2011).
Protein amiloidogenik, seperti
alpha-synuclein
,
tau
, dan
Aβ
diduga
berperan menyebabkan degerasi akson pada beberapa kelainan neurodegeneratif,
yaitu melalui hambatan pada mekanisme transport akson.Agregasi protein tidak
terjadi pada seluruh jenis degerasi akson, tetapi dapat menyebabkan gangguan
akson.Overekspresi
human wild-type alpha-synuclein
oleh lentivirus
menyebabkan agregasi dan degenerasi akson SSP (Decressac et al., 2011).
Agregasi protein amiloidogenik tidak dapat dianggap suatu proses yang berdiri
sndiri, karena penelitian telah membuktikan peningkatan konsentrasi kalsium
akan menyebabkan agregasi
alpha-synuclein
pada kultur sel (Nath et al., 2011).
Karena itu, kita dapat mengeluarkan hipotesis bahwa lesi akson dengan influks
kalsium dapat menyebabkan agregasi protein amyloid. Sebaliknya, adanya
alpha-synuclein
sendiri akan memengaruhi degenerasi akson akibat trauma. Tikus
transgrenik dengan over ekspresi
human alpha-synuclein (Thy1-
αSynWT)
akan
memiliki agregat
alpha-synuclein
pada akson saraf sciatic. Hewan ini akan
mengalami peningkatan degenerasi Wallerian setelah
axotomy
saraf sciatic.
Sementara itu, degenerasi yang terjadi setelah
axotomy
pada tikus tanpa
ekspresi
alpha synuclein (C57BL/6-Ola-hsd strain by Harlan B6)
,
degenerasi akson akan terjadi dalam kecepatan yang lebih lambat (Siebert
et al., 2010). Hasil penelitian ini cukup membingungkan karena
alpha-synuclein
selama ini dianggap sebagai kunci pada kelainan
neurodegeneratif sel saraf pusat.Meskipun demikian, penelitian ini
mekanisme kerusakan akson yang lebih luas, termasuk lesi akibat trauma.
Proses yang terjadi pada kelainan neurodegeneratif kemungkinan besar
merupakan suatu proses yang terpisah. Mekanisme kerja yang tepat belum
dipecahkan sepenuhnya, tetapi data yang ada mengarahkan kita pada
kemungkinan adanya suatu interaksi langsung dengan sitoskeleton, seperti
neurofilamen, tau, dan tubulin (Kanazawa et al., 2008) serta interaksi
dengan protein transport, seperti
dynein dan kinesin-1
(Utton et al., 2005).
i. Transport Akson
Jika kita berasumsi bahwa degenrasi akson pada trauma merupakan suatu
proses mekanis, gangguan transport akson sesudah trauma merupakan suatu hal
yang sangat mungkin terjadi. Karena akson memiliki hubungan yang kompleks
dengan inti sel, dibutuhkan suatu transport yang efektif pada tujuan, seperti sinaps
terminal atau
nodes of Ranvier.
Gangguan transport akson sudah terbukti terjadi
pada beberapa kelainan degeneratif, seperti
Parkinson, Alzheimer’s,
dan
Huntington
(Morfini et al., 2009).
Transport akson dimediasi oleh dua kelompok proten utama. Kelompok
pertama adalah kinein.Kinein berperan dalam memediasi transport
anterograde.Sementara itu, kelompok kedua adalah dynein, yang berperan dalam
transport retrograd. Tikus percobaan dengan mutasi gen KIF1Bβ, gen pengkode
kinesin menunjukkan gangguan vesikel sinaps dan kelemahan otot progresif
akibat neuropati perifer. Pada manusia, mutasi KIF1Bβ ditemukan pada penderita
Gangguan transport akibat mutasi kinesin light chain-1 juga akan mengaktivasi
stress kinase, seperti c-Jun-N terminal kinase. Ini akan menyebabkan fosforilasi
yang abnormal dan agregasi tau (Falzone et al., 2009). Gangguan transport
retrograd sendiri tidak memberikan efek klinis yang relevan.
Missense point
mutation
pada
dynein heavy-chain
menyebabkan degenerasi motoneuron pada
tikus
heterozygous
dan pembentukan
inclusion body
pada binatang
homozygous
(Hafezparast, 2003).Mutasi dynein juga mungkin dapat dihubungkan dengan
degenerasi akson pada penyakit motoneuron (Ravikumar et al., 2005).
Gangguan transport akson pada akhirnya akan menyebabkan gangguan,
mulai dari distrofi akson sampai degenerasi akson. Gangguan ini potensial
menjadi target terapi pada trauma dan kelainan neurodegenerative.Namun, proses
yang terjadi sangat kompleks dan tidak dapat ditangani hanya dengan satu
intervensi spesifik.
j. Aktivasi Kinase
Kinase berperan dalam eksekusi destruksi akson. JNK dikenal sebagai
protein kinase yang dipicu stress karena aktivitasya meningkat pada stress seluler
akibat lingkungan, seperti
osmotic stress, redox stress
, atau
irradiation
(Weston
dan Davis, 2007). Begitu diaktivasi, kinase akan mempropagasi sinyal yang
memacu apoptosis sel. Cedera juga dapat mengaktivasi JNK pada akson dan
menyebabkan gangguan transport akson. Activated pospho-JNK terdapat dalam
nantinya akan memperbaiki fungsional tikus (Yoshimura et al., 2011). Sebaliknya,
mutasi
kinesin-light-chain 1
terjadi akibat aktivasi JNK, bersamaan dengan
tau
protein
yang mengalami proses hiperfosforilasi pada akson yang mengalami
proses distrofi (Falzone et al., 2009).
k. Autofagi dan sistem ubiquitin-proteasome
Degradasi protein atau organela terjadi dengan berbagai jalur
degradasi.Salah satu di antaranya adalah autofagi.Setelah kerusakan akson
mekanis, dijumpai peningkatan autofagosom pada lesi yang bergantung influks
kalsium (Koch et al., 2010). Autofagi juga diinduksi oleh neurit simpatis yang
mengalami degenerasi, seperti setelah aksotomi.
Sistem ubiquitin-proteason juga terbukti berhubungan dengan degenerasi
akson. Pada binatang percobaan dengan cedera nervus optikus, inhibisi sistem
ubiquitin-proteasome akan menghambat degenerasi Wallerian. Fragmenasi
mikrotubuli yang terjadi segera setelah trauma dapat dikurangi dengan pemberian
proteasome inhibitor MG132
(Zhai et al., 2003).
B.
Sistem Ubiquitin Proteasome (SUP)
Sistem
Ubiquitin Proteasome
(SUP) merupakan jalur seluler utama pada
eukariotik yang mengontrol pergantian protein melalui
proteasome 26 S
. Banyak
proses dasar sel, seperti siklus sel, transduksi sinyal, apoptosis, dan pengontrolan
kualitas protein diatur oleh SUP (Cardozo dan Pagano, 2004). Induksi proses ini
penandaan protein yang menjadi target dengan molekul kecil, yang dikenal
dengan nama
ubiquitin
.
Ubiquitin
akan berikatan secara kovalen dengan residu
lisin pada protein yang menjadi target. Proses ini dikenal dengan nama
ubiquinisasi.
Ubiquinisasi merupakan proses bertahap yang memerlukan aktivitas tiga
kelas enzim, yaitu
ubiquitin-activating enzyme
(E1),
ubiquitin-conjugating
enzyme
(E2), dan
ubiquitin-protein ligase
(E3) (Pickart, 2004).
Ubiquitin-activating enzyme
bekerja dengan bergantung ATP dengan mengikat
active site
enzim ini (residu
cysteine
) pada residu
C-terminal glycine
pada
ubiquitin
, yang
pada akhirnya akan mengaktifkan
ubiquitin. Ubiquitin
yang sudah aktif kemudian
akan ditransfer pada E2, yang relatif sedikit terdapat pada eukariot, melalui suatu
reaksi esterifikasi. Akhirnya,
ubiquitin
ditempelkan pada residu
lysine
pada
protein target dengan ikatan isopeptida. Langkah ini memerlukan aktifitas salah
satu dari ratusan sistem E3.E3 bertindak sebagai pengenal substrat dan mampu
berinteraksi baik dengan E2 maupun dengan substrat protein. (Ardley dan
Robinson, 2005)
Ada dua jenis domain pada Ubiquitine – protein ligase (E3). Suatu E3
hanya akan memiliki salah satu di antara keduanya. Kategori pertama memiliki
domain zinc-binding RING (Really Interesting New Gene)
yang memacu
ubiquitinisasi dengan berikatan pada E2 dan substrat (6).Kategori kedua memiliki
selesai dengan sempurna, meninggalkan protein yang menjadi target hanya
berikatan dengan satu
ubiquitin
. Namun, ini akan sering berlanjut melalui
pengikatan molekul
ubiquitin
bebas lainnya pada residu
lysine
spesifik pada
ubiquitin terakhir, membentuk suatu rantai
ubiquitin
. Pada cara ini, protein target
akan berikatan dengan banyak
ubiquitin
dan beberapa protein memerlukan kerja
ubiquitin-chain elongation-factor
(E4) supaya proses ubiquitinisasi dapat berjalan
dengan efisien (Koegi et al., 1999).
Rantai
ubiquitin
bekerja sebagai penanda proses yang secara fungsional
berbeda, tidak hanya terbatas pada degradasi proteasom. Awalnya, protein dengan
banyak
ubiquitin
dianggap hanya memiliki target berupa proteasom 26S. Namun,
saat ini diketahui bahwa proses poliubiquitinisasi dapat terjadi dalam dua bentuk,
yaitu
homopolymeric
dan
heteropolymeric
. Pada bentuk
homopolymeric
, molekul
ubiquitin
berikatan satu sama lain menggunakan donor residu
lysine
yang sama.
Rantai
homopolymeric ubiquitin
akan memanjang melalui ikatan beberapa residu
lysine
yang terdapat pada molekul
ubiquitin
, yaitu Lys
48, Lys
63, Lys
6, Lys
11, Lys
29,
Lys
27and Lys
33. Setiap jenis ikatan ini akan menyebabkan kaskade sinyal yang
spesifik. Sebagai contoh, penambahan rantai yang berikatan dengan Lys
48pada
protein akan menyebabkan munculnya sinyal degradasi, sementara Lys
63akan
menyebabkan sinyal perbaikan DNA, endositosis, atau transduksi sinyal. Rantai
polyubiquitin heteropolymeric
ditandai dengan ikatan molekul
ubiquitin
dengan
lebih dari satu jenis pengikat (Koegi et al., 1999).Fungsi jenis ini belum diteliti
1. Enzim Deubiquitinating (DUB)
Ubiquitinisasi merupakan suatu proses yang reversibel. Terdapat
sekelompok enzim yang dapat melepaskan ikatan
ubiquitin
dengan protein yang
sudah mengalami ubiquitinisasi.Kelompok enzim ini dikenal dengan enzim
deubiquitinating
(DUB). DUB merupakan protease yang dapat menghidrolisis
ikatan isopeptida antara
ubiquitin
dengan substratnya melalui suatu proses yang
bergantung ATP (Yao dan Cohen, 2002). Hampir mirip dengan ubiquitinisasi,
proses deubiquitinisasi merupakan suatu proses yang sangat dikontrol dengan
ketat dan berperan dalam regulasi siklus sel (Song dan Rape, 2008), ekspresi gen
(Daniel dan Grant, 2007), degradasi protein bergantung
lysosome
dan
proteasome
(Schmidt, 2005),
DNA repair
(Kennedy dan D’Andrea, 2005), aktivasi kinase
(Komada, 2008), dan proses seluler lainnya. DUB memegang peranan pada jalur
ubiquitin. Pertama, DUB akan berperan sebagai antagonis dalam proses
ubiquitinisasi dengan melepaskan
ubiquitin
dari protein yang sudah mengalami
ubiquitinisasi pada suatu sel (Nijman, 2005). Kedua, DUB berperan dalam
aktivasi proprotein
ubiquitin
. Pada dasarnya, ubiquitin selalu diekspresikan
sebagai proprotein yang berikatan pada protein ribosomal atau diekspresikan
sebagai rantai poliubiquitin linear yang harus menjalani proses hidrolisis agar
monomeric
bebas dari rantai poliubiquitin yang tidak berikatan dalam sel
(Piotrowski, 1997). Kelompok ini juga mencakup poliubiquitin bebas yang
dihasilkan oleh enzim konjugasi yang sudah dilepaskan dari protein lain yang
sudah menjalani deubiquitinisasi).
Pada sel eukariot, DUB dibagi lagi menjadi lima kelompok berdasarkan
struktur dan fungsinya, yaitu
ubiquitin-specific protease (USP), ubiquitin
C-terminal hydrolase (UCH), Otubain protease (OTU), Machado-Joseph disease
protease (MUD),
dan suatu kelompok
metalloprotease
(Nijman et al., 2005).
2. Fungsi Sistem
Ubiquitin Proteasome
pada Neuron
SUP sudah dianggap menjadi protein kunci dalam proses modifikasi dan
degradasi yang penting dalam perkembangan, pemeliharaan, dan remodeling
koneksi sinaps pada otak (Kuo et al., 2006). Karena proses seluler dan fisiologi
yang melibatkan
ubiquiti
n sangat banyak, tidak mengejutkan ada banyak fungsi
saraf yang sangat bergantung pada SUP yang baik. Pada sistem saraf
Drosophila
,
SUP terlibat dalam pruning dendrit.
Pruning
merupakan suatu proses yang terjadi
saat metamorphosis Drosophila. Saat metamorphosis, sekelompok neuron sensori
larva tumbuh kembali membentuk suatu jaringan dendrit yang baru, Pada tahun
2006, sekelompok peneliti dari UCSF menunjukkan bahwa proses pruning
berhubungan dengan aktivitas enzin E2/E3 yang spesifik (Kuo et al., 2006).
Banyak penelitian menunjukkan bahwa UPS akan terlibat dalam
terbukti meningkatkan jumlah percabangan sinaps pada neuromuscular
junction.Ukuran sinaps juga diatur oleh SUP (van Roessel, 2004).
Modifikasi efikasi sinaps juga disertai dengan perubahan komposisi
protein pada sinaps (Mallnow, 2003).Perubahan komposisi protein ini dapat
terjadi dengan mengikutkan protein yang baru disintesis atau melalui degradasi
sebagian komponen protein terebut. Proses sintesis dan degradasi ini sangat diatur
oleh SUP.
Lebih lanjut lagi, aktivitas yang bergantung degradasi protein diperlukan
untuk plastisitas saraf pada hippocampus mammalia. Inhibisi proteasome dapat
memacu
long term potentiation (LTP)
dan
long term depression (LTD).
Keduanya
membentuk plastisitas sinaps pada
hippocampus
yang terlibat dalam pada proses
belajar dan memori. Inhibisi
proteasome
terbukti menghambat depresi sinaps
yang bergantung NMDA dan mGluR (Deng dan Lei, 2007).
3. Penyakit Neurodegeneratif dan SUP
SUP berperan pada banyak kelainan sistem saraf, terutama kelainan
neurodegenerative, seperti
Alzheimer’s
(Gong et al., 2006),
Parkinson’s
(Upadhya
dan Hedge, 2005),
Amylotrophic Lateral Sclerosis (ALS),
dan
Huntington’s
(Rubinsztein, 2006). Seluruh penyakit ini sekarang dianggap sebagai
“proteinopati” sistem saraf, yang ditandai dengan akumulasi
misfolded
protein
yang menumpuk dan tidak dapat didegradasi. Tumpukan
misfolded
protein ini
dapat berupa
plaque
dan
tangle (Alzheimer’s), Lewy bodies (Parkinson’s),
dan
sudah menjalani proses ubiquitinisasi ditemukan pada tumpukan protein ini. Ini
menggambarkan suatu kemungkinan bahwa protein ini sudah ditandai untuk
proses degradasi, tetapi proses degradasi itu sendiri belum berjalan dengan efisien.
Pada penderita
Dementia
dengan
Lewy bodies
dan penderita Parkinson, hanya
ditemui satu sampai tiga rantai
ubiquitin
, sementara proses pengenalan dan
degradasi yang sempurna terjadi bila ikatan
ubiquitin
berjumlah lebih dari empat
(Anderson et al., 2006). Aktivitas
proteasome
menurun pada penderita
Alzheimer’s
dan
Parkinson’s.
Karena itu, disfungsi SUP kemungkinan terjadi
karena peningkatan jumlah
misfolded
dan agregat protein serta ketidakmampuan
untuk mengenali dan mendegradasinya (Dahlmann, 2007).
Bagian penting pemahaman disfungsi SUP pada proses degenerasi saraf
adalah identifikasi gen yang terlibat, baik secara familial maupun sporadik. Ada
sepuluh lokus gen yang sudah diidentifikasi pada penderita
Parkinson’s
. Dua di
antaranya merupakan bagian dari SUP.Pertama, gen PARK2 mengkode parkin
(
E2-dependent E3 ligase
) (Shimura, 2000). Mutasi PARK2 menyebabkan
terjadinya
Parkinson’s
pada usia muda dengan hilangnya neuron dopaminergik
pada substansia Nigra tanpa adanya
Lewy Bodies
(van Coelln, 2004). Gen kedua
adalah PARK5 yang mengkode UCH-L1, sejenis DUB yang memiliki fungsi
hydrolase dan ligase
(Osaka et al., 2003). Mutasi PARK5 berhubungan dengan
4.
Ubiquitin Carboxyl-Terminal Hydrolase (UCH-L1)
dan Hubungannya dengan
Degenerasi Saraf
UCH-L1, juga dikenal dengan PGP9.5 merupakan ikatan 223 asam amino
yang memegang peranan penting pada SUP. Ini termasuk ke dalam kelompok
DUB. Ada lima jenis kelompok ini, yaitu UCH-L1 – UCH-L5. UCH-L1 hanya
selektif ditemui pada neuron dalam jumlah banyak, sekitar 1-2% di antara seluruh
soluble protein
otak (Wilkinson, 1989).Awalnya, UCH-L1 dianggap hanya
berperan sebagai DUB.Namun, beberapa penelitian terakhir menunjukkan bahwa
protein ini juga memegang peranan besar pada SUP. UCH-L1 diketahui akan
menciptakan
ubiquitin
bebas monomeric, baik dari bentuk prekursor (
ubiquitin
polypeptide
) maupun dari
ubiquitin
yang bergabung dengan protein ribosomal
(Larsen et al., 1998). Pada penelitian in vitro, UCH-L1 juga terbukti memiliki
aktifitas ubiquitin-ligase (Liu et al., 2002). Selain itu, UCH-L1 bersama dengan
ubiquitin dapat menghambat degradasi lisosomal, sehingga dapat
mempertahankan kadar
ubiquitin monomeric
pada neuron (Osaka et al., 2003).
Banyak penelitian membuktikan hubungan antara UCH-L1 dengan
kelainan neurodegeneratif, seperti
Alzheimer’s
dan
Parkinson’s
.Ini diawali
dengan penemuan mutasi gen UCH-L1 pada dua saudara di keluarga Jerman yang
menderita Parkinson terkait autosomal dominan (Leroy et al., 1998).Isoleusin
pada posisi 93 diganti dengan methionine.Produk gen yang dihasilkan dinamai
UCH-L1 193M. Hanya satu dari kedua alel UCH-L1 yang berubah pada mutasi
Namun, orang tua kedua anak tersebut tidak menderita Parkinson. Ini
menggambarkan kemungkinan mutasi 193M mungkin merupakan suatu
polimorfisme yang sangat jarang terjadi yang tidak berkaitan dengan suatu
kelainan atau mutasi pada orang tua kedua anak itu bersifar inkomplit (Liu et al.,
2002). Pada penelitian
in vitro,
mutasi ini menyebabkan penurunan fungsi katalis
sampai 50%. Hilangnya fungsi UCH-L1 akan mengurangi ketersediaan
ubiquitin
bebas dan menyebabkan gangguan degradasi protein melalui SUP. Lebih lagi,
tikus transgrenik dengan mutasi 193M mengalami penurunan neuron
dopaminergik pada substansia nigra yang signifikan (Setsuie et al., 2007).
Seperti yang disebutkan sebelumnya, UCH-L1 dipercaya memiliki
aktiivtas
ubiquitin ligase.
Pada percobaan invitro, aktivitas ligase ini bergantung
pada proses dimerisasi dan berperan dalam ubiquitinisasi
a-synuclein,
kemungkinan dengan ikatan rantai K63.
A-synuclein
merupakan protein yang
banyak terdapat pada ujung pre-sinaps dan terlibat dalam pelepasan
neurotransmiter (Kahle et al., 2000). Protein ini merupakan komponen utama dari
Lewy bodies
yang banyak ditemui pada penderita Parkinson. Pada tikus
percobaan, UCH-L1 berada dalam vesikel pada sinaps dan terlibat dalam
presipitasi
a-synuclein
. Ubiquitinisasi
a-synuclein
dengan UCH-L1 dipercaya
akan menghambat degradasi proteasome, sehingga menyebabkan penumpukan
Lewy bodies.
Selain itu, ada polimorfisme UCH-L1 lain yang tampaknya
mengurangi kemampuan ligase. Mutasi ini ditemui pada populasi tertentu di Asia
Timur, berupa perubahan
serine 18
menjadi
serine.
Mutasi ini dinamai UCH-L1
S18Y. Mutasi S18Y dari UCH-L1 menyebabkan penurunan kadar
a-synuclein
yang mengalami ubiquitinisasi dan berhubungan dengan penurunan resiko
terjadinya
Parkinson’s.
UCH-L1 juga terlibat dalam plastisitas sinaps pada tikus percobaan. Tikus
transgenic percobaan ini dinamai
gracile axonal dystrophy (gad
), berupa tikus
yang sudah mengalami delesi ekson 7 dan 8 pada gen Uch-l1. Ekspresi UCH-L1
pada susunan saraf tikus-tikus ini tidak dapat dideteksi. Tikus gad mengalami
paresis motorik akibat degenerasi aksonal dari neuron spinal cord dan traktus
spinoserebelar
.
Tikus gad ini juga menunjukkan penurunan kadar
ubiquitin
monomeric
(Gong et al., 2006).
C. Biomarker pada Cedera Kepala
Aplikasi penggunaan biomarker pada cedera kepala akan sangat
memberikan manfaat sebagai tambahan alat diagnosis berbagai macam cedera
kepala. Sebagai contoh, pelatih pada sepak bola dapat menggunakan biomarker
sebagai penentu keputusan yang objektif untuk penghentian permainan saat terjadi
sport concussion.
Biomarker yang tervalidasi akan merevolusi penatalaksanaan
dan diagnosis cedera kepala, bahkan sekaligus dapat membantu menilai prognosis
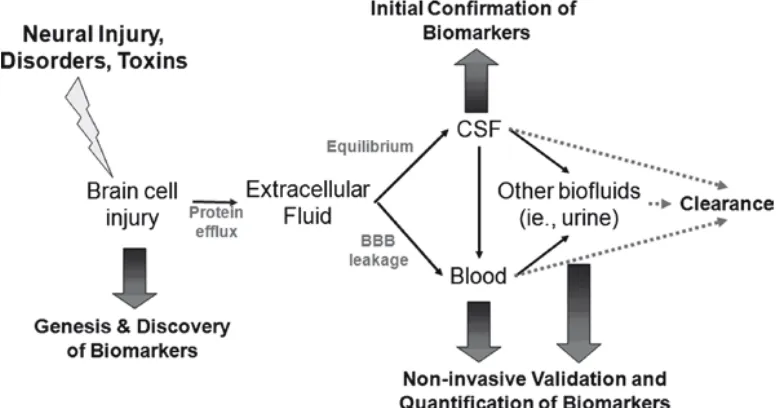
Gambar 2 menggambarkan jalur mulai terbentuknya biomarker cedera
kepala pada jaringan otak sampai deteksinya pada CSF dan darah.Selama cedera
otak, protein neural terlepas ke lingkungan ekstrasel, dan kemudian
CSF.Biomarker ini muncul dalam konsentrasi tinggi pada CSF. Protein-protein ini
kemudian akan mencapai aliran darah melalui sawar darah otak yang terganggu
atau melalui filtrasi CSF. Karena volume CSF manusia adalah sekitar 30-40 kali
lebih sedikit dibandingkan volume darah (CSF 125-150 mL, darah 4-5 L),
konsentrasi biomarker akan jauh lebih tinggi pada CSF dibandingkan darah.
Gambar 2. Terbentuknya biomarker setelah trauma dan penyebarannya pada darah (Kobeissy et al., 2008)
Sampai saat ini, kebanyakan penelitian biomarker cedera kepala
berfokus pada profil protein.Namun, genom manusia diperkirakan
mengandung 23.000 gen. Separuh di antaranya ada dalam jumlah sangat
sedikit.Akibatnya, mendata seluruh
proteasome
yang ada menjadi sangat
proses cedera kepala.
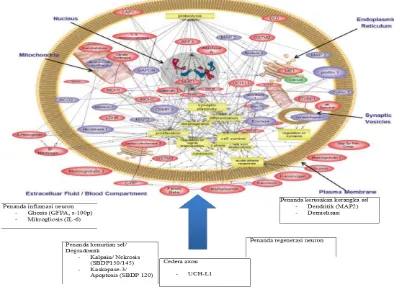
Sebuah
review
tahun 2008 oleh Kobeissy et al mencoba mendata
seluruh
proteasome
yang kemungkinan besar terlibat dalam cedera kepala
manusia (Gambar3 )
Gambar 3 Proses cedera kepala dengan protein yang potensial menjadi biomarker
(Kobeissy et al, 2008)
Penanda yang ada dapat dikelompokkan ke dalam beberapa
penanda inflamasi, metabolit, neurotransmitter, metabolit lipid, dan
biomarker secondary insult
(tabel 1)
Tabel 1
Penanda cedera kepala (Dash et al., 2010)
Biomarker Kegunaan Diagnosis Sumber sampel Keterangan
α II-Spectrin BDPs Meningkat pada cedera kepala, berhubungan dengan ukuran lesi dan keparahan cedera
CSF, jaringan otak, serum Banyak ditemui pada otak, meningkat pada kematian sel
α –Synuclean C-tau Meningkat pada cedera kepala, peningkatan C-tau berhubungan dengan perburukan hasil akhir dan peningkatan TIK
Jaringan otak, CSF, serum Spesifik pada SSP, memerlukan validasi lagi pada manusia, alat diagnosis yang jelek pada CKR
3’,5’cAM/2’,3’cAMP Kadar pada CSF berkorelasi dengan derajat koma
CSF Second messenger
Ceruloplasmin/Cu Menurun pada peningkatan TIK Serum CK-BB Gangguan blod brain barrier
mempermudah penetrasi serum, tidak berhubungan dengan volume kontusio
CSF, serum Waktu paruh pendek, dieliminasi dengan cepat
CRP & SAA Marker cedera secara umum Serum Diinduksi dengan cepat. Mengindikasikan politrauma
CRMP-2 Menurun pada penderita mesial temporal lobe epilepsy. Berhubungan dengan Alzheimer’s
Jaringan otak Mungkin dapat digunakan sebagai marker epilepsy post trauma
FABP Meningkat pada cedera kepala ringan
Serum/plasma, jaringna otak Akurasi tinggi untuk cedera kepala ringan F2-Isoprostane Meningkat pada penyakit
multipel
CSF, serum Penanda kerusakan oksidatif
4-HNE Meningkat pada jaringan otak penderita cedera kepala
Jaringan otak Marker peroksidasi lemak
5-HIAA Meningkat pada CSF, indikator trauma
CSF Marker gangguan
neurotransmisi GFAP Prediktif untuk peningkatan ICP,
penurunan MAP, penurunan CPP, dan GOS
CSF, serum Spesifik untuk SSP. Mungkin kurang sensitif pada cedera kepala ringan
HVA Meningkat pada CSF CSF Marker gangguan
neurotransmisi ICAM Meningkat pada gangguan sawar
darah otak
CSF, serum Penanda disfungsi neurovaskuler IL-1b Peningkatannya berhubungan
dengan hasil akhir yang buruk
CSF Muncul saat terjadi jaringan parut astroglia IL-6 Hasil yang meragukan.
Peningkatan IL-6 berhubungan dengan hasil akhir yang membaik
CSF, serum, dialisat intrakranial
Penanda cedera organ multipel
IL-8 Meningkat pada CSF penderita cedera kepala berat
CSF, serum Penanda inflamasi
mortalitas
Biomarker Kegunaan Diagnosis Sumber Sampel Keterangan
IL-12p70 Meningkat pada CSF penderita CKB
CSF Marker inflamasi
Laktat Berhubungan dengan keparahan cedera
CSF Marker gangguan
metabolism otak Magnesium Penurunan Mg dalam lima hari
pertama berhubungan dengan keparahan cedera
Serum
MBP Peningkatan MBP berhubungan dengan hasil akhir yang lebih jelek pada anak-anak
CSF, serum Penanda cedera white matter
MCP-1 Peningkatan pada otak tikus dalam empat jam pertama setelah trauma
Jaringan otak Belum diuji pada manusia
MIP-1a Meningkat pada CSF penderita CKB
CSF Penanda invasi sel inflamasi
Phospho-neurofilament
Meningkat pada penderita cedera kepala
Serum Perlu divalidasi manusia NSE Gangguan neuropsikologi pada
lesi intrakranial
CSF, Serum, jaringan otak Penanda small cell lung cancer, neuroendocrine bladder tumor, stroke, dan neuroblastoma NE Meningkat pada penderita dalam
keadaan koma dan penderita politrauma
Serum Penanda gangguan neurotransmisi
S100b Meingkat pada penderita cedera kepala ringan
CSF, Serum Tidak spesifik untuk cedera kepala. Dapat digunakan sebagai penanda gangguan Sawar darah otak TGF-b Meningkat pada cedera kepala,
tetap meningkat selama 3 minggu
CSF Pertumbuhan dan
diferensiasi sel, angiogenesis, fungsi imun, apoptosis TNF-a Meningkat pada penderita cedera
kepala berat.
CSF, serum Penanda inflamasi
UCH-L1 Meningkat pada CSF,
berhubungan dengan mortalitas, komplikasi, dan hasil akhir dalam 6 bulan pertama
CSF Marker yang banyak ditemukan pada otak