ABSTRAK
ISMI WILLYSIA BILLIRANTAU. Konstruksi Gen Penyandi Kitinase pada
Vektor Ekspresi dengan Metode Gateway dan Transformasi pada
Agrobacterium
sp. Dibimbing oleh EDY DJAUHARI PK dan TETTY CHAIDAMSARI.
ABSTRACT
ISMI WILLYSIA BILLIRANTAU. Construction of Gene Chitinase in Expression
Vector by Method Gateway and Transformation to
Agrobacterium
sp. Under the
direction of EDY DJAUHARI PK and TETTY CHAIDAMSARI.
PENDAHULUAN
Kelapa sawit merupakan tumbuhan industri penting penghasil minyak goreng, minyak industri, maupun bahan bakar. Komoditas kelapa sawit menduduki peringkat ketiga penyumbang devisa nonmigas setelah karet dan kopi bagi Indonesia (Lubis 1992). Usaha peningkatan produksi perkebunan kelapa sawit tersebut secara terus menerus ternyata tidak terlepas dari masalah hama dan penyakit. Hama dan penyakit yang sering menyerang kelapa sawit umumnya disebabkan oleh cendawan patogen Ganoderma spp (Santoso et al. 2001). Penyakit yang disebabkan oleh Ganoderma spp menyerang bagian akar dan gejala penyakit yang ditimbulkannya tersebut hanya terlihat pada akhir infeksi sehingga tanaman tidak dapat diselamatkan. Penyakit tersebut dikenal dengan nama busuk akar (Singh 1991).
Pengendalian Ganoderma spp. sulit dilakukan karena ketika gejala dan tanda serangannya dapat diamati, tingkat serangannya sudah parah dan tanaman sudah tidak mungkin diselamatkan lagi sehingga perlu dilakukan pendekatan untuk menanggulangi masalah tersebut. Pengendalian penyakit busuk akar dapat dilakukan dengan pendekatan umum yaitu cara pendekatan kimia maupun cara hayati menggunakan biofungisida Trichoderma dan Penicilium, akan tetapi ini kurang efektif karena daya bunuh relatif lebih lama serta menimbulkan dampak negatif terhadap lingkungan seperti pencemaran tanah (Lubis 1992). Program pemuliaan tanaman dengan menemukan gen yang tahan terhadap Ganoderma spp. menjadi alternatif bagi pengendalian penyakit busuk akar, namun hingga saat ini tanaman yang dimaksud belum pernah ada sehingga untuk memecahkan masalah ini diperlukan pendekatan baru yaitu mendapatkan tanaman kelapa sawit yang tahan terhadap Ganoderma spp. dengan cara menyisipkan gen yang menghasilkan zat yang berfungsi sebagai antijamur, seperti enzim kitinase, glukanase maupun stilbena sintase yang dapat melisis dinding sel hifa jamur patogen.
Gen yang yang akan disisipkan dalam rekayasa genetika kelapa sawit diharapkan dapat diekspresikan secara terus-menerus sepanjang hidup tanaman kelapa sawit khususnya pada bagian perakaran, maka dari itu pemilihan promotor yang bersifat konstitutif serta cocok pada kelapa sawit memiliki peran yang sangat penting terhadap
pengendalian penyakit Ganoderma spp. Upaya yang dapat dilakukan adalah dengan menyisipkan gen penghasil zat yang berfungsi sebagai antijamur salah satunya adalah gen penyandi kitinase. Gen penyandi kitinase dapat diisolasi dari jamur Trichoderma harzianum yang telah terbukti memiliki agen pengendali biologis terhadap jamur patogen tanaman (Schirmbock et al. 1994; Lorito et al. 1996).
Penyisipan gen penyandi kitinase lengkap ke dalam vektor ekspresi dengan metode Gateway dilakukan untuk menguji apakah tanaman kelapa sawit dapat tahan terhadap Ganoderma spp. Gen penyandi kitinase yang telah tersisipkan dalam vektor ekspresi selanjutnya ditransformasikan ke dalam Agrobacterium tumefaciens. Metode Gateway digunakan untuk pengklonan karena merupakan metode yang masih baru, cepat dan efisien.
Penelitian ini bertujuan untuk membuat konstruksi gen penyandi kitinase dalam vektor ekspresi dengan metode Gateway yang cepat dan terarah. Hipotesis penelitian ini adalah gen penyandi kitinase dapat dikonstruksikan ke dalam vektor ekspresi menggunakan metode Gateway dan ditransformasikan pada Agrobacterium tumefaciens. Hasil penelitian ini diharapkan dapat menghasilkam bibit transgenik kelapa sawit yang mengekspresikan gen penyandi kitinase sehingga resisten terhadap Ganoderma dan memberikan informasi mengenai metode Gateway sebagai salah satu metode pengklonan.
TINJAUAN PUSTAKA
Kelapa Sawit
PENDAHULUAN
Kelapa sawit merupakan tumbuhan industri penting penghasil minyak goreng, minyak industri, maupun bahan bakar. Komoditas kelapa sawit menduduki peringkat ketiga penyumbang devisa nonmigas setelah karet dan kopi bagi Indonesia (Lubis 1992). Usaha peningkatan produksi perkebunan kelapa sawit tersebut secara terus menerus ternyata tidak terlepas dari masalah hama dan penyakit. Hama dan penyakit yang sering menyerang kelapa sawit umumnya disebabkan oleh cendawan patogen Ganoderma spp (Santoso et al. 2001). Penyakit yang disebabkan oleh Ganoderma spp menyerang bagian akar dan gejala penyakit yang ditimbulkannya tersebut hanya terlihat pada akhir infeksi sehingga tanaman tidak dapat diselamatkan. Penyakit tersebut dikenal dengan nama busuk akar (Singh 1991).
Pengendalian Ganoderma spp. sulit dilakukan karena ketika gejala dan tanda serangannya dapat diamati, tingkat serangannya sudah parah dan tanaman sudah tidak mungkin diselamatkan lagi sehingga perlu dilakukan pendekatan untuk menanggulangi masalah tersebut. Pengendalian penyakit busuk akar dapat dilakukan dengan pendekatan umum yaitu cara pendekatan kimia maupun cara hayati menggunakan biofungisida Trichoderma dan Penicilium, akan tetapi ini kurang efektif karena daya bunuh relatif lebih lama serta menimbulkan dampak negatif terhadap lingkungan seperti pencemaran tanah (Lubis 1992). Program pemuliaan tanaman dengan menemukan gen yang tahan terhadap Ganoderma spp. menjadi alternatif bagi pengendalian penyakit busuk akar, namun hingga saat ini tanaman yang dimaksud belum pernah ada sehingga untuk memecahkan masalah ini diperlukan pendekatan baru yaitu mendapatkan tanaman kelapa sawit yang tahan terhadap Ganoderma spp. dengan cara menyisipkan gen yang menghasilkan zat yang berfungsi sebagai antijamur, seperti enzim kitinase, glukanase maupun stilbena sintase yang dapat melisis dinding sel hifa jamur patogen.
Gen yang yang akan disisipkan dalam rekayasa genetika kelapa sawit diharapkan dapat diekspresikan secara terus-menerus sepanjang hidup tanaman kelapa sawit khususnya pada bagian perakaran, maka dari itu pemilihan promotor yang bersifat konstitutif serta cocok pada kelapa sawit memiliki peran yang sangat penting terhadap
pengendalian penyakit Ganoderma spp. Upaya yang dapat dilakukan adalah dengan menyisipkan gen penghasil zat yang berfungsi sebagai antijamur salah satunya adalah gen penyandi kitinase. Gen penyandi kitinase dapat diisolasi dari jamur Trichoderma harzianum yang telah terbukti memiliki agen pengendali biologis terhadap jamur patogen tanaman (Schirmbock et al. 1994; Lorito et al. 1996).
Penyisipan gen penyandi kitinase lengkap ke dalam vektor ekspresi dengan metode Gateway dilakukan untuk menguji apakah tanaman kelapa sawit dapat tahan terhadap Ganoderma spp. Gen penyandi kitinase yang telah tersisipkan dalam vektor ekspresi selanjutnya ditransformasikan ke dalam Agrobacterium tumefaciens. Metode Gateway digunakan untuk pengklonan karena merupakan metode yang masih baru, cepat dan efisien.
Penelitian ini bertujuan untuk membuat konstruksi gen penyandi kitinase dalam vektor ekspresi dengan metode Gateway yang cepat dan terarah. Hipotesis penelitian ini adalah gen penyandi kitinase dapat dikonstruksikan ke dalam vektor ekspresi menggunakan metode Gateway dan ditransformasikan pada Agrobacterium tumefaciens. Hasil penelitian ini diharapkan dapat menghasilkam bibit transgenik kelapa sawit yang mengekspresikan gen penyandi kitinase sehingga resisten terhadap Ganoderma dan memberikan informasi mengenai metode Gateway sebagai salah satu metode pengklonan.
TINJAUAN PUSTAKA
Kelapa Sawit
2
warna buah. Tiga tipe dikenal berdasarkan varietas tebal tipisnya cangkang, yaitu: Dura, Pisifera, dan Tenera. Varietas berdasarkan warna buahnya juga terdapat tiga tipe, yaitu: Nigrescens, Virescens, dan Albescens (Setyamidjaja 2006).
Upaya peningkatan produksi kelapa sawit memiliki hambatan, salah satu hambatan tersebut adalah adanya gangguan penyakit yang disebabkan oleh hama dan penyakit. Terdapat pelbagai cara untuk mengklasifikasikan hama dan penyakit pada tananam kelapa sawit. Klasifikasi tersebut berdasarkan bagian tanaman yang diserang dan jenis hama yang menyerang. Klasifikasi berdasarkan bagian tanaman yang diserang dikenal dengan hama perusak (pemakan) daun, perusak bunga dan buah, perusak akar dan batang. Ditinjau dari jenis hama yang menyerang maka hama yang menyerang dapat dibedakan yaitu hama serangga, nematoda, mamalia dan lain-lain (Lubis 1992).
Penyebab penyakit busuk pangkal batang atau akar pada kelapa sawit adalah Ganoderma spp. Gejala awal penyakit ini adalah pelepah daun yang berada di pucuk berwarna merah pucat seperti kekurangan hara selanjutnya daun mengalami nekrosis yang dimulai dari daun yang lebih tua sampai ke daun yang lebih muda.
Pelepah daun tersebut akan patah dan menggantung pada tanaman kelapa sawit. Umumnya tanaman akan mati setelah 6-12 bulan sejak gejala terakhir, hal ini disebabkan infeksi yang terjadi karena kontak akar yang sakit (Lubis 1992).
Gambar 1 Tanaman kelapa sawit.
Ganodermaspp.
Menurut Alexopoulos (1996), Ganoderma spp. diklasifikasikan ke dalam dunia fungi yang merupakan anggota Basidiomycetes penyebab penyakit pada tanaman keras dengan kemampuan mendekomposisi lignin, selulosa, dan polisakarida. Jamur ini pertama
kali dideskripsikan Karsten pada 1881 dengan nama GanodermaP Karsten. Ganoderma spp. sendiri berasal dari bahasa latin yaitu gan yang berarti berkilauan dan dermyang berarti kulit. Arti tersebut menggambarkan bahwa tubuh buah Ganoderma agak keras dan mengkilap (Widyastuti 2009). Adapun klasifikasi lengkap dari Ganoderma spp. menurut Alexopoulos (1996) adalah filum Fungi, kelas Basidiomycetes, subkelas Holobasidiomycetes, seri Hymenomycetes, ordo Agaricales, family Polyporaceae, genus Ganoderma, dan speciesGanodermaspp.
Widyastuti (2009) menyatakan bahwa Ganoderma spp. merupakan fungi penyebab penyakit busuk akar yang dapat membentuk tubuh buah yang merupakan hasil pembuahan seksual. Permukaan tubuh buahnya berpori, berwarna cokelat kemerahan seperti terlihat pada Gambar 2. Posisi tubuh buahnya ada yang duduk (sessile) dan bertangkai (stipitate). Ganoderma spp. memiliki ciri-ciri diantaranya yaitu adanya struktur yang disebut basidium (suatu sel berbentuk tabung atau seperti pemukul bola yang mempunyai empat buah basidiospora di bagian luarnya). Himenium yang dimiliki dapat menutupi permukaan berpori, tubuh buah berkayu, keras dan ulet, serta mempunyai lapisan-lapisan membran, permukaan atas tubuh buah (konus) rata dan halus, dan spora pipih di bagian bawahnya.
Ganoderma tumbuh pada daerah dengan ketinggian 300 meter di atas permukaan laut sampai dataran tinggi dengan suhu rata-rata 26-32oC. Di Indonesia Ganoderma tersebar di hutan-hutan Sumatera, Kalimantan dan beberapa hutan tropis lainnya. Ganoderma dapat tumbuh pada kayu yang masih hidup dan kayu yang sudah mati (Martawijaya 1986). Serangan Ganoderma pada tanaman keras sulit dideteksi karena gejalanya mirip
dengan gejala serangan penyakit perakaran lainnya termasuk juga mirip gejala
kekeringan. Tanaman meskipun sudah menunjukkan gejala sakit, namun tubuh buah Ganodermakadang-kadang belum terbentuk.
3
Di lain pihak, pada tanaman yang tampak sehat dapat ditemukan tubuh buah Ganoderma di pangkal batangnya (Bassett & Peters 2003).
Gen Penyandi Kitinase sebagai Gen Ketahanan
Kitinase merupakan enzim yang mempunyai kemampuan mendegradasi kitin, sebagai komponen utama dinding sel jamur (Datta et al. 2000). Kitinase selain memiliki kemampuan tersebut juga melepaskan oligo-N-asetil-glukosamin yang berfungsi sebagai substansi yang telah terbukti berperan penting dalam mengaktifkan respon ketahanan (Ren & West 1992). Faath (1994) menyatakan bahwa kitinase juga dapat diproduksi oleh bakteri untuk memenuhi kebutuhan nutrisinya dengan mendegradasi kitin karena kitin merupakan sumber utama karbon dan nitrogen untuk beberapa bakteri. Hal ini menunjukkan bahwa kitinase merupakan enzim ekstrasel. Beberapa bakteri yang diketahui dapat menghasilkan kitinase diantaranya adalah Bacillus circulans, Serratia marcescens, Pseudomonas aeruginosa, Streptomyces sp., Aeromonas hydrophyla, Vibrio furnissi (Kupiec & Ilan 1998) dan bakteri lain yang mampu menghasilkan kitinase adalah Aeromonas cavae, Bacillus licheneformis, Enterobacter agglomerans, dan Xanthomonas sp. (Patil et al. 2000).
Beberapa publikasi hasil penelitian melaporkan bahwa tanaman yang mengekspresikan gen penyandi kitinase terbukti mempunyai ketahanan terhadap cendawan tertentu, seperti tanaman padi yang tahan terhadap Rhizoctonia solani (Lin et al. 1995; Datta et al.2000), tanaman mentimun yang tahan terhadap serangan Botrytis cinerea (Tabei et al. 1998) dan tanaman tembakau yang tahan terhadap cendawan Sclerotinia sclerotiorum(Terekawa et al.1997). Kitinase diduga dapat digunakan sebagai perlindungan terhadap jamur patogen. Pendugaan tersebut didukung oleh pengamatan tumbuhan tingkat tinggi yang tidak mepunyai kitinase, ternyata setelah diberikan kitinase tumbuhan tersebut menunjukkan aktivitas antijamur secara in vitro(Lin et al. 1995).
Metode Pengklonan Gateway
Metode pengklonan Gateway dikembangkan oleh peneliti dari perusahaan Life Technologies (Hartley et al. 2000). Metode Gateway merupakan metode biologi molekular yang memungkinkan peneliti untuk memindahkan fragmen DNA dari suatu
plasmid ke plasmid lain secara efisien dengan menggunakan suatu set urutan rekombinasi yang disebut situs”att Gateway”. Metode pengklonan Gateway pada dasarnya bergantung pada dua reaksi yakni, reaksi Bacteriophage (BP) danLeft Right(LR).
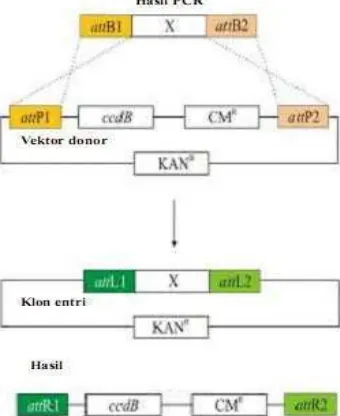
Reaksi BP (Gambar 3) pada metode pengklonan Gateway terjadi penggabungan situs attP yang berukuran 242 pb dari faga lamda dan situs attB yang berukuran 25 pb dari rekombinasi E. coli dan genom faga lamda disambungkan ke genom E. coli. Hasilnya genom faga diikat oleh attL yang berukuran 100 pb dan attR yang berukuran 168 pb sedangkan kebalikanya faga lamda dipotong dari genom E. coli dengan rekombinasi di antara situs attL dan attR dalam reaksi LR seperti terlihat pada Gambar 4. Reaksi ini merekombinasikan gen sisipan yang diikat oleh situs attL dengan vektor destinasi yang membawa situs attR. Hasilnya gen sisipan diikat oleh dua situs yakni situs attB1 dan attB2 yang selanjutnya disebut klon ekspresi.
Reaksi BP terjadi ketika gen yang akan dikloning di desain dengan menggunakan situs att lalu direaksikan dengan vektor donor yang mengandung ccdB. Reaksi tersebut menggunakan bantuan BP clonase sehingga menghasilkan klon entri dimana gen yang akan dikloning bertukar tempat dengan ccdB pada vektor donor seperti yang terlihat pada Lampiran 5. Klon entri yang sudah mengandung gen yang akan di klon kemudian direaksikan dengan vektor donor pada reaksi LR menggunakan LR klonase sehingga gen yang terdapat pada klon entri bertukar tempat
4
Gambar 4 Reaksi LR dalam metode Gateway.
dengan ccdB pada vektor destinasi menghasilkan vektor ekspresi yang telah mengandung gen yang ingin di klon (Lampiran 6)
Tujuan utama dari modifikasi pengklonan Gateway adalah untuk memperkirakan Open Reading Frame(ORF) yang menyandi protein dalam vektor entri untuk mencegah keberadaan atau penambahan muatan sekuens yang sering kali tinggi setelah rekombinasi yang dapat mempengaruhi fungsi protein (Dubin et al.2008).
Polymerase Chain Reaction(PCR)
Teknik PCR merupakan teknik yang berguna dalam amplifikasi suatu sekuen DNA spesifik dengan bantuan enzim, dan dilakukan secara in vitro. Sampai sekarang telah terdapat beberapa pengembangan dari teknik PCR dan telah banyak digunakan untuk pelbagai macam manipulasi dan analisis genetik (Yuwono 2006). Instrumen PCR dapat digunakan untuk menggandakan jumlah molekul DNA pada target tertentu dengan mensintesis molekul (Muladno 2002)
DNA baru yang berkomplemen dengan molekul DNA target tersebut dengan bantuan enzim dan oligonukleotida sebagai primer dalam suatu termocycler. Primer yang berada sebelum daerah target disebut sebagai primer forward sedangkan primer yang berada setelah daerah target disebut primer reverse. Enzim polimerase merupakan enzim yang digunakan sebagai pencetak rangkaian molekul DNA baru.
Menurut Yuwono (2006) komponen yang dibutuhkan dalam reaksi PCR adalah (1) DNA target (template), yaitu fragmen DNA yang
akan dilipatgandakan, (2) oligonukleotida primer, yaitu suatu sekuen oligonukleotida pendek (15-25 basa nukleotida) yang digunakan untuk mengawali sintesis rantai DNA, (3) deoksiribonukleotida trifosfat (dNTP), terdiri atas dATP, dCTP, dGTP, dTTP, dan (4) enzim DNA polimerase, yaitu enzim yang melakukan katalis reaksi sintesis rantai DNA. Komponen lain yang juga penting adalah senyawa buffer.
Reaksi PCR merupakan tiruan dari proses replikasi DNA, yaitu dengan adanya pembukaan rantai DNA utas ganda, penempatan primer dan perpanjangan dari arah 5’ ke 3’, hanya saja pada teknik PCR tidak menggunakan enzim ligase dan primer RNA. Secara ringkas, teknik PCR dilakukan dengan cara mencampurkan sampel DNA dengan primer oligonukleotida, deoksiribonukleotida trifosfat, enzim termostabil Taq DNA polimerase dalam larutan yang sesuai, kemudian menaikkan dan menurunkan suhu campuran secara berulang selama beberapa jam sampai diperoleh jumlah sekuen DNA yang diinginkan (Saiki et al. 1989).
Menurut Saiki et al. (1989) satu siklus yang terjadi pada teknik PCR terdiri atas tiga tahap (Gambar 5), yaitu denaturasi, annealing dan ekstensi. Denaturasi dilakukan pada suhu 90-95oC sehingga terjadi pemisahan utas ganda DNA menjadi dua utas tunggal DNA yang menjadi cetakan (template) tempat penempelan primer dan tempat kerja DNA polimerase. Suhu kemudian diturunkan pada tahap annealinguntuk penempelan primer
5
oligonukleotida pada sekuens yang komplementer dengan molekul DNA cetakan. Suhu annealing tiap sekuens DNA bersifat spesifik dan merupakan faktor penentu keberhasilan suatu reaksi PCR. Tahap terakhir adalah ekstensi, pada tahap ekstensi dilakukan pada suhu 72oC. Suhu ini merupakan suhu optimum untuk kerja enzim Taq DNA polimerase. Pada tahap ini terjadi sintesis DNA komplemen dengan DNA cetakan. Ketiga tahap PCR dilakukan berulang kali dalam mesin PCR, pada umumnya antara 25-30 kali (siklus) bergantung dari jumlah DNA yang diinginkan sehingga pada akhir siklus akan didapatkan molekul-molekul DNA rantai ganda yang baru hasil polimerasi dalam jumlah yang jauh lebih banyak dibandingkan dengan jumlah DNA cetakan yang digunakan (Yuwono 2006).
Elektroforesis
Elektroforesis merupakan teknik yang digunakan untuk memisahkan dan memurnikan fragmen-fragmen DNA ataupun RNA yang memiliki muatan listrik di bawah pengaruh medan listrik (Clark & Christopher 2008). Mobilitas fragmen DNA pada gel elektroforesis sangat dipengaruhi oleh komposisi dan kelarutan ion bufer elektroforesis. Konsentrasi ion-ion sangat sedikit dapat menyebabkan migrasi DNA menjadi lambat sedangkan konsentrasi ion yang berlebih akan mengakibatkan gel mencair dan DNA terdenaturasi (Sambrook & Russell 2001).
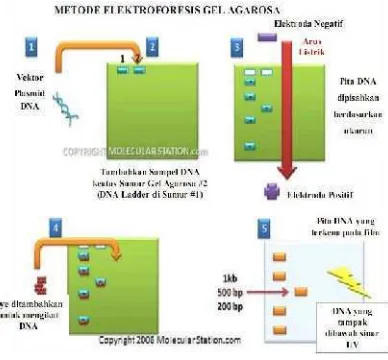
Prinsip elektroforesis adalah memisahkan molekul berdasarkan muatannya (Gambar 6). DNA yang bermuatan negatif akan bergerak ke arah kutub positif selama elektroforesis karena adanya gugus fosfat. Fragmen DNA mempunyai muatan negatif yang sama untuk tiap-tiap ukuran panjang, sehingga pergerakan DNA ini akan memiliki kecepatan yang sama untuk mencapai kutub positif (Clark & Christopher 2008). Gel yang digunakan biasanya berupa polimer bertaut silang (crosslinked) dengan porositas yang diatur sesuai kebutuhan. Gel yang umum digunakan untuk memisahkan protein atau asam nukleat berukuran kecil adalah poliakrilamida. Pemisahan molekul yang lebih besar (lebih dari beberapa ratus basa) menggunakan gel agarosa yang dibuat dari ekstrak rumput laut yang sudah dimurnikan (Khopkar 1990).
Konsentrasi agarosa yang sering dipakai yaitu berkisar antara 0.8-1.5%. Konsentrasi gel yang sangat encer (0.1-0.2%) dapat meningkatkan daya pisah elektroforesis tetapi
hal tersebut sulit dilakukan karena gel yang encer sangat mudah pecah. Larutan bufer yang digunakan dalam elektroforesis sama dengan yang digunakan untuk membuat gel. Bufer tersebut dapat dibuat seperti tris-asetat-EDTA (TAE) atau tris-borat-tris-asetat-EDTA (TBE). Gel agarosa dicampur dengan etidium bromida (EtBr) dan dicetak dengan sisir yang dibuat sumur-sumur tempat memasukkan sampel DNA (Yuwono 2005). Menurut Sambrook & Russell (2001) teknik elektroforesis DNA juga memerlukan loading buffer selain bufer elektroforesis. Bufer tersebut berfungsi untuk meningkakan densitas sampel sehingga fragmen tersebut berada di dasar sumur gel (well) dan tidak menyebar.
Hasil elektroforesis dapat teramati secara visual dengan menambahkan etidium bromide (EtBr) pada gel sebelum dicetak. Etidium bromida akan menyisip ke dalam DNA sehingga apabila dilihat di bawah sinar UV pita-pita DNA menjadi terlihat karena etidium bromida akan memendarkan sinar UV (Yuwono 2005).
Gambar 6 Prinsip elektroforesis.
Agrobacterium tumefaciens
Transformasi ke tanaman dengan Agrobacterium tumefaciens memerlukan keberadaan dua komponen genetik yang terdapat pada plasmid Ti yaitu DNA T, bahan genetik yang dipindahkan ke genom tanaman dan virulen (vir), daerah yang tersusun atas beberapa tempat yang menyandikan protein DNA T dan pemindahan DNA T ke tanaman (Tzafira & Citovsky 2002).
BAHAN DAN METODE
Bahan dan Alat
Bahan-bahan yang digunakan dalam proses amplifikasi menggunakan kit platinum dari Invitrogen dan memerlukan bahan-bahan seperti gen penyandi kitinase, primer spesifik Gateway KTN-F, primer spesifik Gateway KTN-R, M13-R, M13-F, larutan bufer, MgCl2, dNTPs, Taq polimerase, dan
molecular water (aquabides). Bahan yang digunakan untuk elektroforesis yaitu gel agarosa (Sigma), bufer Tris-Borate-EDTA (TBE) 0.5x, Etidium bromida (EtBr) 5 µg/100ml, loading buffer (Bromfenol biru 2.5%, sukrosa 40%), dan marker 1 kb plus DNA Ladder (Invitrogen). Ekstraksi dan Pemurnian menggunakan kit platinum dari Invitrogen (PureLinkTM Quick Gel Extraction). Isolasi DNA plasmid menggunakan kit dari Fermentas (GeneJETTM Plasmid MiniPrep Kit). Bahan lainnya yang digunakan yaitu Gateway® Technology, Invitrogen (vektor pDONRTM, enzim BP clonaseTM, proteinase K, enzim LR clonaseTM), sel kompeten Escherichia coli galur XL-1 Blue, sel kompeten Agrobacterium tumefaciensgalur AGL-0, media Luria Bertani (LB), media Luria Agar (LA), nitrogen cair, media YEP (Yeast Extract Pepton), larutan bufer 10x dream Taq, larutan bufer TE, kanamisin 100.000 ppm, dan rifampisin 25.000 ppm.
Alat yang digunakan untuk elektroforesis adalah sisir dan cetakan agar, bak elektroforesis, mikropipet, microwave, tabung mikro, adaptor 100 Volt, transluminator ultraviolet (UV) T2201 (Sigma). Alat lain yang digunakan adalah mesin PCR (ESCO Swift max), DNA speed vacum 110 savant, inkubator bergoyang, laminar air flow cabinet, segitiga penyebar, pinset, pisau potong (scalpel), autoklaf, Eppendorf sentrifus 5417R, neraca analitik, dan peralatan-peralatan gelas seperti cawan petri, gelas piala, labu Erlenmeyer, labu takar, dan gelas ukur.
Metode
Penelitian sebelumnya oleh Novianthy (2009) telah dilakukan sampai kemudian didapatkan pengurutan basa nukleotida gen penyandi kitinase. Penelitian ini dimulai dari amplifikasi gen penyandi kitinase dengan primer Gateway sampai transformasi ke Agrobacterium sp.
Amplifikasi Gen Penyandi Kitinase dengan Primer Gateway (Invitrogen 2010)
Amplifikasi gen penyandi kitinase menggunakan kit dari Invitrogen (Platinum® Taq DNA Polimerase). Amplifikasi ini menggunakan sepasang primer spesifik Gateway yakni, Gateway KTN-Forward (KTN-F) dan Gateway KTN-Reverse (KTN-R). Proses amplifikasi dimulai dengan menyiapkan campuran reaksi yang terdiri atas 2.5 µL bufer, 1 µL MgCl2, 1 µL dNTPs, 0.2
µL Taq polimerase, dan 14.3 µL aquabides. Siapkan tabung mikro kemudian tambahkan 3 µL aquabides. DNA cetakan (template) dimasukkan ke dalam tabung mikro sebanyak 1 µL. Primer Forward(KTN-F) ditambahkan sebanyak 1 µL ke dalam tabung mikro kemudian ditambahkan pula sebanyak 1 µL Primer Reverse (KTN-R) ke dalam tabung. Campuran reaksi yang telah dipersiapkan sebelumnya ditambahkan sebanyak 19 µL ke dalam larutan DNA.
Gen penyandi kitinase diamplifikasi dengan mesin PCR sebanyak 35 siklus dengan program PCR diantaranya adalah predenaturasi pada suhu 94oC selama 7 menit, denaturasi pada suhu 94oC selama 45 detik, penempelan primer (annealing) pada suhu 55oC selama 45 detik, pemanjangan primer (extension) pada suhu 72oC selama 2 menit, dan pascapemanjangan pada suhu 72oC selama 5 menit. Verifikasi produk PCR pada gel agarosa 1%.
Elektroforesis Hasil Amplifikasi
Membuat Gel Agarosa Konsentrasi 1%.
Larutkan serbuk agarosa sebanyak 0.3 gram dalam 30 ml larutan TBE 0.5x. Larutan agarosa kemudian dipanaskan dalam microwavesampai larut ± 1 menit. Diamkan larutan sampai terasa hangat, lalu tambahkan larutan EtBr sebanyak 1.5 µL. Larutan yang telah ditambahkan EtBr dituang ke dalam cetakan dan dibiarkan hingga mengeras membentuk gel. Gel agarosa yang telah mengeras digunakan untuk elektroforesis gen hasil amplifikasi.
Transformasi ke tanaman dengan Agrobacterium tumefaciens memerlukan keberadaan dua komponen genetik yang terdapat pada plasmid Ti yaitu DNA T, bahan genetik yang dipindahkan ke genom tanaman dan virulen (vir), daerah yang tersusun atas beberapa tempat yang menyandikan protein DNA T dan pemindahan DNA T ke tanaman (Tzafira & Citovsky 2002).
BAHAN DAN METODE
Bahan dan Alat
Bahan-bahan yang digunakan dalam proses amplifikasi menggunakan kit platinum dari Invitrogen dan memerlukan bahan-bahan seperti gen penyandi kitinase, primer spesifik Gateway KTN-F, primer spesifik Gateway KTN-R, M13-R, M13-F, larutan bufer, MgCl2, dNTPs, Taq polimerase, dan
molecular water (aquabides). Bahan yang digunakan untuk elektroforesis yaitu gel agarosa (Sigma), bufer Tris-Borate-EDTA (TBE) 0.5x, Etidium bromida (EtBr) 5 µg/100ml, loading buffer (Bromfenol biru 2.5%, sukrosa 40%), dan marker 1 kb plus DNA Ladder (Invitrogen). Ekstraksi dan Pemurnian menggunakan kit platinum dari Invitrogen (PureLinkTM Quick Gel Extraction). Isolasi DNA plasmid menggunakan kit dari Fermentas (GeneJETTM Plasmid MiniPrep Kit). Bahan lainnya yang digunakan yaitu Gateway® Technology, Invitrogen (vektor pDONRTM, enzim BP clonaseTM, proteinase K, enzim LR clonaseTM), sel kompeten Escherichia coli galur XL-1 Blue, sel kompeten Agrobacterium tumefaciensgalur AGL-0, media Luria Bertani (LB), media Luria Agar (LA), nitrogen cair, media YEP (Yeast Extract Pepton), larutan bufer 10x dream Taq, larutan bufer TE, kanamisin 100.000 ppm, dan rifampisin 25.000 ppm.
Alat yang digunakan untuk elektroforesis adalah sisir dan cetakan agar, bak elektroforesis, mikropipet, microwave, tabung mikro, adaptor 100 Volt, transluminator ultraviolet (UV) T2201 (Sigma). Alat lain yang digunakan adalah mesin PCR (ESCO Swift max), DNA speed vacum 110 savant, inkubator bergoyang, laminar air flow cabinet, segitiga penyebar, pinset, pisau potong (scalpel), autoklaf, Eppendorf sentrifus 5417R, neraca analitik, dan peralatan-peralatan gelas seperti cawan petri, gelas piala, labu Erlenmeyer, labu takar, dan gelas ukur.
Metode
Penelitian sebelumnya oleh Novianthy (2009) telah dilakukan sampai kemudian didapatkan pengurutan basa nukleotida gen penyandi kitinase. Penelitian ini dimulai dari amplifikasi gen penyandi kitinase dengan primer Gateway sampai transformasi ke Agrobacterium sp.
Amplifikasi Gen Penyandi Kitinase dengan Primer Gateway (Invitrogen 2010)
Amplifikasi gen penyandi kitinase menggunakan kit dari Invitrogen (Platinum® Taq DNA Polimerase). Amplifikasi ini menggunakan sepasang primer spesifik Gateway yakni, Gateway KTN-Forward (KTN-F) dan Gateway KTN-Reverse (KTN-R). Proses amplifikasi dimulai dengan menyiapkan campuran reaksi yang terdiri atas 2.5 µL bufer, 1 µL MgCl2, 1 µL dNTPs, 0.2
µL Taq polimerase, dan 14.3 µL aquabides. Siapkan tabung mikro kemudian tambahkan 3 µL aquabides. DNA cetakan (template) dimasukkan ke dalam tabung mikro sebanyak 1 µL. Primer Forward(KTN-F) ditambahkan sebanyak 1 µL ke dalam tabung mikro kemudian ditambahkan pula sebanyak 1 µL Primer Reverse (KTN-R) ke dalam tabung. Campuran reaksi yang telah dipersiapkan sebelumnya ditambahkan sebanyak 19 µL ke dalam larutan DNA.
Gen penyandi kitinase diamplifikasi dengan mesin PCR sebanyak 35 siklus dengan program PCR diantaranya adalah predenaturasi pada suhu 94oC selama 7 menit, denaturasi pada suhu 94oC selama 45 detik, penempelan primer (annealing) pada suhu 55oC selama 45 detik, pemanjangan primer (extension) pada suhu 72oC selama 2 menit, dan pascapemanjangan pada suhu 72oC selama 5 menit. Verifikasi produk PCR pada gel agarosa 1%.
Elektroforesis Hasil Amplifikasi
Membuat Gel Agarosa Konsentrasi 1%.
Larutkan serbuk agarosa sebanyak 0.3 gram dalam 30 ml larutan TBE 0.5x. Larutan agarosa kemudian dipanaskan dalam microwavesampai larut ± 1 menit. Diamkan larutan sampai terasa hangat, lalu tambahkan larutan EtBr sebanyak 1.5 µL. Larutan yang telah ditambahkan EtBr dituang ke dalam cetakan dan dibiarkan hingga mengeras membentuk gel. Gel agarosa yang telah mengeras digunakan untuk elektroforesis gen hasil amplifikasi.
7
selanjutnya dielektroforesis untuk mengetahui gen tersebut telah teramplifikasi atau tidak. Hasil amplifikasi diambil sebanyak 5 µL, kemudian dicampurkan dengan loading buffer sebanyak 1 µL. Campuran tersebut kemudian dimasukkan dimasukkan ke dalam sumur gel agarosa dan dielektroforesis pada 75 volt selama ± 1 jam.
Ekstraksi dan Pemurnian DNA (Invitrogen 2010)
Elektroforegram positif ditunjukkan dengan adanya pita pada gel setelah diamati dengan transluminator UV. Proses ekstraksi dan pemurnian gen penyandi kitinase dari gel dilakukan dengan menggunakan kit dari Invitrogen. Pita yang terang pada gel agarosa dipotong dengan scalpel di bawah transluminator UV. Gel hasil potongan ditambahkan bufer pelarut (Gel solubilisation buffer) sebanyak 3x volume kemudian diinkubasi pada suhu 500C selama 10 menit sampai gel tersebut larut lalu inkubasi lagi pada suhu ruang selama 5 menit. Gel yang telah larut dipindahkan ke kolom (Quick Gel Extraction) dan sentrifus 1 menit dengan kecepatan 12000 rpm pada suhu ruang. Supernatan yang terbentuk dibuang, lalu sebanyak sebanyak 500 µL bufer pencuci (wash buffer) ditambahkan ke dalam kolom dan disentrifus 1 menit pada 12000 rpm, suhu ruang. Supernatan dibuang dan disentrifus kembali dalam keadaan kosong pada kecepatan 12000 rpm, suhu 25oC selama 1 menit. Bufer elusi selanjutnya ditambahkan sebanyak 30 µL tepat ditengah kolom dan inkubasi selama 1 menit pada suhu ruang. Sentrifus kembali larutan tersebut pada kecepatan 12000 rpm, suhu 25oC selama 2 menit. Hasil ekstraksi dan pemurnian gel dilihat dengan elektroforesis gel agarosa.
Rekombinasi Gen Penyandi Kitinase pada Vektor Donor dan Vektor Destinasi (Invitrogen 2003)
Rekombinasi gen penyandi kitinase pada vektor donor dimulai dengan menyiapkan sebanyak 5 µL bufer TE kemudian tambahkan 2 µL DNA hasil pemurnian, lalu vektor donor (pDONR 221TM) sebanyak 1 µL. Penambahan BP ClonaseTM sebanyak 2 µL setelah semuanya telah siap kemudian diinkubasi selama 2 jam pada suhu 250C. Larutan kemudian ditambahkan sebanyak 1 µL proteinase K dan diinkubasi lagi pada suhu 370C selama 15 menit. Hasil rekombianasi pada vektor destinasi kemudian ditransformasikan ke dalam E. coli. yang
tumbuh pada media LA dikonfirmasi dengan metode PCR. Koloni bakteri rekombinan ditandai adanya pita setelah pengujian dengan metode PCR. Koloni bakteri rekombinan kemudian diisolasi DNA plasmidnya untuk direkombinasikan ke vektor destinasi.
Plasmid rekombinan sebanyak 1 µL dimasukkan ke dalam tabung mikro dan ditambahkan 1 µL vektor destinasi, kemudian tambahkan bufer TE hingga volume larutan mencapai 8 µL. LR ClonaseTM ditambahkan sebanyak 2 µL ke dalam tabung mikro kemudian diinkubasi pada suhu 250C selama 1 jam. Larutan kemudian ditambahkan 1 µL proteinase K setelah inkubasi yang berfungsi menghilangkan protein yang ada pada DNA. Larutan yang telah ditambahkan proteinase K diinkubasi kembali pada suhu 370C selama 15 menit. Hasil kombianasi kemudian ditransformasikan ke dalam E. coli. Koloni bakteri yang tumbuh diisolasi DNA plasmidnya kemudian dikonfirmasi dengan metode PCR. Plasmid rekombinan hasil rekombinasi selanjutnya ditransformasikan ke Agrobacterium sp.
Transformasi Plasmid Rekombinan ke dalam Escherichia coli XL-1 Blue
Konfirmasi Koloni Transforman dengan Teknik PCR Koloni
Koloni bakteri yang tumbuh setelah transformasi dianalisis dengan metode PCR koloni. Koloni yang tumbuh pada media LA diambil dengan menggunakan tusuk gigi kemudian dipindahkan ke dalam tabung Eppendorf. Pemindahan koloni dilakukan secara steril dalam laminar air flow cabinet. Persiapan PCR koloni dilakukan dengan menyiapkan campuran reaksi sebanyak total tabung yang berisi koloni yang terdiri atas, aquabides 2.75 µL, buffer complete 1.5 µL, dNTPs 0.3 µL, M13-F 0.15 µL, M13-R 0.15 µL, Taq polimerase 0.15 µL. Tahap pertama PCR adalah program lisis 96oC selama 5 menit, 50oC selama 1 menit 30 detik, 96oC selama 1 menit 30 detik, 45oC selama 1 menit 30 detik, 96oC selama 1 menit, dan 40oC selama 1 menit. Program dihentikan sejenak untuk penambahan sebanyak 5 µL campuran reaksi ke dalam masing-masing tabung Eppendorf. Program PCR kemudian dilanjutkan kembali dengan program PCR yakni, 94oC selama 30 detik, 55oC selama 1 menit, 72oC selama 2 menit. Koloni transforman setelah perbanyakan dengan teknik PCR selesai kemudian dielektroforesis dengan gel agarosa 1%.
Isolasi DNA Plasmid Rekombinan (Fermentas 2006)
Isolasi DNA plasmid dilakukan berdasarkan hasil PCR DNA koloni yang diketahui mengandung plasmid terinsersi fragmen yang diinginkan. Plasmid diisolasi dengan GeneJetTM Plasmid Miniprep Kit. Koloni bakteri yang tumbuh pada media LA dikulturkan ke media LB yang telah ditambahkan antibiotik kanamisin 50 ppm (bakteri yang ditransformasikan dengan vektor donor). Bakteri diinkubasi pada inkubator bergoyang pada suhu 370C selama semalam untuk pembiakan bakteri. Sampel yang positif mengandung plasmid terinsersi disentrifus dengan kecepatan 12000 rpm terlebih dahulu. Pelet dihomogenisasi dengan penambahan 250 µL larutan resuspensi kemudian divortex. Pelet yang telah dilarutkan tersebut ditambahkan larutan lisis sebanyak 250 µL dan dikocok bolak-balik. Sebanyak 350 µL larutan netralisasi ditambahkan ke dalam larutan tersebut kemudian disentrifugasi pada kecepatan dan suhu yang sama selama 5 menit. Kecepatan sentrifugasi pada isolasi DNA plasmid dengan kit Fermentas seluruhnya pada 12000 rpm dan
suhu 250C. Setiap penambahan larutan dalam tabung dibolak-balik sebanyak 6x.
Supernatan dipindahkan ke dalam kolom dan disentrifugasi selama 1 menit. Kolom dicuci dengan 500 µL larutan pencuci dan disentrifus selama 1 menit (dilakukan sebanyak 2x). Kolom dalam keadaan kosong disentrifus kembali selama 1 menit. Kolom dipindahkan ke dalam tabung mikro baru dan ditambahkan 30 µL elution buffer tepat di tengah membran kolom. Kolom diinkubasi selama 2 menit dan disentrifugasi kembali selama 2 menit. DNA plasmid diverifikasi pada gel agarosa 1%.
Transformasi ke dalam Agrobacterium tumefaciensGalurAGL-0
Sebanyak 10 µL DNA plasmid dimasukkan ke dalam Agrobacterium galur AGL-0lalu didiamkan di dalam es selama 15 menit. Inkubasi kembali di dalam nitrogen cair selama 5 menit dan pada suhu 37oC selama 5 menit. Tambahkan 1 ml YEP (Yeast Exctract Pepton) lalu dikocok dengan inkubator bergoyang selama 3 jam pada suhu 28oC. Larutan kemudian disentrifugasi pada kecepatan 6000 rpm selama 3 menit.
Supernatan yang dihasilkan sebagian dibuang dan sebanyak ± 200 µL supernatan yang tersisa diresuspensikan dengan pelet yang terbentuk lalu disebar ke dalam media LB yang telah berisi antibiotik kanamisin 50 ppm dan rifampisin 50 ppm. Inkubasi selama 2 hari pada suhu 280C dalam kondisi gelap.
HASIL DAN PEMBAHASAN
Hasil Amplifikasi Gen Penyandi Kitinase dengan Primer Spesifik Gateway
Konfirmasi Koloni Transforman dengan Teknik PCR Koloni
Koloni bakteri yang tumbuh setelah transformasi dianalisis dengan metode PCR koloni. Koloni yang tumbuh pada media LA diambil dengan menggunakan tusuk gigi kemudian dipindahkan ke dalam tabung Eppendorf. Pemindahan koloni dilakukan secara steril dalam laminar air flow cabinet. Persiapan PCR koloni dilakukan dengan menyiapkan campuran reaksi sebanyak total tabung yang berisi koloni yang terdiri atas, aquabides 2.75 µL, buffer complete 1.5 µL, dNTPs 0.3 µL, M13-F 0.15 µL, M13-R 0.15 µL, Taq polimerase 0.15 µL. Tahap pertama PCR adalah program lisis 96oC selama 5 menit, 50oC selama 1 menit 30 detik, 96oC selama 1 menit 30 detik, 45oC selama 1 menit 30 detik, 96oC selama 1 menit, dan 40oC selama 1 menit. Program dihentikan sejenak untuk penambahan sebanyak 5 µL campuran reaksi ke dalam masing-masing tabung Eppendorf. Program PCR kemudian dilanjutkan kembali dengan program PCR yakni, 94oC selama 30 detik, 55oC selama 1 menit, 72oC selama 2 menit. Koloni transforman setelah perbanyakan dengan teknik PCR selesai kemudian dielektroforesis dengan gel agarosa 1%.
Isolasi DNA Plasmid Rekombinan (Fermentas 2006)
Isolasi DNA plasmid dilakukan berdasarkan hasil PCR DNA koloni yang diketahui mengandung plasmid terinsersi fragmen yang diinginkan. Plasmid diisolasi dengan GeneJetTM Plasmid Miniprep Kit. Koloni bakteri yang tumbuh pada media LA dikulturkan ke media LB yang telah ditambahkan antibiotik kanamisin 50 ppm (bakteri yang ditransformasikan dengan vektor donor). Bakteri diinkubasi pada inkubator bergoyang pada suhu 370C selama semalam untuk pembiakan bakteri. Sampel yang positif mengandung plasmid terinsersi disentrifus dengan kecepatan 12000 rpm terlebih dahulu. Pelet dihomogenisasi dengan penambahan 250 µL larutan resuspensi kemudian divortex. Pelet yang telah dilarutkan tersebut ditambahkan larutan lisis sebanyak 250 µL dan dikocok bolak-balik. Sebanyak 350 µL larutan netralisasi ditambahkan ke dalam larutan tersebut kemudian disentrifugasi pada kecepatan dan suhu yang sama selama 5 menit. Kecepatan sentrifugasi pada isolasi DNA plasmid dengan kit Fermentas seluruhnya pada 12000 rpm dan
suhu 250C. Setiap penambahan larutan dalam tabung dibolak-balik sebanyak 6x.
Supernatan dipindahkan ke dalam kolom dan disentrifugasi selama 1 menit. Kolom dicuci dengan 500 µL larutan pencuci dan disentrifus selama 1 menit (dilakukan sebanyak 2x). Kolom dalam keadaan kosong disentrifus kembali selama 1 menit. Kolom dipindahkan ke dalam tabung mikro baru dan ditambahkan 30 µL elution buffer tepat di tengah membran kolom. Kolom diinkubasi selama 2 menit dan disentrifugasi kembali selama 2 menit. DNA plasmid diverifikasi pada gel agarosa 1%.
Transformasi ke dalam Agrobacterium tumefaciensGalurAGL-0
Sebanyak 10 µL DNA plasmid dimasukkan ke dalam Agrobacterium galur AGL-0lalu didiamkan di dalam es selama 15 menit. Inkubasi kembali di dalam nitrogen cair selama 5 menit dan pada suhu 37oC selama 5 menit. Tambahkan 1 ml YEP (Yeast Exctract Pepton) lalu dikocok dengan inkubator bergoyang selama 3 jam pada suhu 28oC. Larutan kemudian disentrifugasi pada kecepatan 6000 rpm selama 3 menit.
Supernatan yang dihasilkan sebagian dibuang dan sebanyak ± 200 µL supernatan yang tersisa diresuspensikan dengan pelet yang terbentuk lalu disebar ke dalam media LB yang telah berisi antibiotik kanamisin 50 ppm dan rifampisin 50 ppm. Inkubasi selama 2 hari pada suhu 280C dalam kondisi gelap.
HASIL DAN PEMBAHASAN
Hasil Amplifikasi Gen Penyandi Kitinase dengan Primer Spesifik Gateway
9
al. 1996) dengan ukuran gen penyandi kitinase yang diperoleh adalah 1500 pb.
Amplifikasi gen penyandi kitinase bertujuan menggandakan gen tersebut secara in vitro menggunakan metode PCR. Amplifikasi dilakukan menggunakan kit dari Invitrogen (2003) dengan primer gateway yang spesifik, yaitu Gateway Kitinase-Forward (KTN-F) dan Gateway Kitinase-Reverse (KTN-R). Kedua primer tersebut dirancang berdasarkan aturan perancangan sistem Gateway, yaitu terdapat empat basa nukleotida GGGG yang diikuti situs attB dan ditambahkan 18-25 urutan basa nukleotida spesifik gen penyandi kitinase. Urutan situs attB dan urutan basa nukleotida gen penyandi kitinase dapat dilihat pada lampiran 4.
Hasil amplifikasi gen penyandi kitinase selanjutnya dielektroforesis dengan konsentrasi gel agarosa 1% untuk mengetahui gen tersebut teramplifikasi atau tidak. Hasil pengujian dengan elektroforesis gel agarosa menunjukkan pita berukuran lebih sedikit dari 1500 pb (Gambar 7). Ukuran pita yang terlihat setelah elektroforesis selanjutnya dibandingkan tingkat kehomologian ukuran pita dengan berbagai gen yang terdapat dalam situs http://blast.ncbi.nlm.nih.gov/Blast.cgi. dan gen tersebut merupakan gen penyandi kitinase. Ukuran ini mirip dengan cds berbagai gen yang mempunyai fungsi sejenis dari spesies tanaman lainnya seperti 1318 pb pada kitinase Allium sativum (Van Damme 1993), 1320 pb pada kitinase Poa pratensis (Du & Ha 1999), 1321 pb pada kitinase Zea diploperennis(Tiffin 2004), dan 1333 pb pada kitinase C dari Ananas comosus (Taira & Akimoto 2007). Gen penyandi kitinase yang sudah teramplifikasi kemudian diekstraksi dan dimurnikan.
Gambar 7 Elektroforegram amplikon gen penyandi kitinase dengan primer Gateway; (M) marker 1 kb plus DNA Ladder, (a) gen penyandi kitinase berukuran 1500 pb.
Hasil Ekstraksi dan Pemurnian DNA dari Gel Agarosa
Ekstraksi dan pemurnian ini menggunakan kit dari Invitrogen. Penelitian dilakukan sesuai dengan prosedur yang terdapat dalam metode. Ekstraksi dan pemurnian bertujuan untuk memurnikan DNA dari pelbagai pengotor yang tidak diinginkan seperti protein dan RNA yang diperoleh dari pengotor yang mungkin ada pada produk PCR. Hasil pemurnian selanjutnya diuji dengan menggunakan elektroforesis gel agarosa.
Elektroforegram amplikon pada gambar sebelumnya menunjukkan gen tersebut berada pada ukuran 1500 pb. Pita yang terlihat setelah pengujian amplifikasi kemudian akan diekstraksi dan dimurnikan. Gel diletakkan di atas transluminator ultraviolet (UV) untuk melihat pita yang akan dipotong. Pita DNA yang terlihat terang setelah penyinaran kemudian dipotong dengan pisau scalpellalu diekstraksi dan dimurnikan.
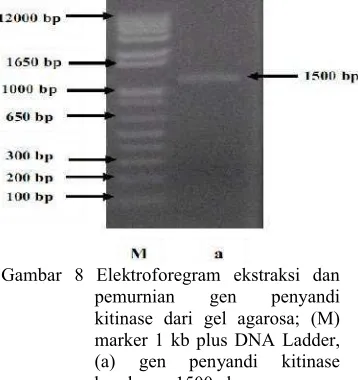
Hasil ekstraksi dan pemurnian diperiksa dengan menggunakan elektroforesis gel agarosa 1% dan menghasilkan pita berukuran 1500 pb seperti yang terlihat pada Gambar 8. Ukuran pita DNA hasil ekstraksi dan pemurnian hampir sama dengan ukuran pita setelah amplifikasi karena esktraksi dan pemurnian hanya menghilangkan pengotor yang ada pada DNA. Hasil ekstraksi dan pemurnian kemudian direkombinasikan ke dalam vektor donor menggunakan metode Gateway.
Gambar 8 Elektroforegram ekstraksi dan pemurnian gen penyandi kitinase dari gel agarosa; (M) marker 1 kb plus DNA Ladder, (a) gen penyandi kitinase berukuran 1500 pb.
Rekombinasi Gen Penyandi Kitinase pada Vektor Donor dan Vektor Destinasi
10
teknik pengklonan. Pengklonan bertujuan memperbanyak DNA yang tersisip pada sel kompeten. Pengklonan gen penyandi kitinase sampai pada vektor destinasi dilakukan dengan menggunakan metode Gateway. Tahapan pengklonan pada metode Gateway terdiri atas dua rekombinasi, yaitu rekombinasi BP dan rekombinasi LR. Tahapan pengklonan dengan metode Gateway tersebut diawali dengan rekombinasi pada vektor donor dan dilanjutkan dengan rekombinasi ke dalam vektor destinasi.
Hasil ekstraksi dan pemurnian disisipkan ke dalam vektor donor (pDONRTM 221) dengan reaksi BP. Gen penyandi kitinase hasil amplifikasi yang memiliki situs attB1 dan attB2 akan direaksikan dengan vektor donor (pDONRTM 221) yang memiliki situs attP1 dan situs attP2 sebagai tempat rekombinasi dengan hasil PCR yang telah mempunyai situs attB1 dan situs attB2 sehingga adanya situs tersebut menyebabkan kemungkinan tidak adanya kesalahan orientasi gen yang direkombinasikan. Reaksi rekombinasi selanjutnya dikatalisis oleh enzim BP ClonaseTM sehingga dalam metode Gateway reaksi rekombinasi pada vektor donor disebut reaksi BP. BP rekombinasi kemudian ditransformasi ke dalam sel kompeten Escherechia coli galur XL-1 Blue untuk dibuktikan sudah terjadi rekombinan. Reaksi BP pada metode Gateway dapat dilihat pada lampiran 5.
Hasil BP rekombinasi selanjutnya ditransformasi ke dalam sel kompeten Escherechia coli galur XL-1 Blue. Sel dapat dibuat kompeten dengan perlakuan garam CaCl2, LiCl2, atau RbCl2. Garam CaCl2 dapat
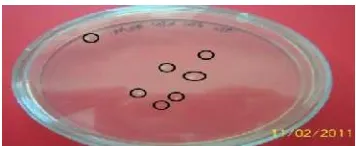
meningkatkan porositas dinding sel sehingga afinitas sel terhadap DNA juga dapat meningkat (Howe 1995). Hasil rekombinasi gen penyandi kitinase pada vektor donor kemudian ditransformasikan ke dalam sel kompeten E. coli galur XL-1 Blue yang ditumbuhkan pada media LA yang telah ditambahkan antibiotik kanamisin 50 ppm. Penambahan kanamisin dilakukan karena vektor donor yang digunakan mempunyai marka seleksi resisten terhadap antibiotik tersebut. Oleh karena itu, seleksi transforman dilakukan dengan mengamati koloni yang tumbuh setelah masa inkubasi seperti yang terlihat pada Gambar 9. Koloni yang tumbuh dengan metode pengklonan Gateway umumnya terdapat dua jenis koloni yaitu koloni putih dan koloni biru. Koloni putih yang tumbuh pada media seleksi diduga kuat klon entri yang membawa gen penyandi
Gambar 9 Koloni yang tumbuh setelah reaksi BP pada metode Gateway.
kitinase,sedangkan by productyang tersisipi ccdB akan mati dan tidak tumbuh sebagai koloni putih. Koloni berwarna putih yang tumbuh diduplikasi dan dikultur untuk memperbanyak jumlah plasmid rekombinan. Koloni hasil duplikasi yang tumbuh diambil dengan menggunakan tusuk gigi steril untuk di konfirmasi menggunakan PCR koloni dengan sepasang primer universal M13 karena peta vektor donor (pDONRTM 221) menunjukkan bahwa amplifikasi dengan PCR koloni memerlukan primer M13 Forwarddan M13 Reverse(Lampiran 7).
Koloni yang terbukti rekombinan setelah pengujian dengan PCR koloni diisolasi DNA plasmidnya kemudian direkombinasikan ke dalam vektor destinasi (vektor ekspresi). Vektor destinasi yang digunakan ada 3, yaitu pGD625, pARC983, dan pDEST. Plasmid rekombinan yang telah diikat oleh situs attL1 dan attL2 direkombinasikan ke vektor destinasi yang membawa situs attR1 dan attR2. Reaksi ini dikatalisis oleh enzim LR ClonaseTM sehingga dalam metode Gateway reaksi rekombinasi ini disebut reaksi LR. Mekanisme reaksi LR dapat dilihat pada lampiran 6. Vektor detinasi yang telah mengandung gen penyandi kitinase (vektor rekombinan) ditransformasikan ke E. coli galur XL-1 Blueuntuk pembiakan sel.
11
Gambar 10 Koloni yang tumbuh setelah reaksi LR pada metode Gateway menggunakan vektor destinasi: (a) pGD625, (b) pARC983, dan (c) pDEST.
Hasil Konfirmasi Koloni Transforman setelah Reaksi BP dengan Teknik PCR dan
Isolasi DNA Plasmid Rekombinan (Fermentas 2006)
Hasil rekombinasi ke dalam vektor donor ditransformasikan ke dalam E.coli untuk pembiakan sel. Koloni yang tumbuh setelah transformasi ke dalam E.coli diuji dengan metode PCR koloni untuk memastikan koloni tersebut mengandung plasmid rekombinan. Pengujian koloni tersebut menggunakan sepasang primer universal M13. Penggunaan primer ini karena pada peta pDONRTM 221 terlihat bahwa amplifikasi dengan PCR koloni memerlukan primer M13 Forward dan M13 Reverse(Lampiran 7).
Elektroforegram PCR koloni pada Gambar 11 menunjukkan bahwa dari tujuh koloni yang diujikan hanya ada lima koloni yang rekombinan yang ditunjukkan dengan adanya pita setelah elektroforesis. Keberhasilan proses PCR koloni dapat diamati dari ukuran klon transforman. Ukuran pita yang terdapat pada elektroforegram tersebut menunjukkan ukuran sekitar 1500 pb yaitu merupakan hasil koloni rekombinan.
Koloni rekombinan yang telah diuji dengan metode PCR koloni selanjutnya dilakukan pengkulturan ke dalam media Luria Bertani (LB) untuk dijadikan sumber nutrisi yang membantu pertumbuhan bakteri. Hasil kultur bakteri yang ditumbuhkan dari media LB cair kemudian diisolasi DNA plasmidnya untuk rekombinasi ke vektor destinasi. Isolasi DNA plasmid dilakukan berdasarkan hasil PCR DNA koloni yang diketahui mengandung plasmid terinsersi fragmen yang diinginkan. Plasmid diisolasi menggunakan Fermentas GeneJet Plasmid Miniprep Kit. Plasmid kemudian dielektroforesis menggunakan gel agarosa 1%.
Gambar 11 Elektroforegram PCR koloni gen penyandi kitinase pada vektor donor; (M) marker 1 kb plus DNA Ladder, (1-7) koloni bakteri.
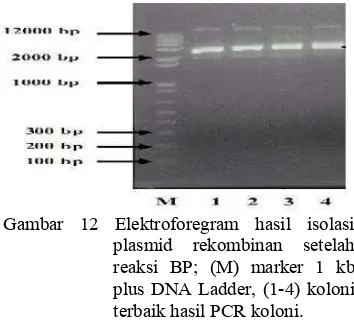
Elektroforegram pada Gambar 12 menunjukkan hasil gen sisipan yang berada pada plasmid rekombinan. Elektroforesis hasil isolasi plasmid sebenarnya bertujuan untuk mengecek secara kualitatif keberhasilan isolasi plasmid. DNA plasmid yang telah diisolasi selanjutnya direkombinasikan ke dalam vektor destinasi.
Gambar 12 Elektroforegram hasil isolasi plasmid rekombinan setelah reaksi BP; (M) marker 1 kb plus DNA Ladder, (1-4) koloni terbaik hasil PCR koloni.
Hasil Konfirmasi Koloni Transforman setelah Reaksi LR dengan Teknik PCR dan
Isolasi DNA Plasmid Rekombinan (Fermentas 2006)
12
koloni. PCR koloni dapat mengkonfirmasi sel transforman yang sudah tersisipi DNA rekombinan. Melalui tahapan ini, gen penyandi kitinase yang disisipkan ke dalam vektor destinasi diamplifikasi menggunakan primer spesifik.
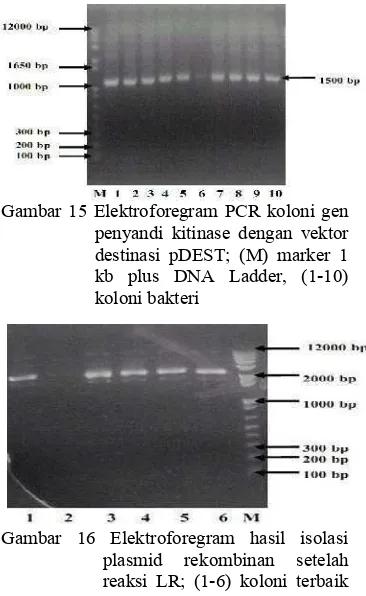
Elektroforegram PCR koloni setelah reaksi LR pada gen penyandi kitinase yang berukuran 1500 pb menunjukkan bahwa dari sembilan koloni yang menggunakan vektor destinasi pGD625, semua koloni mengandung gen penyandi kitinase. Hal ini ditunjukkan dengan pita yang terang pada ukuran sekitar 1500 pb (Gambar 13). Hasil PCR koloni pada sembilan koloni yang menggunakan vektor destinasi pARC983 menunjukkan semua koloni mengandung gen penyandi kitinase berukuran 1500 pb (Gambar 14), dan hasil PCR koloni pada sepuluh koloni yang menggunakan pDEST menunjukkan semua koloni berukuran ukuran 1500 pb seperti yang terlihat pada Gambar 15.
Tujuan menggunakan tiga vektor yang berbeda pGD625, pARC983, dan pDEST yaitu untuk mempelajari gen sesuai kebutuhan dalam penelitian dan sebagai cadangan jika tidak terdapat vektor yang berhasil disisipi. Koloni E. coliyang mengandung sisipan gen penyandi kitinase kemudian dilakukan proses isolasi DNA plasmid dari koloni yang positif pada masing-masing vektor. Koloni nomer 3 dan 4 diambil dari koloni yang menggunakan vektor destinasi pGD625, koloni nomer 4 dan 6 diambil dari koloni yang menggunakan vektor destinasi pARC983 dan koloni nomer 1 dan 2 diambil dari koloni yang menggunakan vektor destinasi pDEST. Secara umum, isolasi plasmid mirip dengan isolasi DNA total, yaitu melibatkan proses pembiakan sel pembawa plasmid, pemanenan sel, pembuatan ekstrak sel, penghilangan protein dan RNA, dan pemekatan DNA dengan presipitasi etanol.
Gambar 13 Elektroforegram PCR koloni gen penyandi kitinase dengan vektor pGD625; (M) marker 1 kb plus DNA Ladder, (1-9) koloni bakteri.
Gambar 14 Elektroforegram PCR koloni gen penyandi kitinase dengan vektor destinasi pARC983; (M) marker 1 kb plus DNA Ladder, (1-9) koloni bakteri.
Isolasi DNA plasmid setelah reaksi LR menggunakan kit dari Fermentas. Elektroforegram pada Gambar 16 menunjukkan gen sisipan yang berada pada plasmid rekombinan. Pengujian plasmid rekombinan yang telah diisolasi dilakukan dengan metode PCR plasmid. Elektroforegram hasil isolasi plasmid rekombinan setelah reaksi LR menunjukkan dari keenam koloni yang diambil dari ketiga vektor mengandung gen yang telah disisipkan pada plasmid rekombinan dan selanjutnya siap di transformasi ke dalam A. tumefaciens.
Gambar 15 Elektroforegram PCR koloni gen penyandi kitinase dengan vektor destinasi pDEST; (M) marker 1 kb plus DNA Ladder, (1-10) koloni bakteri
Hasil Transformasi ke dalam
Agrobacterium tumefaciensGalur AGL-0 Transformasi ke dalam Agrobacterium tumefaciens (A. tumefaciens) bertujuan untuk menguji ekspresi pengaruh gen tersebut ke tanaman. Sel Agrobacterium yang membawa plasmid rekombinan digunakan untuk menginfeksi protoplast tanaman. Penggunaan Agrobacteriumgalur AGL-0dilakukan karena galur ini mempunyai virulensi yang tinggi dibandingkan galur Agrobacterium lainnya.
DNA plasmid setelah rekombinasi pada vektor destinasi diisolasi kemudian ditransformasikan ke Agrobacterium tumefaciensdengan metode kejut dingin (cool shock). Hal ini dikarenakan terjadi lonjakan suhu es 0oC ke nitrogen cair yang bersuhu sekitar -196oC. Lonjakan suhu tersebut akan membuat membrane sel Agrobacterium menjadi tidak selektif terhadap molekul asing sehingga DNA sisipan dapat masuk. Hasil transformasi ke dalam Agrobacterium menunjukkan bahwa koloni bakteri yang tumbuh selama inkubasi ± 2 hari terdapat 1-5 koloni (Gambar 17). Pada saat proses inkubasi, koloni dalam cawan petri dibungkus dengan kertas koran agar kondisi menjadi gelap. Hal ini dilakukan agar koloni Agrobacterium tumefaciens dapat menyesuaikan kondisinya selama hidup di dalam tanah (akar).
Seleksi transforman yang sudah ditransformasikan ke dalam Agrobacterium tumefaciens dilakukan dengan cara menumbuhkannya di dalam media LA. Koloni yang ditumbuhkan diambil berdasarkan elektroforegram hasil isolasi plasmid rekombinan setelah reaksi LR, yaitu plasmid rekombinan nomer 1 (pGD625), plasmid rekombinan nomer 3 (pARC983), dan plasmid rekombinan nomer 5 (pDEST). Media LA yang digunakan mengandung antibiotik kanamisin dan rifampisin. Sel transforman tersebut akan tumbuh membentuk koloni berwarna putih. Kanamisin pada media LA digunakan karena vektor destinasi yang digunakan membawa marka seleksi berupa gen resistensi terhadap antobiotik kanamisin, sedangkan rifampisin digunakan untuk membunuh E. coli. Koloni yang tumbuh dimedia seleksi lebih sedikit dibandingkan koloni yang tumbuh saat transformasi ke E. coli. Hal ini dikarenakan A. tumefaciens memiliki jumlah salinan yang lebih sedikit (low copy number) dibandingkan E. coli.
Koloni yang tumbuh kemudian di PCR koloni untuk menguji gen penyandi kitinase telah berhasil ditransformasi atau tidak. Hasil
PCR koloni menunjukkan pita yang terlihat berukuran sekitar 1500 pb seperti yang terlihat pada Gambar 18.
Gambar 17 Koloni A. tumefaciens yang tumbuh setelah reaksi LR pada gen penyandi kitinase menggunakan vektor destinasi (a) pGD625, (b) pARC983, dan (c) pDEST.
Gambar 18 Elektroforegram hasil PCR koloni A. tumefaciens; (1-2) dengan vektor destinasi pGD625, (3-7) dengan vektor destinasi pARC983, (8) dengan vektor destinasi pDEST, (M) marker 1 kb plus DNA Ladder.
SIMPULAN DAN SARAN
Simpulan
Hasil Transformasi ke dalam
Agrobacterium tumefaciensGalur AGL-0 Transformasi ke dalam Agrobacterium tumefaciens (A. tumefaciens) bertujuan untuk menguji ekspresi pengaruh gen tersebut ke tanaman. Sel Agrobacterium yang membawa plasmid rekombinan digunakan untuk menginfeksi protoplast tanaman. Penggunaan Agrobacteriumgalur AGL-0dilakukan karena galur ini mempunyai virulensi yang tinggi dibandingkan galur Agrobacterium lainnya.
DNA plasmid setelah rekombinasi pada vektor destinasi diisolasi kemudian ditransformasikan ke Agrobacterium tumefaciensdengan metode kejut dingin (cool shock). Hal ini dikarenakan terjadi lonjakan suhu es 0oC ke nitrogen cair yang bersuhu sekitar -196oC. Lonjakan suhu tersebut akan membuat membrane sel Agrobacterium menjadi tidak selektif terhadap molekul asing sehingga DNA sisipan dapat masuk. Hasil transformasi ke dalam Agrobacterium menunjukkan bahwa koloni bakteri yang tumbuh selama inkubasi ± 2 hari terdapat 1-5 koloni (Gambar 17). Pada saat proses inkubasi, koloni dalam cawan petri dibungkus dengan kertas koran agar kondisi menjadi gelap. Hal ini dilakukan agar koloni Agrobacterium tumefaciens dapat menyesuaikan kondisinya selama hidup di dalam tanah (akar).
Seleksi transforman yang sudah ditransformasikan ke dalam Agrobacterium tumefaciens dilakukan dengan cara menumbuhkannya di dalam media LA. Koloni yang ditumbuhkan diambil berdasarkan elektroforegram hasil isolasi plasmid rekombinan setelah reaksi LR, yaitu plasmid rekombinan nomer 1 (pGD625), plasmid rekombinan nomer 3 (pARC983), dan plasmid rekombinan nomer 5 (pDEST). Media LA yang digunakan mengandung antibiotik kanamisin dan rifampisin. Sel transforman tersebut akan tumbuh membentuk koloni berwarna putih. Kanamisin pada media LA digunakan karena vektor destinasi yang digunakan membawa marka seleksi berupa gen resistensi terhadap antobiotik kanamisin, sedangkan rifampisin digunakan untuk membunuh E. coli. Koloni yang tumbuh dimedia seleksi lebih sedikit dibandingkan koloni yang tumbuh saat transformasi ke E. coli. Hal ini dikarenakan A. tumefaciens memiliki jumlah salinan yang lebih sedikit (low copy number) dibandingkan E. coli.
Koloni yang tumbuh kemudian di PCR koloni untuk menguji gen penyandi kitinase telah berhasil ditransformasi atau tidak. Hasil
PCR koloni menunjukkan pita yang terlihat berukuran sekitar 1500 pb seperti yang terlihat pada Gambar 18.
Gambar 17 Koloni A. tumefaciens yang tumbuh setelah reaksi LR pada gen penyandi kitinase menggunakan vektor destinasi (a) pGD625, (b) pARC983, dan (c) pDEST.
Gambar 18 Elektroforegram hasil PCR koloni A. tumefaciens; (1-2) dengan vektor destinasi pGD625, (3-7) dengan vektor destinasi pARC983, (8) dengan vektor destinasi pDEST, (M) marker 1 kb plus DNA Ladder.
SIMPULAN DAN SARAN
Simpulan
Saran
Penelitian lebih lanjut dapat dilakukan yaitu DNA rekombinan yang telah ditransformasikan ke Agrobacterium tumefaciens galur AGL-0 perlu ditransformasikan ke tanaman untuk mengetahui pengaruh gen penyandi kitinase terhadap pangkal batang dan akar pada tanaman kelapa sawit.
DAFTAR PUSTAKA
Abd-Elsalam KA. 2003. Bioinformatics tools and guideline for PCR primer design. African journal of Biotechnology 2:93-95.
Alexopoulus CJ, Mim CW, Black W. 1996. Introduction Mycology. New York: John & Wiley Sons.
Bassett K, Peters RN. 2003. Ganoderma: a significant root pathogen. Texas: Arborilogical services Inc. Publication.
Clark W, Chistpher K. 2008. An Introduction to DNA : Spechtrophotometry, Degradation, and the “Frangekel” Eksperimen. Alberta: University of Alberta.
Datta K, Nicola ZK, Baisakh N, Oliva N, Datta K. 2000. Agrobacterium mediated engineering for sheath blight resistance of indica rice cultivars from different ecosystems. Theor Appl Genet 100: 832-839.
Du M & Ha SB. 1999. Direct Submission. Submitted (22-APR-1997) Plant Biology, USA.
Dubin MJ, Bowler C, Benvenuto G. 2008. A modified Gateway cloning strategy for overexpressing tagged proteins in plants. Plant methods4:1-11.
Faath I. 1994. Isolation of Chitin Degrading Bacteria from Various Habitats. Bonn: Microbial diversity.
Fermentas. 2006. GeneJet TM plasmid miniprep kit. Life Science.
Hartley JL, Temple GF, Brasch MA. 2000. DNA cloning using in vitro site-specific recombination. Genome Res 10:17881795.
Howe C. 1995. Gene Cloning an Manipulation. Cambridge: Cambridge University Press.
Hwang HS, Gelvin SB. 2004. Plant proteinsthat interact with virB2, theAgrobacterium tumefaciens pilin protein,mediate plant transformation. The PlantCell 20:2661-2680.
Invitrogen. 2003. Gateway® technology: a universal technology to clone DNA sequences for functional analysis and expression in multiple systems. California: Life Technologies.
Invitrogen. 2010. Platinum® Taq DNA polymerase. California: Life Technologies.
Invitrogen. 2010. PureLinkTM quick gel extraction kit. California: Life Technologies.
Khopkar SM. 1990. Konsep Dasar Kimia Analitik. Jakarta: UI Press.
Kupiec RC, Ilan C. 1998. The molecular biology of chitin degradation. Current Opinion in Biotech 9: 270-277.
Lin W, Anuartha CS, Datta K, Potrykus L, Muthukrishnan. 1995. Genetic engineering of rice for resistance to sheath blight. Biotechnology 13: 686-691.
Lorito M et al. 1996. Synergistic interaction between cell wall degrading enzymes and membrane affecting compound. Mol. Plant-Microbe Interact9: 206-213.
Lubis. 1992. Kelapa Sawit di Indonesia. Sumatera Utara: Marihat.
Martawijaya A. 1986. Indonesian Wood Atlas Volume 1. Bogor: Departement of Forestry Agency for Forestry Research and Development.
Muladno. 2002. Seputar Teknologi Rekayasa Genetika. Bogor: Pustaka Wirausaha Muda.
Muladno. 2002. Teknologi Rekayasa Genetik Edisi ke-1. Bogor: Pustaka Wirausaha Mura.
KONSTRUKSI GEN PENYANDI KITINASE PADA VEKTOR
EKSPRESI DENGAN METODE GATEWAY DAN
TRANSFORMASI PADA
Agrobacterium
sp.
ISMI WILLYSIA BILLIRANTAU
DEPARTEMEN BIOKIMIA
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM
INSTITUT PERTANIAN BOGOR
Saran
Penelitian lebih lanjut dapat dilakukan yaitu DNA rekombinan yang telah ditransformasikan ke Agrobacterium tumefaciens galur AGL-0 perlu ditransformasikan ke tanaman untuk mengetahui pengaruh gen penyandi kitinase terhadap pangkal batang dan akar pada tanaman kelapa sawit.
DAFTAR PUSTAKA
Abd-Elsalam KA. 2003. Bioinformatics tools and guideline for PCR primer design. African journal of Biotechnology 2:93-95.
Alexopoulus CJ, Mim CW, Black W. 1996. Introduction Mycology. New York: John & Wiley Sons.
Bassett K, Peters RN. 2003. Ganoderma: a significant root pathogen. Texas: Arborilogical services Inc. Publication.
Clark W, Chistpher K. 2008. An Introduction to DNA : Spechtrophotometry, Degradation, and the “Frangekel” Eksperimen. Alberta: University of Alberta.
Datta K, Nicola ZK, Baisakh N, Oliva N, Datta K. 2000. Agrobacterium mediated engineering for sheath blight resistance of indica rice cultivars from different ecosystems. Theor Appl Genet 100: 832-839.
Du M & Ha SB. 1999. Direct Submission. Submitted (22-APR-1997) Plant Biology, USA.
Dubin MJ, Bowler C, Benvenuto G. 2008. A modified Gateway cloning strategy for overexpressing tagged proteins in plants. Plant methods4:1-11.
Faath I. 1994. Isolation of Chitin Degrading Bacteria from Various Habitats. Bonn: Microbial diversity.
Fermentas. 2006. GeneJet TM plasmid miniprep kit. Life Science.
Hartley JL, Temple GF, Brasch MA. 2000. DNA cloning using in vitro site-specific recombination. Genome Res 10:17881795.
Howe C. 1995. Gene Cloning an Manipulation. Cambridge: Cambridge University Press.
Hwang HS, Gelvin SB. 2004. Plant proteinsthat interact with virB2, theAgrobacterium tumefaciens pilin protein,mediate plant transformation. The PlantCell 20:2661-2680.
Invitrogen. 2003. Gateway® technology: a universal technology to clone DNA sequences for functional analysis and expression in multiple systems. California: Life Technologies.
Invitrogen. 2010. Platinum® Taq DNA polymerase. California: Life Technologies.
Invitrogen. 2010. PureLinkTM quick gel extraction kit. California: Life Technologies.
Khopkar SM. 1990. Konsep Dasar Kimia Analitik. Jakarta: UI Press.
Kupiec RC, Ilan C. 1998. The molecular biology of chitin degradation. Current Opinion in Biotech 9: 270-277.
Lin W, Anuartha CS, Datta K, Potrykus L, Muthukrishnan. 1995. Genetic engineering of rice for resistance to sheath blight. Biotechnology 13: 686-691.
Lorito M et al. 1996. Synergistic interaction between cell wall degrading enzymes and membrane affecting compound. Mol. Plant-Microbe Interact9: 206-213.
Lubis. 1992. Kelapa Sawit di Indonesia. Sumatera Utara: Marihat.
Martawijaya A. 1986. Indonesian Wood Atlas Volume 1. Bogor: Departement of Forestry Agency for Forestry Research and Development.
Muladno. 2002. Seputar Teknologi Rekayasa Genetika. Bogor: Pustaka Wirausaha Muda.
Muladno. 2002. Teknologi Rekayasa Genetik Edisi ke-1. Bogor: Pustaka Wirausaha Mura.
15
Ganoderma pada vektor ekspresi pCambia 1303 [skripsi]. Bogor: Fakultas Matematika dan Ilmu Pengetahuan Alam, Institut Pertanian Bogor.
Patil RS, Vandona G, Mukund VD. 2000. Chitinolytic enzymes: a exploration. Enzyme and Microbial Tech 26: 473-483.
Ren Y, West CA. 1992. Elicitation of diterpene biosynthesis in rice (Oryza sativa L.) by chitin. Plant Physiol 99: 1169-1178.
Saiki RK et al. 1989. The Design and Optimization of the PCR. Di dalam: Erlinch HA, editor. PCR Technology Principles and Applications for DNA Amplification. New York: M Stockton Press.
Sambrook J, Russell DW. 2001. Molecular Cloning a Laboratory Manual. Ed ke-3. New York: Cold Spring Harbor Laboratory.
Santoso D, Chaidamsari T, Budiani A, Minarsih H, Siswanto. 2001. Over ekspresi gen penyandi kitinase dengan enhancer sintesis kaya GC pada tanaman tembakau (Nicotiana tabacum L.) [Laporan Penelitian]. Bogor: Unit Penelitian Bioteknologi Perkebunan.
Schirmbock et al.1994. Parallel formation ad synergism of hydrolytic enzymes and peptabiol antibiotics: molecular mechanism involved in the antagonistic action oh Trichoderma harzianum against phytopathogenic fungi. Ppl Environ Microbial 60: 4364-4370.
Setyamidjaja D. 2006. Kelapa Sawit: Teknik Budidaya, Panen, dan Pengolahan. Yogyakarta: Kanisius.
Singh G. 1991. Ganoderma the scourge of oil palm in the coastal areas. Planter 67: 421-444.
Tabei Y et al. 1998. Transgenic cucumber plants harbouring a rice chitinase gene exhibit enhanced resistance to gray mold (Botrytis cinerea). Plant Cell Rep 17: 159-164.
Taira T & Akimoto R. 2007. Molecular cloning of chitinase from Ananas
comosus. Submitted (19-JAN-2007). Department of Bioscience and Biotechnology University of the Ryukyus.
Terekawa N, Takaya N, Horiuchi H, Koike M. 1997. A fungal chitinase gene from Rhizopus oligosporus confers antifungal activity to transgenic tobacco. Plant Cell Rep16: 439-443.
Tiffin P. 2004. Comparative evolutionary histories of chitinase genes in the Genus zea and Family poaceae. Genetics 167(3):1331-1340.
Tzafira T, Citovsky V. 2002. Partners in infection: Host proteins involved in the transformation of plant cells by Agrobacterium. Trends Cell Biol 12:121129.
Van Damme EJM, Willems P, Peumans W. 1993. Isolation and characterization of two different chitinase cDNA clones from garlic (Allium sativum L.) shoots (Unpublished). Physiologia Plantarum 87:177–186.
Widyastuti SM. 2009. Peran Trichoderma spp. dalam Revitalisasi Kehutanan di Indonesia. Yogyakarta: Gadjah Mada University Press.
Yuwono T. 2005. Biologi Molekular. Jakarta: Erlangga.