Synthesis of methanol from methane: Challenges and advances on the
multi-step (syngas) and one-multi-step routes (DMTM)
Article in Fuel Processing Technology · February 2016 DOI: 10.1016/j.fuproc.2016.01.023
CITATIONS
18
READS
3,947
1 author:
Some of the authors of this publication are also working on these related projects:
Ferric sulfate View project
UTILIZAÇÃO DOS CATALISADORES ZIRCÔNIA SULFATADA E ÓXIDO DE ZIRCÔNIA PARA TRANSESTERIFICAÇÃO DA TRIACETINA View project Marcio Jose da Silva
Universidade Federal de Viçosa (UFV)
71PUBLICATIONS 789CITATIONS
SEE PROFILE
Review
Synthesis of methanol from methane: Challenges and advances on the
multi-step (syngas) and one-step routes (DMTM)
Marcio Jose da Silva
Chemistry Department, Federal University of Viçosa, Viçosa, Minas Gerais 36570-000, Brazil
a b s t r a c t
a r t i c l e
i n f o
Article history:
Received 26 October 2015
Received in revised form 13 January 2016 Accepted 19 January 2016
Available online xxxx
To transform the methane oxidation to methanol in a selective, straight, economically attractive, and less energy-intense process is a goal pursued by the industry since its discovery. Methane is the main constituent of shale and natural gas while methanol is either a fuel as feedstock in the chemical industry. Thus, to develop a technology that combines an affordable raw material with a strategic product became pivotally important for the chemical industry. Currently, the industrial route for methanol production from methane is accomplished via a syngas pro-cess, passing by stream catalytic reform of products (i.e., CO and H2). This is a costly route due to its high-energy
consumption. Alternatively, methane partial oxidation to methanol (i.e. DMTM route) in a single step can consti-tute a more economically viable strategy. Toward achieving this goal, different approaches were proposed; none-theless, until now, any was industrially feasible. In this review, we paid particular attention to the methane direct oxidation processes to methanol carried out in a gas phase under homogeneous or heterogeneous conditions. In general, heterogeneous processes are solid-catalyzed in the gas phase, while homogeneous processes occur with-out a catalyst. We too assessed the advances achieved in the traditional rwith-oute to producing methanol from syngas, as well as recent developments of syngas production from methane.
© 2016 Elsevier B.V. All rights reserved.
Keywords:
4.3. DMTM route (i): free-catalyst process of methane direct partial oxidation to methanol by oxygen (i.e. homogeneous process) . . . 50
1. Introduction
1.1. Natural gas
Natural gas is a clean and effective energy source because its com-bustion generates fewer greenhouse gases than coal or petroleum liquid fuels. However, to store and transport natural gas from remote sites
up-holds a great challenge, making it difficult to make the prices of its
de-rivatives (i.e., chemicals or liquid fuels) competitive in relation to
those of fossil oil[1]. Nevertheless, the inevitable depletion of petroleum
reserves and the recent discovery of natural gas sites may make it more
economically attractive, and fulfill society's demand for alternative
en-ergy sources[2].
Different from shale gas, which contains other hydrocarbons, the natural gas comprises only methane as a major component. Currently, the most of the methane is consumed as fuel, resulting in greenhouse
gas (i.e., CO and CO2). Indeed, methane itself is too more deleterious
molecule to the atmosphere, and also contributes to the greenhouse ef-fect. Nonetheless, it can also provide clean fuels, such as hydrogen or methanol.
Methanol itself can be converted to gasoline and valuablefine
chemicals throughout catalytic processes (Fig. 1)[3]. Natural gas
con-sumption in this century has exponentially increased compared to
other energy sources (Fig. 2)[4]. Indeed, countries, where methane is
highly accessible consumed it as fuel for industrial and domestic heating, or electric power generation. Consequently, almost all methane
produced has become an energy source[4].
Rather than be burned as fuel, natural gas is potentially useful as an industrial feedstock. Although there are different approaches to using the methane present in the natural gas as a raw material, the most explored and economically viable is its transformation to
syn-gas (i.e., CO and H2). Industrially, methane has been upgraded to
syngas throughout multistep processes that use steam and/or oxygen (i.e., steam reforming, autothermic reforming, or partial
oxi-dation)[5].
As shown inFig. 1, syngas is a starting material to valuable chemicals
(i.e., dimethyl ether, formaldehyde, acetic acid) or liquid fuels (i.e., via
Fischer–Tropsch catalysis). Nevertheless, the most attractive routes
are those that involve a single step, such as oxidative coupling of meth-ane (i.e., OCM), homologation, aromatization, and methmeth-ane partial oxi-dation to methanol (i.e., DMTM). This latter constitutes the focus of this review.
In this work, we paid special attention to two processes of methanol
production from methane: thefirst, the syngas-based multistep
pro-cess; and the second, the single-step process, which directly transforms methane into methanol (i.e. DMTM route).
2. Syngas-based multistep process for the production of methanol
2.1. A brief overview of methane conversion to syngas
After exhaustive research, Green et al. grouped three main types of catalysts as the more effective for the conversion of methane to syngas: (1) supported nickel, cobalt, or iron catalysts, (2) supported noble metal
and (3) transition metal carbide catalysts[6,7]. However, herein we will
focus only on supported Ni catalysts.
The reforming of methane over Ni catalysts with steam, carbon diox-ide, or oxygen, separately, generates syngas at different stoichiometries
[8].Table 1displays these reactions and their enthalpy obtained at
1173 K[8].
Currently, the syngas route has been the only economically viable process for the conversion of methane to chemicals or high value-Fig. 1.Conversion route of natural gas to chemicals and fuels[3].
Fig. 2.Projection of world energy consumption by fuel type 2000 to 2020. Adapted from U.S. Energy Information Administration/International Energy Outlook, 2005[4].
Table 1
Process for syngas production from natural or shale gasa.
Process Reaction H2:CO molar
added liquid fuels; nevertheless, syngas production is unquestionably
the most expensive step of this production chain (Fig. 3)[8].
The reaction of methane present in the natural or shale gas with steam, oxygen, or a mixture of them, provides syngas at industrial
scale (Fig. 3). Lunsford et al. studied the activity of alumina-supported
nickel on conversion of methane to syngas in the temperature ranging
of 720 to 1173 K[9].
Industrially, in this method, steam reforming of methane over
Ni/−Al2O3catalyst heated to 1173 K occurs in a primary reactor. The
unreacted methane is then reformed with oxygen and steam in a
sec-ondary reactor, producing a mixture of CO and H2in equilibrium
(Fig. 3)[10].
During the next two steps, steam is then added to the syngas gener-ated under milder thermal treatment than earlier (ca. 673 K), over iron oxide or copper catalysts. Throughout these water gas shift stages, the
addition of steam may then adjust the H2:CO molar ratio to ones
required for the further use of syngas[10,11]. In general, the syngas
stoichiometry is handled toward its use as feedstock for diesel synthesis
via Fischer–Tropsch process (Fe2O3catalyst) or methanol synthesis
(Cu–ZnO/Al2O3catalyst)[11]. Although not shown, these reaction
con-ditions produce coke, resulting in catalyst deactivation. The use of Ni supported over rare earth oxides or alkaline metals minimizes this un-desirable reaction, and improves the lifetime of the catalyst restricting
the carbon deposit[12–15].
Alternatively, methane can be reformed with a steam and oxygen mixture heated to a high temperature (ca. 2273 K) without catalyst.
These conditions favor radical reaction commonly called as“
homoge-neous oxidation”(Fig. 4).
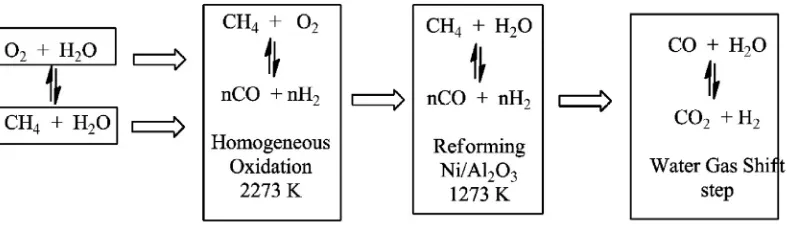
The following step occurs in another reactor, where the mixture ob-tained is then reformed over Ni catalysts resulting in syngas and water, which can be processed under water gas shift reaction conditions.
Significant advances in the use of promoters (i.e., B, La, and Rh) have
outstandingly improved the activity of Ni catalysts on the syngas
gener-ation process[16]. On the other hand, the replacing of alumina supports
by ceria, zirconia, and silica, all doped with rhodium nanoparticles,
sig-nificantly enhanced the process of syngas production through steam
methane reforming[17].
Several alternative routes have tried to circumvent the high costs as-sociated with large-scale methanol plants. The Topsøe Group developed
a technology that employs the concept known as“two-step reforming”
for syngas production (Fig. 5)[1]. This conventional process makes
methanol from syngas since the last century.
As depicted in the layout ofFig. 5, this traditional route includes
adi-abatic pre-reforming, tubular reforming, and oxygen-blown secondary reforming. In this process, oxygen is the source for internal combustion
of hydrocarbons. The two-step reforming process combines afired
tu-bular reforming and oxygen-fired adiabatic reforming allows obtaining
syngas at most suitable composition. On methanol synthesis process, the quantity of hydrogen and carbon oxides in the syngas should be properly balanced, to reduce the consumption per unit methanol pro-duction rate.
In two-step reforming process, the primary reforming produces ex-cess of hydrogen, while secondary reforming has use this exex-cess hydro-gen on step of hydrohydro-genation of CO to produce methanol. The
combination of the two types of reforming creates an energy efficient
creation of synthesis gas.
CO + 2 H2→CH3OH (−ΔH298 K,50 bar= 90.7 kJ mol−1)
On the other hand, the use of stand-alone autothermal reforming (ATR technology) together with a suitable selection of reaction condi-tions and suitable catalysts can produce methanol with high availability Fig. 3.Production of syngas from traditional methane steam reforming.
Adapted from Ref.[10].
conditions and viability of operation. The ATR technology operates at
low steam to carbon molar ratio, thus decreasing theflow through the
plant and diminishing the investment.
Stand-alone autothermal reforming (ATR) technology merges par-tial combustion and catalytic steam reforming in one compact refracto-ry lined reactor to yield syngas for methanol production in large scale.
ATR Technology has great advantageous such as avoidance of the supply
or dissipation of thermal energy to or from the reaction[18]. Arguably,
this characteristic makes ATR suitable to industrially provide syngas with high quality in large-scale plants.
Stand-alone autothermal reforming (ATR) technology comprises a
stand-alone reformer and oxygen-fired reformer. The autothermal
Fig. 5.Methanol process layout with Topsøe's two-step reforming. Adapted from Refs.[18, 19].
Fig. 6.Syngas production equipped with Topsøe's ATR stand-alone reforming. Adapted from Refs.18, 19].
reformer comprises a burner, a combustion zone, and a catalyst bed in a refractory lined pressure vessel. The burner mixes the feed and the oxidant. In the combustion zone, the feed and oxygen are burned.
The catalyst bed brings the steam reforming and shift conversion of reactions to equilibrium. By adjusting of low molar ratio of steam to carbon, ATR plants can run similar to a two-step reforming plant but with a single incoming stream.
Nowadays, ATR technology replaces“two-step reforming process”
by a Topsøe's optimized solution, aiming large-scale methanol
pro-duction. The route based on“stand-alone ATR technology”avoids
the use of a tubular reformer, required in the old two-step process (Fig. 6).
Topsøe's ATR consists of afixed bed reactor (i.e. Ni-based on ceramic
rings and pellets), where the reforming process takes place. Therefore, unlike the two step process, in a stand-alone ATR reforming process the natural gas is pre-reformed and straightly sent to an ATR reformer, where a mixer/burner burning all the hydrocarbons with oxygen and steam. In the combustion chamber, partial combustion reactions occur, followed by methane steam reforming reaction and shifting of equilibrium conversion over the catalyst bed. Because tubular reformer is not required, in a stand-alone reforming process the addition of
steam to the feed stream is drastically decreased[20]. Therefore,
the stand-alone ATR technology operates at low ratio molar steam to carbon.
However, though the syngas generated from stand-alone ATR reforming process is more reactive than that obtained from two-step
reforming, it is less reactive than syngas produced from gasification
route. For this reason, currently the most of industrial units produce
methanol from syngas obtained through gasification process. The low
reactivity is not the main challenge of manufacturing methanol from syngas obtained via stand-alone ATR. Indeed, the integration between the reforming units and the methanol synthesis is the greatest hamper. To overcome these drawbacks, we should considerer some points
[21];
I. The process will operate with syngas having a high CO/CO2molar
ratio;
II. The process is highly exothermal and should be operated at low temperature;
III. Side-product formation should be reduced;
IV. The energy releasing should be well employed;
V. It is required that reactants and residual aftermaths can be ade-quately disposed.
The main positivefigures of the stand-alone ATR reforming process
are the high rates of methanol reaction (i.e. due to syngas with high
CO/CO2proportion), the low steam throughput and low necessity of
steam that reduce investment cost and operation of large scale plants.
3. Oxidation of methane to methanol in a multistep processes
3.1. Brief background
Giulio Natta, who won the Nobel Prize in Chemistry in 1963, stated,
“among the main industrial organic reactions, the synthesis of methanol
is an outstanding example of practical importance of catalytic
process-es”[22]. Undeniably, until the half of the twenty century, most
metha-nol had a natural origin (i.e., waste woods distillation). Conversely, the petrochemical industry today is responsible by the entire methanol consumed.
Although its high toxicity or lower energy content than other liquid hydrocarbon fuels, methanol has employ as a pure liquid fuel or
gaso-line blend[3,23]. Moreover, it is also a feedstock to synthesize
commod-ities, such as methylterc-butyl ether (ca. 28%), formaldehyde (ca. 34%),
acetic acid (ca. 7%), and other chemicals, such as methyl acrylate or
sol-vents (ca. 31%)[24–27]. Moreover, MTO (i.e., methane to olefins) and
MTG (i.e., methane to gasoline) processes that produce light olefins or
gasoline, respectively, start from methanol[28,29].
Industrially, methanol has been produced throughout the syngas
route, which involves two consecutive steps: thefirst, in which
meth-ane is steam reforming over Ni/Al2O3catalysts resulting in syngas
(Fig. 4), and the second, in which the syngas is then converted to
meth-anol over Zn–CuO/Al2O3catalyst (see the next sections).
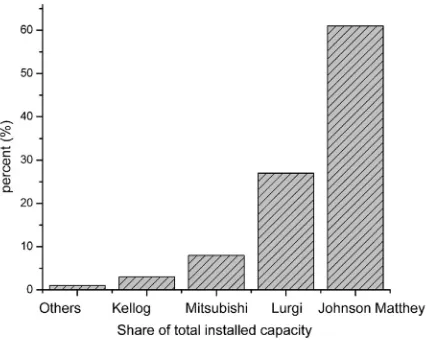
Currently, few industrial processes synthesize methanol (i.e., Jonhson
Matthey, Lurgi, Mitsubishi, or Kellogg processes;Fig. 5). Indeed, all of
them employ Zn–CuO/Al2O3catalyst, and operate under pressures of
50 to 100 atm over a range of temperature of 473 to 573 K. Although are highly intensive-energy routes, this catalytic process produces
meth-anol with 99% selectivity and energy efficiency higher than 70%[30].
Scheme 1.Equations of reactions to produce methanol and syngas. Adapted from Ref.[31].
3.2. Conventional production route of methanol from syngas: from BASF to Synetix process
Three equations describe the methanol production through the
syn-gas route (Eqs. (1)–(3);Scheme 1). Methanol synthesis (Eqs. (1) and
(2)) is an exothermic process that involves a decreasing number of gas-eous mol. Consequently, the reactions carried out at low temperatures
and high pressures reach the maximum conversion[31].
The BASF process (named as a“high pressure process”) convert
syn-gas to methanol over ZnO–Cr2O3catalysts, operating under high
pres-sures (ca. 250 to 350 bar) and in a temperature range of 573 to 673 K
[32]. This catalyst is highly tolerant to sulfur, a poison present in large
amounts in shale gas, which was a feedstock widely used in the begin-ning of the twentieth century.
Nonetheless, the BASF process employs drastic reaction conditions for methanol production. For this reason, several researchers intensively
worked to achieve milder reaction conditions. During thefirst thirty
years of the past century, copper oxide catalysts permitted a lowering of the temperature and pressure of the BASF process. However, copper catalysts possessed a major drawback, i.e., high sensitivity to poison by sulfur.
Imperial Chemical Industries, Ltd. (ICI) developed syngas purifi
ca-tion systems. They discovered that Cu–ZnO catalyst was much more
ac-tive than ZnO–Cr2O3, even though the former continued to be easily
poisoned by sulfur[33,34]. The development of effective purification
systems and active catalysts resulted in the process currently used,
which works over Cu–ZnO/Al2O3 catalyst to convert “metgas”
(i.e., syngas with an adequate molar ratio between CO and H2) to
meth-anol under pressure of 50 to 100 bar and temperatures in the range of
513 to 533 K (Scheme 2)[10].
3.3. Cu–Zn/Al2O3catalyst: Synetix process for methanol production
This process was thefirst route commercialized for methanol
production from syngas at low pressure, and was labeled as the“ICI
process”; however, it is currently named as the“Synetix process”[10].
Initially, it was accepted that the Cu(0) species constituted the active sites. Nonetheless, study showed that other phases also played impor-tant role in the activity and catalyst lifetime. Indeed, Nonneman and Ponec demonstrated that pure Cu is an inactive catalyst in methanol
synthesis[35]. They concluded that Cu(I) ions are formed throughout
the process and are stabilized by promoters (i.e., ZnO, CsCO3) on
the Cu(0) surface, which is supplied by adsorbed hydrogen atoms (i.e., Hads).
The role of zinc oxide on Cu–ZnO/Al2O3-catalyzed methanol
syn-thesis has been goal of study. Zinc oxide alkalinity may minimize the action of the acidic sites on the alumina phase, which would pro-mote methanol conversion to dimethyl ether, and under certain
conditions, hydrogen supply by spillover from ZnO to the CuO[35].
This could be highly desirable for methanol synthesis over Cu–ZnO
catalysts [36]. It is noteworthy that Cu–ZnO/Al2O3 catalyst is
sensitive to poisoning by chloride and mainly sulfur, and thus
re-quires pre-purification steps, mainly when the feedstock is the
shale gas.
From a mechanical perspective, the methanol synthesis over Cu–
ZnO/Al2O3via the syngas route has many issues that are currently the
subject of some controversy. For instance, identifying the nature of
ac-tive sites or the methanol precursor (i.e., CO or CO2) and the main
adsorbed intermediate species, as well as determining the reaction
pathway or the rate-determining step, are actively discussed topics[37].
Kinetic studies, besides investigations about the adsorption of isotopic-labeled species on the surface of different catalysts could be
useful to understand the methanol formation from the CO2and H2
adsorbed in the catalyst surface[38–40]. The combination of these
adsorbed molecules provides the adsorbed format (H2COOads)[38–40].
The most probable rate-limiting step is the hydrogenolysis of the
adsorbed format (i.e., H2COOads) to give methoxy species (i.e., CH3Oads)
that capture a proton, which then generates methanol[16].
On the other hand, determining how to increase the catalysts' toler-ance to the poisons (i.e., chloride or sulfur) or thermal sintering is still a great challenge. The poisoning and sintering of catalyst result in the de-activation and consequent stopping of the process. In this regard, during methanol synthesis, the catalyst undergoes deactivation even in the
ab-sence of a poison; catalyst activity drops to less than 30% after thefirst
thousand hours of work[41]. Studies revealed that even in the absence
of steam or carbon dioxide, Cu/ZnO catalyst was irreversibly deactivated
due to the reduction of CuO catalyst to Cu2O[42]. Therefore, increase the
catalyst lifetime is a key issue that determines economic feasibility of the process.
Although it is easily synthesized by alkaline co-precipitation of metal
nitrates (i.e., Cu, Zn, and Al), it is difficult to reproduce the industrially
used catalyst (i.e. Cu/ZnO). It complicates evaluating of catalyst stability at the laboratory scale. In addition, there is another experimental prob-lem. The heating to 450 to 510 K activates catalyst oxides under a
reduc-ing mixture diluted with inert gas (i.e., natural gas and N2). This mixture
commonly contains hydrogen, generated during the manufacture of syngas. Furthermore, the presence of hydrogen reduces only CuO
oxide to metallic copper, whereas to ZnO or Al2O3that remains as
oxides[27].
The catalyst activity depends on the high copper surface area or on the small crystallite size, which are responsible by the contact between
copper and the promoter zinc oxide on the alumina support[43].
Al-though the true role of zinc and copper oxides in the mechanism of the methanol synthesis remains unclear, Spencer et al. proposed that only when adsorbed hydrogen concentration on copper phase is low,
the hydrogen dissociation on zinc oxide is important[36,44].
The tolerance to the poisoning and the thermal stability are features that affect the activity and lifetime of catalysts. Spencer et al. reported
that when the methanol is synthesized over Cu–ZnO/Al2O3catalysts
containing promoters, the formation of coking or the poisoning of cata-lyst are minimized; therefore, only thermal sintering provokes catacata-lyst
deactivation[45].
Scheme 3.Methane steam reforming coupled tometgassynthesis. Adapted from Ref.[46].
An adequate adjustment of the syngas stoichiometry to generate metgasconstitutes an important stage throughout the conventional pro-duction process. While syngas is produced by steam methane reforming
at a CO:H2molar ratio equal to 3:1, methanol synthesis requires a
2:1 molar proportion between CO and H2(seeScheme 3)[46].
Themetgashas been manufactured from methane after an additional step, in which the homogeneous methane partial oxidation with oxygen (i.e., POX) and metal-catalyzed methane steam reforming (i.e., SR) are
consecutively performed (Scheme 2, step III). This extra step implies
adding additional costs to the process[46].
3.4. Main challenges of the conventional production process of methanol from syngas
Scheme 4presents a block diagram of the conventional production
process of methanol from syngas over Cu–ZnO/Al2O3catalyst[47].
First, it is important to highlight that although methanol synthesis from syngas is exothermic, the overall enthalpy of the reactions 1 to 3
(Eqs. (1)–(3),Scheme 5) is not favorable in terms of feasibility of
meth-anol synthesis from syngas under standard conditions[48].
Replacing steam by CO2 could minimize process costs because
syngas is provided at the equimolar amount of CO and H2(Eq. (4),
Scheme 5). However, the enthalpy variation of this reaction is higher
than that of steam methane reforming (Eq. (1),Scheme 5). Really, this
process is inefficient and requires high temperatures (ca. as high as
1000 K), which promote CO disproportionality and result in the unde-sirable formation of coke. All these aspects become the replacement of
steam by CO2economically infeasible[48,49].
Despite large capital investment to build industrial plants, the syn-gas route constitutes the most important process to convert methane
to clean fuels, such as methanol[49]. Nevertheless, it presents some
serious drawbacks, such as high consumption of energy and requiring expensive infrastructure. Moreover, the large amount of steam needed
results in the reactors' corrosion and difficulty with handling. For
these reasons, the price of liquid methanol (i.e., produced via the syngas route) compared to petroleum derivative liquid fuels tends not to be
competitive[30]. Indeed, the syngas process is responsible for 60 to
70% of methanol production costs[50].
4. Oxidation of methane to methanol in a one-step process
The straight conversion of methane present in natural gas into meth-anol is a process that could avoid the highly energy-dependent conven-tional route, reducing the number of stages, and thus avoiding the large capital investment required to build a syngas industrial plant. This route may constitute an economically viable technology and a more environ-mentally benign process than methanol production via the syngas path. Undeniably, such development could be an achievement that could
notably expand the production of methanol-derivatives and influence
the planet's economy[48]. However, until today, these processes are
far from being considered as established or industrially practical due to numerous reasons.
Extensive research has been devoted to the direct oxidation of meth-ane to methanol, which comprises the following technologies cited in
items (i) to (v)[51].
(i) Free-catalyst homogeneous processes at high temperatures based on radical reactions in the gas phase;
(ii) Solid-catalyzed processes in the gas phase; (iii) Solid-catalyzed processes in the liquid phase;
(iv) Homogeneous catalytic processes in the liquid phase, in the presence of soluble catalysts;
(v) Enzymatic catalytic processes. Scheme 4.Block diagram of the conventional production process of methanol. Adapted from Ref.[47].
Throughout this review, we will attempt to pay special attention to
methane partial oxidation to methanol in“free-catalyst processes”
(i.e., homogeneous processes based on radical chain reactions in the
gas phase), and“catalytic processes”(i.e., activation of C–H bonds by
bioinorganic complexes, solid metal catalyzed-methane partial oxida-tion in the gas phase).
4.1. Main challenges of direct methane partial direct oxidation to methanol routes (DMTM routes)
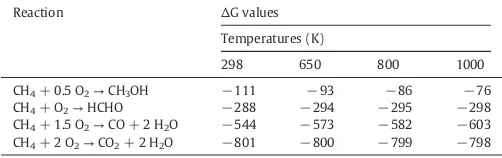
The reaction is thermodynamically spontaneous even at room
tem-perature (Scheme 4, Eq. (3)). However, since methanol is less stable
than the over oxidation products, to develop a direct route to make methanol from methane under room conditions has been referred to
as the“holy grail”due to its immense difficulty[52](Table 2).
Methane reactivity is lower than methanol due to its stronger C–H
bonds. The dissociation energy of the C–H bond of methanol is
393 kJ mol−1; whereas of methane, where it is 440 kJ mol−1. Therefore,
it is easier to oxidize methanol to stable products of over oxidation
(i.e., CO or CO2) than to oxidize methane itself. It becomes difficult to
control the selectivity of the one-step process of methane oxidation to
methanol (Table 2). Thus, because the methanol yield is always low,
the direct conversion route of methane to methanol is commercially
in-feasible[53].Table 2presents the effect of temperature on the straight
oxidation of methane into methanol[48].
Thermodynamically, it is possible to oxidize methane to methanol at
room temperature[48]. Indeed, the highest conversion theoretically
achieved when the equilibrium is reached at room temperature
(i.e., 298 K) is near 33%, calculated based on Gibbs free energy[43].
Con-versely, the best selectivity was obtained after maximum conversion is near 5%. This constitutes a greatest challenge because the carbon yield of the conventional syngas process is quite superior to this value (ca.
70 to 75%)[54].
Deserves highlight that utilizing of molecular oxygen as oxidant is a key aspect to do competitive any commercial DMTM (i.e., direct meth-ane to methanol) process. Molecular oxygen is an inexpensive, environ-mentally benign, and highly affordable reactant, which are essential features for large-scale methane conversion to liquid fuels or chemicals
[55]. However, because the molecules of methanol and methane
possess singlet ground states, their oxidation by oxygen is a spin-forbidden reaction, and thus requires an adequate catalyst to
activate them[52].
In this regard, a solid catalyst could be an attractive option. Nonethe-less, methanol has a dipole moment higher than methane. Therefore, it is preferentially activated over solid metal catalyst surface and is totally oxidized, compromising thus the reaction yield. In addition, this strong adsorption implies in additional recovery steps, where a polar solvent to extract the adsorbed methanol.
4.2. Activation of C–H bonds by enzymatic complexes
Nature has a notable ability in to do that the chemists pursue since long time. For these reasons, an extensively work has been done aiming
improve our understanding about how replicate this reaction at indus-trial scale[56].
Methanotrophic bacteria have two methane monooxygenase (MMO) enzymes that convert methane to methanol at room tempera-ture; soluble MMO (sMMO) and particulate MMO (pMMO) during the
first step of metabolic cycle (Eq.(5))[57].
CH4þO2 NADHþHþ→
MMO
CH3OHþH2O NADþ ð5Þ
Literature describes that pMMO is a metalloenzyme that con-tains multi-copper active sites that are found in the plasma of all methanotrophic bacteria, responsible by its activity. Several au-thors have proposed that active sites might contain copper at a wide range of stoichiometries (i.e., two or three mono- or divalent
copper ions)[58–60]. Indeed, the level of copper ions is
determin-ing to decide which enzyme will be expressed; if low concentration of copper cations is feasible, the enzyme expressed is sMMO; whereas, at high copper concentrations the pMMO enzyme is the
active site[61,62].
Conversely, only the action mechanism of sMMO is well known. A large number of spectroscopy study show that the sMMO enzyme
con-tains a carboxylate-bridged diiron unit[61–63]. The atmosphere oxygen
is the oxidant used by both enzymes to oxidize methane to methanol.
Kinetic parameters suggest that pMMO is more specific for methane
oxidation than sMMO enzyme.
Nowadays, there is a growing interest on the pMMO-mediated oxi-dation reactions which involves the reactivity of copper oxygenase en-zymes. Particularly, the developing of catalysts for oxidation of methane to methanol at industrial scale can be improved by under-standing how pMMO enzyme works. Consequently, several biomimetic studies have been carried out. The protein comprises three distinct sub-units (i.e., PmoA, PmoB, and PmoC).
On this sense, Chan et al. proposed that the catalytic site of pMMO enzyme might be a trinuclear copper cluster, and developed a series of tricopper complexes that are capable of catalytic oxidize the methane
to methanol under ambient conditions[64,65]. In according with their
experiments, those authors reported that two tricopper complexes con-verted methane to methanol: a tricopper-peptide complex derived
from pMMO and a biomimetic model tricopper complex[66].
Chan et al. assessed the oxidation of methane to methanol
medi-ated by the tricopper complex [Cu1 +–Cu1 +–Cu1 +(7-N-Etppz)]1 +
in acetonitrile solutions. The ligand (7-N-Etppz) correspond to the
3,3′
-(1,4-diazepane-1,4-diyl)bis[1-(4-ethyl[piperazine-1-yl)propan-2-ol] is showed inFig. 9, besides catalytic cycle proposed by those authors
[64–66].
Chan et al. reported that a turnover number equal to 0.92 was achieved when tricopper complex was activated by oxygen molecular in the presence of methane excess. Nonetheless, the process becomes catalytic by addition of hydrogen peroxide, which recovery the spent catalyst, after tricopper complex transfer oxygen atom to the methane (Fig. 10).
Chan et al. pointed that because tricopper-peptide complex is insol-uble in aqueous buffer, it was encapsulated in mesoporous carbon. XANES and EPR spectra demonstrated that copper(I) cations on the tricopper peptide were quickly reoxidized by air or oxygen, similarly to the observed on putative tricopper cluster in pMMO. Therefore, it
was clearly established that Cu1 +–Cu1 +–Cu1 +–peptide complex is
able to mediate transfer of oxygen atom from molecular oxygen to
methane at room temperature[64–66]. This biomimetic approach
allows formulating either homogeneous or heterogeneous catalysts to
oxidize methane to methanol[67].
Inspired by thesefindings, a number of researchers have tried to
build active sites similar to those of sMMO or pMMO enzymes into
cavity of zeolites. In the near brief future, Cu–ZSM-5 zeolite could
poten-tially serve as a model for the building of selective active sites to Table 2
Temperature effects on Gibbs free energy for products oxidation of methanea.
Reaction ΔG values
understand the working mechanisms of enzymes[68]. For instance,
re-actions carried out over Cu–ZSM-5 zeolite achieved high methanol
se-lectivity (ca. 98%)[69].
Another highlighted instance was recently described by Lercher et al., which demonstrated that single-site trinuclear copper oxygen
clusters in mordenite was an efficient catalyst for selective conversion
of methane to methanol[70]. Copper-exchanged mordenite zeolite
mimics both nuclearity and reactivity of pMMO enzyme active sites.
Lercher et al. verified that multiple Al framework atoms of the
mordenite microporous stabilize trinuclear copper-oxo clusters (i.e.
[Cu3(μ-O)3]2 +), which are able to activate carbon–hydrogen bond in
the methane, and its conversion to methanol. They demonstrated that reversible rearrangements of the trinuclear clusters occurring during the selective transformations of methane to methanol in both
enzymat-ic and copper-exchanged mordenite catalysts[70]. These results
corrob-orate with those reported by Chan et al.[64–66].
4.3. DMTM route (i): free-catalyst process of methane direct partial oxida-tion to methanol by oxygen (i.e. homogeneous process)
The relevance of homogeneous processes for methane oxidation through the radical chain in the gas phase is undeniable. The radical re-actions can occur as a consequence only of working temperature. Hence, depending on the temperature, its contribution to the reaction yield
even when solid catalysts are present could be significant.
In the absence of catalysts, methane oxidation to methanol can also occur over the reactor wall, in the gas phase, or in both. Since the ener-gies involved are quite close, distinguishing between the two ways
be-comes difficult. After an initiation step, a series of radical reactions
govern entirely the reaction course, which comprises of propagation and termination steps (or removal of active species).
In general, the main topics of interest to research in thefield of
ho-mogenous oxidation in the gas phase are as follows: (i) optimization of high-pressure conditions (ca. higher than 10 bar), aiming an ade-quate conversion; (ii) effect of additives; (iii) modeling mechanistic studies; (iv) design of novel reactors and (v) use of photochemical ini-tiators[69].
The methane partial oxidation to methanol under free-catalyst con-ditions involves more than 1000 elementary reaction steps with
numer-ous reactive species[69,71]. Verma et al. developed a complete kinetic
model for these reactions in the gas phase at a temperature range of
308 to 973 K[72].
Those authors show that the rate-limiting step of the partial
oxida-tion of methane is the abstracoxida-tion of thefirst hydrogen atom to form
methyl radical. Consequently, photochemical initiators could decrease
the activation energy of this step, favoring the overall reaction[72].
The data ofScheme 6reveal that while a great amount of energy is
needed to activate the methane, the succeeding reaction also produces other larger quantities. Clearly, since further steps are highly
exother-mic, it is difficult to stop the reaction where methanol was the only
product. Any attempt to drive the reaction toward high conversions leads to poor selectivity. The energy gap between reactions suggests that it is probable to obtain methanol and formaldehyde individually, al-though from an experimental perspective, this requires a strict reaction control. Therefore, it becomes imperative to adjust the reaction
equilibrium controlling variables such as temperature and reactant pressure, aiming shift selectivity toward methanol. High methane pres-sure can prevent deep oxidation if adequate temperatures and prespres-sure
are employed[72–74].
Zhang et al. described that other process control parameters,
includ-ing reactor type, temperature, feed oxygen concentration, and gasflow
also affect the efficiency of the catalyst-free gas phase reactions[75].
Those authors verified that pressure exerts a pronounced effect on
methanol selectivity. Under methane pressure equal to 5.0 MPa and heating to a temperature of 703 or 743 K, reactions gave 30 and 40% se-lectivity at conversions of 5 and 10%, respectively. They inhibited the
production of CO2, insulating the ringed gap between the inner quartz
line and the reactor tube. Indeed, they realized that both reactants and products must be isolated from contacting the metal wall of the reactor; for this reason, the best result was obtained when the methane partial oxidation to methanol is done in quartz and Pyrex glass-lined reactors
[47,75].
There is experimental and theoretical evidence that demonstrates that it is impossible to achieve high methanol yields in these systems, because an increase on the conversion always produces a decrease on
the selectivity [76–78]. Consequently, the methanol production
throughout homogeneous oxidation of methane will be viable at
indus-trial scale only if the price of raw material (natural gas) is sufficiently
in-expensive to compensate the high investment cost. Arutyunov et al. assessed the technological prospects and applications of the direct con-version of methane to methanol via free-catalyst reactions in the gas phase and concluded that these technologies can be useful if employed at least for low-scale and local applications, mainly if natural gas is abundant and located in remote sites, where storage and transportation
challenges exist[79].
4.3.1. The use of other oxidants in catalyst-free process of methane direct partial oxidation to methanol
Methane oxidation to methanol with nitrogen oxides has been
widely described in the literature[79,80]. While in the absence of NO,
DMTM gave only 1% methane conversion at 966 K and room pressure, the addition of NO (ca. 0.5%) led to a remarkable increase in methane
conversion to 10%, even at 808 K. The selectivity to C1-oxygenates was
high, but methanol, methyl nitrite, and formaldehyde were always in relation near 1:0.5:1. The optimum pressure for methanol production
was 1.0 MPa at 4% conversion of CH4in gas phase reactions with
0.50% NO, which gave 30% methanol selectivity and virtually no
formal-dehyde[79,80]. However, the presence of NO2promoted an increase of
Scheme 6.Enthalpy variation of the reactions involved in methane and methanol oxidation at 298 K[72].
Scheme 7.Process scheme for the net oxidation of methane to methanol (X = OSO3H).
CO selectivity, caused by over oxidation of methane, methanol, and mainly formaldehyde. However, the applicability of this process com-pared to the process that employs only oxygen is considerably limited. Periana et al. described a high-yield system for the low-temperature conversion of methane to methanol based on Pt(II)-bipym catalyst (i.e., bpym = bipyrimidine ligand) that operate in liquid sulfuric acid, which act as oxidant. A positive aspect of this system is stability of
meth-anol formed to suffer overoxidation in this reaction medium[81].
Con-versely, the major shortcoming of this solvent system is the difficulty
of separating the methanol from the sulfuric acid. Periana et al. have
found that platinum(II) catalysts were more efficient than Hg(II)
cata-lysts previously described by them, and oxidize C–H bond of methane
at temperatures as low as 100 °C. Those authors showed that bipyrimidine ligand stabilize the reduced species of platinum avoiding its deactivation.
The positive features of this system are facile hydrolysis of the
meth-yl bisulfate, the use of recyclable oxidant (i.e., SO2is easily oxidized to
SO3), and the use of stable complex catalyst (Scheme 7).
4.4. DMTM route (ii): solid-catalyzed processes of direct oxidation of meth-ane to methanol
It is clear that, in terms of competitiveness, any route of direct oxida-tion of methane to methanol (DMTM route) based on a solid-catalyzed process poses a major challenge to operating at lower costs without a loss of selectivity. Additionally, it is desirable that the solid catalysts to be used in solid-catalyzed DMTM reactions carried out in the gas phase should be active under milder reaction conditions than those
used in the free-catalyst DMTM processes[82].
Compared to the OCM process, which is another solid-catalyzed pro-cess for the direct conversion of methane in chemicals (i.e., ethane and ethylene), the DMTM process has been relatively less explored. The mo-tive for examining DMTM is that, unlike the OCM reactions that are fea-sible only in the gas phase, DMTM route via solid-catalyzed process
competes with other DMTM routes that may also performed in liquid phase (i.e., solid-catalyzed DMTM reactions in the liquid phase or even
with soluble catalysts). We summarized the mainfindings by grouping
the solid catalysts in accordance with the metal employed.
4.4.1. Copper–zinc/alumina catalysts
The high selectivity of Cu–ZnO/Al2O3catalyst when used in
metha-nol production via the syngas route also motivated its use in the DMTM reactions. Tabata et al. investigated the optimization of methanol
yield achieved via DMTM reactions with O2and NO over Cu–ZnO/Al2O3
catalysts [82,83]. They demonstrated that although the reactions
reached conversions of close to 5.5% with or without catalyst (Table 3), in the presence of Cu-ZnO/Al2O3, the selectivity and methanol
yield increased.
The best result was obtained under 4 bar of pressure and CH4/O2=
8.0 (ca. 2.3% methanol yield). Previously, those authors suggested that
the formation of CH3OH in reactions over Cu–ZnO/Al2O3-catalysts
occur through the hydrogenation of HCHO, and the water gas shift
reac-tion (WGSR)[84].
Actually, in a subsequent work, Tabata et al. suggested that three steps: (i) formaldehyde hydrogenation, (ii) formaldehyde dissociation, and (iii) the water gas shift reaction, simultaneously occur in methane
oxidation to methanol over Cu–ZnO/SiO2catalyst, when CH4–O2–NO
feed is used[85]. Moreover, they verified that when heating the Cu–
ZnO/SiO2catalyst bed to 423 to 623 K, the methanol selectivity almost
did not change during the reactions, unlike that observed in reactions
over alumina as support. They suggested that this indicates that SiO2
was a more favorable support to enhance methanol selectivity than
alu-mina at these temperatures[86].
4.4.2. Zeolite catalysts
Currently, studying the support effect, as well as the role of Cu
cata-lyst on DMTM reactions, has assumed great significance due to the
dis-covery of a selective route to oxidize methane to methanol. Groothaert Table 3
Effects of reaction pressure on product selectivity in the presence or absence of Cu–ZnO/Al2O3catalysta.
Adapted from Ref.[82].
aCatalyst, Cu:Zn:Al = 39.7:40.8:19.5;flow rate 120 cm3; reaction time = 180 min; catalyst bed temperature 523 K; feed gas composition = CH
4(77.5%); O2(5.8%); NO (0.5%); Ar
(16.2%).
Table 4
Methane partial oxidation with nitrous oxide over Cu–Fe-ZSM-5 catalystsa.
Adapted from Ref.[97].
Exp. CH4:N2O molar ratio Space velocity × 10−4(h−1) Reaction temperature (K) CH4conversion (%) Product selectivityb(%C)
CO2 CO CH3OH HCHO Olef.
aData taken after 60 min on stream.
b Olef. = mixture of C
2and C3olefins; n.a. = not available.
et al. were thefirst to report the selective oxidation of methane over Cu–
ZSM-5 catalyst (i.e., Zeolite Socony Mobil-5, an aluminosilicate zeolite
produced by Mobil Oil Company)[87–89]. They discovered that after
activating the dried Cu–ZSM-5 zeolite with oxygen or nitrous oxide
(ca. 448 K or room temperature, respectively), andflow methane over
the catalyst, methanol was then selectively formed.
Even though the reaction is not a truly catalytic process (i.e., it is a stoichiometric reaction, the catalyst does not turn over), it has attracted great attention because it was highly selective when working at a mild temperature range (ca. 373 to 473 K), a possible avenue to reach the
previously mentioned“Holy Grail”[52]. In fact, recent studies
demon-strated that methanol formation was directly linked to the in situ forma-tion of tricopper species on the active sites on the cavity of ZSM-5
zeolite. In situ ultraviolet–visible spectroscopy studies allowed
monitor-ing the methane oxidation progress over pre-activated Cu–ZSM-5
zeo-lite (i.e., with O2or N2O)[90]. Throughout the reaction, a decrease of
the band intensity at a wave number close to 22,700 cm−1was
un-equivocally attributed to the copper (II)μ-dioxide active site, which
si-multaneously occurred with an increase in methanol concentration.
Nevertheless, to carry this process over Cu–ZSM-5 zeolite displays
great challenges. First, removing the methanol from the zeolite requires an additional step of solvent extraction, lowering the methanol yield. Moreover, the reaction is not genuinely catalytic; there is a stoichiomet-ric characteristic of the reaction between the methane and the copper of
the active sites of the zeolite activate cavity[90].
Different approaches such as try convert methane to methanol over
Cu–ZSM-5 zeolites even under non-catalytic conditions have
consid-ered. However, as far we know, they have failed[91]. In fact, to
reacti-vate the Cu+sites on the zeolite cavity, the methanol must
first be
removed[92]. Thus, a possible catalytic cycle should consist of three
Recently, a breakthrough in the chemistry of copper zeolites was
de-scribed; the ability of another Cu zeolite (i.e., named as Cu–MOR) to
convert methane to methanol at 473 K through a multistep process
[95]. In that process, the Cu–MOR catalyst is pre-oxidized, and then
after further hydration and methanol recovery, the catalyst is then reactivated at the reaction end. These results suggest that this zeolite
is also able to produce the tricopper oxide active sites[96]. Although
opening a new possibility to oxidize methane to methanol through straight reactions, this work did not achieve a complete catalytic cycle. The success of copper zeolites on methane oxidation reactions to methanol has motivated assessment of the activity of other zeolite
cat-alysts. Indeed, even before thefirst data related methane oxidation to
methanol in the presence of Cu–ZSM-5 at room temperature, Anderson
and Tsai and Anderson described methane oxidation by nitrous oxide
over Fe-ZSM-5 catalysts, synthesized by the exchange of Al3+cations
in the lattice of the Cu–ZSM-5 zeolite by Fe3+cations[97]. Those
au-thors devoted most of their attention to assessing the activity of Cu–
FeZSM-5 on methane oxidation by nitrous oxide at a temperature
range of 500 to 620 K.Table 4summarizes the main results.
Over H–FeZSM-5, the carbon oxides were the major products (not
showed herein). Moreover, over Cu–FeZSM-5, the reaction was highly
selective to methanol, reaching nearly 78% under favorable conditions
(entry 3,Table 4). Those authors attributed the exceptional behavior
of Cu–Fe-ZSM-5 zeolite to probable synergism between the Cu2+and
Fe3+cations[97].
Michalkiewicz performed the DMTM reactions over Fe-ZSM-5 cata-lysts, under room pressure and at temperatures between 623 and 923 K
[98]. He assessed the effects of acidic hydrogen replacement by sodium
and variations of the Si/Fe molar ratio. The best result in terms of
selec-tivity was achieved in the Fe–NaZSM-5-catalyzed reactions, which
re-sulted in 74% methanol selectivity when the Si:Fe molar proportion
was 45:1 (Table 5). However, these reactions achieved only poor
con-versions (ca. 0.06 to 0.10%).
Conversely, high conversions were reached in the presence of
cata-lyst Fe-HZSM-5 (ca. 31.51% for Si/Fe ratio = 22:1) (Table 6).
Neverthe-less, it resulted in poor methanol selectivity for methanol. Those authors concluded that increases of temperature and contact time favored the formation over oxidation products, as suggested by the increase of
se-lectivity to HCHO and CO2.
Panov et al. assessed the methane partial oxidation by nitrous oxide
in the processes in which theα-oxygen sites were created at room
tem-perature or 433 K temtem-perature over FeZSM-5 zeolite[99,100]. In the
first study, they increased the amount ofα-sites to 100μmol/g, and
in-vestigated the methane oxidation byα-oxygen pre-deposited from
ni-trous oxide at room temperature. They proposed that the following
reactions occur over Fe–HZSM-5 or Fe–Na-ZSM-5 catalyst (Scheme 8)
[99]:
3 22 0.10 63.47 14.84 21.68
aReaction conditions: temperature (623 K); contact time (0.5 s); oxygen (15.5 vol.%).
b Temperature (663 K).
Table 6
Methane partial oxidation with oxygen over various Fe–HZSM-5 catalystsa.
Adapted from Ref.[98].
Exp. Si:Fe molar ratio CH4conversion (%) Product selectivity (%)
CH3OH HCHO CO2
1 45 11.22 16.51 25.48 58.01
2 34 19.11 14.17 18.79 67.03
3 22 31.51 10.79 17.06 72.14
aReaction conditions: temperature (903 K); contact time (2.5 s); oxygen (15.5 vol.%).
They discovered that the reactions occur through a hydrogen ab-straction mechanism, making methoxy or hydroxy groups bounded to
theα-sites. When the same reaction is performed with heating to
433 K, they verified that at CH4:N2O molar ratio equal to 1:1, the
reac-tions directly provide methanol (Table 7)[100].
Spectroscopic techniques such as FT-IR (i.e., Fourier
Transform-Infrared Spectroscopy), 13C CP/MAS NMR (i.e., Solid-state
Cross-Polarization Magic Angle Spinning Carbon-13 Nuclear Magnetic Reso-nance spectroscopy) and an extraction method were used to quantify product formation on the zeolite surface. Remarkably, the spillover of
methanol fromα-sites allowed these sites to be reused in new steps
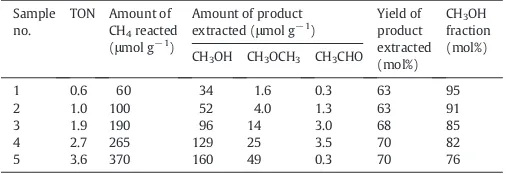
of oxygen deposition, making the reaction a“quasicatalytic”process
(i.e., the turnover number was slightly larger than one). Notably, a max-imum TON (i.e., turnover number) equal to 3.6 was reached after a 19 h
reaction (Table 7). Those authors verified that a fraction of methanol
underwent dehydration on zeolite surface, yielding mainly dimethyl ether, in addition to other minority products that were not extractable
[100].
Wood et al. studied the mechanism of“catalytic”oxidation reactions
of methane by nitrous oxide over Fe-ZSM-5 zeolite, and concluded that the primary products of methane oxidation are methoxy groups
bound-ed to active iron (i.e., Fe–OCH3)[101]. IR spectroscopy data show that
these methoxy groups are formed at 448 K and reach a maximum con-centration of 548 K; whereas, the release of reaction products begins at 523 K.
In accordance with those authors, the hydrolysis reaction by the water generated after over oxidation of methane may then convert these species to methanol. However, poor selectivity (ca. 2%) was ob-tained due to methane combustion, in addition to the strong interaction
of methanol and the zeolite, which leads to the formation Si–OCH3
spe-cies[98].
Panov et al. investigated the“quasicatalytic”reactions of methane
partial oxidation to methanol by nitrous oxide, using IR and13C CP/
MAS NMR spectroscopies[102]. They analyzed the zeolite after the
reac-tions and identified adsorbed species on the catalyst surface. Those
au-thors proposed a possible sequence of equations that describes these
“quasicatalytic”reactions (i.e., reaction of CH4+ N2O +αsites over
Fe-ZSM-5 catalyst), based on kinetic dependences data measured at
433 K (Scheme 9)[102].
As can be seen, the Fe+2cations of
α-sites are activated by nitrous
oxide generating adsorbed oxidant species (i.e., Fe3 +–O•−)α-sites,
which convert methane to adsorbed methanol. Afterwards, adsorbed methanol can be converted to dimethyl ether and water.
Recently, Panov et al. assessed the methane oxidation by N2O at
473 K again and showed that the reaction occurred in a quasicatalytic mode, resulting in product accumulation on the Fe-ZSM-5 surface
[102]. In this work, after 4 h reaction the catalyst achieved a turnover number close to 7. In accordance with these authors, methanol was
formed throughout methane oxidation by oxygen(α) over (Fe3+–O)
α
sites at 473 K. They verified that methanol and dimethyl ether were
the main products extracted from the catalyst surface. Those authors discovered an outstanding result in which water noticeably increased methanol selectivity (ca. 62% at 548 K; including coke). Because DME (i.e. dimethyl ether) was absent in the gas phase, they supposed that it
was converted to coke. That work was thefirst to describe quasicatalytic
and catalytic reactions over a broad temperature range[102].
Beznis et al. assessed the activity of Co–ZSM-5 solid catalysts on
reactions of partial oxidation of methane. They found that methanol production proportionally increased in relation to surface area of catalyst, which can be increased treating the catalyst with NaOH
[103]. Those authors discovered that the active sites (i.e., cobalt oxidic
species, such as Co3O4and CoO, present on catalyst external surface)
were also proportionally formed in relation to surface area of solid catalyst.
In the same work, the authors concluded that the presence of
extra-framework Al+3cations blocked the zeolite pores, hampering the
forma-tion of the Co+2active species, which are crucial for reaction selectivity
[103].
The acidic treatment removed the Al+3cations, triggering the
speci-ation of cobalt oxides, thus affecting their distribution on the catalyst
surface.Table 8presents the main results, which were obtained via
H2-TPR gas desorption analysis.
The highest concentration of isolated Co+2cations was detected in
the Co–ZSM-5 zeolite treated with HNO3during 20 h (Exp. 3,Table 8),
indicating that a high aluminum removal is implied in a high presence of these cations. Furthermore, if carried out after an alkaline treatment,
the acidic treatment did not eliminate Al+3cations, and therefore the
isolated Co+2cation concentration is ca. 25% (Exp. 4,Table 8),
remain-ing close to that of non-treated samples (ca. 30%, Exp. 1,Table 8).
Table 7
Product extraction from the Fe-ZSM-5 surface at different TON of the quasicatalytic reac-tion CH4+ N2O +α-sites at 433 K temperature.
Scheme 9.Equations of reaction CH4+ N2O +αsites over Fe-ZSM-5 catalyst.
Adapted from Ref.[102].
Table 8
Methane partial oxidation with oxygen over various Co–HZSM-5 catalystsa.
Adapted from Ref.[103].
aReaction conditions: metal catalysts prepared by impregnation. CH
4/ O2/ He molar ratio
= 23/ 3/ 5; pressure = 15 bar; GHSV = 5000 h-1; metal catalysts prepared by impregnation.
Those authors evaluated the catalytic performance of these zeolites, and
the main results are displayed inFig. 11 [103].
The zeolite alkaline treatment provoked an improvement in
metha-nol selectivity (Fig. 11). In contrast, an increase in zeolite acidic changes
the selectivity toward the formaldehyde formation, signifying that the
Co+2isolated ions give preferentiality to this reaction.
The zeolite acid treatment minimizes the effect of Al+3cations;
however, it also modifies the amount of H+or Na+cations present in
the zeolites. Consequently, the reaction selectivity is also changed (Fig. 12).
Remarkably, the cation nature (i.e., H+or Na+cations) was essential
to control the reaction selectivity.Fig. 7reveals that the catalyst
obtain-ed after alkaline treatment was much more selective to methanol than other acid or non-treated zeolites. Those authors linked this observation
to the fact that after thefirst alkaline treatment, the Na+–Co–ZSM-5
cat-alysts contain more cobalt on the external surface than H+–Co–ZSM-5
catalyst, which favors the methanol formation[104]. (SeeFig. 8.)
4.4.3. Molybdenum catalysts
For the past 70 years, the methane oxidation reactions to methanol have usually carried out over molybdenum catalysts under high oxygen pressure. Dowden and Walker investigate the activity of a number of supported and unsupported molybdenum catalysts, at pressures of 50 bar, temperatures ranging from 403 to 573 K, and feed gas with 97% methane and 3% oxygen. The most active catalyst was
Fe2O3(MoO3)3, which resulted in 869 g methanol/kg cat h[105].
However, in accordance with these authors, if the desired products were removed from the catalyst surface and cooled to temperature
Nitrous oxide was also an oxidant assessed in methane oxidation
re-actions to methanol over MoO3/SiO2catalyst. The best result was
reached in reactions over 1.7% MoO3/SiO2catalyst, at 1 bar pressure
and 833 K temperature. The reactions under these conditions formed methanol and formaldehyde with total selectivity equal to 84.6%
[107]. However, the same researchers could not reproduce their own
re-sults in their next work[108]. The methanol selectivity obtained by
them was lower; however, they discovered that an addition of steam to the feed gas increased methanol selectivity to ca. 78.1%, an effect that they attributed to the inhibition of carbon oxides.
The partial oxidation of methane to methanol over molybdenum or tungsten trioxide based catalysts containing different metal oxides as dopants was investigated, aiming to identify a catalytic composition that would be able to activate methane without resulting in a loss of
methanol selectivity[109]. Herein, we only display the results of
molyb-denum catalysts (Table 9).
The Cu/MoO3catalyst was more selective than the free catalyst gas
phase oxidation performed in a reactor bed packed with quartz glass
beads, similar to that observed for the Ga2O3/MoO3catalyst (Exps. 5
and 8,Table 9)[109]. Those authors attributed the high methanol
yield to the possible synergism between MoO3and Ga2O3oxides.
An outstanding result was obtained by comparing the activity of all these different molybdenum based catalysts with data obtained from an empty reactor; none of the reactions performed in the presence of catalysts assessed by them reached the conversion and methanol
selec-tivity achieved in reactions made with an empty reactor (Table 9)[109].
We concluded that due to high temperatures employed, the reactions in the gas phase were preferably more accurate rather than those on the catalyst surface.
Zhang et al. studied the activity of MoO3or ZrO2/La–Co–O supported
catalysts in the methane partial oxidation reactions to methanol using oxygen as an oxidant, under high pressures (ca. 4.2 to 5.0 MPa) and
temperatures ranging from 653 to 693 K[110]. They designed a
continuous-flow reactor capable of operating at high pressures, in
which the methane oxidation to methanol by dioxygen over MoOx/
La–Co–O and MoOx/ZrO2catalysts was performed (Table 10). We
here-in highlight only results here-in which methanol selectivity was relevant (Table 10)[110].
Under the studied conditions, ZrO2and MoOx/ZrO2gave only trace
amounts of methanol and for such reasons, omitted inTable 10. The
MoOx/La–Co–O catalyst containing 7 wt.% showed the highest
Fig. 7.Industrial processes installed to synthesize methanol. Adapted from Johnson Matthey.
selectivity and yield to methanol (ca. 60% and 6%, respectively), in
reac-tions performed at 4.2 bar and 693 K[110].
Those authors evaluated the reducibility of the supported catalyst
and verified that 7 wt.% MoOx/La–Co–O catalyst showed higher (O−)/
(O2−) proportion than 12 wt.% MoOx/ZrO2ones. They obtained such
data by measurement of hydrogen uptake of each catalyst. They suggest that this feature can be crucial to obtain high methanol selectivity (Table 10).
The activity, yield, and hydrogen consumption data displayed in
Table 11were calculated considering the catalyst surface area. These
re-sults support the explanation that the reducibility, (O−)/(O
2
−) ratio, as
well as the presence of MoOx are essential conditions for good methanol selectivity.
The (O−)/(O
2
−) ratio was linked by those authors to the higher de
fi
-ciency of lattice sites commonly described for LaCoO3and Co3O4species
compared with other catalysts. These phases are present on MoOx/La–
Co–O catalysts and were identified by XRD spectroscopy analyses
[110]. Such a deficiency contributes to the migration and transformation
of O2from the gas phase to give O2−and O−species on the catalyst
surface.
4.4.4. Iron sodalite catalysts
For the past 90 years, numerous researchers have investigated the activity of iron sodalite based catalysts in methane oxidation reactions
to methanol[111]. Lyions et al. carried out reactions using a feed
com-position equal to 3:1 of methane to air, under 55 bar pressure at Fig. 9.Productive cycling in the oxidation of methane by O2, mediated by the [Cu1+–Cu1+–Cu1+(7-N-Etppz)]1+complex in the presence of H2O2as the sacrificial reductant.
Adapted from Refs.[64–66].
Fig. 10.Abortive cycling in the oxidation of methane by O2, mediated by the Cu1+–Cu1+–Cu1+(7-N-Etppz)1+complex in the presence of H
2O2as the sacrificial reductant.
Adapted from Refs.[64–66]].
framework and extra framework[112]. However, even though iron so-dalite catalysts were successful, Hutchings et al. performed experimen-tal and theoretical studies, and showed that caexperimen-talyst used in a simple
flow reactor was less effective that in an empty reactor[109,113].
Chun and Anthony also assessed the participation of the reactor's
wall in the reaction of methane oxidation to methanol[115]. They
com-pared their results to ones obtained with different metal oxide catalysts, under the same reaction conditions (i.e., high pressure and tempera-ture). Those authors synthesized a number of catalysts employing dif-ferent methods (i.e., sol gel method, ionic exchange, precipitation), and tested them on methane oxidation reactions by oxygen under
high pressure (i.e., 50 atm; 95.65% CH4and 4.35% O2feed composition)
and temperatures varying from 640 to 750 K.Fig. 13shows the main
re-sults as well as the catalyst synthesis conditions.
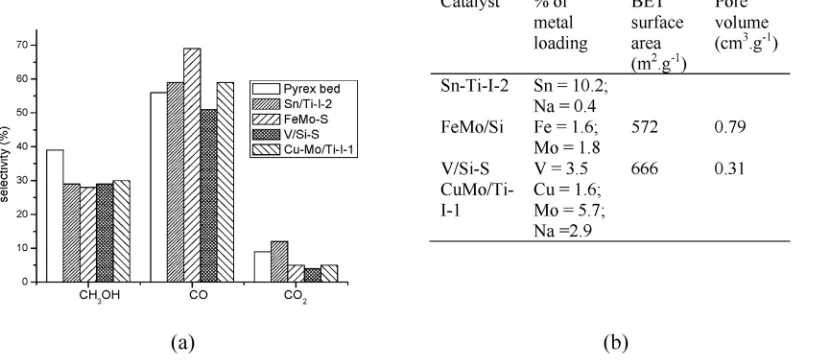
Although the highest methanol selectivity was achieved using a Pyrex bed (ca. 38%), in general all the metal oxides exhibited methanol selectivity close to ca. 30%. In addition, conversions of methane and ox-ygen were almost constant (ca. 95%), regardless of the catalyst used.
Those authors verified that under conditions of high pressure and
tem-perature, the homogeneous reactions were predominant, occurring probably in the empty space present on packed bed of reactor, being
re-sponsible for a large fraction of products formed (Fig. 13)[115].
Chun and Anthony also performed several reactions using an empty Pyrex glass reactor or packed reactor with Pyrex beads at a range of
temperatures from 704 to 757 K. Those authors verified that regardless
of the conditions used, the methanol selectivity ranged from 37 to 42%
[115].
They concluded that the Pyrex glass surface could inhibit the free radicals formation, which promotes the homogeneous oxidation of
methane. Nevertheless, it is worth noting that this effect was insignifi
-cant when the reactions were performed using a low surface:volume ratio. When the reactor bed was packed with larger Pyrex beads (ca.
1.41 to 2.00 mm), at high surface to volume ratio (ca. 300 cm−1) and
temperatures of 724 to 730 K, the conversion of methane was drastically
reduced from 4.0 to 0.7% (Exp. 4,Table 12). Conversely, at temperatures
of 704 to 726 K, and a lower ratio of surface to volume with smaller Pyrex beads (0.125/0.149 mm), methane maximum conversion was Fig. 11.Selectivity toward methanol and formaldehyde, and the total amount of produced
oxygenates of Co–ZSM-5 zeolites for methane activation with O2at 423 K: (A) Co–
ZSM-5-p; (B) Co–ZSM-5-at-1–20; (C) Co–ZSM-5-at-1–20-acid; and (D) Co–ZSM-5-acid. Adapted from Ref.[103].
Fig. 12.Selectivity toward methanol and formaldehyde, and the total amount of produced oxygenates of Co–ZSM-5 zeolites for methane activation with O2at 423 K: (A) Co– ZSM-5-p; (B) Co–ZSM-5-at-1–20; (C) Co–H-ZSM-5; and (D) Co–H-ZSM-5-at-1–20.
Adapted from Ref.[103].
Fig. 13.(a) Comparison of selectivity for heterogeneous reactions (O2= 4.35%; pressure = 50 atm; oxygen conversion = 95–100%; residence time = 4.5 s); (b) properties of catalysts.
reached, limited by total consumption of oxygen (i.e., 4.0 and 100% of
conversions for CH4and O2, respectively). This inhibitor effect is a
con-sequence of the free volume reduction of the reactor that hampers the
gas phase reactions[115].
4.4.5. Other solid catalysts
Stolcova et al. described the methane oxidation to methanol by
ni-trous oxide over Cu–Fe pyrophosphates at high temperatures (ca.
903 K) and at low pressures (ca. 0.65 bar and 0.26 bar for methane
and nitrous oxide, respectively)[116].Table 13summarizes the main
results obtained on assessing the activity of the copper iron pyrophos-phates. We omitted results with methanol selectivity lower than 1.9
for simplification.
Starting from (Cu+2)(Fe+3)
2(P2O7−4)2catalyst, they replaced part of
Fe+3cations by Cu+2, and otherwise replaced part of Cu+2cations by
Fe+3, keeping the neutrality of catalysts constant throughout an
ade-quate adjustment of pyrophosphate anions load. The monometallic Cu
catalyst (i.e., Exp. 4, CuFe-2/0–1023 K catalyst,Table 13) was more
ac-tive than iron ones (i.e., Exp. 9, CuFe-0/3–1023 K catalyst,Table 13).
Conversely, in these reactions, the selectivity was very distinct; whereas, the former was more selective for methanol, the latter was
more selective for formaldehyde (Exps. 6 and 11,Table 13). Moreover,
those authors concluded that oxygenates' yield depends on the molar
ratio of Fe/Fe + Cu in pyrophosphates (Fig. 14)[116].
In the same work, the oxidant effect was investigated at different
temperatures[116]. In the presence of N2O at 673 K, methanol and
formaldehyde were formed at an equal molar ratio, however, with low methane conversion (ca. 4%).
Fig. 15shows that increasing the reaction temperature result in an increase of the methane conversion, reaching a maximum of ca. 6%. However, a concomitant decreasing of the methanol selectivity is also favored, and an increase of selectivity for formaldehyde and carbon ox-ides simultaneously occurs. Using similar reaction conditions, Stolcova
et al. replaced the nitrous oxide by oxygen and verified that under
Fig. 14.Effect of Fe/Fe + Cu molar ratio in the oxygenates' yield. Adapted from Ref.[116].
Fig. 15.Effect of temperature on conversion and selectivity of methane partial oxidation to methanol using N2O over CuFe-1/2–1023 K–4 h catalyst (reaction conditions: pressure =
CH4(0.65 bar), N2O (0.26 bar); temperature range (673–923 K), catalyst weight (0.5 g), flow (3.6 L·h−1)).
Adapted from Ref.[116].
Fig. 16.Effect of NO concentration on the conversion of methane and O2, and on the yield
of C1-oxygenates in reactions over V2O5/SiO2catalyst (reaction conditions: catalyst weight
(V2O5/SiO2catalyst, 0.22 g); totalflow (89.8 mL min−1); feed = O2(29%); CH4(41.5%);
NO (0 to 2.9% in N2)).
Adapted from Ref.[118].
Fig. 17.Effect of temperature and of CH4/O2ratio on the CH4conversion in reactions over
V2O5/SiO2 catalyst (reaction conditions: catalyst weight (0.30 g); total flow
(122 mL min−1); feed (22.9–34.4% CH4, 1.0% NO in N2)). Adapted from Ref.[118].
![Fig. 1. Conversion route of natural gas to chemicals and fuels [3].](https://thumb-ap.123doks.com/thumbv2/123dok/1629635.1556836/3.595.310.563.683.736/fig-conversion-route-natural-gas-chemicals-fuels.webp)
![Fig. 5. Methanol process layout with Topsøe's two-step reforming.Adapted from Refs. [18, 19].](https://thumb-ap.123doks.com/thumbv2/123dok/1629635.1556836/5.595.89.515.482.718/fig-methanol-process-layout-topsoe-reforming-adapted-refs.webp)

![Fig. 9. Productive cycling in the oxidation of methane by O2, mediated by the [Cu1+–Cu1+–Cu1+(7-N-Etppz)]1+ complex in the presence of H2O2 as the sacrificial reductant.Adapted from Refs](https://thumb-ap.123doks.com/thumbv2/123dok/1629635.1556836/15.595.103.502.54.287/productive-oxidation-mediated-complex-presence-sacricial-reductant-adapted.webp)