LAPORAN PRAKTEK KERJA PROFESI FARMASI INDUSTRI
di
PT. PRADJA PHARIN (PRAFA), CITEUREUP, BOGOR Periode 1 Nopember-24 Desember 2010
Disusun Oleh:
Alfan Martina, S.Farm. 093202101
PROGRAM PENDIDIKAN PROFESI APOTEKER FAKULTAS FARMASI
UNIVERSITAS SUMATERA UTARA MEDAN
Lembar Pengesahan
LAPORAN PRAKTEK KERJA PROFESI FARMASI INDUSTRI
di
PT. PRADJA PHARIN (PRAFA), CITEUREUP, BOGOR Periode 1 Nopember-24 Desember 2010
Laporan ini disusun sebagai salah satu syarat untuk memperoleh gelar Apoteker pada Fakultas Farmasi
Universitas Sumatera Utara Medan
Disusun oleh :
Alfan Martina, S.Farm. 093202101
PT. PRADJA PHARIN (PRAFA) Citeureup,
Pembimbing I, Pembimbing II,
Antonius Raino Dymita, S.Si, Apt. Drs. Budi Utomo
Fakultas Farmasi Universitas Sumatera Utara Dekan,
KATA PENGANTAR
Puji syukur kepada Tuhan Yang Maha Kuasa atas rahmat dan karunia-Nya sehingga penulis dapat menyelesaikan kegiatan serta laporan praktek kerja profesi farmasi industri di PT. Pradja Pharin (PRAFA), Citeureup, Bogor, periode 1 Nopember hingga 24 Desember 2010 dengan baik dan lancar.
Selesainya praktek kerja profesi ini juga tidak terlepas dari bantuan berbagai pihak. Dalam kesempatan ini, penulis mengucapkan terima kasih yang setulusnya kepada:
1. Bapak Drs. Antonius Sutaryo, Apt. selaku Plant Manager PT. Pradja Pharin (PRAFA) yang telah memberi izin pelaksanaan praktek kerja profesi farmasi industri ini.
2. Bapak Antonius Raino Dymita, S.Si, Apt., selaku pembimbing I sekaligus validation & calibration supervisor, yang telah memberikan bimbingan serta bantuan kepada penulis dalam menyelesaikan kegiatan dan laporan praktek kerja profesi di PT. Pradja Pharin (PRAFA).
3. Bapak Drs. Budi Utomo selaku pembimbing II sekaligus sterile non Betalaktam supervisordi PT. Pradja Pharin (PRAFA).
4. Bapak Prof. Dr. Sumadio Hadisahputra, Apt. selaku dekan dan Bapak Drs. Wiryanto, MS., Apt. selaku koordinator program pendidikan profesi apoteker Fakultas Farmasi Universitas Sumatera Utara Medan yang telah memberi kesempatan dan fasilitas kepada penulis untuk dapat menjalani praktek kerja profesi ini.
5. Terakhir adalah kepada Ibu Dra. Emi Pujiastuti, Aprilia Primadawaty, S.Farm., Apt., Happy Monda P. N., S.Farm., Apt. beserta seluruh manager, supervisor, section head, dan staf PT. Pradja Pharin (PRAFA) yang telah banyak membantu dan berbagi pengalamannya sehingga penulis mendapat banyak wawasan dan ilmu selama menjalani praktek kerja profesi.
dengan industri farmasi. Namun penulis menyadari sepenuhnya bahwa laporan ini belumlah sempurna. Oleh karena itu, diharapkan saran dan kritik dari para pembaca untuk menyempurnakan laporan ini.
Bogor, 23 Desember 2010 Penulis,
DAFTAR ISI
Halaman
JUDUL ... i
LEMBAR PENGESAHAN ... ii
KATA PENGANTAR ... iii
DAFTAR ISI ... v
DAFTAR TABEL ... viii
DAFTAR GAMBAR ... ix
DAFTAR LAMPIRAN ... x
BAGIAN I. LAPORAN UMUM ... 1
BAB I PENDAHULUAN ... 2
1.1 Latar Belakang ... 2
1.2 Tujuan ... 3
BAB II TINJAUAN UMUM PT. PRADJA PHARIN (PRAFA) ... 4
2.1 Sejarah dan Perkembangan Perusahaan ... 4
2.2 Struktur Organisasi ... 7
2.3 Visi dan Misi ... 7
2.4 Lokasi dan Sarana Produksi ... 8
2.5 Jenis Produksi ... 8
BAB III KEGIATAN INDUSTRI PT. PRADJA PHARIN (PRAFA) ... 11
3.1 Departemen Logistik ... 11
3.1.1 Gudang Bahan Baku ... 15
3.1.2 Gudang Bahan Kemas ... 16
3.1.3 Gudang Obat Jadi ... 16
3.2 Departemen Produksi ... 17
3.2.1 Produksi Sediaan Solid Non Betalaktam ... 17
3.2.2 Produksi Sediaan Steril Non Betalaktam ... 20
3.2.3 Produksi Sediaan Betalaktam dan Cephalosporin .. 21
3.2.4 P&G Line ... 23
3.3.1 Pemeriksaan Kimia ... 26
3.3.2 Pemeriksaan Mikrobiologi ... 27
3.3.3 QA Inspector... 29
3.3.4 Validasi dan Kalibrasi ... 31
3.3.5 QA Compliance ... 33
3.4 Product Development Department(PDD) ... 38
3.5 Departemen Technical Services(TS) ... 39
3.6 Departemen Personnel and General Affairs(PGA) ... 43
BAB IV HASIL DAN PEMBAHASAN ... 48
4.1 Manajemen Mutu ... 48
4.2 Personalia ... 49
4.3 Bangunan dan Fasilitas ... 50
4.4 Peralatan ... 52
4.5 Sanitasi dan Higiene ... 53
4.6 Produksi ... 54
4.7 Pengawasan Mutu ... 56
4.8 Inspeksi Diri dan Audit Mutu ... 56
4.9 Penanganan Keluhan Terhadap Produk, Penarikan Kembali Produk dan Produk Kembalian ... 57
4.10 Dokumentasi ... 58
4.11 Pembuatan dan Analisis Berdasarkan Kontrak ... 59
4.12 Kualifikasi dan Validasi ... 59
BAB V KESIMPULAN DAN SARAN ... 61
5.1 Kesimpulan ... 61
5.2 Saran ... 61
DAFTAR PUSTAKA 1 ... 62
LAMPIRAN ... 63
BAGIAN II. LAPORAN TUGAS KHUSUS ... 86
BAB I PENDAHULUAN ... 87
1.1 Latar Belakang ... 87
BAB II TINJAUAN PUSTAKA ... 89
2.1 Definisi Kualifikasi... 89
2.2 Tingkatan Kualifikasi ... 89
2.2.1 Kualifikasi Desain ... 89
2.2.2 Kualifikasi Instalasi ... 90
2.2.3 Kualifikasi Operasional ... 92
2.2.4 Kualifikasi Kinerja ... 93
BAB III HASIL DAN PEMBAHASAN ... 95
BAB IV KESIMPULAN DAN SARAN ... 100
4.1 Kesimpulan ... 100
4.2 Saran ... 100
DAFTAR TABEL
Halaman Tabel 1 Daftar Contoh Original Productdari PT. PRAFA ... 9 Tabel 2 Daftar Contoh Produk Toll Manufacturingyang Diproduksi di
DAFTAR GAMBAR
DAFTAR LAMPIRAN
Halaman
Lampiran 1 Struktur Organisasi PT. PRAFA ... 63
Lampiran 2 Alur Barang yang Dikelola Departemen Logistik di PT. PRAFA secara Umum ... 65
Lampiran 3 Struktur Organisasi Departemen Produksi ... 67
Lampiran 4 Alur Proses Produksi secara Umum ... 68
Lampiran 5 Alur Proses Produksi Sediaan Solid dengan Metode Granulasi Basah ... 69
Lampiran 6 Alur Proses Produksi Sediaan Solid dengan Metode Spraying ... 70
Lampiran 7 Alur Proses Produksi Sediaan Solid dengan Metode Granulasi Kering ... 71
Lampiran 8 Alur Proses Produksi Sediaan Steril ... 72
Lampiran 9 Alur Proses Produksi Sediaan Solid dengan Metode Cetak Langsung ... 73
Lampiran 10 Alur Proses Produksi Sirup Kering ... 74
Lampiran 11 Alur Proses Produksi Serbuk Injeksi Kering ... 75
Lampiran 12 Alur Proses Pengemasan Sentral ... 76
Lampiran 13 Struktur Organisasi Departemen QA/QC ... 77
Lampiran 14 Struktur Organisasi Product Development Department .. 78
Lampiran 15 Alur Pengembangan Produk Baru oleh Product Development Department... 79
Lampiran 16 Struktur Organisasi DepartemenTechnical Services... 80
Lampiran 17 Alur Pembuatan Purified Waterdi PT. PRAFA ... 81
Lampiran 18 Alur Pembuatan Water For Injection ... 82
Lampiran 19 Struktur Organisasi Departemen Personnel and General Affairs... 83
Lampiran 20 Alur Proses Pretreatment Limbah Cair Betalaktam Dan Cephalosporin ... 84
BAB I PENDAHULUAN
1.1 Latar Belakang
Obat merupakan sarana utama yang digunakan untuk meningkatkan derajat kesehatan masyarakat dan bahkan untuk menyelamatkan jiwa manusia. Oleh karena itu, sebagai industri yang hi-regulated, pabrik obat atau industri farmasi diwajibkan untuk menjamin keamanan, khasiat dan mutu produk obat yang dihasilkannya selama diberikan ijin edar oleh BPOM.
Kriteria aman dan berkhasiat dijamin lewat proses pemilihan bahan awal dari pemasok secara cermat dan hati-hati. Sedangkan mutu ditentukan oleh rangkaian proses desain dan formulasi produk obat, komponen dan proses pengemasan, serta lingkungan produksi dan cara penyimpanan selama masa edarnya. Jika rangkaian proses ini hendak dipertahankan maka diperlukan kendali mutu yang ketat berupa seperangkat sistem manajemen mutu.
Untuk menjamin keamanan dan khasiat serta mengendalikan mutu produk obat yang sedemikian rumit maka sangat diperlukan tenaga-tenaga profesional di industri farmasi. Salah satu tenaga profesional yang dimaksud adalah apoteker. Apoteker merupakan profesi yang memiliki tanggung jawab baik moral maupun legal sebagai pelindung terakhir (last safeguard) bagi pasien atau konsumen pengguna obat.
Pentingnya peran apoteker di industri farmasi disebutkan dalam Keputusan Kepala Badan Pengawas Obat dan Makanan No.HK.00.05.3.02152 tahun 2002 bahwa manajer produksi dan pengawasan mutu hendaklah seorang apoteker. Apoteker yang bersangkutan disyaratkan untuk memiliki pengalaman praktis yang memadai di industri farmasi, cakap dan terlatih. Seiring dengan pemberlakuan harmonisasi pasar ASEAN pada tahun 2015, persyaratan ini akan kian meningkat sehingga diperlukan lulusan apoteker yang memiliki kemampuan mutakhir dan profesional di bidang farmasi industri dalam rangka memenangkan daya saing nasional.
Untuk menjawab tantangan tersebut, sebagai salah satu fakultas farmasi yang menyelenggarakan program pendidikan profesi apoteker, Fakultas Farmasi Universitas Sumatera Utara bekerjasama dengan PT. Pradja Pharin (PRAFA) juga turut membekali mahasiswanya dengan kecakapan dan pengalaman praktis di industri farmasi yang mutakhir dengan menyelenggarakan praktek kerja profesi.
Praktek kerja profesi di industri farmasi mencakup pengetahuan peran dan tanggung jawab apoteker serta pembelajaran berdasarkan pengalaman kerja tentang aspek CPOB yang mutakhir dan terkini di industri farmasi. Sebagai bahan evaluasi pelaksanaan praktek kerja profesi farmasi industri di PT. Pradja Pharin (PRAFA) maka disusunlah laporan ini oleh penulis.
1.2 Tujuan
Adapun tujuan pelaksanaan praktek kerja profesi di industri farmasi adalah sebagai berikut:
a. Mengenal dan memahami fungsi dan tanggung jawab profesi apoteker di industri farmasi.
BAB II
TINJAUAN UMUM PT. PRADJA PHARIN (PRAFA)
2.1 Sejarah dan Perkembangan Perusahaan
PT. PRAFA didirikan pada tahun 1960 oleh Tjipto Pusposuharto, yang berawal dari sebuah industri rumah tangga dengan karyawan berjumlah 20 orang di area berukuran 325 m2.
Pada tahun 1968, dengan semakin luasnya pasar dan semakin kuatnya kepercayaan prinsipal utama, PT. PRAFA ditunjuk sebagai importir dan penyalur tunggal di Indonesia untuk Meiji Seika, Jepang. Kemudian pada tahun 1971, PT. PRAFA menjadi perusahaan Penanaman Modal Dalam Negeri (PMDN) dengan tujuan untuk meningkatkan fasilitas produksi yang lebih besar. Sejak saat itu, pembangunan pabrik dimulai di area seluas 2300 m2 di jalan Bandengan Selatan 58 A Jakarta Utara. Dengan demikian, pabrik dapat memproduksi berbagai jenis sediaan obat yang jumlahnya lebih besar.
Pada tahun 1975, PT. PRAFA semakin intens dalam melibatkan diri dengan prinsipal-prinsipal multinasional dengan maksud untuk memperoleh keahlian manajerial yang lebih baik dan peningkatan teknologi. Antara tahun 1975-1978, PT.PRAFA ditunjuk sebagai wakil tunggal OXOID dan BDH dari Inggris, Cutter Laboratories dari Amerika Serikat dan Flow Laboratories dari Australia.
Sejak tahun 1988, PT. PRAFA tumbuh menjadi suatu industri farmasi dengan ± 1000 karyawan, 200 jenis sediaan obat berkualitas dan total penanaman modal mencapai lebih dari 10 miliar rupiah. Pada tahun itu pula, dibangun pabrik modern di atas area seluas ± 12 hektar, dengan luas bangunan 32.208,52 m2 , yang terletak di daerah Citeureup, kabupaten Bogor.
Pada tahun 1989, PT. PRAFA memperoleh lisensi dari DONG-A Pharmaceutical, Korea, untuk memproduksi minuman tonik Bacchus-D dan juga memperoleh lisensi dari ANDRELON Cosmetic B.V yang merupakan salah satu produsen kosmetika utama di Belanda. Pabrik baru selesai dibangun pada tahun 1990 dan PT. PRAFA resmi pindah ke lokasi tersebut sampai sekarang. Pabrik tersebut dirancang dan dibangun sesuai dengan aspek Cara Pembuatan Obat yang Baik (CPOB). Semua fasilitas dibangun dengan teknologi mutakhir baik dalam produksi sediaan solid, steril dan lain sebagainya.
PT. PRAFA melakukan merger dengan Darya Varia Group dan dibeli oleh First Pacific Investment, Hongkong, pada tahun 1995. Darya Varia Group terdiri dari tiga perusahaan yaitu PT. Darya Varia Laboratoria, PT. Kenrose Indonesia dan PT. Dupa dengan distributor PT. Wigo Distributor Farmasi. Tahun 1998 PT. Dupa dan PT. Kenrose ditutup sebagai upaya restrukturisasi usaha bagi perseroan (Darya Varia Group). Sejak tanggal 21 Desember 2001 hingga sekarang Darya Varia Group diambil alih oleh United Laboratories, Inc. (UNILAB), Filipina. Selain Darya Varia Group yang kini hanya terdiri dari PT. Darya Varia Laboratoria (DVL) dan PT. Pradja Pharin (PRAFA), UNILAB juga memiliki perusahaan farmasi lain di Indonesia yakni PT. Medifarma Laboratories.
72%. Dengan perjuangan dan komitmen yang tinggi, akhirnya hanya dalam waktu satu tahun, PT. PRAFA berhasil menaikkan pointnya menjadi 92% saat diaudit kembali oleh P&G. Sejak saat itu, PT. PRAFA dipercaya oleh perusahaan P&G untuk menerima toll manufacturing hingga kini, yaitu memproduksi Vicks Formula 44, Vicks Vaporub dan Vicks Inhaler. Pada tahun 2008, 2009, dan 2010 P&G memberikan point 100% untuk audit yang dilakukan pada PT. PRAFA. Pada tahun 2008 perusahaan Novartis juga mempercayakan toll manufacturing kepada PT. PRAFA untuk memproduksi tablet effervescent Ca-Sandoz.
Pada tahun 2005, PT. PRAFA memperoleh sertifikat industri farmasi kelas A dari hasil mapping Badan POM dalam menilai kesiapan industri farmasi menghadapi harmonisasi pasar ASEAN. Hal ini berarti bahwa PT. PRAFA diijinkan untuk melakukan produksi di fasilitas sendiri dan menerima toll manufacturing dari industri farmasi lain. Hingga kini, PT. PRAFA senantiasa berusaha untuk meningkatkan kualitas sarana dan sumber daya manusianya, terutama dalam mematuhi standar PIC/S dan FDA Regulation.
Pada tahun 2009, Darya Varia Group melakukan project spesialization yakni PT. Medifarma Laboratories dikhususkan dalam produksi high volume solid order dan obat-obat bebas (Over The Counter/OTC), PT. Darya Varia Laboratories untuk produksi kapsul gelatin lunak (soft gelatin capsules), sediaan cair dan semisolid serta PT. PRAFA diarahkan pada produksi low volume solid order, produk etikal (solid dan injeksi), antibiotik betalaktam dan cephalosporin (solid dan injeksi), serta produk toll manufacturing. Oleh karena banyaknya prinsipal lokal dan multinasional yang melakukan toll manufacturing, PT. PRAFA lalu dikhususkan sebagai Centre of Excellent Toll Manufacturing.
Segitiga pada gambar tersebut mengimplikasikan lambang huruf awal nama perusahaan. Sementara, bentuk segitiga itu sendiri melambangkan kemajuan dan budaya perusahaan yang modern. Pertemuan antar segitiga pada logo tersebut melambangkan kerjasama, kebersamaan dan komitmen. Sisi sama panjang mencerminkan bahwa PRAFA terdiri dari elemen yang memiliki kepentingan bersama serta saling menunjang dan mendukung sehingga tidak ada yang dapat berdiri sendiri tanpa dukungan kekuatan elemen yang lain. Warna biru pada logo PRAFA melambangkan semangat, rasa aman, bersih dan kepercayaan melalui produk-produk yang dihasilkannya sehingga akan memberikan kesan yang berlangsung lama di hati para konsumennya.
2.2 Struktur Organisasi
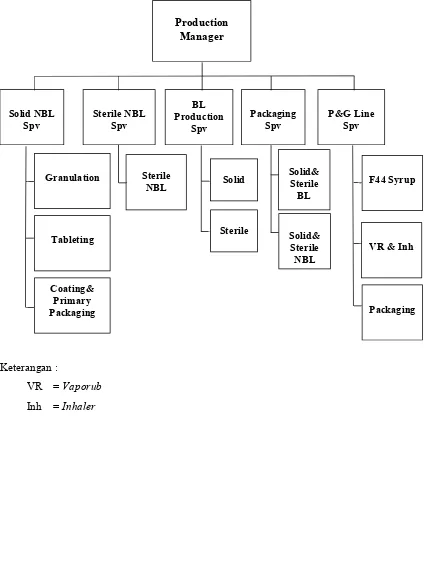
PT. PRAFA dipimpin oleh seorang Plant Manager yang membawahi lima departemen. Masing-masing departemen dipimpin oleh seorang manager yang dibantu oleh beberapa supervisor. Struktur organisasi PT. PRAFA per 1 Juni 2010, dapat dilihat pada Lampiran 1.
2.3 Visi dan Misi
Visi dan Misi PT. PRAFA tertuang dalam motto “We Commit to Speed, Quality, Cost and Safety”. Dengan motto ini, PT. PRAFA selalu berusaha menghasilkan produk bermutu tinggi dan terjangkau oleh masyarakat dengan mengutamakan keselamatan kerja. Untuk dapat menembus pangsa pasar internasional, PT. PRAFA juga berusaha untuk meningkatkan kualitas sarana dan sumber daya manusianya dengan turut mempedomani standar PIC/S dan FDA Regulation.
2.4 Lokasi dan Sarana Produksi
Kantor pusat PT. PRAFA berada di Talavera Office Park, 8th-10th Floor, Jl. Letjen Simatupang no. 22-26, Jakarta. Sedangkan lokasi pabrik berada di Desa Karang Asem Barat, Kecamatan Citeureup, Kabupaten Bogor, Jawa Barat. Pabrik ini menempati areal seluas 12 hektar dengan bangunan produksi seluas 17.208 m2 yang relatif terpisah dari lokasi pemukiman penduduk.
Sarana yang dimiliki pabrik PT. PRAFA antara lain: a. Bangunan utama. Terdiri dari tiga gedung utama, yaitu:
i. Gedung pertama, digunakan sebagai lokasi kantor, produksi non betalaktam, ruang produksi dan kemas P&G, departemen Product Development Department(PDD) dan lokasi central packaging.
ii. Gedung kedua, digunakan sebagai lokasi kantor departemen Quality Assurance(QA)/Quality Control(QC), departemen logistik ,kantor PPIC, gudang bahan baku dan bahan kemas.
iii.Gedung ketiga, digunakan untuk lokasi produksi betalaktam dan cephalosporin.
b. Bangunan penunjang, meliputi gedungTechnical Service, instalasi listrik,Air Handling Unit (AHU),steam unit, compress air unit, waste water treatment, water system unit, masjid, kantin, area parkir, pos satpam, dan unitlaundry. c. Bangunan lain, seperti gudang obat jadi, gudang api untuk penyimpanan
bahan-bahan yang mudah terbakar, pengolahan limbah dan insinerator.
2.5 Jenis Produksi
Sehubungan dengan adanya project spesializationpada Darya Varia Group pada tahun 2009, PT. PRAFA saat ini hanya memproduksi sediaan tablet dan kapsul dalam skala kecil, infus 100 ml, larutan injeksi, suspensi injeksi, injeksi kering serta sirup kering untuk obat etikal, antibiotik betalaktam dan cephalosporin.
disebut sebagai original product dan produk keluaran prinsipal lain yang diproduksi oleh PT. PRAFA lewat kerjasama toll manufacturing. Daftar contoh original product dari PT. PRAFA dapat dilihat pada Tabel 1 dan daftar contoh produk toll manufacturing yang diproduksi di PT. PRAFA dapat dilihat pada Tabel 2.
Tabel 1. Daftar Contoh Original Productdari PT. PRAFA
Jenis Bentuk Sediaan Contoh Produk
[image:18.612.131.508.219.454.2]Sediaan Non Steril Non Betalaktam
Tablet
Vicee, Mediamer SCT, Moloco SCT, Spasmal, Ossopan SCT 200 mg & Ossopan FCT 800 mg, H2Bloc 10 mg dan 20 mg, Paratusin
Sediaan Steril Non Betalaktam
Infus Fortagyl 100 ml
Suspensi injeksi Injeksi Cortison acetat
Larutan injeksi Injeksi Paramidon 15 ml, Paradryl 15 ml
Sediaan Non Steril Betalaktam & Cephalosporin
Tablet Penicillin V, Griseofulvin 500 mg
Kapsul Bannoxillin 500 mg, Urticef 50
mg Sediaan Steril
Betalaktam & Cephalosporin
Injeksi kering Cefurox
Tabel 2. Daftar Contoh Produk Toll Manufacturing yang Diproduksi di PT. PRAFA
No. Nama Prinsipal Toller Contoh Produk Toll Manufacturing 1 PT. Actavis Indonesia Dumozol infus
2 PT. Armoxindo Farma Kloramidina dry inj., Kanarco dry inj.,Kloramixin eye drop
3 PT. Darya Varia
[image:18.612.131.508.502.712.2]Stop Cold SCT, Hobat soft capsul, Blood Care soft capsul, Agulan, Alanox kaplet, Otopraf tetes telinga, Delsoralen, Disurdin, trifluoperazine, Fordiamil FCT, Degirol tablet hisap, Isoprinosine, Livercare kapsul, Lifezar, Kendaron, Theravask, Metsel kaplet, Odace
4 PT. Dipa Pharmalab
Intersains Ditranex FCT dan injeksi
6 PT. Kalbe Farma Clavamox injeksi, Kalitake puyer, Bactesyn
7 PT. Lapi Lapimox, Lapicef, Cravox, Neuciti amp
8 PT. Medifarma
Enervon C tablet effervescent, Gastran tablet, Obimin FCT, Fundamin E, Norizec, Unagen, Urdafalk
9 PT. Nufarindo Nufamox kaplet dan injeksi
10 PT. Novartis Ca-Sandoz tablet effervescent
11 PT. Novell Pharmaceutical Cefixime kapsul
12 Nycomex Cedocard, Cedona
13 P&G
Vicks Formula 44child,adult , DT, Vicks F44 DT sachet, Vicks Vaporub, dan Vicks Inhaler
14 PT. Pharos Ranin injeksi, Narfoz, Polysilane tablet, Zemyc infus
15 PT. Pyridam Pyricef 500 mg kapsul dan sirup kering
16 PT. Sandoz Indonesia Surpas FCT, Ospamox FCT, Banadoz kaplet, Biotriax inj., Baxima inj. 17 PT. Servier Indonesia Ardium, Diamicron MR tab, Arcalion
SCT, Natrilix SR tab, Prexum, Stablon
BAB III
KEGIATAN INDUSTRI PT. PRADJA PHARIN (PRAFA)
3.1 Departemen Logistik
Departemen logistik terdiri dari gudang bahan baku, gudang bahan kemas, dan gudang obat jadi. Departemen ini menggunakan program komputer ERIC/Enterprise Resource Information & Controlsebagai sistem pencatatan stok.
Adapun tugas dan tanggung jawab departemen ini adalah sebagai berikut: a. Menerima, menyimpan dan mengeluarkan barang serta mengelola semua
inventaris yang meliputi bahan baku/raw material, bahan kemas dan obat jadi/finished goods.
b. Menjaga kualitas dan kuantitas bahan baku, bahan kemas dan obat jadi di dalam gudang sesuai dengan syarat dan ketentuan CPOB yang berlaku.
c. Memonitor persediaan bahan baku, bahan kemas dan obat jadi.
Berdasarkan uraian tugas dan tanggungjawabnya, kegiatan departemen logistik secara mendasar meliputi kegiatan penerimaan, penyimpanan, pengeluaran dan penimbangan barang baik itu bahan baku, kemas maupun obat jadi. Kegiatan ini akan diuraikan sebagai berikut.
a. Penerimaan barang.
tidak tembus air. Hal ini dilakukan untuk memastikan tidak terjadi kontaminasi terhadap barang yang diangkut sehingga kualitasnya tidak terjamin.
Kondisi fisik barang diperiksa dengan mengamati kondisi kemasan, jumlah barang dan lain-lain, yang mana kemudian dituliskan dalam Incoming Material Checklist/IMC. Bila sudah sesuai dengan persyaratan, barang akan diterima dan dilanjutkan dengan penanganan surat jalannya. Setelah itu, barang baru kemudian dapat disusun di atas pallet yang sudah bersih dan pihak gudang lalu menempelkan label quarantine (warna kuning) pada tiap pallet serta mencatatnya ke dalam bincard. Selanjutnya petugas bagian gudang akan mengisi Incoming Material List/IML dan membuat Purchase Receipt Local/PRL atau Purchase Receipt Import/PRI (untuk barang impor) sebagai bukti penerimaan bahan baku atau bahan kemas yang kemudian akan diperiksa oleh pihak departemen QA/QC.
Selain berasal dari pihak departemen produksi, pihak gudang juga menerima barang retur yang berasal dari kiriman distributor, berupa obat jadi yang sudah mendekati masa kadaluarsa atau yang kemasannya rusak. Dokumen yang menyertainya adalah berupa dokumen retur yang mana isi dokumen ini sama seperti dokumen PHP sehingga dapat ditelusuri nomor bets produk yang diretur. Proses penerimaan ini disertai pemeriksaan fisik oleh petugas gudang serta oleh petugas QA Inspector. Dalam hal ini, pihak departemen QA/QC berwenang membuat disposisi untuk produk retur terkait masa kadaluarsa apakah akan dimusnahkan jika memang sudah melewati masa kadaluarsa atau jika ternyata belum maka produk tersebut dapat ditolak dan dikembalikan ke distributor bersangkutan. Sedangkan untuk produk yang kemasannya rusak, disposisi dari pihak departemen QA/QC dapat berupa pengemasan ulang/repacking. Jika kerusakan ternyata dikarenakan kelalaian distributor misalnya kondisi penyimpanan yang tidak sesuai dengan persyaratan, maka biaya repacking ini akan ditanggung oleh pihak distributor.
b. Penyimpanan barang.
Penyimpanan barang baik itu bahan baku maupun bahan kemas, diwajibkan mematuhi persyaratan kondisi penyimpanan yang baik sesuai dengan rekomendasi pemasok terkait. Hal ini terutama terkait dengan suhu penyimpanan dikarenakan suhu penyimpanan umumnya sangat mempengaruhi kualitas barang. Berdasarkan alasan inilah, tiap lokasi gudang umumnya memiliki beberapa area penyimpanan seperti AC area, non AC areadan cool storage area.
Penyimpanan barang-barang yang fast movingdiposisikan dekat dengan pintu keluar dengan maksud untuk memudahkan pengambilan, serta masing-masing stok barang memiliki kartu rak/bincardtersendiri dengan tujuan untuk mencatat kegiatan keluar-masuk barang, jumlah dan tanggal transaksi serta untuk memudahkan pengambilan barang dari rak gudang.
c. Pengeluaran barang.
sistem First Expired First Out/FEFO dan First In First Out/FIFO untuk bahan baku dan sistem First In First Out/FIFO untuk bahan kemas, sementara itu pengurangan stok barang dilakukan di sistem ERIC. Sebagai tanda bukti pengeluaran barang, pihak gudang akan mengeluarkan dokumen yang bernama Manufacturing Issue/MI. MI akan dikeluarkan setelah penimbangan yang mana mengacu pada Batch Production Record/BPR untuk bahan baku atau setelah dikirim atau diterima oleh divisi pengemasan sentral/central packaginguntuk bahan kemas.
d. Penimbangan barang/Dispensary.
Kegiatan penimbangan yang dilakukan umumnya sudah terjadwal dan disesuaikan dengan jadwal kegiatan produksi. Dokumen penimbangan yang terkait antara lain Manufacturing Order/MO, Material Requirement Document/MRD dan Batch Production Record/BPR.
Proses penimbangan diawali ketika bahan baku beserta bincard-nya yang sudah disiapkan sesuai dengan MRD dibawa ke ruang antara untuk kemudian dibuka kemasan terluarnya dan dimasukkan ke dalam ruang dispensaryuntuk ditimbang. Bincard digunakan untuk mencatat hasil penimbangan dan nantinya akan disesuaikan dengan pencatatan stok di ERIC. Setelah penimbangan selesai, bagian gudang akan mengeluarkan Manufacturing Issue/MI dimana waktu pemotongan stoknya di sistem ERIC paling lama 16 jam setelah ditimbang. Hasil penimbangan selanjutnya akan diberi label penimbangan dan diserahkan ke departemen produksi beserta dokumen-dokumen terkait.
Adapun fasilitas gudang yang menjadi tanggungjawab departemen ini meliputi gudang bahan baku, bahan kemas, obat jadi, dan gudang api.
3.1.1 Gudang Bahan Baku
Gudang bahan baku ditujukan sebagai tempat penyimpanan semua bahan baku, baik untuk kegiatan produksi PRAFA maupun toll manufacturing dimana area penyimpanan bahan P&G dan bahan toll manufacturing ditempatkan di lokasi tersendiri, sementara bahan baku berupa zat aktif untuk kegiatan produksi betalaktam dan cephalosporin disimpan di area gudang yang terpisah secara fisik dari gudang lain. Gudang ini sendiri terdiri dari beberapa area penyimpanan yakni: a. AC Area, merupakan area gudang dengan suhu ≤ 25°C dan kelembaban RH ≤
75% yang mana ditujukan untuk menyimpan bahan baku yang tidak stabil pada suhu > 25°C.
b. Non AC Area, merupakan gudang dengan suhu 32°C dan kelembaban RH ≤ 75% yang mana ditujukan untuk menyimpan bahan yang stabil pada suhu tersebut
3.1.2 Gudang Bahan Kemas
Gudang bahan kemas ditujukan sebagai tempat penyimpanan semua bahan yang diperlukan pada proses pengemasan untuk menghasilkan obat jadi. Gudang ini memiliki dua ruang penyimpanan yakni :
a. AC Areaatau ruang AC merupakan area gudang yang mana ditujukan untuk penyimpanan label dan alu-foil.
b. Non AC Area atau ruang non AC merupakan area gudang yang mana ditujukan untuk penyimpanan box, botol, ampul, vial danrubber stopper dan leaflet.
3.1.3 Gudang Obat Jadi
Gudang obat jadi digunakan untuk menyimpan hasil produksi berupa obat jadi yang siap dikirimkan ke distributor. Gudang ini memiliki beberapa fasilitas ruangan yakni:
a. AC Area, merupakan ruangan yang mana ditujukan untuk menyimpan obat jadi yang memerlukan penyimpanan pada suhu ≤ 25°C dan kelembaban RH≤ 75%.
b. Non AC Area, merupakan ruangan yang mana ditujukan untuk menyimpan obat jadi yang tidak memerlukan persyaratan khusus dalam penyimpanannya. c. Cool Storage Area, merupakan ruangan dengan suhu 2-8°C yang mana
ditujukan untuk menyimpan produk injeksi.
d. Quarantine Area, merupakan ruangan yang mana ditujukan untuk menyimpan obat jadi yang masih dalam status pemeriksaan QC. Obat jadi ini yang disimpan di sini terutama merupakan obat-obat kembalian dari distributor.
3.2 Departemen Produksi
Departemen ini dipimpin oleh seorang apoteker sebagai production manager, yang terdiri dari lima divisi yaitu solid non betalaktam, steril non betalaktam, betalaktam dan cephalosporin, P&G line serta pengemasan sentral/central packaging. Masing-masing divisi dipimpin oleh seorang supervisor yang dibantu oleh beberapasection head. Adapun struktur organisasi departemen produksi dapat dilihat pada Lampiran 3.
Departemen produksi melaksanakan kegiatan produksi berdasarkan dokumen perintah produksi (Manufacturing Order/MO) yang dilengkapi dengan dokumen permintaan bahan baku dan bahan kemas ke gudang (Manufacturing Requirement Document/MRD), bukti pengeluaran bahan baku dan bahan kemas dari gudang (Manufacturing Issue/MI), catatan pengolahan bets (Batch Production Record/BPR), dokumen pengemasan primer (Packaging Direction Record Primary/PDRP) dan dokumen pengemasan sekunder (Packaging Direction Record Secondary/PDRS) dari PPIC. Setelah itu, proses produksi dimulai dengan penyiapan jalur produksi/line clearance untuk memastikan kesesuaian jenis dan jumlah bahan baku, kesiapan peralatan dan kondisi ruangan. Selama proses produksi berlangsung, juga dilakukan In Process Control/IPC oleh bagian produksi sesuai dengan SOP pembuatan masing-masing sediaan dan oleh QA inspector dengan di bawah pengawasan departemen QA/QC. Setelah proses produksi selesai, dilakukan pembersihan/sanitasi terhadap semua mesin dan ruangan yang dipakai, kemudian diberi label “bersih” lengkap dengan nama operator dan tanggal pembersihan. Produk ruahan yang dihasilkan lalu dikirim ke divisi pengemasan sentral. Setelah proses pengemasan sekunder selesai, obat jadi akan dikirim ke gudang obat jadi dilengkapi dengan dokumen Pengiriman Hasil Produksi (PHP) untuk kemudian disalurkan ke distributor . Alur proses produksi secara umum dapat dilihat pada Lampiran 4.
3.2.1 Produksi Sediaan Solid Non Betalaktam
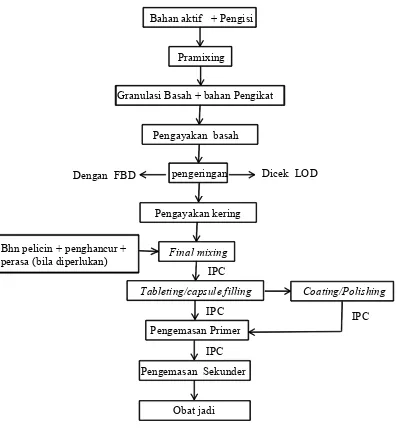
Sebagian besar produksi solid non betalaktam menggunakan metode granulasi basah (wet granulation) dan hanya sebagian kecil saja dengan metode granulasi kering (dry granulation).
a. Granulasi basah
dilanjutkan ke tahap penyalutan. Proses penyalutan yang dilakukan ada dua jenis yaitu penyalutan gula/sugar coating dan penyalutan lapis tipis/film coating. Tahap berikutnya dilakukan pengemasan primer (blistering/stripping). Selama proses blistering/stripping, juga dilakukan pemeriksaan secara visual yang meliputi penampilan hasil blistering/stripping, penandaan nomor bets dan tanggal kadaluarsa serta uji kebocoran/leakage test oleh petugas produksi dan QA inspector. Setelah melalui proses ini dan mendapat status QA released, produk selanjutnya dapat dikirim ke divisi pengemasan sentral untuk dilakukan pengemasan sekunder. Alur proses produksi sediaan solid dengan metode granulasi basah dapat dilihat pada Lampiran 5.
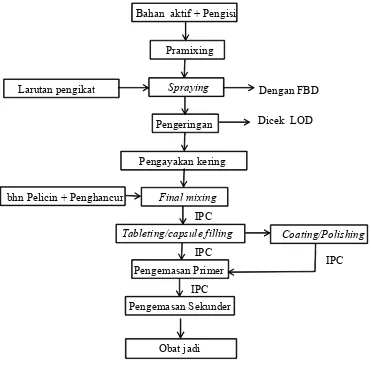
Sekarang ini telah dilakukan modifikasi prosedur proses produksi yang lebih efisien yaitu proses premixing dan pengadukan basah dilakukan sekaligus dengan menggunakan FBD. Larutan pengikat dibuat terlebih dahulu dengan menggunakan premier mixer lalu disemprotkan melalui sprayer (peristaltic pump) disertai dengan pengeringan hingga terbentuk granul sesuai dengan spesifikasi yang diinginkan. Setelah dilakukan pemeriksaan kadar air granul, tahapan dilanjutkan dengan granulasi kering menggunakan mesin comminutor, lalu dilakukan final mixing dan pencetakan. Proses ini dinamakan dengan istilah spraying. Pada dasarnya semua bahan yang digunakan dalam proses sprayingsama halnya dengan proses granulasi basah, hanya saja jumlah larutan pengikat/binder yang diperlukan lebih banyak. Modifikasi prosedur ini memberikan beberapa keuntungan antara lain penghematan man hours (granulasi dan pengadukan basah tidak dilakukan), lead time produksi menjadi lebih singkat, biaya produksi berkurang, penghematan ruang produksi serta dari segi formulasi diperoleh waktu hancur tablet yang lebih baik. Alur proses produksi sediaan solid dengan metode spraying dapat dilihat pada Lampiran 6.
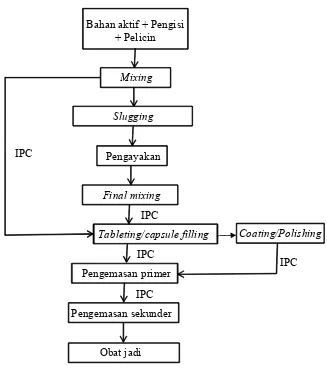
b. Granulasi kering
ditambahkan sebagian maka terlebih dahulu dilakukan slugging. Setelah proses slugging, dilakukan pengayakan/sieving untuk menghasilkan granul dengan ukuran mesh tertentu, kemudian dilanjutkan ke tahap final mixingdan pencetakan tablet atau pengisian kapsul. Alur proses produksi sediaan solid dengan metode granulasi kering dapat dilihat pada Lampiran 7.
3.2.2 Produksi Sediaan Steril Non Betalaktam
Divisi ini menangani produksi sediaan ampul, vial, infus (100 ml), tetes mata serta tetes telinga. Kegiatan produksi steril non betalaktam terdiri dari dua jenis yaitu produksi sediaan secara aseptis dan produksi sediaan yang disterilisasi akhir. Untuk bahan aktif yang tidak tahan panas diproses secara aseptis sedangkan bahan aktif yang tahan panas dilakukan sterilisasi akhir.
Tahapan proses produksi sediaan steril non betalaktam akan dijelaskan secara singkat sebagai berikut:
a. Pencucian/Washing
Pencucian wadah (ampul/vial/botol infus) dilakukan sehari sebelum produksi berjalan dengan menggunakan mesin pencuci ampul/vial/botol infus. Proses pencucian ini dilakukan di bawah LAF (Laminar Air Flow). Setelah dicuci, wadah gelas berupa ampul/vial/botol infus disterilisasi dengan menggunakan oven pada suhu 215°C selama 2 jam sedangkan alat-alat non gelas disterilisasi menggunakan autoklaf pada suhu 121°C selama 1 jam.
b. Pencampuran/Mixing
Proses mixingdilakukan di zona kelas A di bawah LAF dengan menggunakan mixing tank yang dilengkapi dengan mixer dan kompresor. Proses mixing terdiri dari proses pencampuran bahan awal yang telah ditimbang dan pelarutan.
c. Penyaringan/Filtration
d. Pengisian/Filling
Proses filling berupa pengisian larutan obat ke dalam wadah kemasan primer steril dilakukan di zona kelas A dengan latar belakang zona kelas B (yakni di bawah LAF di ruang white area). Sampling IPC dilakukan oleh petugas dari produksi dan QA inspector guna dilakukan pemeriksaan keseragaman volume. Setelah memperoleh status QA released, proses baru dilanjutkan ke tahap flame sealing (untuk sediaan ampul) dan sealing cap (untuk sediaan vial dan botol infus). Selanjutnya, QA inspector melakukan pemeriksaan berupa uji kebocoran.
e. Sterilisasi akhir
Untuk produk yang tahan pemanasan dilakukan proses sterilisasi akhir dengan menggunakan autoklaf sedangkan untuk produk yang diproses secara aseptis tidak dilakukan sterilisasi akhir. Setelah tahapan ini, QA inspector akan melakukan samplinguntuk uji sterilisasi.
f. Inspeksi
Proses terakhir adalah inspeksi. Inspeksi yang dilakukan adalah secara visual yakni dengan melihat ada tidaknya partikel-partikel pengotor berupa serat, pecahan kaca atau black spot. Sedangkan inspeksi yang lain meliputi penyeleksian terhadap seal cap yang rusak, kemasan primer yang bocor, mulut vial yang pecah ketika proses sealing cap dan vial yang permukaan luarnya kotor sebelum dilakukan pengemasan sekunder. Alur proses produksi sediaan steril dapat dilihat pada Lampiran 8.
3.2.3 Produksi Sediaan Betalaktam dan Cephalosporin
memiliki tingkat sensitisasi yang tinggi sehingga dikhawatirkan dapat membahayakan individu yang alergi terhadap antibiotik jenis ini. Pencegahan kontaminasi ini juga tercermin dari kebijakan manajemen yaitu setiap karyawan yang akan meninggalkan bangunan tersebut diharuskan mandi terlebih dahulu sebelum keluar. Selain itu, pengolahan limbah terhadap sisa produksi betalaktam juga dilakukan secara terpisah dari limbah sisa produksi lainnya.
Sediaan-sediaan yang diproduksi bagian betalaktam dan cephalosporin antara lain tablet, kapsul, sirup kering dan serbuk injeksi kering. Proses produksi solid betalaktam dan non solid betalaktam sesungguhnya hampir sama namun yang membedakan adalah pada metode granulasi. Proses produksi solid betalaktam tidak ada yang menggunakan metode granulasi basah tetapi hanyalah granulasi kering dan cetak langsung. Hal ini disebabkan sifat bahan aktif golongan betalaktam yang mudah terhidrolisis. Proses produksi diawali dengan penimbangan zat aktif di unit dispensary betalaktam ataupun cephalosporin sedangkan bahan pembantu sudah ditimbang sebelumnya di bagian dispensary dari departemen logistik. Untuk metode cetak langsung, proses dilanjutkan ke tahap mixing menggunakan drum mixer. Setelah bulk yang dihasilkan telah mendapat status QA released, bulk sudah bisa langsung dicetak (tableting). Sedangkan untuk proses produksi dengan metode granulasi kering, proses yang dikerjakan sama seperti proses produksi sediaan solid non betalaktam. Alur proses produksi sediaan solid dengan metode granulasi kering dapat dilihat pada Lampiran 7 sedangkan dengan metode cetak langsung dapat dilihat pada Lampiran 9.
Proses produksi sirup kering diawali dengan penimbangan bahan aktif kemudian dilanjutkan dengan proses mixing menggunakan drum mixer. Hasilnya berupa bulkyang selanjutnya diisikan ke dalam botol-botol kering di ruangan low humidity dengan tingkat RH lebih rendah dari 40% dan suhu lebih rendah dari 25°C. Setelah selesai, proses langsung dilanjutkan ke tahap sealing. Alur proses produksi sirup kering dapat dilihat pada Lampiran 10.
dilakukan secara manual menggunakan mesin vakum sedangkan di bagian cephalosporin menggunakan sistem automatic line jadi semua proses dilakukan secara kontinu mulai dari pencucian, sterilisasi dan pendinginan vial, pengisian bulk, penutupan dengan rubber stopper dan flip off serta sealing. Alur proses produksi serbuk injeksi kering dapat dilihat pada Lampiran 11.
3.2.4 P&GLine
Proses produksi P&G (P&G Line) dilakukan secara terpisah dari produksi PT. PRAFA dikarenakan kegiatan produksinya berskala besar sehingga produk-produk P&G memiliki jalur produk-produksi sendiri. Ada 3 jenis item yang diproduk-produksi di P&Glineyaitu:
a. Vicks Formula 44, dengan tiga variannya yakni F44 adult, F44 anak-anak dan F44 DT (Day Time). Masing-masing diproduksi dalam kemasan botol berukuran 27 ml, 54 ml dan 100 ml serta dalam kemasan sachet berukuran 7 ml khusus untuk F44 DT.
b. Vicks Vaporub, yakni dalam kemasan ukuran 10 g dan 50 g. c. Vicks Inhaler.
Kegiatan di jalur ini menggunakan sistem produksi secara in line atau automatic lineuntuk meningkatkan efektivitas dan efisiensi proses produksi. Hal ini berarti proses dilakukan secara kontinu mulai dari pencampuran bahan awal, pengisian, pengemasan primer hingga ke pengemasan sekunder. Proses produksi ini berbeda dengan sebagian besar produk PRAFA yang mana setelah dilakukan pengemasan primer, produk harus dikirim dahulu ke divisi pengemasan sentral untuk dilakukan pengemasan sekunder.
3.2.5 Pengemasan Sentral/Central Packaging
Divisi pengemasan sentral dibagi lagi menjadi 3 area yaitu pengemasan produk non betalaktam, pengemasan produk betalaktam dan produk cephalosporin. Divisi ini dipimpin oleh seorang supervisor yang membawahi section head, group leaderdan line leader. Tahapan pengemasan yaitu:
a. Persiapan bahan kemas dengan meminta bahan pengemas (master box, primary box, label serta leaflet) dari gudang bahan kemas serta melakukan coding.
b. Pengemasan produk ruahan dari semua divisi produksi. Pengemasan yang dilakukan disesuaikan dengan jenis produk dalam kemasan (botol, vial, strip, sachet, blister, ampul) dari primary box sampai master box. Setelah selesai pengemasan, produk jadi (produk ruahan yang sudah dikemas sekunder) dikirim ke gudang obat jadi.
Secara garis besar, divisi pengemasan sentral melakukan dua kegiatan utama yakni coding dan secondary packaging. Alur prosesnya dimulai ketika departemen PPIC mengeluarkan MO, MRD dan PDRS beserta jadwal kegiatan guna dilakukan pengemasan sekunder. MRD terkait juga akan diberikan ke bagian gudang bahan kemas paling lambat tiga hari sebelum jadwal pengemasan yang telah ditentukan (H-3). Pada H-2, gudang akan mengirimkan bahan kemas sekunder beserta MI terkait ke divisi pengemasan sentral. Setelah itu, pada hari H-1 baru dilakukan coding yang meliputi penomoran bets, manufacturing date, expired date, dan harga eceran tertinggi/HET pada kemasan sekunder (box, label, master box) dengan menggunakan mesin coding. Untuk menghindari terjadinya kesalahan coding maka proses diawali terlebih dahulu dengan coding satu buah kemasan kemudian diverifikasi dan ditandatangani oleh group leader, petugas QA inspector dan operatornya. Bila tahapan ini sudah selesai dan tidak terjadi masalah, proses coding baru dapat dilanjutkan untuk semua kemasan. Setelah selesai, hasil coding ini lalu dimasukkan ke dalam suatu kotak penyimpanan terkunci. Pada H-0, pengemasan sekunder dimulai dan dilakukan berdasarkan PDRS (Packaging Direction Record Secondary).
untuk dikemas sekunder. Produk yang tidak memenuhi syarat ini lalu dicatat dan dilaporkan kepada supervisor pengemasan sentral, yang kemudian akan mengembalikan produk tersebut ke divisi kemas primer bersangkutan untuk dilakukan pengemasan ulang.
Selama pengemasan sekunder berlangsung, juga dilakukan IPC internal setiap jamnya dan inspeksi akhir oleh petugas QA inspector baru dilakukan setelah proses pengemasan sekunder selesai (sebelum masuk master box). Setelah itu dilakukan penimbangan menggunakan alat timbang yang senantiasa diverifikasi setiap hari seperti yang terdapat dalam SOP (dalam proses penimbangan tiap hari walau nomor betsnya sama). Untuk setiap batch produk operator membuat standar bobot/box dengan cara operator mengambil 10 box berikut isinya dan ditimbang, kemudian mengambil isinya dan menimbang kembali box-nya saja untuk mengetahui variasi berat box yang digunakan. Penyimpangan penimbangan master box ini tidak boleh lebih dari berat ½ unit primary box.Setelah tahapan ini selesai, divisi pengemasan sentral akan membuat dokumen Pengiriman Hasil Produksi/PHP untuk kemudian diserahkan ke gudang obat jadi dan dimasukkan datanya ke dalam stok gudang di sistem informasi ERIC. Hingga tahapan ini, produk jadi tersebut belum dinyatakan released atau dalam status karantina dengan penempelan label karantina pada setiap palet oleh pihak QA/QC sampai semua dokumen lengkap yang terkait dengan produk ini telah terkumpul dan tidak ada laporan penyimpangan yang signifikan. Alur proses pengemasan sentral dapat dilihat pada Lampiran 12.
3.3 Departemen Quality Assurance/Quality Control(QA/QC)
Departemen ini bertanggung jawab atas kualitas produk yang dihasilkan oleh PT. PRAFA dan dipimpin oleh QA/QC manager. Adapun struktur organisasi departemen QA/QC dapat dilihat pada Lampiran 13. Secara fungsional, peran departemen ini mencakup baik fungsi QA maupun QC.
penyimpangan bets, pengendalian perubahan, penanganan penarikan kembali obat jadi, pelulusan obat jadi serta mengkoordinasi peninjauan produk tahunan.
Sedangkan fungsi QC adalah pemeriksaan bahan awal, pengelolaan sampel pertinggal/retained sample, pembuatan spesifikasi dan metode pemeriksaan, pengelolaan standar pembanding, pemeriksaan produk antara, ruahan dan obat jadi, pemeriksaan stabilitas, kalibrasi alat, pengelolaan pengambilan sampel/sampling, statistical process control dan statistical quality control, pemantauan lingkungan, pemeriksaan In Process Control/IPC.
Departemen QA/QC terdiri dari lima divisi yaitu pemeriksaan kimia, pemeriksaan mikrobiologi, QA inspector, validasi dan kualifikasi serta divisi QA Compliance.
3.3.1 Pemeriksaan Kimia
Bagian ini melakukan pemeriksaan sifat fisika dan kimia di laboratorium kimia, mulai dari bahan awal sampai dihasilkan produk jadi.
Pemeriksaan kimia terhadap bahan awal dilakukan untuk memastikan bahwa bahan awal yang dikirim pemasok sesuai dengan spesifikasi yang ditentukan pada saat pemesanan. Departemen QA/QC akan memeriksa bahan baku yang ada di gudang setelah menerima Surat Permintaan Pemeriksaan Bahan Baku (SPPBB). Sampling dilakukan pada setiap kontainer kecuali untuk bahan yang sudah dikualifikasi dapat dilakukan reduced sampling menjadi √n+1. Parameter uji yang dilakukan terhadap bahan baku berpedoman pada SOP pemeriksaan bahan baku dan juga dicantumkan dalam RMAR (Raw Material Analitycal Report). Selain pemeriksaan, sampling juga dilakukan untuk menyimpan contoh bahan baku sebagai retained sample. Bila hasil pemeriksaan bahan awal sesuai dengan spesifikasi yang ditentukan maka akan diberikan label QA released. Tetapi bila tidak memenuhi spesifikasi, bahan awal tersebut di-reject dengan membuat laporan penyimpangan mutu dan dikembalikan ke pemasok. Sementara menunggu pengiriman kembali oleh pemasok, bahan awal diberi label tidak diluluskan (rejected)dan disimpan di area khusus yang terkunci.
dengan metode PTA (Pertama, Tengah, Akhir) waktu produksi. Metode analisa diadopsi dari buku-buku standard resmi yang dituliskan ke dalam SOP untuk masing-masing item obat yang tervalidasi. Selain itu, bagian ini juga bertanggung jawab atas pemeriksaan sampel uji stabilitas untuk kontrol stabilitas produk yang beredar di pasaran. Uji stabilitas dapat dilakukan pada dua kondisi, yaitu long term (30° ± 2°C, RH 75 ± 5%) dan accelerated(40° ± 2°C, RH 75 ± 5%). Untuk proses ini, sampel disimpan di inkubator dengan lama pengujian stabilitas adalah n + 1 tahun (n = expired date). Penambahan satu tahun dilakukan dengan maksud untuk dapat memperpanjang masa kadaluarsa produk.
Terakhir, divisi ini memeriksa air yang digunakan untuk kegiatan produksi seperti air untuk injeksi (water for injection/WFI) dan purified water secara harian. Pemeriksaan yang dilakukan antara lain meliputi pemeriksaan konduktivitas, pH serta kandungan klor dalam air. Sampling dilakukan pada setiap titik pengambilan sampel yang terdapat di masing-masing proses pengelolaan air kemudian dari sampel tersebut diperiksa berdasarkan parameter yang ada sehingga air yang digunakan dalam kegiatan produksi selalu merupakan air yang memenuhi persyaratan. Jika ada masalah seperti pH air lebih kecil 5 atau lebih besar dari 7 (syarat pH air yang dimurnikan/purified water adalah 5-7) maka bagian QC akan mengirimkan Laporan Penyimpangan Mutu/LPM kepada pihak produksi dan TS sementara dilakukan investigasi. Selanjutnya departemen TS memperbaiki alat agar menghasilkan air yang memenuhi persyaratan.
3.3.2 Pemeriksaan Mikrobiologi
Ruang yang terdapat di bagian mikrobiologi terdiri dari enam ruangan yaitu: a. Ruang potensi; digunakan untuk pemeriksaan yang menggunakan kuman
(antara lain potensi antibiotik dan uji kelayakan media/growth promotion test). Aliran udara LAF pada ruang ini berupa laminar dan tidak diijinkan mengalir keluar. Hal ini ditujukan untuk menjaga keamanan personil dan mencegah udara dari ruangan ini keluar sehingga mengkontaminasi udara luar ruang.
b. Ruang TPC (Total Plate Count); digunakan untuk pemeriksaan yang tidak menggunakan kuman (antara lain TPC, uji sterilitas dan uji bioburden). Aliran udara LAF pada ruang ini mengalir keluar untuk menjaga kebersihan ruang dan mencegah kontaminasi udara dari luar ruang.
c. Ruang steril; merupakan ruang yang dikondisikan sama seperti ruang produksi sediaan steril (white area) dan digunakan untuk uji sterilisasi. Sebelum memasuki ruangan ini, terdapat ruang antara, ruang gowning offdan air shower.
d. Ruang preparasi media; merupakan ruang pembuatan media pertumbuhan mikroba yang akan digunakan untuk menginkubasi mikroba pada uji potensi antibiotik.
e. Ruang inkubasi. f. Ruang pencucian.
Jika tidak tumbuh mikroba di media tersebut, berarti media yang digunakan sudah dalam keadaan steril. Selain itu, setiap media dikontrol dengan uji kelayakan media dengan tujuan untuk memastikan bahwa media yang digunakan merupakan media pertumbuhan yang baik untuk mikroba.
Pemeriksaan udara di ruang produksi menggunakan 2 metode yaitu: metode settling plate dan metode air sampler. Pemeriksaan udara terbuka dilakukan dengan pemaparan media menggunakan cawan papar/settling plate di udara terbuka selama 4 jam. Sedangkan metode air sampler dilakukan dengan menggunakan suatu alat dispossible yang cara kerjanya adalah menghisap udara sebanyak 1000 L menuju suatu media, kemudian media-media tersebut diinkubasi dan diperiksa jumlah mikrobanya.
Pemeriksaan sanitasi ruang produksi dilakukan dengan metode apus/swab, caranya adalah menggunakan suatu batang kecil seperti cotton bud yang telah dinetralisir dengan lesitin dan tween untuk menetralkan desinfektan yang mungkin masih menempel pada dinding sel mikroba, kemudian diapuskan seluas 5 x 5 cm pada permukaan dinding ruangan secara horizontal dan vertikal. Setelah itu, hasil apusan diinokulasi ke dalam media pertumbuhan mikroba lalu diinkubasi.
Selain pemeriksaan kimia, PW dan WFI juga diperiksa secara mikrobiologi. Untuk PW dilakukan pemeriksaan TPC yakni ada tidaknya mikroba E.coli dan Pseudomonas. Untuk WFI, selain pemeriksaan TPC, juga dilakukan uji endotoksin. Uji endotoksin yang serupa juga dilakukan terhadap sediaan injeksi dengan menggunakan Limulus Amebocyte Lysate/LAL.
3.3.3 QA Inspector
diharapkan, sedangkan IPC yang dilakukan olehQA inspectoradalah usaha untuk memastikan bahwa produk tersebut telah memenuhi spesifikasi sekaligus sebagai double checkterhadap IPC yang dilakukan oleh pihak produksi.
Pemeriksaan IPC produksi meliputi pemeriksaan kesiapan jalur produksi/line clearance dengan tujuan untuk memastikan bahwa semua peralatan, jenis dan jumlah bahan baku obat telah siap dan kondisi ruang produksi telah sesuai dengan yang seharusnya, lalu juga pemeriksaan keseragaman bobot, ketebalan, diameter sediaan solid, waktu hancur, kekerasan, kerapuhan (friabilitas) dan uji kebocoran. Untuk sediaan cair dilakukan pemeriksaan keseragaman volume, kebocoran, viskositas, dan lainnya. IPC juga melakukan pemeriksaan obat jadi yang meliputi coding, jumlah isi, keadaan kemasan, warna kemasan, ukuran kemasan, dan lainnya. Seluruh hasil pemeriksaan tersebut lalu didokumentasikan.
Pemeriksaan terhadap bahan kemas juga menjadi tanggung jawab dari divisi inspeksi. Metode sampling yang digunakan untuk mengambil sampel dari bahan pengemas seperti vial dan ampul adalah metode sampling millitary standard, tetapi untuk aluminium foil digunakan metode sampling √N + 1. Jika ada masalah seperti salah cetak, perbedaan warna, perbedaan nomor bets pada kemasan serta perbedaan lainnya, maka divisi inspeksi bertanggung jawab penuh dan mempunyai hak untuk menolak seluruh bahan pengemas dari pemasok yang bermasalah.
3.3.4 Validasi dan Kalibrasi
Jenis-jenis validasi yang dilaksanakan oleh divisi ini antara lain: a. Kualifikasi
Kualifikasi bertujuan untuk menjamin mesin/peralatan, sistem, sarana penunjang, bangunan yang digunakan dalam proses produksi sesuai dengan spesifikasi dan tujuan penggunaan yang telah ditentukan sebelumnya. Kualifikasi yang dilakukan meliputi kualifikasi desain (Design Qualification), kualifikasi instalasi (Installation Qualification), kualifikasi operasional (Operational Qualification), kualifikasi kinerja (Performance Qualification) dan kualifikasi bangunan (Building Qualification). Kualifikasi tidak hanya dilakukan pada alat atau mesin baru tetapi dapat juga terhadap alat atau mesin lama yang telah mengalami modifikasi sehingga mempengaruhi output atau produk yang dihasilkan (kualifikasi ulang).
b. Validasi metode analisis
Validasi dilakukan terhadap metode analisis fisika, kimia dan juga mikrobiologi. Hal ini bertujuan untuk menjamin ketelitian, ketepatan dan keterulangan hasil analisis. Parameter metode analisis yang divalidasi meliputi kesesuaian sistem/system suitability, spesifisitas, linearitas, rentang/range, akurasi, presisi, ketegaran (robustness/ruggedness), limit deteksi dan limit kuantitasi.
c. Validasi proses
Kebijakan perusahaan mengharuskan dilakukannya kualifikasi dan validasi terhadap peralatan dan sistem yang menunjang proses produksi, termasuk di antaranya adalah validasi proses. Validasi proses bertujuan untuk memastikan dan menyediakan bukti terdokumentasi bahwa proses yang dilakukan mampu selalu dan dapat diandalkan untuk menghasilkan obat jadi dengan kualitas yang diinginkan. Validasi proses yang dilakukan meliputi validasi proses pengolahan dan pengemasan.
d. Validasi pembersihan dan proses sanitasi
mikroba. Parameter kritis validasi pembersihan dan proses sanitasi berupa batas residu baik bahan baku, bahan tambahan, residu bahan pembersih/detergent maupun mikroba. Penentuan batas residu ini dapat dilakukan baik dengan uji kimia maupun uji mikrobiologi.
e. Validasi media fill
Merupakan validasi simulasi proses produksi sediaan larutan injeksi dan serbuk injeksi kering yang dilakukan secara aseptis. Validasi media fill dilakukan dengan menggunakan media Trypticase Soy Broth/TSB 3% (untuk sediaan larutan injeksi) dan media berupa campuran TSB dan laktosa dengan perbandingan 3:7 yang telah disterilkan dengan sinar gamma 25 kiloGray (untuk serbuk injeksi kering). Selain itu, uji GPT (Growth Promotion Test) juga dilakukan pada media yang digunakan baik sebelum dan sesudah proses filling.
Kriteria pemilihan proses produksi yang akan divalidasi media fill yaitu produk injeksi dengan jalur proses produksi yang rumit dan diproduksi dalam jumlah bets yang paling besar. Validasi media fill dilakukan pada 3 bets berturut-turut.
Proses validasi media fill diawali dengan mengisi larutan media TSB 3% (untuk sediaan larutan injeksi) atau campuran TSB dan laktosa dengan perbandingan 3:7 (untuk serbuk injeksi kering) ke dalam wadah sesuai dengan jalur proses produksi aseptis yang biasa dilakukan. Selanjutnya dilakukan uji GPT dan diinkubasi pada suhu 22,5±2,5°C selama 7 hari dan pada 32,5±2,5°C selama 7 hari, lalu selanjutnya diperiksa pertumbuhan mikrobanya.
Kriteria penerimaan untuk keberhasilan validasi media fill adalah sebagai berikut:
i. Jika pengisian kurang dari 5000 unit maka tidak boleh ada kontaminasi. ii. Jika pengisian antara 5000-10000 unit, maka:
1 (satu) unit terkontaminasi harus dilakukan investigasi termasuk mempertimbangkan pengulangan validasi media fill.
iii. Jika pengisian lebih dari 10.000 unit, maka:
1 (satu) unit terkontaminasi harus dilakukan investigasi.
2 (dua) unit terkontaminasi dipertimbangkan untuk revalidasi diikuti investigasi.
f. Validasi sistem informasi (komputer)
Sistem informasi/komputer juga divalidasi sebelum digunakan pada sistem mutu misalnya pada pencatatan stok material, pendataan masalah mutu dan kontrol proses. Hal ini dilakukan melalui proses pengambilan data, kemudian evaluasi ketepatan rumus serta keamanan data.
Selain kegiatan validasi dan kualifikasi, divisi ini juga bertanggungjawab melakukan kalibrasi terhadap alat ukur seperti neraca timbang, thermohygrometer, gelas ukur dan lain-lain. Kalibrasi dilakukan sesuai dengan jadwal program yang telah dibuat dengan menggunakan prosedur kalibrasi yang spesifik untuk setiap alat. Kalibrasi dilakukan dengan menggunakan kalibrator tertelusur yang setiap tahun dikalibrasi oleh badan atau lembaga lain yang telah terakreditasi/tersertifikasi. Setiap alat ukur yang telah dikalibrasi akan diberi label calibratedyang memuat waktu dilakukan kalibrasi dan waktu jatuh tempo untuk dilakukan kalibrasi ulang. Data hasil kalibrasi kemudian dicatat dalam Laporan Hasil Kalibrasi.Validation officer atau validation coordinator bertanggung jawab penuh atas validasi seluruh sistem, kualifikasi seluruh peralatan dan kalibrasi seluruh alat ukur yang digunakan di pabrik.
3.3.5 QACompliance
Divisi ini mempunyai tanggungjawab dalam menangani hal-hal sebagai berikut:
a. Dokumentasi atau Document Control Centre/DCC
menyimpan back-up-nya dalam bentuk CD. Dokumen lain yang disimpan di DCC antara lain daftar pemasok yang disetujui/approved supplier, laporan obat jadi, protokol dan laporan validasi.
Salah satu dokumen asli yang disimpan oleh DCC adalah SOP. Kontrol peredaran SOP dilakukan dengan memberikan stempel pada setiap SOP yaitu SOP yang asli akan diberi stempel “original” dan duplikatnya diberi stempel “copy”. Pada stempel “copy” juga dituliskan kode angka yang mengindikasikan nomor urut duplikat yang beredar serta bagian dan personil yang memiliki duplikat SOP tersebut. SOP akan di-review setiap 2 tahun sekali dan dinyatakan berlaku bila personil terkait telah diberi pelatihan mengenai SOP bersangkutan.
b. Proses kontrol perubahan (change control process)
c. Program inspeksi diri (Self Inspection Program/SIP)
Ada empat jenis SIP yang dilaksanakan di PT. PRAFA yaitu:
i. Self quality audit, merupakan audit yang dilakukan oleh perusahaan itu sendiri. Ada 3 macam audit internal, yaitu:
General plant quality audit.Audit ini dilakukan oleh satu tim gabungan dari internal PT. PRAFA setiap satu tahun sekali untuk semua area. Area quality audit. Audit ini dilaksanakan oleh tim dari departemen
QA/QC ke seluruh area tiap enam bulan sekali untuk masing-masing area.
Area self inspection. Audit ini dilaksanakan oleh GMP coordinator terhadap area sendiri dengan frekuensi sesering mungkin minimal tiap sebulan sekali.
ii. Cross audit, merupakan audit yang dilakukan oleh bagian QA dari pihak PT. Darya Varia Laboratoria Tbk. atau PT. Medifarma Laboratories Inc. untuk mengaudit pihak PRAFA atau sebaliknya.
iii. Third party audit, merupakan audit yang dilakukan oleh pihak ketiga seperti P&G, perusahaan pemberitoll manufacturing atau BPOM.
iv. Vendoraudit
QA manager melakukan penilaian (assesment) kepada vendor atau material supplier (raw material dan packaging material). Assesment dilakukan dengan mengirimkan kuisioner yang akan diisi vendor tersebut dan juga melakukan kunjungan langsung ke lokasi vendor.
Penelusuran terhadap proses SIP juga dilakukan untuk menentukan:
i. Persentase audit excecution, menghitung persentase pelaksanaan audit yang dilakukan setiap bulan.
ii. Persentase CAPA excecution and ontime yakni menghitung persentase CAPA yang dilaksanakan tepat waktu.
d. Behavior Observation System(BOS)
Tujuan dari BOS adalah untuk memperbaiki kebiasaan buruk personil yang berkaitan dengan CPOB. BOS dilakukan oleh tiap personil secara harian dengan melakukan pemeriksaan mandiri lalu mengisi suatu daftar periksa/checklist singkat, seperti pemeriksaan kebersihan ruang timbang, reagen belum expired date. Program BOS bersifat dynamic checklist yang berarti jika suatu kebiasaan buruk sudah diperbaiki maka dapat diganti dengan pemeriksaan kebiasaan buruk yang lain. QA compliance kemudian akan menghitung persentase BOS yang dilakukan dan dilanggar, menindaklanjuti dan memperbaiki rencana tindak lanjut.
e. Penanganan keluhan produk (product complain)
Formulir keluhan/complain ditandatangani oleh pihak QA compliance. Formulir keluhan ini meliputi catatan produk/product record yang berisi nama produk dan nomor bets, catatan konsumen/consumer recordberisi asal produk, nama konsumen, umur, nomor telepon dan keadaan kesehatan, jenis keluhan (apakah life threatening, critical atau general). Setelah pengisian formulir, keluhan akan ditindak lanjuti oleh departemen QA/QC dengan membuat investigation notification dan permintaan investigasi. Di samping itu, terdapat keluhan yang tidak perlu diinvestigasi seperti ketika produk tidak ada di pasaran.
sama, QDR dan APR terkait bets tersebut. Setelah melalui proses investigasi secara keseluruhan, selanjutnya akan ditarik kesimpulan penyebab keluhan dan CAPA yang direkomendasikan. Laporan hasil investigasi ini kemudian dikirim kepada pihak terkait termasuk pelapor.
f. Penarikan kembali produk (product recall)
Recall dapat dilakukan atas inisiatif industri farmasi jika produk memiliki cacat mutu dan beresiko membahayakan konsumen. Recall juga dapat dilakukan atas permintaan BPOM. Terdapat dua SOP yang berhubungan dengan product recall di PT. PRAFA yaitu SOP recall itu sendiri dan SOP simulasi jika terjadi recall atau disebut juga mock recall. SOP simulasi yang dimaksudkan di sini hanya bersifat administrasi bukan recall produk sebenarnya.
Mock recall dilakukan satu tahun sekali dan dikoordinasikan oleh QA/QC manager. Manajer akan membuat protokol produk dan bets yang akan ditarik kembali dari distributor utama dan subdistributor. Setelah proses ini selesai maka kemudian akan dibuatkan laporannya kepada plant manager. Adapun tujuan dilakukannya mock recallini adalah menelusuri catatan distribusi serta sarana pelatihan rutin untuk penanganan product recall. Sistem distribusi dinilai baik jika 98% dari produk yang ditarik kembali/recall terlacak.
g. Laporan penyimpangan mutu (Quality Deviation Report/QDR)
h. Tinjauan produk tahunan (Annual Product Review/APR)
Peninjauan produk tahunan merupakan rangkaian peninjauan terhadap tiap produk yang diproduksi selama satu tahun yang berisi antara lain jumlah bets yang diproduksi selama satu tahun dan status produk (apakah QA rejected atau QA released), bahan baku dan bahan kemas yang digunakan, status kalibrasi dan validasi, QDR yang ada, pengontrolan pengubahan yang dilakukan, CAPA yang sudah dan yang belum diselesaikan, product recall yang muncul, jumlah retur produk/returned productdari distributor, keluhan dari publik atau distributor, data pemasok serta hasil analisis sifat fisik dan kimia produk tersebut. Tujuan APR adalah untuk membuktikan konsistensi proses, kesesuaian dari spesifikasi bahan baku, bahan pengemas, obat jadi, untuk melihat kecenderungan/trend yang timbul dan mengidentifikasi perbaikan yang diperlukan baik untuk produk maupun proses produksinya. Jika dalam satu tahun tersebut terdapat banyak perubahan maka diperlukan revalidasi.
3.4 Product Development Department(PDD)
Product Development Department (PDD) adalah suatu departemen yang berperan dalam pengembangan produk-produk Darya Varia Group yakni PT. Darya Varia Laboratories, PT. Pradja Pharin (PRAFA) dan PT. Medifarma Laboratories yang mana berlokasi di pabrik PRAFA. Departemen ini dibagi menjadi tiga divisi yakni natural product, cosmeceutical product dan generic product development. Adapun struktur organisasi PDD dapat dilihat pada Lampiran 14.
Secara garis besar, tanggung jawab PDD meliputi: a. Mengembangkan formulasi produk baru:
- Produk Darya Varia
- Produk lisensi (transfer teknologi)
b. Reformulasi (menyempurnakan formulasi produk yang sudah ada). c. Evaluasi bahan baku alternatif
PDRF ini berisi spesifikasi produk yang akan dikembangkan seperti kandungan aktifnya, bentuk sediaan yang dikehendaki, pengemasan primernya dan desired time launch, forecast. Setelah PDRF diterima, pihak PDD melakukan studi literatur antara lain dari buku, internet, produk inovator dan kompetitor. Adapun tujuan dilakukannya studi literatur adalah untuk mencari berbagai kemungkinan formula yang sesuai dengan bentuk sediaan yang akan diproduksi nantinya.
Selanjutnya PDD melakukan laboratory trial untuk mendapatkan minimal dua formula yang optimal, di mana besarnya jumlah bets disesuaikan dengan kapasitas peralatan yang ada di laboratorium PDD. Pada tahap laboratory trial, dilakukan uji prestability.
Setelah laboratory trial dan formula disetujui, PDD kemudian melangsungkan pilot batch trial. Pada tahap ini akan dilakukan proses produksi dengan menggunakan peralatan/mesin produksi sebanyak 100.000 tablet/kapsul atau minimal 10% dari bets produksi yang direncanakan. Produk yang dihasilkan selanjutnya dikirim ke departemen QA/QC untuk dilakukan uji stabilitas dan marketing untuk approval. Secara pararel sambil menunggu hasil pengujian stabilitas dan nomor registrasi, PDD akan menyusun manufacturing procedure. Setelah nomor registrasi keluar maka dilakukan scale updimana obat diproduksi dengan skala yang lebih besar, sesuai dengan batch size yang direncanakan dalam commercial production batch. Alur pengembangan produk baru oleh departemen PDD dapat dilihat di Lampiran 15.
3.5 Departemen Technical Services(TS)
Departemen TS bertanggung jawab atas kelancaran kegiatan pabrik terutama pemeliharaan mesin/peralatan produksi dan sarana penunjang produksi yakni electricity, clean compressed air, water system, HVAC (Heating, Ventilating and Air Conditioning) dansteam boiler, agar selalu dalam keadaan siap pakai. Struktur organisasi departemen TS dapat dilihat pada Lampiran 16.
Adapun tugas dan tanggung jawab departemen TS adalah sebagai berikut: a. Memelihara semua mesin produksi dan sarana penunjang agar siap dipakai
b. Melakukan maintenance schedule. Mesin produksi harus dirawat oleh departemen TS dua minggu sebelum jadwal produksi atau MPS (Master Production Schedule) dikeluarkan. MPS merupakan dokumen yang berisi jadwal penggunaan mesin produksi dan jadwal kegiatan produksi yang dikeluarkan oleh bagian PPC setiap bulannya.
c. Memodifikasi mesin produksi sehingga bekerja lebih optimal.
d. Menangani proyek pembangunan. Rencana pembangunan fasilitas produksi terlebih dahulu diajukan kepada departemen TS, kemudian ditentukan material dan bahan kontruksi yang diperlukan serta anggaran belanja dalam Bill of Quantity.
Perawatan rutin dan modifikasi yang dilakukan oleh departemen TS bertujuan agar mesin produksi tidak rusak pada saat dipakai sehingga jadwal produksi yang telah disusun PPIC tidak mengalami downtime. Ada dua jenis downtimeyang dapat terjadi yaitu:
a. Unschedule downtime, merupakandowntime yang terjadi karena berhentinya mesin secara tiba-tiba selama proses produksi sedang berlangsung. Hal ini menyebabkan mesin harus diperbaiki sehingga memperpanjang lead time produksi.
b. Schedule downtime, merupakandowntimeyang terjadi karena mesin berhenti beroperasi akibat suatu kegiatan yang tidak dapat ditolak, misalnya gulungan aluminium foil habis pada saat proses stripping sehingga membutuhkan waktu untuk memasang gulungan aluminium foil yang baru pada mesin stripping, saat sanitasi ruangan atau pelaksanaan preventive maintenance.
Sedangkan sarana penunjang produksi (utility) di PT. PRAFA yang dikelola departemen TS antara lain:
a. Electricity
b. Clean compressed air
Terdapat dua buah kompressor yang digunakan untuk menghasilkan clean compressed air atau udara bersih bertekanan di pabrik. Mesin ini bekerja dengan cara memampatkan udara hingga bertekanan maksimum 10 bar dengan kualitas 1-1-1, yaitu mengandung partikel 0,1 mg/m3, residual water content ≤ 0,003 mg/m3 dan residual oil content ≤ 0,01 mg/m3. Clean compressed airini digunakan baik untuk yang contact product seperti proses spraying pada FBD maupun yang non-c