PERBANDINGAN EKSTRAKSI CIRI
K-MERS
DAN
SPACED
K-MERS
PADA KLASIFIKASI FRAGMEN METAGENOME
DENGAN
NAÏVE BAYES CLASSIFIER
VIANI RAHMAWATI
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM INSTITUT PERTANIAN BOGOR
PERNYATAAN MENGENAI SKRIPSI DAN
SUMBER INFORMASI SERTA PELIMPAHAN HAK CIPTA
Dengan ini saya menyatakan bahwa skripsi berjudul Perbandingan Ekstraksi Ciri K-Mers dan Spaced K-Mers pada Klasifikasi Fragmen Metagenome dengan Naïve Bayes Classifier adalah benar karya saya dengan arahan dari komisi pembimbing dan belum diajukan dalam bentuk apa pun kepada perguruan tinggi mana pun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir skripsi ini.
Dengan ini saya melimpahkan hak cipta dari karya tulis saya kepada Institut Pertanian Bogor.
Bogor, 15 Agustus 2013
ABSTRAK
VIANI RAHMAWATI. Perbandingan Ekstraksi Ciri K-Mers dan Spaced K-Mers pada Klasifikasi Fragmen Metagenome dengan Naïve Bayes Classifier. Dibimbing oleh WISNU ANANTA KUSUMA, TOTO HARYANTO.
Metagenom adalah material genetis yang diperoleh dari sampel yang diambil langsung dari lingkungan, misalnya tanah, air laut, atau isi perut manusia. Fragmen-fragmen yang diperoleh dari metagenome ini mengandung berbagai organisme sehingga proses binning diperlukan untuk mengelompokkannya sebelum perakitan genom dilakukan. Penelitian ini menggunakan metode klasifikasi naïve Bayes dengan metode ekstraksi ciri spaced k-mers untuk mengklasifikasikan fragmen ke takson genus. Hasilnya dibandingkan dengan teknik klasifikasi menggunakan ekstraksi ciri k-mers dan naïve Bayes classifier. K-mers adalah ciri umum yang banyak digunakan untuk pengklasifikasian fragmen DNA. Hasil perbandingan menunjukkan bahwa hasil pengklasifikasian dengan menggunakan naïve Bayes classifier dan spaced k-mers menghasilkan nilai akurasi yang lebih tinggi dibandingkan dengan menggunakan naïve Bayes classifier dan ekstraksi ciri k-mers.
Kata kunci: metagenome, naïve Bayes Classifier, k-mers, spaced k-mers
ABSTRACT
Metagenome is a genetic material obtained from a simple which is taken directly from the environment such as soil, marine, or human entrails. These metagenome fragments contain a variety of organism , so that a binning process required to classify them before conducting genome assembly. This research employs naïve Bayes classifier (NBC) and spaced k-mers as a feature extraction to classify these fragments into genus level. The results will be compared to those of method which uses NBC and k-mers feature extraction. K-mers feature is a common feature in the DNA fragments classification problem. The comparison results show that the accuracy of classifier using NBC and spaced k-mers is higher than that of classifier using NBC and k-mers.
Skripsi
sebagai salah satu syarat untuk memperoleh gelar Sarjana Ilmu Komputer
pada
Departemen Matematika dan Ilmu Pengetahuan Alam
PERBANDINGAN EKSTRAKSI CIRI
K-MERS
DAN
SPACED
K-MERS
PADA KLASIFIKASI FRAGMEN METAGENOME
DENGAN
NAÏVE BAYES CLASSIFIER
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM INSTITUT PERTANIAN BOGOR
BOGOR 2013
Judul Skripsi : Perbandingan Ekstraksi Ciri K-Mers dan Spaced K-Mers pada Klasifikasi Fragmen Metagenome dengan Naïve Bayes Classifier Nama : Viani Rahmawati
NIM : G64090058
Disetujui oleh
Diketahui oleh
Dr Ir Agus Buono, MSi MKom Ketua Departemen
Tanggal Lulus:
Dr Ir Wisnu Ananta Kusuma, ST, MT Pembimbing I
PRAKATA
Puji dan syukur penulis panjatkan kepada Allah subhanahu wa ta’ala atas segala karunia-Nya sehingga karya ilmiah ini berhasil diselesaikan. Tema yang dipilih dalam penelitian yang dilaksanakan sejak bulan Desember 2012 ini ialah klasifikasi fragmen metagenome, dengan judul Perbandingan Ekstraksi Ciri K-Mers dan Spaced K-Mers pada Klasifikasi Fragmen Metagenome dengan Naïve Bayes Classifier.
Penghargaan penulis sampaikan kepada ayah, ibu, serta seluruh keluarga, atas segala doa dan kasih sayangnya. Di samping itu, terima kasih penulis ucapkan kepada Bapak Dr Ir Wisnu Ananta Kusuma, ST MT dan Bapak Toto Haryanto, SKom MSi selaku pembimbing yang telah banyak memberi saran dan kritik yang membangun. Terimakasih juga penulis ucapkan kepada Bapak Aziz Kustiyo, SSi MKom selaku penguji yang juga telah memberikan saran yang bermanfaat. Serta kepada seluruh rekan yang telah memberi semangat kepada penulis.
Semoga karya ilmiah ini bermanfaat.
Bogor, 15 Agustus 2013
DAFTAR ISI
DAFTAR TABEL ix
DAFTAR GAMBAR ix
PENDAHULUAN 1
Latar Belakang 1
Perumusan Masalah 2
Tujuan Penelitian 2
Manfaat Penelitian 2
Ruang Lingkup Penelitian 2
TINJAUAN PUSTAKA 3
Metagenome 3
Naïve Bayes Classifier (NBC) 3
Sensitifity 3
Imbalanced Data 3
K-fold cross-validation 4
METODE 4
Pengumpulan Data 4
Pembagian Data 6
Praproses Data 8
Ekstraksi Ciri 9
Reduksi Data dengan PCA 10
K-Fold Cross Validation 11
Naïve Bayes Classifier (NBC) 11
Model 12
Pengujian NBC 13
Analisis 13
HASIL DAN PEMBAHASAN 13
Praproses Data 13
Ekstraksi Ciri 14
Reduksi Data dengan PCA 16
Naïve Bayes Classifier (NBC) 16
Model 17
Pengujian NBC dan Analisis 17
Percobaan Dataset Besar 20
SIMPULAN DAN SARAN 21
Simpulan 21
Saran 21
DAFTAR PUSTAKA 21
RIWAYAT HIDUP 22
DAFTAR TABEL
1 Data Latih 7
2 Data Uji 7
3 Ilustrasi hasil perhitungan frekuensi k-mers pada data 3 genus
dengan 10000 pembacaan 9
4 Hasil perhitungan frekuensi spaced k-mers dengan 10 000
pembacaan 15
DAFTAR GAMBAR
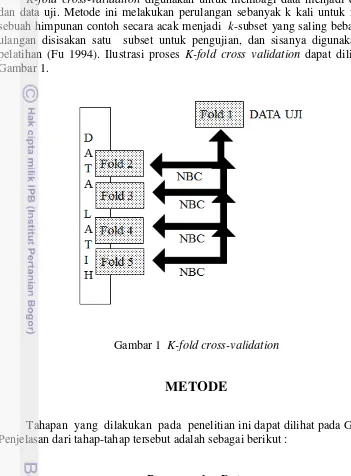
1 K-fold cross-validation 4
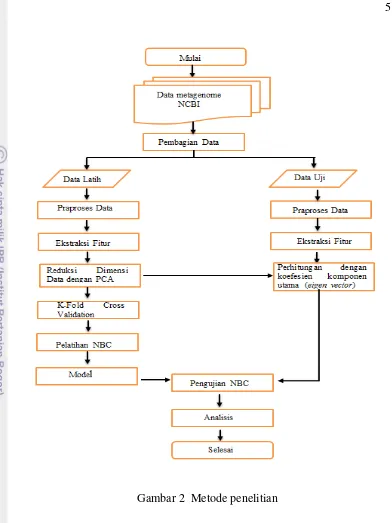
2 Metode penelitian 5
3 Proses memasukkan data metagenome pada MetaSim 6
4 Databasesequences DNA mikroorganisme pada MetaSim 6
5 Screenshoot fail FastA untuk data set kecil dengan panjang 500 bp 8
6 Ekstraksi ciri k-mers 9
7 Spaced k-mers (Kusuma, 2012) 10
8 Proses klasifikasi fragmen metagenome menggunakan NBC 11
9 Sequence DNA Bacillus amyloliquefaciens FZB42 pada pembacaan
1 untuk panjang fragmen 1000 bp 13
10 K-Mers 14
11 Jumlah kombinasi pada spaced k-mersk = 3 15
12 Contoh perhitungan NBC untuk atribut numerik dengan Gaussian (normal) density function 17
13 Confusion matrix untuk data latih untuk pembacaan 10K panjang
fragmen 10 K pada data set kecil 18
14 Akurasi data latih pada data set kecil 18
15 Sensitivity data latih pada data set kecil 19
16 Akurasi data uji baru data set kecil 19
1
PENDAHULUAN
Latar Belakang
Metagenom adalah material genetis yang diperoleh dari sampel yang diambil langsung dari lingkungan, misalnya tanah, air laut, atau isi perut manusia. Fragmen-fragmen yang diperoleh dari metagenome ini mengandung berbagai organisme sehingga proses binning diperlukan untuk mengelompokkannya sebelum perakitan genom dilakukan. Ada dua pendekatan binning, salah satunya adalah berdasarkan komposisi. Binning berdasarkan komposisi memiliki beberapa keunggulan dibandingkan pendekatan binning lainnya yang berdasarkan homologi. Binning berdasarkan komposisi merupakan jalan pintas (by pass) kebutuhan akan penjajaran sequences, vektor masukan yang dihasilkan dari ekstraksi ciri berupa pasangan basa (base pair) akan dihitung sebagai ciri komposisi, kemudian ciri tersebut akan digunakan sebagai masukan pada pembelajaran dengan contoh (supervised learning) atau pada pembelajaran secara observasi (unsupervised learning). Contoh metode yang menggunakan binning berdasarkan komposisi dengan supervised learning adalah Phylopythia (McHardy et al. 2007), naïve bayessian classification (Rosen et al. 2008), dan Phymm (Brady dan Salzberg 2009). Adapun contoh metode menggunakan binning berdasarkan komposisi dengan unsupervised learning adalah TETRA (Teeling et al. 2004), growing self organizing map (Hsu dan Halgamuge 2002; Chan et al. 2007), dan self organizing clustering (Amano et al. 2003; Amano et al. 2007).
Supervised learning banyak digunakan pada proses binning. Metode klasifikasi dan prediksi dinamakan supervised learning karena ada proses supervisi, yaitu data latih disertai dengan label yang menunjukkan kelas observasi dan data baru diklasifikasikan berdasarkan training set. Langkah pertama dalam klasifikasi adalah membangun model untuk mendeskripsikan predetermined set kelas data atau konsep. Langkah kedua, pemakaian model untuk klasifikasi. Model akan dipakai setelah dilakukan pembuatan estimasi keakuratan model dengan teknik holdout. Jika keakuratan model dapat diterima, model dapat digunakan untuk mengklasifikasikan data baru yang label kelasnya belum diketahui. Salah satu proses yang paling krusial sebelum pembuatan model tersebut adalah ekstraksi ciri. Ekstraksi ciri merupakan proses untuk mengambil ciri penting suatu objek.
Gail Rosen et al (2008) melakukan pengklasifikasian pada suatu komunitas yang mengandung 635 mikroorganisme. Metode yang digunakan untuk ekstraksi ciri adalah k-mers dan pengklasifikasian menggunakan naïve Bayes classifier (NBC). Penelitian tersebut menghasilkan akurasi 38% untuk fragmen dengan panjang 500 base pair (bp) setelah menggunakan k = 3, serta akurasi tertingginya 88.8% setelah menggunakan k = 15.
2
sampai dengan 92%, serta akurasi untuk data uji baru yaitu antara 78% sampai dengan 87%. Penelitian tersebut menghasilkan akurasi yang tinggi hanya dengan menggunakan k = 3. Begitu pun klasifikasi fragmen dengan panjang 500 bp menghasilkan akurasi sebesar 78% hanya dengan penggunaan k = 3 dengan spaced k-mers. Untuk itu, pada penelitian ini metode klasifikasi yang digunakan adalah NBC dengan metode ekstraksi ciri spaced k-mers untuk mengklasifikasikan fragmen ke takson genus. Hasilnya dibandingkan dengan teknik klasifikasi menggunakan ekstraksi ciri k-mers dan NBC.
Hal ini didasari ingin membandingkan pengaruh metode ekstraksi ciri k-mers dan spaced k-mers pada hasil klasifikasi yang menggunakan NBC. Dengan demikian dapat ditentukan metode ekstraksi ciri spaced k-mers atau k-mers yang dapat meningkatkan akurasi hasil klasifikasi fragmen metagenome jika metode klasifikasi yang digunakan adalah NBC.
Perumusan Masalah
Berdasarkan latar belakang penelitian yang telah diuraikan sebelumnya, masalah yang akan diteliti antara lain:
1. Seberapa besar tingkat akurasi yang dapat diperoleh bila digunakan metode NBC pada penelitian ini?
2. Bagaimana pengaruh metode ekstrasi ciri yang dipakai untuk hasil akurasi pengklasifikasian?
Tujuan Penelitian
Tujuan penelitian ini adalah mengklasifikasikan fragmen metagenome ke dalam tingkat genus (sebagai kelasnya) dengn metode NBC. Selain itu juga ingin membandingkan pengaruh penggunaan metode ekstrasi ciri k-mers dan spaced k-mers terhadap hasil akurasi yang dihasilkan jika classifier yang digunakan adalah NBC.
Manfaat Penelitian
Sebagai acuan untuk membantu para peneliti biologi dalam mengatasi masalah perakitan genom melalui proses binning.
Ruang Lingkup Penelitian
Ruang lingkup penelitian ini terbatas pada:
1. Penggunaan data latih yang sama dengan penelitian Kusuma dan Akiyama (2011) yang menggunakan data set kecil dengan 10 mikroorganisme yang termasuk ke dalam 3 genus (sebagai kelas)
2. Data uji yang digunakan 9 mikroorganisme yang termasuk dalam kelompok genus (sebagai kelas) yang sama dengan data latih.
3
TINJAUAN PUSTAKA
Metagenome
Metagenome ialah genom dari mikrob tanpa pengulturan mikrob. Istilah tersebut berasal dari konsep statistik meta-analisis (proses yang secara statistik mengombinasikan metode-metode analisi yang terpisah), serta genomik (analisis menyeluruh dari materi genetika suatu organisme). Metagenomik dikembangkan berdasarkan kemajuan terkini bidang biologi molukuler dan bioinformatika. Bioinformatika ini mempunyai peranan yang penting diantaranya adalah untuk manajemen data biologi molekul, terutama sekuen DNA dan informasi genetika (Thontowi 2009).
.
Naïve Bayes Classifier (NBC)
Metode NBC adalah salah satu metode klasifikasi yang mengasumsikan seluruh atribut dari contoh yang bersifat independen satu sama lain pada konteks kelas (McCallum dan Nigam, 1998). Meskipun secara umum asumsi tersebut merupakan asumsi yang buruk, pada praktiknya metode NBC menunjukkan kinerja yang sangat baik. Menurut Manning et al. (2008), peluang Bayes dapat digunakan untuk menghitung peluang bersyarat, yaitu peluang kejadian apabila suatu kejadian diketahui. Metode ini dapat memprediksi kemungkinan anggota suatu kelas berdasarkan sampel yang berasal dari anggota kelas tersebut (Rosen et al, 2008). Karena yang diasumsikan sebagai variabel independent, maka hanya varians dari suatu variabel dalam sebuah kelas yang dibutuhkan untuk menentukan klasifikasi, bukan keseluruhan dari matriks kovarians.
Sensitifity
Sensitifity atau true positive rate mengukur untuk menghitung akurasi dari tiap kelas.
Imbalanced Data
Sebuah himpunan data dikatakan menjadi tidak seimbang (imbalanced) jika terdapat satu kelas yang direpresentasikan dalam jumlah instance yang kecil bila dibandingkan dengan jumlah instance kelas lainnya (Barandela et al. 2002).
4
K-fold cross-validation
K-fold cross-validation digunakan untuk membagi data menjadi data latih dan data uji. Metode ini melakukan perulangan sebanyak k kali untuk membagi sebuah himpunan contoh secara acak menjadi k-subset yang saling bebas. Setiap ulangan disisakan satu subset untuk pengujian, dan sisanya digunakan untuk pelatihan (Fu 1994). Ilustrasi proses K-fold cross validation dapat dilihat pada Gambar 1.
METODE
Tahapan yang dilakukan pada penelitian ini dapat dilihat pada Gambar 2. Penjelasan dari tahap-tahap tersebut adalah sebagai berikut :
Pengumpulan Data
Data yang digunakan pada penelitian ini adalah data metagenome yang diunduh dari situs National Centre for Biotechnology Information (NCBI). NCBI merupakan suatu institusi yang fokus sebagai sumber informasi perkembangan biologi molekuler. Data metagenome ini merupakan sequences DNA mikroorganisme. Data yang telah diunduh dari situs NCBI akan di-generate meggunakan perangkat lunak MetaSim. Fail yang berisi sequences DNA mikroorganisme yang telah diunduh dari NCBI dimasukkan ke dalam perangkat lunak tersebut.
5
Setelah memasukkan data dari NCBI ke dalam perangkat lunak MetaSim, proses selanjutnya adalah memilih beberapa sequences DNA mikroorganisme yang telah tersedia pada database sesuai dengan kebutuhan penelitian. Tahapan pengumpulan data dapat dilihat pada Gambar 3 dan Gambar 4.
6
Gambar 3 Proses memasukkan data metagenome pada MetaSim (V.009)
7 Pembagian Data
Data yang digunakan dalam penelitian ini terdiri atas data set kecil sebagai bahan analisis utama. Data set kecil sama dengan penelitian Kusuma (2011) yang menggunakan 10 mikroorganisme yang termasuk ke dalam 3 genus sebagai organisme yang diketahui. Data tersebut dilatih dengan menggunakan 5-fold cross validation untuk mendapatkan model naïve Bayes. Adapun untuk data uji yang digunakan adalah 9 mikroorganisme sebagai organisme baru. Daftar organisme untuk data set kecil, data latih pada Tabel 1 dan data uji pada Tabel 2. Adapun daftar 381 organisme yang termsuk ke dalam 48 genus dapat dilihat pada Lampiran 1.
Tabel 1 Data latih
Spesies Genus
Agrobacterium radiobacter K84
chromosome 2
Agrobacterium Agrobacterium tumefaciens str. C58
chromosome circular
Agrobacterium vitis S4 chromosome 1
Bacillus amyloliquefaciens FZB42
Bacillus Bacillus anthracis str. Ames Ancestor
Bacillus cereus 03BB102
Bacillus pseudofarmus OF4 chromosome
Staphylococcus aureus subsp. Aureus JH
Staphylococcus Staphylococcus epidermidis ATCC 12228
Staphylococcus haemolyticus JCSC1435
Tabel 2 Data uji
Spesies Genus
Agrobacterium radiobacter K84
chromosome 1
Agrobacterium Agrobacterium tumefaciens str. C58
chromosome linear
Agrobacterium vitis S4 chromosome 2
Bacillus thuringiensis str Al Hakam
Bacillus Bacillus subtilis subsp. Subtilis str 16B
Bacillus pumilus SAFR-032
Staphylococcus carnosus
Staphylococcus Staphylococcus saprophyticus subsp.
saprophyticus ATCC 1530 S
8
Praproses Data
Pada tahap praproses data, sequences DNA metagenome yang sudah dipilih akan diuraikan fragmennya menggunakan perangkat lunak MetaSim. Data yang akan diproses akan dibaca berkali-kali sesuai dengan kebutuhan penelitian. Pada penelitian ini, data yang dipersiapkan dibaca sebanyak 10 000 kali untuk keperluan data latih pada data set kecil. Hal ini berarti ada 10 000 baris fragmen mikroorganisme. Panjang fragmen yang digunakan adalah 500 base pair (bp), 1 kbp, 5 kbp, dan 10 kbp. Maksud dari panjang fragmen 1 bp dapat diwakili oleh adenine (A), cytosine (C), guanine (G) atau thymine (T). Data uji pada data set kecil akan dibaca sebanyak 5 000 kali. Untuk data uji, fragmen yang digunakan adalah fragmen dengan panjang 500 bp. Keluaran dari pengolahan MetaSim ini adalah fail FastA yang berisi sequence DNA yang sudah terfragmen sesuai dengan kriteria parameter yang diinginkan. Berikut screen shoot hasil keluaran fail FastA untuk data set kecil dengan pembacaan panjang 500 bp yang dapat dilihat pada Gambar 5.
9 Ekstraksi Ciri
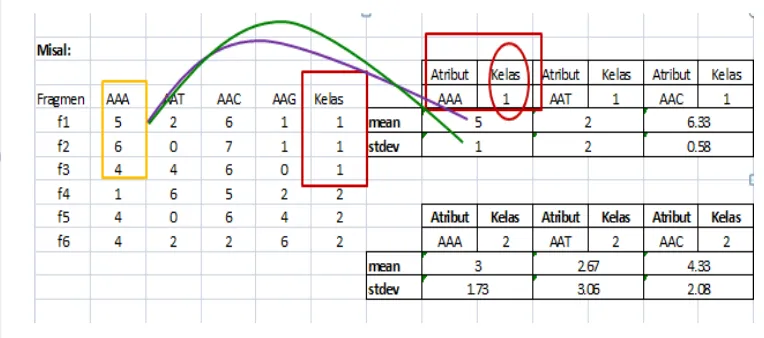
Tahap selanjutnya dari praproses data adalah ekstraksi ciri. Ekstraksi ciri dilakukan dengan membaca frekuensi dari kombinasi nukleotida yang mungkin terbentuk dengan menggunakan k-mers untuk k = 3. Pola kemunculan k adalah pola yang menampilkan k pada suatu waktu dalam suatu sequences. Pola kemunculan dalam sequences dihitung menggunakan empat basa utama (A, C, G, dan T) dipangkat dengan rangkaian pasangan basa yang ingin digunakan (pola kemunculan : , dengan ). Pada penelitian ini k = 3 berarti akan ada pola kemunculan yang terbentuk. Ilustrasi perhitungan frekuensi pola kemunculan dengan ekstraksi ciri k-mers dapat dilihat pada Gambar 6 dan Tabel 3.
K-mers yang digunakan adalah k = 3 maka ada 43 = 64 pola kemunculan yang terbentuk. Pola kemunculan tersebut direpresentasikan oleh jumlah atribut pada data yaitu X1, X2, …, X64. Jumlah pembacaan yang digunakan pada data latih yaitu 10 000 pembacaan yang direpresentasikan oleh jumlah baris fragmen metagenome pada data. Adapun untuk atribut kelas terdiri atas 3 genus yang berbeda.
Gambar 6 Ekstraksi ciri k-mers
10
Selain menggunakan ekstraksi ciri k-mers, digunakan juga spaced k-mers frequency yang memperhitungkan kondisi d ’ ca dengan nilai w = 3 dan d = 0,1,2. Gambar 8 mendeskripsikan penggunaan spaced k-mers untuk k = 3 di mana w = weight of pattern adalah banyaknya posisi yang cocok (1’s) adapun d = pada suatu waktu dalam suatu rangkaian DNA. Mengacu pada penelitian Kusuma (2012), pola terbaik yang menghasilkan akurasi tinggi dari hasil pencarian lengkap adalah dengan pola w = 3 dan d = 0, 1, 2. Ilustrasi perhitungan frekuensi pola kemunculan dengan ekstraksi ciri spaced k-mers dapat dilihat pada Gambar 7
Metode ini akan memeriksa frekuensi nukleotida dari fragmen DNA mulai dari AAA – CCC, A*AA - C*CC, dan A**AA - C**CC. Pengertian dari simbol * (d ’ ca ) pada fragmen DNA yang diperiksa adalah dapat merupakan basa apapun, baik A, T, G, maupun C. Adapun untuk simbol **, berarti diperbolehkan basa manapun mengisi 2 bit tersebut. Sehingga kondisi itu dapat diisi oleh 24 pasang basa mulai dari AA, AC, AT, AG, dan seterusnya hingga CC.
Reduksi Data dengan PCA
Teknik reduksi dimensi data yang digunakan pada penelitian ini adalah Principal Component Analysis (PCA). PCA adalah teknik yang digunakan untuk menyederhanakan suatu data dengan cara mentransformasi linier sehingga terbentuk sistem koordinat baru dengan varian maksimum. Analisis komponen utama merupakan teknik statistik yang dapat digunakan untuk menjelaskan struktur variansi-kovariansi dari sekumpulan variabel melalui variabel baru dimana variabel baru ini saling bebas, dan merupakan kombinasi linier dari variabel asal (Johnson dan Wichern, 2002). Selanjutnya variabel baru ini dinamakan komponen utama. Salah satu tujuan dari analisis komponen utama adalah mereduksi dimensi data asal yang semula terdapat p variabel bebas menjadi q komponen utama (dimana q < p). Adapun kriteria pemilihan q menurut Johnson (2002) yaitu proporsi kumulatif keragaman data asal yang dijelaskan oleh q komponen utama minimal 80 %, dan proporsi total variansi populasi bernilai cukup besar.
Proses PCA didapatkan dengan cara menentukan eigenvalue dari matriks kovariannya. Pada penelitian ini perlu dijelaskan bahwa fungsi princomp adalah statistics toolbox MATLAB yang melakukan analisis komponen utama pada matriks data X dan menghasilkan suatu matriks yang dinamakan COEFF, SCORE,
11 dan LATENT . Matriks COEFF adalah matriks p-by-p yang berisi eigen vector, masing-masing kolom berisi koefisien untuk satu komponen utama. Matriks SCORE adalah data yang dibentuk dengan mengubah data asli ke dalam ruang dari komponen utama yang sudah dikalikan dengan COEFF. LATENT adalah eigenvalue yang nilai-nilainya telah diurutkan secara menurun. Perhitungan eigen vector hanya dilakukan pada data latih. Adapun untuk data uji baru hanya dikalikan dengan koefesien komponen utama dari data latih.
K-Fold Cross Validation
Pelatihan data set dilakukan dengan menggunakan k-fold cross validation. K-fold cross-validation digunakan untuk membagi data menjadi data latih dan data uji. Pada penelitian ini k yang digunakan adalah 5. Data akan dibagi menjadi 5 bagian berukuran sama dimana 4 bagian akan menjadi data latih, dan 1 bagian sisanya akan digunakan untuk validasi. Data yang digunakan pada 5-fold cross-validation ini adalah data latih dengan jumlah 10 000. Hal ini berarti dari 10000 fragmen tersebut, 8000 fragmen digunakan sebagai data latih dan 2000 fragmen sebagai data uji.
Naïve Bayes Classifier (NBC)
NBC berfungsi untuk menghitung peluang dari suatu kelas dalam masing-masing kelompok atribut yang ada dan menentukan kelas mana yang paling optimal. Proses klasifikasi data dengan NBC diilustrasikan pada Gambar 8.
12
Ekspresi ini menjamin kesalahan minimum di seluruh ruang yang direntang oleh ciri k di W. Peluang posterior, P (Ci | w), dapat dihitung dengan menggunakan aturan Bayes:
( | ( |
NBC mengasumsikan kondisi independen antara spaced k-mer ciri dan menghitung peluang bersyarat dari kelas sebagai produk K individual probabilitas.
Peluang bersyarat individu, P (Wj | Ci), dapat diestimasi dari data latih, numerik, maka diasumsikan mempunyai distribusi Gaussian. Jadi, dalam kasus ini berlaku: nilai (frekuensi) dari masing-masing atribut (dalam hal ini adalah spaced K-mer) dari semua sampel dalam kelas yang sama (Han & Kamber, 2001).
Untuk mengklasifikasikan suatu sample , ( | ) ( i) dievaluasi data latih dengan akurasi tinggi akan divalidasi dengan data uji menggunakan NBC.
13 Pengujian NBC
Pengujian akan mengklasifikasikan data uji sebanyak 9 mikroorganisme ke dalam kelas dalam genusnya masing-masing. Setiap mikroorganisme akan dikelaskan dan hasil pengkelasan tersebut akan dihitung berapa persen mikroorganisme yang telah dikelaskan dengan benar.
Analisis
Dari hasil pelatihan dan pengujian NBC akan didapatkan hasil untuk kinerja pengujian NBC dalam klasifikasi fragmen metagenome ini. Matriks konvolusi akan merepresentasikan hasil klasifikasi jumlah fragmen dari tiap kelas. Akurasi untuk hasil klasifikasi dapat dicari dengan:
Akurasi = ∑data u i b na
∑data u i 1
HASIL DAN PEMBAHASAN
Praproses Data
Sequences DNA metagenome yang sudah dipilih akan diuraikan fragmennya menggunakan perangkat lunak MetaSim. Data yang diproses akan dibaca berkali-kali sesuai dengan kebutuhan penelitian. Pada data set kecil, data yang dipersiapkan akan dibaca sebanyak 10 000 kali untuk keperluan data latih sehingga jumlah pembacaan untuk masing-masing organisme ini adalah 1 000 kali pembacaan. Panjang fragmen yang digunakan adalah 500 bp, 1 kbp, 5 kbp, dan 10 kbp. Data uji pada data set kecil akan dibaca sebanyak 5 000 kali. Untuk data uji, fragmen yang digunakan adalah fragmen dengan panjang 500 bp. Keluaran dari pengolahan MetaSim ini adalah fail FASTA yang berisi sequences DNA yang sudah terfragmen sesuai dengan kriteria parameter yang diinginkan. Berikut screenshoot sequences DNA yang dapat dilihat pada Gambar 9.
14
Ekstraksi Ciri
Ekstraksi Ciri pada penelitian ini adalah dengan melakukan pembacaan frekuensi nukleotida dengan k-mer dan spaced k-mer pada sequences DNA yang telah di-generate menggunakan MetaSim. K-mer akan menampilkan pola kemunculan k pada suatu waktu dalam suatu sequences. Contoh, jika hendak menghitung trinukleotida, dihitung empat base utama (A, T, G, C) dipangkat dengan jumlah k. Hasilnya, untuk trinukleotida adalah 43 = 64 base pair (bp).
Pada penelitian ini k-mers yang digunakan adalah k = 3 maka ada 43 = 64 pola kemunculan yang terbentuk. Pola kemunculan tersebut direpresentasikan oleh jumlah atribut pada data yaitu X1, X2, …, X64. Jumlah pembacaan yang digunakan pada data latih yaitu 10 000 pembacaan mewakili jumlah baris fragmen metagenome pada data, serta 5000 pembacaan pada data uji baru. Adapun untuk atribut kelas terdiri atas 3 genus yang berbeda. Perhitungan frekuensi k-mers pada sequences DNA diilustrasikan pada Gambar 10.
Selain menggunakan frekuensi k-mer, digunakan spaced k-mer yang memperhitungkan kondisi d ’ ca . Spaced k-mer dikemukakan oleh Kusuma (2012) yang mencari akurasi terbaik dari dan , dengan adalah weight of pattern yang merepresentasikan banyaknya posisi yang sesuai atau matching positions (nilai 1) adapun adalah posisi dari kondisi d ’ ca (*). Dari hasil percobaan, didapatkan hasil akurasi terbaik adalah pada pola 111 1*11 1**11. Metode ini akan memeriksa frekuensi nukleotida dari fragmen DNA mulai dari AAA - CCC, A*AA - C*CC, dan A**AA - C**CC. Pengertian dari simbol * (d ’ ca ) pada fragmen DNA yang diperiksa adalah dapat merupakan basa apapun, baik A, T, G, maupun C. Adapun untuk symbol **, berarti diperbolehkan basa manapun mengisi 2 bit tersebut. Sehingga kondisi itu dapat diisi oleh 24 pasang basa mulai dari AA, AC, AT, AG, dan seterusnya.
Oleh karena itu, banyaknya pola kemunculan yang terbentuk pada perhitungan frekuensi spaced k-mers adalah sebanyak 192 pola kemunculan. Pola kemunculan tersebut mewakili jumlah atribut pada data yaitu X1, X2, …, X192. Jumlah pembacaan yang digunakan pada data latih yaitu 10 000 pembacaan mewakili jumlah baris fragmen metagenome pada data, serta 5000 pembacaan pada data uji baru. Adapun untuk atribut kelas terdiri atas 3 genus yang berbeda. Perhitungan frekuensi spaced k-mers pada sequences DNA diilustrasikan pada Gambar 11.
15
Hasil dari ekstraksi ciri tersebut adalah vektor masukkan yang besarnya dimensi data set D adalah , dengan baris m adalah jumlah pembacaan data yang di generate dan kolom n adalah jumlah kombinasi dari k yang digunakan. Jadi jumlah kombinasi yang terbentuk pada k-mers untuk k = 3 (trinukleotida) adalah 43 = 64 kombinasi sedangkan pada spaced k-mers untuk k = 3 adalah 192 kombinasi. Hasil perhitungan frekuensi oligonukleotida yang berupa array m x n akan digunakan dalam proses klasifikasi. Hasil perhitungan frekuensi spaced k-mers pada sequences DNA diilustrasikan pada Tabel 4.
Gambar 11 Jumlah kombinasi pada spaced k-mersk = 3
16
Reduksi Data dengan PCA
Analisis komponen utama bertujuan untuk mereduksi dimensi data asal yang semula terdapat p variabel bebas menjadi q komponen utama (dimana q < p). Penggunaan metode ekstraksi ciri spaced k-mers dengan pola w = 3 dan d = 0, 1, 2 menghasilkan array m jumlah pembacaan data x 192 kombinasi. Di samping itu, ekstraksi ciri K-mers trinukleotida menghasilkan arraym jumlah pembacaan data x 64 kombinasi. Dimensi data tersebut perlu direduksi tanpa adanya pengurangan karakteristik data secara signifikan sehingga lebih mudah untuk menginterpretasikannya. Pada penelitian ini proporsi kumulatif keragaman data asal yang dipilih adalah sebesar 97%. Pemilihan tersebut berdasarkan teknik mencoba-coba setelah mencoba proporsi yang lain yakni 95% hingga 99%.
K -fold cross validation
Setelah mereduksi data menggunakan PCA dengan threshold 0.97, data set akan dilatih dengan menggunakan k-fold cross validation yang digunakan untuk membagi data menjadi data latih dan data uji. Pada penelitian ini k yang digunakan adalah 5. Data akan dibagi menjadi 5 bagian di mana 4 bagian akan menjadi data latih, dan 1 bagian sisanya akan digunakan untuk validasi. Pada data set kecil, dari 10 000 fragemen tersebut, 8000 fragmen sebagai data latih dan 2000 fragmen menjadi data uji untuk validasi.
Naïve Bayes Classifier (NBC)
Jika nilai atribut dari data adalah continuous-valued atau data numerik, maka diasumsikan mempunyai distribusi Gaussian. Dalam kasus ini berlaku:
( | i) g ( i i) 1 penelitian ini genus sebagai kelas) dihitung mean dan standar deviasinya. Mean dan standar deviasi dari masing-masing atribut seluruh fragmen yang berasal dari kelas yang sama akan digunakan untuk menghitung peluang dalam Gaussian (normal) density function. Perhitungan mean dan standard deviasi pada data diilustrasikan pada Gambar 12. Untuk mengklasifikasikan suatu sample ,
( | i) ( i) dievaluasi untuk tiap kelas i. Sample diklasifikasikan ke
dalam kelas i jika dan hanya jika:
17
Model
Pada proses pelatihan NBC sebelumnya, model yang berupa bagian dari data latih dengan akurasi tinggi akan divalidasi dengan data uji baru yang masih belum digunakan pada data latih menggunakan NBC.
Pengujian NBC dan Analisis
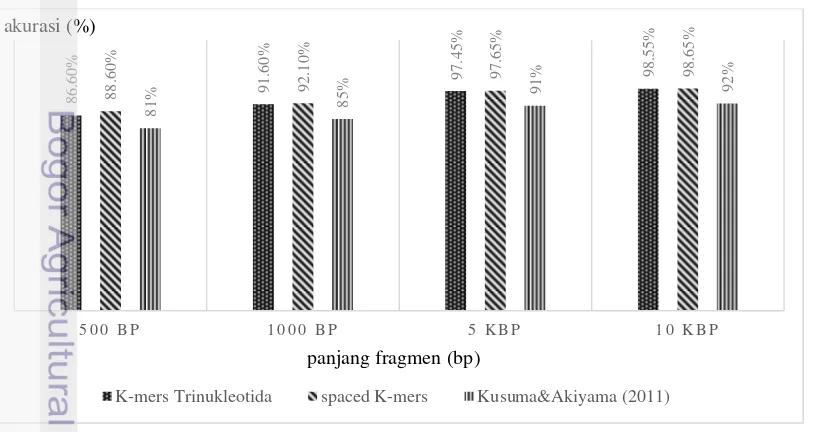
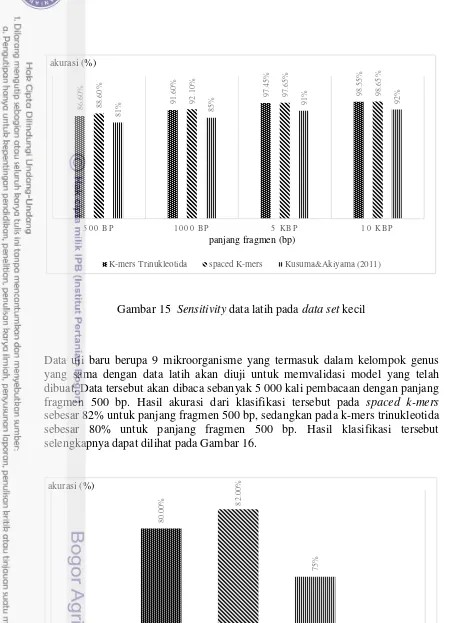
Pada data set kecil, data latih yang di-generate ada sebanyak 10 000 fragmen dengan rincian jumlah pembacaan untuk masing-masing organisme adalah 1 000 kali pembacaan sehingga informasi yang terdapat dalam data lebih banyak. Matriks konfusion akan merepresentasikan hasil klasifikasi untuk masing-masing kelas. Akurasi akan dihitung berdasarkan jumlah data uji yang benar diklasifikasikan ke dalam kelasnya dibagi dengan jumlah seluruh data uji kemudian dikalikan 100%. Sensitifity digunakan untuk menghitung akurasi dari tiap kelas. Sebagai contoh perhitungan, dari 528 fragmen yang seharusnya masuk kelas genus Agrobacterium, 523 fragmen berhasil dikelaskan ke dalam kelas genus Agrobacterium, sedangkan 5 fragmen dianggap masuk ke kelas genus Bacillus. Sensitifity untuk kelas Agrobacterium adalah 523 fragmen yang diklasifikasikan dengan benar dibagi dengan 528 fragmen yang seharusnya diklasifikasikan ke dalam kelas genus Agrobacterium kemudian dikalikan 100 %. Begitu juga sensitifity untuk kelas genus Bacillus dan Staphylococcus dihitung dengan cara yang sama. Sensitifity keseluruhan adalah dengan mencari rata-rata dari sensitifity seluruh kelas. Confusion matrix data latih pembacaan 10K panjang fragmen 10 K pada data set kecil dapat dilihat pada Gambar 13. Adapun akurasi dan sensitifity untuk data latih pada data set kecil selengkapanya dapat dilihat pada Gambar 14 dan Gambar 15.
18
Gambar 13 menunjukkan bahwa jumlah data uji yang digunakan adalah 2000 fragmen. Hal ini dapat diketahui dengan menjumlahkan angka-angka yang tertera pada matriks tersebut. Pada baris pertama menunjukkan bahwa dari 528 fragmen pada kelas genus Agrobacterium, 523 fragmen benar diklasifikasikan ke dalam kelas genus Agrobacterium sedangkan 5 fragmen salah diklasifikasikan ke dalam kelas genus Bacillus. Pada baris kedua menunjukkan bahwa dari 1014 fragmen yang sebenarnya adalah kelas genus Bacillus, 1006 fragmen benar diklasifikasikan ke dalam kelas Bacillus sedangkan 8 fragmen salah diklasifikasikan ke dalam kelas genus Staphylococcus. Adapun pada baris ketiga menunjukkan bahwa dari 458 fragmen yang sebenarnya adalah kelas genus Staphylococcus, 444 fragmen benar diklasifikasikan ke dalam kelas genus Staphylococcus sedangkan 14 salah diklasifikasikan ke dalam kelas Bacillus. Akurasi yang dihasilkan adalah sebesar 98.65 %.
Gambar 13 Confusion matrix untuk data latih untuk pembacaan 10K panjang fragmen 10 K pada data set kecil
Gambar 14 Akurasi data latih pada data set kecil
8
19
Gambar 15 Sensitivity data latih pada data set kecil
Data uji baru berupa 9 mikroorganisme yang termasuk dalam kelompok genus yang sama dengan data latih akan diuji untuk memvalidasi model yang telah dibuat. Data tersebut akan dibaca sebanyak 5 000 kali pembacaan dengan panjang fragmen 500 bp. Hasil akurasi dari klasifikasi tersebut pada spaced k-mers sebesar 82% untuk panjang fragmen 500 bp, sedangkan pada k-mers trinukleotida sebesar 80% untuk panjang fragmen 500 bp. Hasil klasifikasi tersebut selengkapnya dapat dilihat pada Gambar 16.
Gambar 16 Akurasi data uji baru data set kecil
K-mers Trinukleotida spaced K-mers Kusuma&Akiyama (2011)
8
20
Dari hasil tersebut, dapat diketahui bahwa metode ektraksi ciri spaced k-mers menghasilkan akurasi dan sensitifity yang lebih tinggi dibandingkan dengan ekstraksi ciri k-mers trinukleotida. Pereduksian data sebelum proses klasifikasi juga dapat memberikan hasil yang lebih baik karena hanya informasi yang penting saja yang digunakan. Selain itu dapat dilihat juga bahwa panjang fragmen mempengaruhi hasil klasifikasi. Semakin panjang fragmen yang digunakan, semakin banyak juga informasi dari organisme tersebut, maka hasil klasifikasi akan semakin baik. Untuk klasifikasi pada data latih, penelitian ini juga menghasilkan akurasi yang lebih tinggi jika dibandingkan dengan hasil penelitian Kusuma & Akiyama (2011). Hasil klasifikasi pada data uji organisme baru juga tidak terlampau jauh jika dibandingkan dengan penelitian Ananta & Akiyama (2011).
Percobaan Dataset Besar
Data set besar terdiri atas 381 organisme yang termasuk ke dalam 48 genus sebagai data latihnya. Data tersebut dibaca 9600 pembacaan sehingga banyaknya record pada data adalah 9600 fragmen dan panjang fragmen yang digunakan adalah 500 bp. Dengan menggunakan 5-fold cross validation maka dari 9600 fragmen, 7680 digunakan sebagai data latih dan 1920 fragmen digunakan sebagai data uji. Data tersebut diekstraksi menggunakan metode k-mers dan spaced k-mers.
Metode penelitian yang diterapkan pada data set besar sama dengan metode yang diterapkan pada data set 10 organisme. Hanya saja percobaan pada data set besar ini belum mempertimbangkan kondisi imbalanced data dimana jumlah fragmen yang mewakili masing – masing genus (sebagai kelas) tidak sama rata. Hal ini dilakukan untuk mengetahui pengaruh kondisi imbalanced data pada hasil klasifikasi. Hasil akurasi untuk dataset besar selengkapnya dapat dilihat pada Gambar 17.
Gambar 17 Akurasi hasil klasifikasi data set besar
21
SIMPULAN DAN SARAN
Simpulan
Berdasarkan hasil yang diperoleh dari penelitian yang telah dilakukan, dapat disimpulkan bahwa:
1. Metode klasifikasi dengan menggunakan NBC dan ekstraksi ciri spaced k-mers maupun k-mers berhasil mengklasifikasikan fragmen metagenome berukuran pendek (500 bp) pada level genus.
2. Akurasi hasil klasifikasi menggunakan NBC dapat lebih ditingkatkan dengan menggunakan metode ekstraksi ciri spaced K-mers. Selain itu, metode ekstraksi ciri spaced K-mers dapat memberikan hasil pengklasifikasian dengan menggunakan NBC lebih baik dibandingkan dengan menggunakan metode ekstraksi ciri k-mers.
3. Hasil klasifikasi fragmen metagenome dengan menggunakan metode ekstraksi ciri spaced k-mers secara konsisten menunjukkan hasil yang lebih tinggi baik pada data set kecil maupun data set besar dibandingkan dengan menggunakan metode ekstraksi ciri k-mers.
Saran
Akurasi hasil klasifikasi dari data set besar mungkin masih dapat ditingkatkan dengan mempertimbangkan kondisi imbalanced data serta meningkatkan jumlah pembacaan sehingga akan semakin banyak informasi yang dapat meningkatkan hasil klasifikasi.
DAFTAR PUSTAKA
Amano K, Nakamura H, Ichikawa H. 2003. Self-organizing clustering: non-hierarchical method for clustering large amountof sequence DNAs. Genome Informatics. 14: 575-576
Amano K, Nakamura H, Ichikawa H, Numa H, Kobayashi KF, Nagamura Y, Onodera N. 2007. Self-organizing clustering: non-hierarchical clustering for large-scale sequence DNA data. IPSJ Digital Courier. 2(2):523-527.
Brady A, Salzberg SL. 2009. Phymm and PhymmBL: Metagenomic phylogenetic classification with interpolated markov models. Nat Methods. 6(9):673– 676. doi : 10.1038/nmeth.1358
Chan CK, Hsu AL, Tang SL, Halgamuge SK. 2007. Using growing self-organizing maps to prove the binning process in environmental whole-genome shotgun equencing. Journal of Biomedicine and Biotechnology. 2008. doi:10.1155/2008/513701
22
Harayama S, Kasai Y, Hara A. 2004. Microbial Communities in Oil-contaminated Seawater. Current Opinion in Biotechnology. 15:205-214.
Hsu AL, Halgamuge SK. 2002. Enhancement of topology preseration and hierarchical dynamic self-organising maps for data visualisation. International Journal of Approximate Reasoning. 32(2003):259-279.
Johnson RA, Wichern DW. 2002. Applied Multivariate Statistical Analysis, 5th End. New Jersey: Prentice Hall.
Kusuma WA, Akiyama Y. 2011. Metagenome fragment binning based on characterization vectors. Di dalam: Proceeding International Conferences on Bioinformatics and Biomedical Technology; 2011; Sanya, China.
Kusuma WA. 2012. Combined approaches for improving the performance of de novo dna sequence assembly and metagenomic classification of short fragments from next generation sequencer [disertasi]. Tokyo (JP): Tokyo Institute of Technology.
Manning CD, Raghavan P, Schutze H. 2009. An Introduction to Information Retrieval. Cambridge(UK): Cambridge University Pr.
McHardy AC, Martin HG, Tsirigos A, Hugenholtz P, Rigoutsos I. 2007. Accurate phylogenetic classification of variable-lenght dna fragments. Nat Methods. 4(1):63-72
Rosen G, Garbarine E, Caseiro D, Polikar R, Sokhansanj. 2008. Metagenome fragment classification using n-mer frequency profiles. Advances in Bioinformatics. doi:10.1155/2008/205969.
Teeling H, Waldmann J, Lombardot T, Bauer M, Glockner FO. 2004. TETRA : a web service and stand-alone program for the analysis and comparison of tetranucleotide usage pattern in sequence DNAs. BMC Informatics. 5(163). doi:10.1186/1471-2105-5-163.
Thontowi A. 2009. Pendekatan metagenomik dan bioinformatika untuk menganalisis komunitas mikroba laut Indonesia. SIGMA. 12(1):15-22.
RIWAYAT HIDUP
Penulis dilahirkan di Jakarta pada tanggal 08 Desember 1990. Penulis merupakan anak kedua dari 4 bersaudara pasangan Bapak Djaelani dan Ibu Alfianti. Pada tahun 2009 penulis lulus dari SMA Negeri 6 Bogor. Pada bulan Juli 2009 penulis resmi menjadi mahasiswa Institut Pertanian Bogor melalui jalur PMDK. Setelah menyelesaikan Tingkat I (Tingkat Persiapan Bersama) di IPB pada tahun 2010, penulis diterima sebagai mahasiswa Departemen Ilmu Komputer, Fakultas Matematika dan Ilmu Pengetahuan Alam.
24
LAMPIRAN
Lampiran 1 Daftar 381 organisme yang termasuk ke dalam 48 genus
Spesies Genus Jumlah
Fragmen
Bacillus amyloliquefaciens FZB42'
Bacillus 587
Bacillus anthracis str. 'Ames Ancestor'' Bacillus anthracis str. Ames chromosome' Bacillus anthracis str. Sterne chromosome' Bacillus cereus ATCC 10987 chromosome' Bacillus cereus ATCC 14579'
Bacillus cereus E33L'
Bacillus cereus subsp. cytotoxis NVH 391-98' Bacillus clausii KSM-K16'
Bacillus halodurans C-125 chromosome' Bacillus licheniformis ATCC 14580'
Bacillus subtilis subsp. subtilis str. 168 chromosome'
Bacillus thuringiensis serovar konkukian str. 97-27 chromosome' Bacillus thuringiensis str. Al Hakam chromosome'
Bacillus weihenstephanensis KBAB4'
Bacteroides fragilis NCTC 9343 chromosome' Bacteroides fragilis YCH46 chromosome'
Bacteroides thetaiotaomicron VPI-5482 chromosome'
Bacteroides vulgatus ATCC 8482 chromosome' Bacteroides 178
Bartonella bacilliformis KC583' Bartonella henselae str. Houston-1' Bartonella quintana str. Toulouse'
Bartonella tribocorum CIP 105476' Bartonella 76
Bordetella avium 197N chromosome' Bordetella bronchiseptica RB50' Bordetella parapertussis 12822' Bordetella pertussis Tohama I'
Bordetella petrii DSM 12804' Bordetella 187
Borrelia afzelii PKo' Borrelia duttonii Ly'
Borrelia garinii PBi chromosome chromosome linear' Borrelia hermsii DAH chromosome'
Borrelia recurrentis A1'
Borrelia turicatae 91E135 chromosome' Borrelia 43
Bradyrhizobium japonicum USDA 110 chromosome' Bradyrhizobium sp. BTAi1 chromosome'
Bradyrhizobium sp. ORS278 chromosome' Bradyrhizobium 188
Brucella abortus S19 chromosome 1'
25 Brucella canis ATCC 23365 chromosome I'
Brucella melitensis biovar Abortus 2308 chromosome I Brucella melitensis bv. 1 str. 16M chromosome chromosome I' Brucella ovis ATCC 25840 chromosome chromosome I' Brucella suis 1330 chromosome chromosome I'
Brucella suis ATCC 23445 chromosome I' Brucella 139
Burkholderia ambifaria AMMD chromosome chromosome 1' Burkholderia ambifaria MC40-6 chromosome chromosome 1' Burkholderia cenocepacia AU 1054 chromosome 3'
Burkholderia cenocepacia HI2424 chromosome chromosome 1' Burkholderia cenocepacia J2315 chromosome chromosome 1' Burkholderia cenocepacia MC0-3 chromosome chromosome 1' Burkholderia mallei ATCC 23344 chromosome chromosome 1' Burkholderia mallei NCTC 10229 chromosome I'
Burkholderia mallei NCTC 10247 chromosome I' Burkholderia mallei SAVP1 chromosome I'
Burkholderia multivorans ATCC 17616 chromosome chromosome 1' Burkholderia phymatum STM815 chromosome chromosome 1' Burkholderia phytofirmans PsJN chromosome chromosome 1' Burkholderia pseudomallei 1106a chromosome I'
Burkholderia pseudomallei 1710b chromosome chromosome I' Burkholderia pseudomallei 668 chromosome I'
Burkholderia pseudomallei K96243 chromosome chromosome 1' Burkholderia sp. 383 chromosome 1'
Burkholderia sp. 383 chromosome chromosome 2'
Burkholderia thailandensis E264 chromosome chromosome I' Burkholderia vietnamiensis G4 chromosome chromosome 1'
Burkholderia xenovorans LB400 chromosome 1' Burkholderia 612
Campylobacter concisus 13826'
Campylobacter curvus 525.92 chromosome' Campylobacter fetus subsp. fetus 82-40' Campylobacter hominis ATCC BAA-381' Campylobacter jejuni RM1221'
Campylobacter jejuni subsp. doylei 269.97'
Campylobacter jejuni subsp. jejuni NCTC 11168 chromosome' Campylobacter 94
Candidatus Phytoplasma australiense' Candidatus Phytoplasma mali'
Onion yellows phytoplasma OY-M' Candidatus 17
26
Chlamydophila pneumoniae TW-183' Chlamydophila 74
Chlorobium chlorochromatii CaD3 chromosome' Chlorobium limicola DSM 245 chromosome' Chlorobium luteolum DSM 273 chromosome' Chlorobium phaeobacteroides BS1 chromosome' Chlorobium phaeobacteroides DSM 266 chromosome' Chlorobium phaeovibrioides DSM 265 chromosome'
Chlorobium tepidum TLS' Chlorobium 164
Clostridium acetobutylicum ATCC 824'
Clostridium beijerinckii NCIMB 8052 chromosome' Clostridium botulinum A str. ATCC 19397'
Clostridium botulinum A str. ATCC 3502' Clostridium botulinum A str. Hall'
Clostridium botulinum A3 str. Loch Maree' Clostridium botulinum B str. Eklund 17B' Clostridium botulinum B1 str. Okra' Clostridium botulinum E3 str. Alaska E43' Clostridium botulinum F str. Langeland' Clostridium difficile 630 chromosome'
Clostridium thermocellum ATCC 27405 chromosome' Clostridium 511
Corynebacterium diphtheriae NCTC 13129 chromosome' Corynebacterium efficiens YS-314'
Corynebacterium glutamicum ATCC 13032' Corynebacterium glutamicum R chromosome' Corynebacterium jeikeium K411'
Corynebacterium urealyticum DSM 7109' Corynebacterium 137
Cupriavidus metallidurans CH34 chromosome' Cupriavidus necator N-1 chromosome chromosome 1'
Cupriavidus taiwanensis LMG 19424 chromosome 1' Cupriavidus 81
Dehalococcoides ethenogenes 195' Dehalococcoides sp. BAV1'
Dehalococcoides sp. CBDB1 chromosome' Dehalococcoides 36
Ehrlichia canis str. Jake'
Ehrlichia chaffeensis str. Arkansas' Ehrlichia ruminantium str. Gardel'
Ehrlichia ruminantium str. Welgevonden' Ehrlichia 35
Francisella philomiragia subsp. philomiragia ATCC 25017 chromosome' Francisella tularensis subsp. holarctica FTNF002-00
27 Francisella tularensis subsp. holarctica LVS chromosome'
Francisella tularensis subsp. holarctica OSU18' Francisella tularensis subsp. mediasiatica FSC147' Francisella tularensis subsp. novicida U112' Francisella tularensis subsp. tularensis FSC198' Francisella tularensis subsp. tularensis SCHU S4'
Francisella tularensis subsp. tularensis WY96-3418' Francisella 117
Frankia alni ACN14a chromosome' Frankia sp. CcI3 chromosome'
Frankia sp. EAN1pec chromosome' Frankia 183
Geobacter bemidjiensis Bem chromosome' Geobacter lovleyi SZ chromosome'
Geobacter metallireducens GS-15 chromosome' Geobacter sulfurreducens PCA chromosome'
Geobacter uraniireducens Rf4 chromosome' Geobacter 171
Haemophilus ducreyi 35000HP'
Haemophilus influenzae 86-028NP chromosome' Haemophilus influenzae PittEE chromosome' Haemophilus influenzae PittGG chromosome' Haemophilus influenzae Rd KW20 chromosome' Haemophilus somnus 129PT chromosome'
Haemophilus somnus 2336 chromosome' Haemophilus 116
Helicobacter acinonychis str. Sheeba chromosome' Helicobacter hepaticus ATCC 51449 chromosome' Helicobacter pylori 26695'
Helicobacter pylori G27 chromosome' Helicobacter pylori HPAG1 chromosome' Helicobacter pylori J99'
Helicobacter pylori P12 chromosome'
Helicobacter pylori Shi470 chromosome' Helicobacter 102
Lactobacillus acidophilus NCFM chromosome' Lactobacillus brevis ATCC 367'
Lactobacillus casei ATCC 334' Lactobacillus casei BL23 chromosome'
Lactobacillus delbrueckii subsp. bulgaricus ATCC 11842'
Lactobacillus delbrueckii subsp. bulgaricus ATCC BAA-365 chromosome' Lactobacillus fermentum IFO 3956'
Lactobacillus gasseri ATCC 33323' Lactobacillus helveticus DPC 4571' Lactobacillus johnsonii NCC 533' Lactobacillus plantarum WCFS1'
Lactobacillus reuteri DSM 20016 chromosome' Lactobacillus reuteri JCM 1112'
28
Lactobacillus salivarius UCC118' Lactobacillus 250
Leptospira biflexa serovar Patoc strain 'Patoc 1 (Ames)' chromosome chromosome I' Leptospira biflexa serovar Patoc strain 'Patoc 1 (Paris)' chromosome chromosome I' Leptospira borgpetersenii serovar Hardjo-bovis L550 chromosome 1'
Leptospira interrogans serovar Copenhageni str. Fiocruz L1-130 chromosome chromosome I' Leptospira interrogans serovar Lai str. 56601 chromosome
chromosome I'
Leptospira 161
Listeria innocua Clip11262' Listeria monocytogenes EGD-e'
Listeria monocytogenes serotype 4b str. F2365 chromosome'
Listeria welshimeri serovar 6b str. SLCC5334' Listeria 78
Methanococcus maripaludis C5 chromosome' Methanococcus maripaludis C6 chromosome' Methanococcus maripaludis C7 chromosome'
Methanococcus maripaludis S2 chromosome' Methanococcus 51
Methanosarcina acetivorans C2A chromosome' Methanosarcina barkeri str. Fusaro chromosome'
Methanosarcina mazei Go1 chromosome' Methanosarcina 100
Methylobacterium extorquens PA1 chromosome' Methylobacterium populi BJ001 chromosome'
Methylobacterium radiotolerans JCM 2831 chromosome'
Methylobacterium sp. 4-46 chromosome' Methylobacterium 196
Mycobacterium abscessus ATCC 19977 chromosome chromosome 1' Mycobacterium avium 104'
Mycobacterium avium subsp. paratuberculosis K-10' Mycobacterium bovis AF2122/97'
Mycobacterium bovis BCG str. Pasteur 1173P2' Mycobacterium gilvum PYR-GCK chromosome' Mycobacterium leprae TN chromosome'
Mycobacterium marinum M'
Mycobacterium smegmatis str. MC2 155' Mycobacterium sp. JLS chromosome'
Mycobacterium vanbaalenii PYR-1 chromosome' Mycobacterium 723
Mycoplasma agalactiae PG2' Mycoplasma arthritidis 158L3-1'
29 Mycoplasma hyopneumoniae 232'
Mycoplasma hyopneumoniae 7448 chromosome' Mycoplasma hyopneumoniae J chromosome' Mycoplasma mobile 163K'
Mycoplasma mycoides subsp. mycoides SC str. PG1 chromosome'
Mycoplasma penetrans HF-2' Mycoplasma pneumoniae M129' Mycoplasma pulmonis UAB CTIP'
Mycoplasma synoviae 53' Mycoplasma 112
Pseudomonas aeruginosa PA7'
Pseudomonas syringae pv. phaseolicola 1448A chromosome' Pseudomonas syringae pv. syringae B728a'
Pseudomonas syringae pv. tomato str. DC3000 chromosome' Pseudomonas syringae pv. tomato str. DC3000 plasmid pDC3000A'
Pseudomonas 600
Psychrobacter arcticus 273-4'
Psychrobacter cryohalolentis K5 chromosome'
Psychrobacter sp. PRwf-1 chromosome' Psychrobacter 71
Pyrobaculum aerophilum str. IM2 chromosome' Pyrobaculum arsenaticum DSM 13514'
Pyrobaculum calidifontis JCM 11548 chromosome'
Pyrobaculum islandicum DSM 4184 chromosome' Pyrobaculum 52
Pyrococcus abyssi GE5 chromosome' Pyrococcus furiosus DSM 3638'
Pyrococcus horikoshii OT3 chromosome' Pyrococcus 40
Rickettsia akari str. Hartford' Rickettsia bellii OSU 85-389' Rickettsia bellii RML369-C' Rickettsia canadensis str. McKiel' Rickettsia conorii str. Malish 7' Rickettsia felis URRWXCal2' Rickettsia massiliae MTU5'
Rickettsia prowazekii str. Madrid E chromosome' Rickettsia rickettsii str. 'Sheila Smith''
Rickettsia rickettsii str. Iowa chromosome'
30
Shewanella pealeana ATCC 700345 chromosome' Shewanella putrefaciens CN-32 chromosome' Shewanella sediminis HAW-EB3'
Shewanella sp. ANA-3 chromosome chromosome 1' Shewanella sp. MR-4 chromosome'
Shewanella sp. MR-7 chromosome' Shewanella sp. W3-18-1 chromosome'
Shewanella woodyi ATCC 51908 chromosome' Shewanella 574
Shigella boydii CDC 3083-94 chromosome' Shigella boydii Sb227'
Shigella dysenteriae Sd197' Shigella flexneri 2a str. 2457T'
Shigella flexneri 2a str. 301 chromosome' Shigella flexneri 5 str. 8401 chromosome'
Shigella sonnei Ss046 chromosome' Shigella 248
Staphylococcus aureus RF122'
Staphylococcus aureus subsp. aureus COL chromosome' Staphylococcus aureus subsp. aureus JH1'
Staphylococcus aureus subsp. aureus JH9'
Staphylococcus aureus subsp. aureus MRSA252 chromosome' Staphylococcus aureus subsp. aureus MSSA476 chromosome' Staphylococcus aureus subsp. aureus MW2'
Staphylococcus aureus subsp. aureus Mu3' Staphylococcus aureus subsp. aureus Mu50' Staphylococcus aureus subsp. aureus N315'
Staphylococcus aureus subsp. aureus NCTC 8325 chromosome' Staphylococcus aureus subsp. aureus USA300_FPR3757 chromosome' Staphylococcus aureus subsp. aureus USA300_TCH1516 chromosome' Staphylococcus aureus subsp. aureus str. Newman chromosome' Staphylococcus epidermidis ATCC 12228 chromosome' Staphylococcus epidermidis RP62A'
Staphylococcus haemolyticus JCSC1435 chromosome' Staphylococcus saprophyticus subsp. saprophyticus ATCC 15305'
Staphylococcus 438
Streptococcus agalactiae 2603V/R' Streptococcus agalactiae A909' Streptococcus agalactiae NEM316'
31 Streptococcus gordonii str. Challis substr. CH1'
Streptococcus mutans UA159 chromosome' Streptococcus pyogenes M1 GAS chromosome' Streptococcus pyogenes MGAS10270 chromosome'
Streptococcus thermophilus LMG 18311 chromosome' Streptococcus 473
Streptomyces avermitilis MA-4680'
Streptomyces coelicolor A3(2) chromosome'
Streptomyces griseus subsp. griseus NBRC 13350' Streptomyces 198
Sulfolobus acidocaldarius DSM 639 chromosome' Sulfolobus solfataricus P2 chromosome'
Sulfolobus tokodaii str. 7 chromosome' Sulfolobus 62
Synechococcus elongatus PCC 6301 chromosome' Synechococcus elongatus PCC 7942 chromosome' Synechococcus sp. CC9311'
Synechococcus sp. CC9605'
Synechococcus sp. CC9902 chromosome' Synechococcus sp. JA-2-3B'a(2-13)' Synechococcus sp. JA-3-3Ab'
Synechococcus sp. PCC 7002 chromosome' Synechococcus sp. RCC307'
Synechococcus sp. WH 7803'
32
Thermoanaerobacter pseudethanolicus ATCC 33223 chromosome'
Thermoanaerobacter sp. X514 chromosome'
Thermoanaerobacter tengcongensis MB4' Thermoanaerobacter 64
Thermotoga lettingae TMO chromosome' Thermotoga maritima MSB8 chromosome' Thermotoga petrophila RKU-1 chromosome'
Thermotoga sp. RQ2 chromosome' Thermotoga 62
Vibrio cholerae O1 biovar El Tor str. N16961 chromosome chromosome I' Vibrio cholerae O395 chromosome 1'
Vibrio fischeri ES114 chromosome I' Vibrio fischeri MJ11 chromosome I'
Vibrio vulnificus CMCP6 chromosome chromosome I'
Vibrio vulnificus YJ016 chromosome I' Vibrio 120
Wolbachia endosymbiont of Culex quinquefasciatus Pel' Wolbachia endosymbiont of Drosophila melanogaster'
Wolbachia endosymbiont strain TRS of Brugia malayi' Wolbachia 28
Xanthomonas axonopodis pv. citri str. 306 chromosome' Xanthomonas campestris pv. campestris str. 8004 chromosome' Xanthomonas campestris pv. campestris str. ATCC 33913' Xanthomonas campestris pv. campestris str. B100'
Xanthomonas campestris pv. vesicatoria str. 85-10 chromosome' Xanthomonas oryzae pv. oryzae KACC10331 chromosome' Xanthomonas oryzae pv. oryzae MAFF 311018'
Xanthomonas oryzae pv. oryzae PXO99A' Xanthomonas 295
Yersinia enterocolitica subsp. enterocolitica 8081' Yersinia pestis Angola'
Yersinia pestis Antiqua chromosome' Yersinia pestis CO92 chromosome' Yersinia pestis KIM 10 chromosome' Yersinia pestis Nepal516'
Yersinia pestis Pestoides F chromosome' Yersinia pseudotuberculosis IP 31758' Yersinia pseudotuberculosis IP 32953'
Yersinia pseudotuberculosis PB1/+ chromosome'