TESIS
PENGEMBANGAN DAN VALIDASI METODE
SPEKTROFOTOMETRI ULTRAVIOLET UNTUK ESTIMASI CAMPURAN PARASETAMOL, ASETOSAL, DAN KOFEIN
DALAM SEDIAAN TABLET
OLEH:
ISMAYUNI NIM 117014011
PROGRAM STUDI MAGISTER ILMU FARMASI FAKULTAS FARMASI
UNIVERSITAS SUMATERA UTARA MEDAN
2020
PENGEMBANGAN DAN VALIDASI METODE
SPEKTROFOTOMETRI ULTRAVIOLET UNTUK ESTIMASI CAMPURAN PARASETAMOL, ASETOSAL, DAN KOFEIN
DALAM SEDIAAN TABLET
TESIS
Diajukan sebagai salah satu syarat untuk memperoleh Gelar Magister dalam Ilmu Farmasi pada Fakultas Farmasi
Universitas Sumatera Utara
OLEH:
ISMAYUNI NIM 117014011
PROGRAM STUDI MAGISTER FARMASI FAKULTAS FARMASI
UNIVERSITAS SUMATERA UTARA MEDAN
2020
PERSETUJUAN TESIS
Nama Mahasiswa : Ismayuni Nomor Induk Mahasiswa : 117014011
Program Studi : Magister Ilmu Farmasi
Judul Tesis : Pengembangan dan Validasi Metode
Spektrofotometri Ultraviolet untuk Estimasi Campuran Parasetamol, Asetosal, dan Kofein dalam Sediaan Tablet
Telah diuji dan dinyatakan LULUS didepan Komisi Penguji Tesis pada hari Jum’at tanggal delapan bulan Februari tahun dua ribu sembilan belas.
Menyetujui:
Komisi Penguji Tesis
Ketua : Prof. Dr. Muchlisyam, M.Si., Apt.
Sekretaris : Prof. Dr. rer. nat. Effendy De Lux Putra, S.U., Apt.
Anggota : Prof. Dr. Siti Morin Sinaga, M.Sc., Apt.
Anggota : Prof. Dr. Ginda Haro, M.Sc., Apt.
KATA PENGANTAR
Bismillahirrahmanirrahim,
Puji dan syukur penulis ucapkan kehadirat Allah SWT, karena atas berkat rahmat dan karunia-Nya sehingga penulis dapat menyelesaikan tesis dengan judul
“Pengembangan dan Validasi Metode Spektrofotometri Ultraviolet untuk Estimasi Campuran Parasetamol, Asetosal, dan Kofein dalam Sediaan Tablet”. Serta sholawat dan salam untuk Rasulullah Muhammad SAW sebagai suri tauladan dalam kehidupan.
Selama menyelesaikan penelitian dan tesis ini, penulis telah banyak mendapatkan bantuan dan dorongan dari berbagai pihak baik moril maupun materil. Untuk itu penulis ingin mengucapkan terima kasih yang sebesar-besarnya kepada:
1. Bapak Rektor Prof. Dr. Runtung, S.H., M.Hum., selaku Rektor Universitas Sumatera Utara, yang telah memberikan kesempatan dan fasilitas kepada penulis untuk mengikuti Program Studi Magister Ilmu Farmasi.
2. Ibu Prof. Dr. Masfria, M.S., Apt., selaku Dekan Fakultas Farmasi Universitas Sumatera Utara, yang telah menyediakan fasilitas dan kesempatan bagi penulis menjadi mahasiswa untuk mengikuti dan menyelesaikan Program Studi Magister Ilmu Farmasi di Fakultas Farmasi.
3. Bapak Prof. Dr. Urip Harahap, Apt., selaku Ketua Program Studi Magister Farmasi dan Ibu Prof. Dr. Rosidah, M.Sc., Apt., selaku Sekretaris Program Studi Magister Ilmu Farmasi yang telah banyak memberikan motivasi dan bimbingan sehingga penulis dapat menyelesaikan pendidikan ini.
PENGEMBANGAN DAN VALIDASI METODE SPEKTROFOTOMETRI ULTRAVIOLET UNTUK ESTIMASI CAMPURAN PARASETAMOL,
ASETOSAL, DAN KOFEIN DALAM SEDIAAN TABLET
ABSTRAK
Kombinasi dari dua atau lebih obat analgetik sering kali digunakan, karena terjadi efek potensiasi. Spektofotometri derivatif merupakan metode transformasi dari spektrofotometri konvensional yang dikembangkan untuk analisis kuantitatif multikomponen senyawa aktif pada suatu sediaan. Tujuan penelitian ini adalah untuk mengembangkan dan menguji validasi dengan spektrofotometri derivatif untuk menetapkan kadar parasetamol, asetosal, dan kofein pada sediaan tablet tanpa adanya tahap pemisahan.
Penelitian dilakukan terhadap campuran tablet parasetamol, asetosal, dan kofein dengan spektrofotometri derivatif metode zero-crossing dalam pelarut etanol.
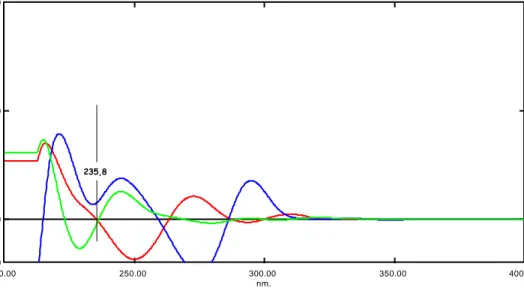
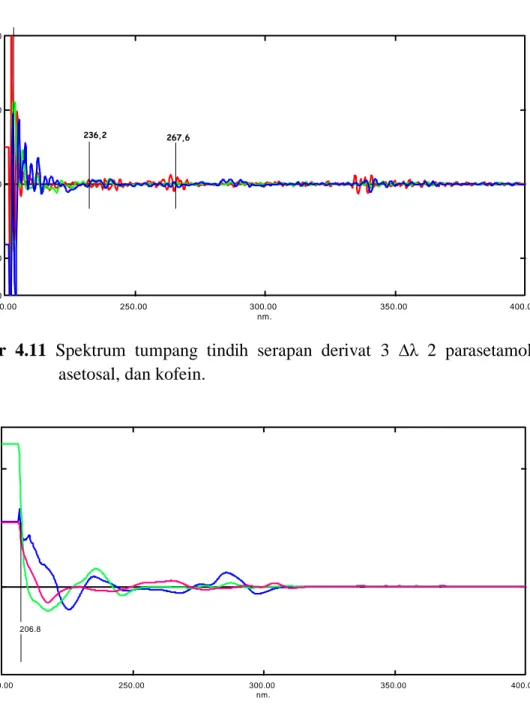
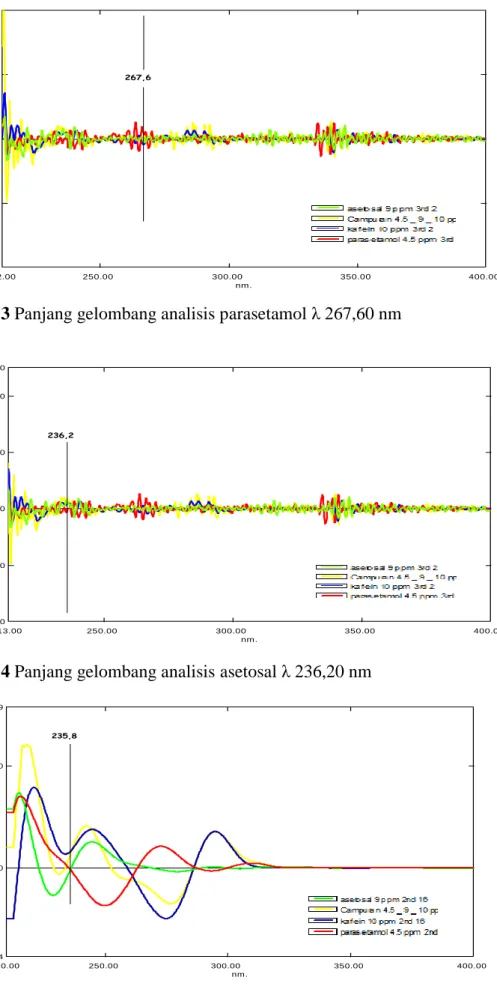
Hasil penelitian menunjukkan aplikasi teknik zero-crossing pada penetapan kadar parasetamol dilakukan pada derivat ketiga dengan λ 267,6 nm (Δλ 2), untuk asetosal dilakukan pada derivat ketiga dengan λ 236,2 nm (Δλ 2), sedangkan kofein pada derivat kedua dengan λ 235,8 nm (Δλ 16) menghasilkan kadar 101,198%, 105,78% dan 107,74% secara berturut-turut untuk parasetamol, asetosal, dan kofein.
Berdasarkan hasil penelitian yang dilakukan maka spektrofotometri derivatif yang digunakan memenuhi persyaratan akurasi dan presisi dan dapat digunakan menetapkan kandungan parasetamol, asetosal, dan kofein dalam sediaan tablet.
Kata kunci: parasetamol, asetosal, kofein, spektrofotometri derivatif.
THE DEVELOPMENT AND VALIDATION OF ULTRAVIOLET SPECTROPHOTOMETRIC METHOD FOR ESTIMATED
MIXTURE OF PARACETAMOL, ACETOSAL AND CAFFEINE IN TABLET DOSAGE FORM
ABSTRACT
Combinations of two or more analgesic drugs are often used because there are potentiation effects. Derivative spectrophotometry is a method transformed from conventional spectrophotometry which is developed for quantitative analysis of multi-components of active compounds in a preparation.
The aims of this research were to develop and test the validation of derivative spechtrophotometry method to determine the level of paracetamol, acetosal, and caffeine in tablet dosage form without prior separation.
The study was conducted with tablet mixtures of paracetamol, acetosal, and caffeine using derivative spectrophotometry with a zero-crossing technique.
Ethanol was used as a solvent for the analysis.
The results showed that the application of derivative spectrophotometry method on the determination of paracetamol level carried out on the third derivative at λ 267.6 nm (Δλ 2), determination of acetosal level performed on the third derivative at λ 236.2 nm (Δλ 2), and determination of caffeine level performed on the second derivative at λ 235.8 nm (Δλ 16) resulted in the level of 101.198%, 105.78%, and 107.74% respectively for paracetamol, acetosal, and caffeine.
Based on the research results, the derivative spectrophotometric method fulfilled the requirements of accuracy and precision, so it can be used to determinate the level of paracetamol, acetosal, and caffeine in tablets.
Keywords: paracetamol, acetosal, caffeine, derivative spectrophotometry.
DAFTAR ISI
HALAMAN JUDUL ... i
HALAMAN PENGESAHAN TESIS ... iii
HALAMAN PERSETUJUAN TESIS ... iv
HALAMAN PERNYATAAN ORISINALITAS ... ... v
KATA PENGANTAR ... vi
ABSTRAK ... viii
ABSTRACT ... xi
DAFTAR ISI ... x
DAFTAR TABEL ... xiii
DAFTAR GAMBAR ... xiv
DAFTAR LAMPIRAN ... xv
BAB I PENDAHULUAN ... 1
1.1 Latar Belakang ... 1
1.2 Perumusan Masalah ... 5
1.3 Hipotesis ... 5
1.4 Tujuan Penelitian ... 6
1.5 Manfaat Penelitian ... 6
1.6 Kerangka Pikir Penelitian ... 7
BAB II TINJAUAN PUSTAKA ... 8
2.1 Parasetamol ... 8
2.2 Asetosal ... 9
2.3 Kofein ... 10
2.4 Stabilitas obat ... 11
2.5 Spektrofotometri ... 13
2.5.1 Spektrofotometri Sinar Ultra Violet ... 13
2.5.2 Spektrofotometri Derivatif ... 14
2.5.3 Komponen Spektrofotometri Derivatif ... 17
2.6 Validasi Metode Analisis... 17
2.6.1 Akurasi (Kecermatan) ... 17
2.6.2 Presisi (Keseksamaan) ... 18
2.6.3 Batas Deteksi dan Batas Kuantitasi ... 18
2.6.4 Liniearitas ... 18
BAB III METODE PENELITIAN ... 19
3.1 Alat dan Bahan ... 19
3.1.1 Alat ... 19
3.1.2 Bahan ... 19
3.2 Pengambilan Sampel ... 19
3.3 Prosedur Penelitian ... 20
3.3.1 Pembuatan pelarut ... 20
3.3.2 Pembuatan larutan induk baku parasetamol ... 20
3.3.3 Pembuatan larutan induk baku asetosal ... 20
3.3.4 Pembuatan larutan induk baku kofein ... 20
3.3.5 Pembuatan larutan standar parasetamol ... 21
3.3.6 Pembuatan larutan standar asetosal ... 21
3.3.7 Pembuatan larutan standar kofein ... 21
3.3.8 Pembuatan spektrum serapan maksimum parasetamol .. 22
3.3.9 Pembuatan spektrum serapan maksimum asetosal ... 22
3.3.10 Pembuatan spektrum serapan maksimum kofein ... 22
3.3.11 Pembuatan spektrum serapan campuran parasetamol, asetosal, dan kofein ... 22
3.3.12 Pembuatan spektrum serapan derivatif parasetamol ... 23
3.3.13 Pembuatan spektrum serapan derivatif asetosal ... 23
3.3.14 Pembuatan spektrum serapan derivatif kofein ... 23
3.3.15 Pembuatan spektrum serapan derivatif campuran parasetamol, asetosal, dan kofein ... 24
3.3.16 Pembuatan dan Penentuan panjang gelombang (λ) analisis untuk metode zero crossing ... 24
3.3.17 Penentuan panjang gelombang (λ) analisis untuk metode zero crossing derivatif pada parasetamol ... 25
3.3.18 Penentuan panjang gelombang (λ) analisis untuk metode zero crossing derivatif pada asetosal ... 25
3.3.19 Penentuan panjang gelombang (λ) analisis untuk metode zero crossing derivatif pada kofein ... 25
3.3.20 Pembuatan kurva kalibrasi metode zero crossing parasetamol ... 26
3.3.21 Pembuatan kurva kalibrasi metode zero crossing asetosal ... 26
3.3.22 Pembuatan kurva kalibrasi metode zero crossing kofein ... 26
3.4 Validasi metode ... 27
3.4.1 Liniearitas, batas deteksi (Limit of Detection, LOD) dan batas deteksi (Limit of Quantitation, LOQ) ... 27
3.4.2 Uji akurasi ... 28
3.4.3 Uji presisi ... 29
3.5 Penentuan kadar campuran parasetamol, asetosal, dan kofein dalam sediaan tablet ... 29
3.5.1 Perhitungan kadar parasetamol, asetosal, dan kofein dalam sediaan tablet ... 30
3.6 Analisis data penetapan kadar secara statistik ... 30
BAB IV HASIL DAN PEMBAHASAN ... 32
4.1 Hasil penentuan spektrum serapan maksimum ... 32
4.2 Hasil penentuan spektrum serapan ... 33
4.3 Penentuan zero-crossing pada serapan derivat ... 34
4.3.1. Penentuan zero crossing pada serapan derivat pertama ... 34
4.3.2. Penentuan zero crossing pada serapan derivat kedua ... 37
4.3.3. Penentuan zero crossing pada serapan derivat ketiga ... 38
4.4 Hasil penentuan panjang gelombang analisis parasetamol, asetosal, dan kofein ... 40
4.5 Validasi metode ... 46
4.5.1 Liniearitas, batas deteksi (Limit of Detection, LOD), dan batas deteksi (Limit of Quantitation, LOQ) ... 46
4.5.2 Penetapan kadar campuran parasetamol, asetosal, dan
kofein pada sediaan tablet ... 49
4.6 Metode zero-crossing ... 50
BAB V KESIMPULAN DAN SARAN ... 51
5.1 Kesimpulan ... 51
5.2 Saran ... 51
DAFTAR PUSTAKA ... 52
LAMPIRAN ... 56
DAFTAR TABEL
4.1 Nilai absorbansi pada titik zero-crossing spektrum parasetamol, asetosal, dan kofein dan spektrum campuran parasetamol,
asetosal, dan kofein pada derivat 2 Δλ 16 ... 43 4.2 Nilai absorbansi pada titik zero-crossing spektrum parasetamol,
asetosal, dan kofein dan spektrum campuran parasetamol,
asetosal, dan kofein pada derivat 3 Δλ 2 ... 43 4.3 Nilai linearitas, akurasi, presisi, LOD dan LOQ untuk
parasetamol, asetosal, dan kofein menggunakan metode zero-
crossing ... 48 4.4 Kadar parasetamol, asetosal, dan kofein dalam sediaan tablet ... 50
DAFTAR GAMBAR
1.1 Kerangka pemikiran penelitian ... 7
2.1 Struktur parasetamol (Depkes RI, 2014) ... 8
2.2 Struktur asetosal (Ditjen POM, 1995) ... 9
2.3 Struktur kofein (Depkes RI, 2014) ... 10
4.1 Spektrum serapan maksimum parasetamol konsentrasi 4,5 µg/mL .. 32
4.2 Spektrum serapan maksimum asetosal konsentrasi 9 µg/mL ... 32
4.3 Spektrum serapan maksimum kofein konsentrasi 10 µg/mL ... 33
4.4 Overlay spektrum serapan parasetamol, asetosal, dan kofein dan spektrum campuran parasetamol, asetosal, dan kofein ... 34
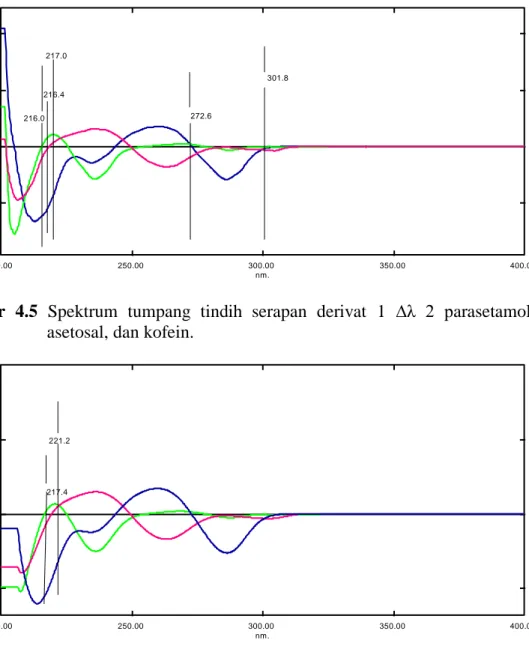
4.5 Spektrum tumpang tindih serapan derivat 1 ∆λ 2 parasetamol, asetosal, dan kofein ... 35
4.6 Spektrum tumpang tindih serapan derivat 1 ∆λ 8 parasetamol, asetosal, dan kofein ... 35
4.7 Spektrum tumpang tindih serapan derivat 1 ∆λ 16 parasetamol, asetosal, dan kofein ... 36
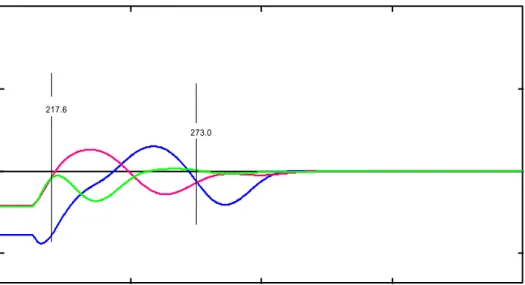
4.8 Spektrum tumpang tindih serapan derivat 2 ∆λ 2 parasetamol, asetosal, dan kofein ... 37
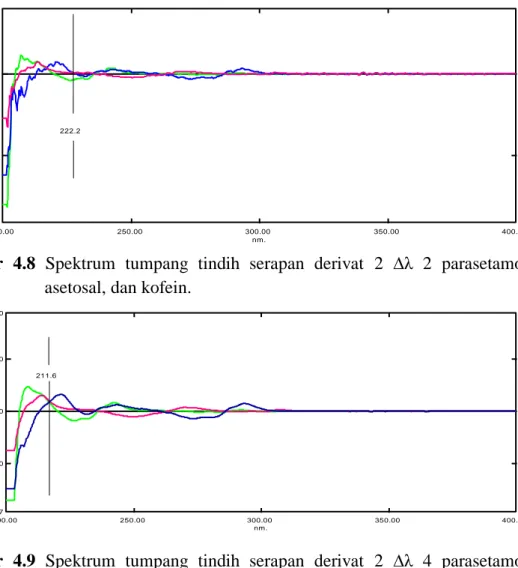
4.9 Spektrum tumpang tindih serapan derivat 2 ∆λ 4 parasetamol, asetosal, dan kofein ... 37
4.10 Spektrum tumpang tindih serapan derivat 2 ∆λ 16 parasetamol, asetosal, dan kofein ... 38
4.11 Spektrum tumpang tindih serapan derivat 3 ∆λ 2 parasetamol, asetosal, dan kofein ... 39
4.12 Spektrum tumpang tindih serapan derivat 3 ∆λ 8 parasetamol, asetosal, dan kofein ... 39
4.13 Panjang gelombang analisis parasetamol λ 267,60 nm ... 41
4.14 Panjang gelombang analisis asetosal λ 236,20 nm ... 41
4.15 Panjang gelombang analisis kofein λ 235,80 nm ... 41
4.16 Kurva kalibrasi parasetamol pada panjang gelombang 267,60 nm ... 46
4.17 Kurva kalibrasi asetosal pada panjang gelombang 236,20 nm ... 47
4.18 Kurva kalibrasi kofein pada panjang gelombang 235,80 nm ... 47
DAFTAR LAMPIRAN
1 Sampel PoldanMig® (Sanbe) ... 56
2 Alat ... 57
3 Titik zero-crossing untuk metode zero crossing ... 58
4 Panjang gelombang analisis untuk parasetamol, asetosal, dan kofein ... 65
5 Kurva dan perhitungan kalibrasi parasetamol dengan menggunakan metode zero crossing pada derivat ketiga panjang gelombang 267,6 nm dengan Δλ 2 ... 66
6 Kurva dan perhitungan kalibrasi asetosal dengan menggunakan metode zero crossing pada derivat ketiga panjang gelombang 236,2 nm dengan Δλ 2 ... 68
7 Kurva dan perhitungan kalibrasi kofein dengan menggunakan metode zero crossing pada derivat kedua panjang gelombang 235,8 nm dengan Δλ 16 ... 70
8 Perhitungan batas deteksi (LOD) dan batas kuantitasi (LOQ) parasetamol ... 72
9 Perhitungan batas deteksi (LOD) dan batas kuantitasi (LOQ) asetosal ... 73
10 Perhitungan batas deteksi (LOD) dan batas kuantitasi (LOQ) kofein ... 74
11 Hasil perhitungan batas deteksi dan batas kuantitasi ... 75
12 Contoh perhitungan kadar parasetamol, asetosal, dan kofein pada sediaan tablet ... 76
13 Kadar parasetamol, asetosal, dan kofein dalam sediaan tablet ... 81
14 Perhitungan statistik kadar parasetamol dengan menggunakan metode zero crossing ... 82
15 Perhitungan statistik kadar asetosal dengan menggunakan metode zero crossing ... 84
16 Perhitungan statistik kadar kofein dengan menggunakan metode zero crossing ... 86
17 Contoh perhitungan persentase perolehan kembali (%recovery) ... 88
18 Spektrum uji perolehan kembali ... 95
19 Data hasil persen perolehan kembali parasetamol, asetosal, dan kofein dengan menggunakan metode zero crossing ... 96
20 Perhitungan rata-rata, standar deviasi, dan relatif standar deviasi perolehan kembali parasetamol ... 97
21 Perhitungan rata-rata, standar deviasi, dan relatif standar deviasi perolehan kembali asetosal ... 98
22 Perhitungan rata-rata, standar deviasi, dan relatif standar deviasi perolehan kembali kofein ... 99
23 Tabel distribusi t ... 100
24 Sertifikat analisis bahan baku parasetamol ... 101
25 Sertifikat analisis bahan baku asetosal ... 102
26 Sertifikat analisis bahan baku kofein ... 103 27 Publikasi Jurnal
BAB I
PENDAHULUAN
1.1 Latar Belakang
Parasetamol, asetosal, dan kofein sering dikombinasikan sebagai obat antipiretik dan analgetik. Analgetik adalah zat-zat yang dapat mengurangi rasa nyeri tanpa menghilangkan kesadaran. Kofein dalam kombinasi obat tersebut berfungsi sebagai efikasi dari analgetik. Campuran ini bertujuan untuk meningkatkan efek terapi dan kemudahan dalam pemakaian (Tjay dan Rahardja, 2013; Derry, et al., 2012; Damayanti, et al., 2003).
Dalam bidang industri obat untuk menganalisis obat multikomponen perlu pemisahan atau ekstraksi. Analisis obat ini menggunakan peralatan canggih seperti high performance liquid chromatography (HPLC), elektroforesis kapiler, atau liquid chromatography-mass spectrometry (LC-MS) membutuhkan biaya tinggi, memerlukan waktu, dan pekerjaan tambahan (Ozdemir et al., 2004).
Penggunaan teknik spektrofotometri dikombinasikan dengan algoritma matematika telah membawa metodologi baru, cepat, dan lebih murah untuk penentuan analit dalam sampel. Untuk penentuan simultan dari dua atau lebih senyawa aktif dalam campuran yang sama tanpa langkah pemisahan, ada beberapa metode spektrofotometri yang dapat menganalisis campuran tersebut, salah satunya adalah spektrofotometri derivatif (Hajian dan Soltaninezhad, 2012).
Metode spektrofotometri derivatif atau metode kurva turunan adalah salah satu metode spektrofotometri yang dapat digunakan untuk analisis campuran beberapa zat secara langsung tanpa harus melakukan pemisahan terlebih dahulu
walaupun dengan panjang gelombang yang berdekatan. Penggunaan spektrofotometri derivatif sebagai alat bantu analisis meningkat seiring dengan perkembangan dunia elektronik yang pesat terutama teknologi mikrokomputer dalam tiga puluh tahun terakhir. Akhir-akhir ini penggunaan spektrofotometri derivatif makin mudah dengan meningkatnya daya pisah instrumen analitik yang dilengkapi mikrokomputer dengan perangkat lunak yang sesuai sehingga mampu menghasilkan spektra derivatif secara cepat. Beberapa keuntungan dari spektrum derivatif antara lain: spektrum derivatif memberikan gambaran struktur yang terinci dari spektrum serapan dan gambaran ini makin jelas dari spektra derivatif pertama ke derivatif keempat (Nurhidayati, 2007).
Berbagai penelitian yang telah dilakukan peneliti menggunakan
spektrofotometri derivatif untuk menetapkan kadar binary ataupun ternary mixtures, yaitu dengan metode zero-crossing dan metode derivative ratio zero-crossing. Penelitian Hajian dan Soltaninezhad (2012) tentang spektrofotometri multikomponen tiga campuran parasetamol, aspirin, dan kofein menggunakan metode double divisor ratio spectra derivative. Pada penelitian ini menggunakan Britton-Robinson yang terdiri dari asam fosfat, asam borat, asam asetat dan natrium hidroksida. Buffer Britton-Robinson tersebut dibuat pH 11.
Penelitian Ozdemir et al., (2004) teknik kalibrasi multivariat parasetamol,
aspirin, dan kofein pada formulasi farmasi menggunakan metode classical least squares (CLS) dan inverse least squares (ILS) pada alat
spektrofotometri yang menggunakan HCl 0,1M.
Penelitian Tsvetkova et al., (2012) untuk analisa campuran parasetamol
dan kofein pada formulasi tablet menggunakan metode validasi
liquid chromatography (LC) dengan fase gerak buffer fosfat pH 3.
Penelitian Ali et al., (2012) validasi spektrofotometri dan spektrodensitimetri pada penentuan tiga campuran obat analgetik pada dosis yang berbeda (parasetamol, aspirin, dan kofein). Pada metode spektrofotometri menggunakan aplikasi ratio spectra derivative dengan pelarut metanol. Metode thin layer chromatography (TLC)-densitometri mengunakan silica gel 60 F254 dengan pelarut kloroform:metanol:asam-asetat:ammonia (95:5:0,5:0,2) untuk pemisahan tiga campuran obat. Penelitian Wijaningtyas (2015) kombinasi spektrofotometri Ultra Violet (UV) dan kalibrasi multivariat untuk analisis parasetamol, asetosal, dan kofein dalam sediaan tablet. Penelitian tersebut mengevaluasi kemampuan metode spektrofotometri Ultra Violet (UV) dikombinasikan dengan kalibrasi multivariate partial least square (PLS) yang menggunakan pelarut etanol.
Pemeriksaan mutu suatu sediaan obat mutlak diperlukan untuk menjamin bahwa sediaan obat mengandung bahan dengan mutu dan jumlah yang ditetapkan dan mengikuti prosedur analisis standar, sehingga menunjang efek terapeutik yang diharapkan. Pengujian stabilitas mencoba untuk memastikan kualitas produk tidak hanya selama manufaktur, produksi, dan kemasan bahkan selama penyimpanan sampai pasien mengkonsumsi obat tersebut. Selama penyimpanan produk obat mungkin bisa terkena kondisi lingkungan yang merugikan seperti suhu yang ekstrim, kelembaban udara yang bervariasi, dan cahaya yang intensif.
Mempertimbangkan aspek-aspek ini, banyak pengawas International Conference on Harmonization (ICH) dan World Health Organization (WHO) membuat pedoman untuk menjamin kualitas produk obat selama kondisi penyimpanan (Wijayaningtyas, 2015; Kabra, et al., 2013).
Para peneliti telah melakukan penelitian pengujian stabilitas dengan
tekanan degradasi pada kondisi yang berbeda direkomendasikan ICH (International Conference on Harmonization), yaitu hidrolisis, oksidasi,
fotolisis, dan degradasi termal (Sayyed, et al., 2015; Pathak dan Rajput, 2009;
Kabra, et al., 2013; Walash, et al., 2013; Wajiha, et al.,2015; Kamble dan Singh, 2011).
Pengujian stabilitas dan tekanan degradasi merupakan komponen penting dari strategi pengembangan obat. Metode ini membantu kita memahami mekanisme dari obat dekomposisi, yang selanjutnya membantu dan memperoleh informasi pada faktor fisik dan kimia yang mengakibatkan ketidakstabilan (Pathak dan Rajput, 2009).
Dalam penelitian ini, peneliti menggunakan spektrofotometri derivatif untuk menetapkan kadar campuran parasetamol, asetosal, dan kofein dengan metode zero-crossing yaitu penetapan panjang gelombang analisis pada titik dimana komponen lain dari campuran melewati garis nol dan penggunaan teknik spektrofotometri dikombinasikan dengan algoritma matematika telah membawa metodologi baru, cepat, dan lebih murah untuk penentuan analit dalam sampel.
Untuk penentuan simultan dari dua atau lebih senyawa aktif dalam campuran yang sama tanpa langkah pemisahan yang dapat menganalisis campuran tersebut.
Metode ini sederhana, akurat, tepat, dan cepat dikembangkan untuk estimasi campuran parasetamol, asetosal, dan kofein dalam bentuk sediaan tablet.
Metode ini divalidasi untuk campuran parasetamol, asetosal, dan kofein dalam
bentuk sediaan tablet harus memenuhi persyaratan validasi metode analisis.
Dengan demikian, validasi metode analisis ditujukan untuk menjamin bahwa
metode analisis memenuhi spesifikasi yang dapat diterima sesuai dengan tujuan yang diharapkan (Abdel-Hay, 2008 dan Ganjar-Rohman, 2012).
1.2 Perumusan Masalah
Berdasarkan latar belakang di atas, maka perumusan masalah penelitian adalah sebagai berikut:
a. Apakah pengembangan dan validasi dengan metode spektrofotometri derivatif zero-crossing dapat dilakukan untuk menetapkan kadar campuran parasetamol, asetosal, dan kofein dalam bentuk sedian tablet ?
b. Apakah kadar parasetamol, asetosal, dan kofein dalam sediaan tablet yang ditentukan dengan metode spektrofotometri derivatif zero-crossing memenuhi persyaratan kadar zat yang ditetapkan dalam Farmakope Indonesia Edisi V ?
c. Apakah hasil uji validasi terhadap metode spektrofotometri derivatif zero-crossing untuk menganalisa kadar campuran parasetamol, asetosal, dan kofein pada sediaan tablet memenuhi syarat pengujian validasi ?
1.3 Hipotesis
Berdasarkan perumusan masalah di atas, maka hipotesis penelitian ini adalah:
a. Adanya pengembangan dan validasi metode spektrofotometri derivatif zero-crossing dapat untuk menetapkan kadar campuran parasetamol, asetosal, dan kofein dalam sediaan tablet.
b. Kadar parasetamol, asetosal, dan kofein dalam sediaan tablet yang ditentukan dengan metode spektrofotometri derivatif zero-crossing
memenuhi persyaratan kadar zat yang ditetapkan dalam Farmakope Indonesia Edisi V.
c. Hasil uji validasi terhadap metode spektrofotometri derivatif untuk menganalisa kadar campuran parasetamol, asetosal, dan kofein pada sediaan tablet dapat memenuhi syarat pengujian validasi.
1.4 Tujuan Penelitian
Berdasarkan hipotesis, maka diperoleh tujuan penelitian ini antara lain:
a. Untuk mengetahui kadar pada pengembangan dan validasi dengan metode spektrofotometri derivatif zero-crossing pada campuran parasetamol, asetosal, dan kofein dalam sediaan tablet.
b. Untuk mengetahui kadar campuran parasetamol, asetosal, dan kofein dalam sediaan tablet yang ditentukan dengan metode spektrofotometri derivatif zero-crossing memenuhi persyaratan kadar zat yang ditetapkan dalam Farmakope Indonesia Edisi V.
c. Untuk mengetahui hasil uji validasi terhadap metode spektrofotometri derivatif dalam menganalisa kadar campuran parasetamol, asetosal, dan kofein pada sediaan tablet dapat memenuhi syarat pengujian validasi.
1.5 Manfaat Penelitian
Berdasarkan tujuan penelitian, maka manfaat penelitian ini adalah untuk memberikan informasi sebagai alternatif untuk analisis multikomponen dan diharapkan metode ini dapat diaplikasikan dari hasil pengembangan dan validasi dengan metode spektrofotometri derivatif zero-crossing untuk estimasi campuran parasetamol, asetosal, dan kofein pada sediaan tablet.
1.6 Kerangka Penelitian
Penelitian ini dilakukan dengan metode zero-crossing, selanjutnya diaplikasikan dengan memilih delta lambda dan derivat yang digunakan untuk mendapatkan panjang gelombang analisis parasetamol, asetosal, dan kofein, kemudian metode dan kondisi diaplikasikan terhadap sampel yang beredar di pasaran, selanjutnya metode divalidasi dan analisis secara statistik dengan menggunakan uji T.
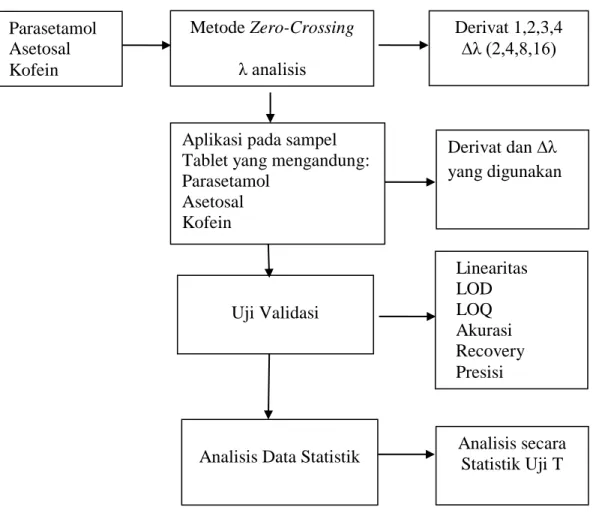
Variabel bebas dalam penelitian ini adalah kadar parasetamol, asetosal, dan kofein sedangkan variable terikatnya adalah metode zero-crossing. Secara ringkasnya kerangka pikir penelitian dapat dilihat pada Gambar 1.1.
Variabel Bebas Variabel Terikat Parameter
Gambar 1.1 Kerangka Penelitian Parasetamol
Asetosal Kofein
Metode Zero-Crossing λ analisis
Derivat 1,2,3,4
∆λ (2,4,8,16)
Aplikasi pada sampel Tablet yang mengandung:
Parasetamol Asetosal Kofein
Uji Validasi
Analisis secara Statistik Uji T Linearitas LOD LOQ Akurasi Recovery Presisi
Analisis Data Statistik
Derivat dan ∆λ yang digunakan
BAB II
TINJAUAN PUSTAKA
2.1 Parasetamol
Parasetamol memiliki nama kimia 4’-Hidroksiasetanilida, dengan rumus molekul C8H9NO2 dan berat molekul sebesar 151,16. Parasetamol mengandung tidak kurang dari 98,0% dan tidak lebih dari 101,0% C8H9NO2, dihitung terhadap zat anhidrat (Depkes RI, 2014). Struktur parasetamol dapat dilihat pada Gambar 2.1.
HO NHCOCH3
Gambar 2.1 Struktur Parasetamol (Depkes RI, 2014)
Pemerian serbuk hablur, putih, tidak berbau, rasa sedikit pahit. Larut dalam air mendidih dan dalam natrium hidroksida 1 N; mudah larut dalam etanol.
Parasetamol merupakan senyawa dimana kelarutannya cenderung tetap dengan perubahan pH (1-8) yaitu sekitar 20,3 mg/ml (Shaw et al., 2005). Penetapan kadar dilakukan dengan cara Kromatografi cair kinerja tinggi, dengan menggunakan fase gerak campuran air:metanol (3:1) sedangkan fase diam yang digunakan adalah oktadesil silana dengan diameter 5µm atau 10µm (L1) (Depkes RI, 2014).
Menurut Auterthoff dan Kovar (1987), parasetamol dalam etanol memberikan serapan maksimum pada panjang gelombang 250 nm ( = 913) dan dalam metanol memberikan serapan maksimum pada panjang gelombang 250 nm ( = 900).
2.2 Asetosal
Memiliki nama kimia asam asetilsalisilat, dengan rumus molekul C9H8O4 dan berat molekul 180,16. Asetosal mengandung tidak kurang dari 99,5% dan tidak lebih dari 100,5% C9H8O4, dihitung terhadap zat yang telah dikeringkan (Depkes RI, 2014). Struktur asetosal dapat dilihat pada Gambar 2.2.
COOH
OCOCH3
Gambar 2.2 Struktur Asetosal (Depkes RI, 2014)
Pemerian asetosal hablur, umumnya seperti jarum atau lempengan tersusun, atau serbuk hablur, putih, tidak berbau atau berbau lemah. Stabil di udara kering, di dalam udara lembab secara bertahap terhidrolisa menjadi asam salisilat dan asam asetat. Asetosal memiliki kelarutan sukar larut dalam air; mudah larut dalam etanol; larut dalam kloroform dan dalam eter; agak sukar larut dalam eter mutlak.
Penetapan kadar asetosal dalam tablet dilakukan menggunakan Kromatografi cair kinerja tinggi, dimana fase gerak yang digunakan yaitu dengan melarutkan 2 g natrium 1-heptansulfonat dalam campuran 850 ml air dan 150 ml asetonitril dan
ditambahkan asam asetat glasial hingga pH 3,4 (Depkes RI, 2014).
Menurut Moffat (2011), asetosal dalam larutan asam memberikan serapan maksimum pada panjang gelombang 230 nm ( = 466), 278 nm ( = 68) dan dalam larutan basa memberikan serapan maksimum pada panjang gelombang 231 nm ( = 409), 298 nm ( = 190).
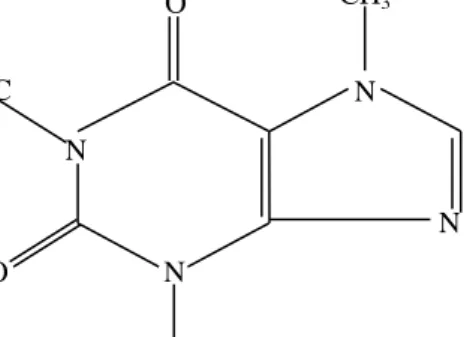
2.3 Kofein
Memiliki nama kimia 1,3,7-Trimetilxantin, dengan rumus molekul C8H10N4O2 dan berat molekul 194,19. Kofein berbentuk anhidrat atau hidrat yang mengandung satu molekul air. Mengandung tidak kurang dari 98,5% dan tidak lebih dari 101,0% C8H10N4O2, dihitung terhadap zat anhidrat (Depkes RI, 2014).
Struktur kofein dapat dilihat pada Gambar 2.3.
Gambar 2.3 Struktur Kofein (Depkes RI, 2014)
Pemerian kofein adalah serbuk putih, atau bentuk jarum mengkilat, biasanya menggumpal; tidak berbau; rasa pahit; larutan bersifat netral terhadap kertas lakmus; bentuk hidratnya mengembang di udara. Kofein memiliki kelarutan agak sukar larut dalam air, dalam etanol; mudah larut dalam kloroform; sukar larut dalam eter. Penetapan kadar kofein dilakukan dengan cara Kromatografi cair kinerja tinggi, dimana fase gerak yang digunakan campuran 1,64 g natrium asetat anhidrat, ditambahkan 50 ml asetonitril dan 40 ml tetrahidrofuran dengan
penambahan asam asetat glasial hingga pH 4,5 (Depkes RI, 2014).
Menurut Moffat (2011), dalam larutan asam memberikan serapan maksimum pada panjang gelombang 273 nm ( = 504).
N
N
CH3
N N CH3
O H3C
O
2.4 Stabilitas Obat
Stabilitas obat adalah kemampuan suatu produk untuk mempertahankan sifat dan karakteristiknya agar sama dengan yang dimilikinya saat dibuat (identitas, kekuatan, kualitas, dan kemurnian) dalam batasan yang ditetapkan sepanjang periode penyimpanan dan penggunaan. Stabilitas obat akan memberikan efek teraupetik yang baik jika obat tersebut dalam keadaan baik.
Stabilitas yang baik mempengaruhi mutu obat, mutu semua obat yang boleh beredar harus terjamin baik dan diharapkan obat akan sampai ke pasien dalam keadan yang baik. Penyimpanan obat yang kurang baik merupakan salah satu masalah dalam upaya peningkatan mutu obat. Selama penyimpanan obat mungkin bisa terkena kondisi lingkungan yang merugikan seperti suhu, kelembaban udara
dan cahaya yang intensif (Waney et al., 2012; Luawo et al., 2012;
Kabra et al.,2013).
Stabilitas sediaan farmasi merupakan salah satu kriteria yang amat penting untuk suatu hasil produksi yang baik. Ketidakstabilan produk obat dapat mengakibatkan terjadinya penurunan sampai dengan hilangnya khasiat obat, obat dapat berubah menjadi toksik atau terjadinya perubahan penampilan sediaan (warna, bau, rasa, konsistensi dan lain-lain) yang akibatnya merugikan bagi pemakai. Ketidakstabilan suatu sediaan farmasi dapat dideteksi melalui perubahan sifat fisika, kimia serta penampilan dari suatu sediaan farmasi. Besarnya perubahan kimia sediaan farmasi ditentukan dari laju penguraian obat melalui hubungan antara kadar obat dengan waktu, atau berdasarkan derajat degradasi dari suatu obat yang jika dipandang dari segi kimia, stabilitas obat dapat diketahui dari
ada atau tidaknya penurunan kadar selama penyimpanan (Ansel, 1989;
Lachman et al.,1994).
Uji stabilitas merupakan bagian penting pada uji bahan obat karena ketidakstabilan obat ditentukan oleh tiga syarat yaitu kualitas, efikasi, dan keamanan.
Tujuan dari pengujian stabilitas adalah untuk memberikan bukti tentang bagaimana kualitas zat aktif atau produk farmasi dengan waktu yang bervariasi juga dibawah pengaruh berbagai faktor lingkungan seperti suhu, kelembaban dan cahaya. Selain itu faktor terkait dalam stabilitas suatu produk misalnya sifat kimia dan fisik dari zat aktif atau tambahan atau eksipien, bentuk sediaan dan komposisi, proses manufaktur, sifat wadah dan penutup, dan sifat-sifat kemasan bahan. Selain itu stabilitas eksipien yang mungkin mengandung atau membentuk produk degradasi reaktif, harus dipertimbangkan (WHO, 2009)
Uji stabilitas dimaksudkan untuk menjamin kualitas produk yang telah diluluskan dan beredar di pasaran. Menurut Carstensen and Rhodes (2000), adapun alasan umum untuk pengujian stabilitas produk farmasi, adalah :
a. hilangnya zat aktif obat (seperti hidrolisis atau oksidasi) b. konsentrasi zat aktif meningkat
c. perubahan bioavaibilitas
d. hilangnya keseragaman kandungan
e. perubahan penampilan (seperti perubahan warna) f. pembentukan hasil penguraian beracun dan iritasi g. adanya aktifitas mikrobiologi
2.5 Spektrofotometri
Spektrofotometri adalah metode pengukuran spektrum cahaya dimana sumber energinya berupa sinar/cahaya dan sistem detektornya menggunakan sel fotolistrik (Noerdin, 1985).
Spektofotometri serapan merupakan pengukuran suatu interaksi antara radiasi elektromagnetik dan molekul atau atom dari suatu zat kimia. Teknik yang sering digunakan dalam analisis farmasi meliputi spektroskopi serapan ultraviolet, cahaya tampak, inframerah, dan serapan atom. Pengukuran spektrofotometri di dalam daerah cahaya tampak, semula disebut kolorimetri, tetapi istilah
“kolorimetri” lebih tepat digunakan untuk persepsi tentang warna (Depkes RI, 2014).
2.5.1 Spektrofotometri Sinar Ultraviolet
Spektrum ultraviolet adalah suatu grafik yang menyatakan hubungan antara panjang gelombang terhadap intensitas serapan (absorbansi) (Sastrohamidjojo, 1985).
Pelarut yang banyak digunakan untuk spektrofotometri UV adalah etanol 95% karena kebanyakan golongan senyawa larut dalam pelarut tersebut.
Alkohol absolut harus dihindari karena mengandung benzena yang dapat menyerap di daerah sinar UV pendek. Pelarut yang sering digunakan ialah air, etanol, metanol, n-heksan dan eter (Harborne, 1987).
Untuk sebagian besar bahan farmasi pengukuran spektrum dalam daerah ultraviolet dan cahaya tampak dapat dilakukan dengan ketelitian dan kepekaan yang lebih baik daripada dalam daerah inframerah. Apabila larutan diamati dalam
sel µg per ml contoh, sering menghasilkan serapan sebesar 0,2-0,8 di daerah ultraviolet dan sinar tampak (Depkes RI, 2014).
Spektrum ultra violet dan cahaya tampak suatu zat pada umumnya tidak mempunyai derajat spesifikasi yang tinggi. Walaupun demikian, spektrum tersebut sesuai untuk pemeriksaan kuantitatif dan untuk berbagai zat, spektrum tersebut bermanfaat sebagai tambahan untuk zat identifikasi (Depkes RI, 2014).
2.5.2 Spektrofotometri Derivatif
Spektofotometri derivatif bersangkutan dengan transformasi spektrum serapan menjadi spektrum derivatif pertama, kedua atau spektrum derivatif dengan order yang lebih tinggi. Spektrum derivat pertama dibuat dengan memplotkan dA/dλ dengan panjang gelombang (Ditjen POM, 1995).
Metode spektrofotometri derivatif merupakan metode manipulatif terhadap spektra pada spektrofotometri ultraviolet dan cahaya tampak (UV-vis). Beberapa keuntungan dari spektrum derivatif antara lain: spektrum derivatif memberikan gambaran struktur yang terinci dari spektrum serapan dan gambaran ini makin jelas dari spektra derivatif pertama ke derivatif keempat. Selain itu, dapat dilakukan analisis kuantitatif suatu komponen dalam campuran dengan bahan yang panjang gelombangnya saling berdekatan. Penentuan panjang gelombang serapan maksimum yang lebar akan lebih akurat menggunakan derivatisasi spektra. Proses yang terjadi dalam derivatisasi data spektra adalah pendiferensialan kurva secara matematis yang tak lain adalah menentukan kemiringan/gradien serapan antara panjang gelombang tertentu secara menyeluruh (Nurhidayati, 2007).
Metode spektrofotometri derivatif dapat digunakan untuk analisis kuantitatif zat dalam campuran yang spektrumnya mungkin tersembunyi dalam suatu bentuk spektrum besar yang saling tumpang tindih dengan mengabaikan proses pemisahan zat yang bertingkat-tingkat.
Dasar perhitungan kuantitatif spektrofotometri derivatif mengikuti hukum Lambert-Beer, dimana serapan derivatif ke-n adalah:
=
× 1 × cDi mana: A = serapan
ε = daya serap molar ( M-1 cm-1) c = konsentrasi molar (M)
l = tebal sel (cm) (Nurhidayati, 2007).
Ada tiga metode spektrofotometri derivatif yang sering digunakan dalam analisa kuantitatif antara lain metode zero crossing, metode peak to peak dan metode multivariate spectrophotometric calibration. Panjang gelombang zero crossing adalah panjang gelombang dimana senyawa tersebut mempunyai serapan nol dan menjadi panjang gelombang analisis untuk zat lain dalam campurannya (Hayun, 2006).
Panjang gelombang serapan maksimum pada suatu senyawa akan menjadi panjang gelombang zero-crossing pada spektrogram derivatif pertama. Metode zero-crossing memisahkan campuran biner dari spektrum derivatifnya pada panjang gelombang pada saat komponen pertama tidak ada sinyal. Pengukuran pada zero-crossing tiap komponen dalam campuran merupakan fungsi tunggal konsentrasi dari yang lainnya. Bila panjang gelombang zero crossing masing-
masing senyawa tidak sama, maka penetapan kadar campuran dua senyawa dapat dilakukan tanpa pemisahan terlebih dahulu. Bila kedua pita serapan mempunyai panjang gelombang yang hampir sama akan terjadi pelebaran pita, maka kurva derivatif pertama tidak akan membantu pemisahan spektranya. Metode zero-crossing memisahkan campuran biner dari spektrum derivatifnya di panjang gelombang pada saat satu komponen tidak ada sinyal. Pengukuran zero-crossing tiap komponen dalam campuran merupakan fungsi tunggal konsentrasi dari yang lainnya (Nurhidayati, 2007).
Untuk pengukuran lebih dari dua analit dilakukan penentuan zero-crossing berturutan. Atau, kurva kalibrasi dapat dibuat pada panjang gelombang yang sinyal rata-ratanya adalah jumlah atau selisih sinyal individual dari dua atau lebih analit. Bila campuran biner memiliki panjang gelombang zero-crossing lebih dari satu, maka yang dipilih untuk dijadikan panjang gelombang analisis adalah panjang gelombang zero-crossing yang serapan pasangannya dan campurannya persis sama, karena pada panjang gelombang tersebut dapat secara selektif mengukur serapan senyawa pasangannya dan memiliki serapan yang paling besar.
Pada serapan yang paling besar, serapannya lebih stabil sehingga kesalahan analisis dapat diperkecil.
Umumnya masalah kuantitatif dapat dibagi dua, yaitu:
1. Senyawa tunggal atau analisis multikomponen dengan puncak serapan saling tumpang tindih. Konsentrasi analit memiliki hubungan linier dengan absorban pada panjang gelombang tertentu. Pada spektra derivatif, konsentrasi analit memiliki hubungan linier dengan amplitudo pada puncak derivat ke-n pada panjang gelombang tertentu.
2. Analisis zat tunggal atau multikomponen dengan terdapatnya serapan matrik background. Terdapatnya serapan matrik background dapat terjadi terutama pada laboratorium klinis, biologi, biokimia dan makanan (Nurhidayati, 2007).
2.5.3 Komponen Spektrofotometer Derivatif
Komponen-komponen pada spektrofotometer UV/Vis biasa sama dengan komponen pada spektrofotometer derivatif. Alat spektrofotometer harus dilengkapi dengan peralatan sedemikian rupa untuk dapat menghasilkan spektrum derivatif (Ditjen POM, 1995). Biasanya spektrofotometer telah mempunyai software untuk mengolah data yang dapat dioperasikan malalui komputer yang telah terhubung dengan spektrofotometer (Moffat, 2007).
2.6 Validasi Metode Analisis
Validasi metode adalah suatu proses yang menunjukkan bahwa prosedur analitik telah sesuai dengan penggunaan yang dikehendaki. Validasi merupakan persyaratan mendasar yang diperlukan untuk menjamin kualitas dan hasil dari semua aplikasi analitik (Ermer, 2005). Adapun karakteristik dalam validasi metode menurut USP (United States Pharmacopeia) XXX yaitu akurasi/kecermatan, presisi/keseksamaan, spesifisitas, batas deteksi, batas kuantitasi, linieritas, rentang dan kekuatan/ketahanan.
2.6.1 Akurasi (Kecermatan)
Akurasi adalah kedekatan antara nilai hasil uji yang diperoleh melalui metode analitik dengan nilai sebenarnya. Akurasi dinyatakan dalam persen perolehan kembali (% recovery) (USP XXX, 2007; Ermer, 2005; Harmita, 2004).
2.6.2 Presisi (Keseksamaan)
Presisi adalah ukuran keterulangan metode analitik, termasuk di antaranya kemampuan instrumen dalam memberikan hasil analitik yang reprodusibel.
Berdasarkan rekomendasi ICH (International Conference on Harmonization), karakteristik presisi dilakukan pada 3 tingkatan, yakni keterulangan (repeatibility), presisi antara (intermediate precision) dan ketertiruan (reproducibility). Keterulangan dilakukan dengan cara menganalisis sampel yang sama oleh analis yang sama menggunakan instrumen yang sama dalam periode waktu singkat. Presisi antara dikerjakan oleh analis yang berbeda. Sedangkan reprodusibilitas dikerjakan oleh analis yang berbeda dan di laboratorium yang berbeda (USP XXX, 2007).
2.6.3 Batas Deteksi dan Batas Kuantitasi
Batas deteksi adalah konsentrasi analit terendah dalam sampel yang masih dapat dideteksi, meskipun tidak selalu dapat dikuantifikasi. Sedangkan batas kuantitasi adalah konsentrasi analit terendah dalam sampel yang dapat ditentukan dengan presisi dan akurasi yang dapat diterima pada kondisi operasional metode yang digunakan (USP XXX, 2007).
2.6.4 Linieritas
Linieritas adalah kemampuan suatu metode untuk memperoleh hasil uji yang secara langsung proposional dengan konsentrasi analit pada kisaran yang diberikan. Linieritas dapat ditentukan secara langsung dengan pengukuran sampel (analit) yang ditambahkan baku pada sekurang-kurangnya lima titik konsentrasi yang mencakup seluruh rentang konsentrasi kerja (Ermer, 2005).
BAB III
METODE PENELITIAN
Penelitian ini merupakan penelitian eksperimental dan dilakukan di Laboratorium Penelitian Fakultas Farmasi Universitas Sumatera Utara.
3.1 Alat dan Bahan 3.1.1 Alat
Spektrofotometer UV-Vis 1800 (Shimadzu) serta seperangkat Personal Computer (PC) yang dilengkapi dengan software UV-Probe 2.42, kuvet 1 cm, oven, alat-alat gelas, lumpang dan alu, neraca analitik (Boeco), sonikator (Branson 1510).
3.1.2 Bahan
Semua pereaksi yang digunakan adalah grade analysis kecuali dinyatakan lain. Parasetamol (Brataco), Asetosal, Kofein (Brataco), Etanol (E-Merck), Akuabidestilata (PT. Ika Pharmindo), kertas saring whatman no. 41, kertas perkamen, tablet Poldan Mig®.
3.2 Pengambilan Sampel
Metode pengambilan sampel yang dilakukan adalah sampling purposif, yaitu sampel dipilih dengan pertimbangan sesuai dengan tujuan penelitian.
Pengambilan sampel secara purposif, yaitu ditentukan tanpa membandingkan sampel antara satu tempat dengan tempat yang lain, karena pengambilan sampel dianggap homogen. Sampel yang digunakan dalam penelitian ini adalah tablet Poldan Mig® yang mengandung parasetamol 400 mg, asetosal 250 mg dan kofein 65 mg.
3.3 Prosedur Penelitian 3.3.1 Pembuatan Pelarut
Digunakan etanol absolut; murni pereaksi. (Ditjen, POM., 1995).
3.3.2 Pembuatan Larutan Induk Baku Parasetamol
Ditimbang dengan seksama 50 mg baku pembanding parasetamol kemudian dimasukkan ke dalam labu tentukur 50 mL, dilarutkan dengan etanol hingga larut, dicukupkan volume dengan etanol sampai garis tanda sehingga didapatkan Larutan Induk Baku (LIB) I dengan konsentrasi 1000 μg/mL. Dari larutan LIB I dipipet 5 mL dimasukkan ke dalam labu tentukur 100 mL, dicukupkan dengan etanol sampai garis tanda sehingga didapatkan larutan dengan konsentrasi 50 μg/mL (LIB II).
3.3.3 Pembuatan Larutan Induk Baku Asetosal
Ditimbang dengan seksama 50 mg baku pembanding asetosal kemudian dimasukkan ke dalam labu tentukur 50 mL, dilarutkan dengan etanol hingga larut, dicukupkan volume dengan etanol sampai garis tanda sehingga didapatkan larutan dengan konsentrasi 1000 μg/mL (LIB I). Dari larutan LIB I dipipet 5 mL dimasukkan ke dalam labu tentukur 100 mL, dicukupkan dengan etanol sampai garis tanda sehingga didapatkan larutan dengan konsentrasi 50 μg/mL (LIB II).
3.3.4 Pembuatan Larutan Induk Baku Kofein
Ditimbang dengan seksama 50 mg baku pembanding kofein kemudian dimasukkan ke dalam labu tentukur 50 mL, dilarutkan dengan etanol hingga larut, dicukupkan volume dengan etanol sampai garis tanda sehingga didapatkan larutan dengan konsentrasi 1000 μg/mL (LIB I). Dari larutan LIB I dipipet 5 mL
dimasukkan ke dalam labu tentukur 100 mL, dicukupkan dengan etanol sampai garis tanda sehingga didapatkan larutan dengan konsentrasi 50 μg/mL (LIB II).
3.3.5 Pembuatan Larutan Standar Parasetamol
Diambil sebanyak 0,5 mL; 0,7 mL; 0,9 mL; 1,1 mL; dan 1,3 mL dari LIB II parasetamol. Kemudian masing-masing dimasukkan ke dalam 5 labu tentukur 10 mL. Dilarutkan dengan pelarut etanol. Kemudian dicukupkan dengan pelarut yang sama untuk membuat larutan standar dengan konsentrasi 2,5 μg/mL;
3,5 µg/mL; 4,5 μg/mL; 5,5 µg/mL; dan 6,5 μg/mL.
3.3.6 Pembuatan Larutan Standar Asetosal
Diambil sebanyak 1 mL; 1,4 mL; 1,8 mL; 2,2 mL; dan 2,6 mL dari LIB II asetosal. Kemudian masing-masing dimasukkan ke dalam 5 labu tentukur 10 mL.
Dilarutkan dengan pelarut etanol. Kemudian dicukupkan dengan pelarut yang sama untuk membuat larutan standar dengan konsentrasi 5 μg/mL; 7 µg/mL;
9 μg/mL; 11 µg/mL; dan 13 μg/mL.
3.3.7 Pembuatan Larutan Standar Kofein
Diambil sebanyak 1 mL; 1,5 mL; 2 mL; 2,5 mL; dan 3 mL dari LIB II kofein. Kemudian masing-masing dimasukkan ke dalam 5 labu tentukur 10 mL.
Dilarutkan dengan pelarut etanol. Kemudian dicukupkan dengan pelarut yang sama untuk membuat larutan standar dengan konsentrasi 5 μg/mL; 7,5 µg/mL;
10 μg/mL; 12,5 µg/mL; dan 15 μg/mL.
3.3.8 Pembuatan Spektrum Serapan Maksimum Parasetamol
Diambil sebanyak 0,9 mL dari LIB II parasetamol (konsentrasi=50 μg/mL) kemudian dimasukkan ke dalam labu tentukur 10 mL untuk kemudian dilarutkan dengan etanol. Selanjutnya larutan diencerkan dengan pelarut yang sama hingga
garis tanda, lalu dikocok sampai homogen untuk memperoleh larutan parasetamol dengan konsentrasi 4,5 μg/mL. Diukur serapannya pada panjang gelombang 200-400 nm.
3.3.9 Pembuatan Spektrum Serapan Maksimum Asetosal
Diambil sebanyak 1,8 mL dari LIB II asetosal (konsentrasi = 50 μg/mL) kemudian dimasukkan ke dalam labu tentukur 10 mL untuk kemudian dilarutkan dengan etanol. Selanjutnya larutan diencerkan dengan pelarut yang sama hingga garis tanda, lalu dikocok sampai homogen untuk memperoleh larutan asetosal dengan konsentrasi 9 μg/mL. Diukur serapannya pada panjang gelombang 200-400 nm.
3.3.10 Pembuatan Spektrum Serapan Maksimum Kofein
Diambil sebanyak 2,0 mL dari LIB II kofein (konsentrasi = 50 μg/mL) kemudian dimasukan ke dalam labu tentukur 10 mL untuk kemudian dilarutkan dengan etanol. Selanjutnya larutan diencerkan dengan pelarut yang sama hingga garis tanda, lalu dikocok sampai homogen untuk memperoleh larutan kofein dengan konsentrasi 10 μg/mL. Diukur serapannya pada panjang gelombang 200-400 nm.
3.3.11 Pembuatan Spektrum Serapan Campuran Parasetamol, Asetosal dan Kofein
Dibuat larutan campuran parasetamol, asetosal dan kofein dengan cara dipipet 0,9 mL LIB II parasetamol, 1,8 mL LIB II asetosal, dan 2 mL LIB II kofein kedalam labu ukur 10 ml, kemudian dicukupkan volumenya menggunakan pelarut etanol sampai garis tanda untuk mendapatkan larutan yang mengandung campuran parasetamol, asetosal dan kofein dengan konsentrasi 4,5 µg/mL;
9µg/mL dan 10 µg/mL secara berurutan. Dari larutan tersebut dibuat spektrum serapannya.
3.3.12 Pembuatan Spektrum Serapan Derivatif Parasetamol
Dibuat spektrum serapan (tanpa diderivatkan) dari larutan standar parasetamol dengan konsentrasi 2,5 μg/mL; 3,5 µg/mL; 4,5 μg/mL; 5,5 µg/mL;
dan 6,5 μg/mL pada panjang gelombang 200-400 nm. Kemudian spektrum ditransformasikan menjadi spektrum serapan derivat ke-1, 2, 3, dan 4 dengan Δλ 2, 4, 8, dan 16 dengan bantuan software UV Probe 2.42 dimana spektrum serapan parasetamol yang telah disimpan dimanipulasi dengan tipe transformation kemudian dipilih derivat dan Δλ yang diinginkan.
3.3.13 Pembuatan Spektrum Serapan Derivatif Asetosal
Dibuat spektrum serapan (tanpa diderivatkan) dari larutan standar asetosal dengan konsentrasi 5 μg/mL; 7 µg/mL; 9 μg/mL; 11 µg/mL; dan 13 μg/mL pada panjang gelombang 200 - 400 nm. Kemudian spektrum ditransformasikan menjadi spektrum serapan derivat ke-1, 2, 3, dan 4 dengan Δλ 2, 4, 8, dan 16 dengan bantuan software UV Probe 2.42 dimana spektrum serapan asetosal yang telah disimpan dimanipulasi dengan tipe transformation kemudian dipilih derivat dan Δλ yang diinginkan.
3.3.14 Pembuatan Spektrum Serapan Derivatif Kofein
Dibuat spektrum serapan (tanpa diderivatkan) dari larutan standar kofein dengan konsentrasi 5 μg/mL; 7,5 µg/mL; 10 μg/mL; 12,5 µg/ml; dan 15 μg/mL pada panjang gelombang 200-400 nm. Kemudian spektrum ditransformasikan menjadi spektrum serapan derivat ke-1, 2, 3, dan 4 dengan Δλ 2, 4, 8, dan 16 dengan bantuan software UV Probe 2.42 dimana spektrum serapan kofein yang
telah disimpan dimanipulasi dengan tipe transformation kemudian dipilih derivat dan Δλ yang diinginkan.
3.3.15 Pembuatan Spektrum Serapan Derivatif Campuran Parasetamol, Asetosal dan Kofein
Spektrum serapan campuran parasetamol, asetosal dan kofein dengan konsentrasi 4,5 µg/mL; 9 µg/mL dan 10 µg/mL. Kemudian spektrum ditransformasikan masing-masing menjadi spektrum derivat ke-1, 2, 3, dan 4 dengan Δλ 2, 4, 8, dan 16 dengan bantuan software UV Probe 2.42 dimana spektrum serapan campuran yang telah disimpan dimanipulasi dengan tipe transformation kemudian dipilih derivat dan Δλ yang diinginkan
3.3.16 Pembuatan dan Penentuan Panjang Gelombang (λ) Analisis untuk Metode Zero Crossing
Dibuat larutan parasetamol dengan konsentrasi 4,5 μg/mL, larutan asetosal dengan konsentrasi 9 μg/mL, larutan kofein dengan konsentrasi 10 μg/mL dan larutan campuran parasetamol 4,5 μg/mL, asetosal 9 μg/mL dan kofein 10 μg/mL.
Kemudian ketiga larutan ini diukur serapannya pada panjang gelombang 200-400 nm. Selanjutnya ditransformasikan menjadi spektrum serapan derivat pertama dan kedua dari masing-masing zat tunggal dan dari campuran parasetamol, asetosal dan kofein. Spektrum serapan derivat kedua dari larutan zat tunggal dan campuran keduanya ditumpangtindihkan. Yang dipilih untuk menjadi panjang gelombang analisis adalah yang pada panjang gelombang tertentu, serapan tunggal salah satu senyawa nol sedangkan serapan tunggal senyawa pasangannya dan campuran keduanya hampir sama atau persis sama. Karena pada panjang gelombang tersebut dapat secara selektif mengukur serapan salah satu senyawa tanpa diganggu oleh serapan senyawa pasangannya.
3.3.17 Penentuan Panjang Gelombang (λ) Analisis untuk Metode Zero Crossing Derivatif pada Parasetamol
Spektrum parasetamol dan spektrum campuran derivat ke-1, 2, 3, dan 4 dengan Δλ 2, 4, 8, dan 16 yang saling ditumpangtindihkan, dicari λ dimana pada λ tersebut memberikan absorbansi yang bernilai untuk parasetamol dan memberikan absorbansi yang bernilai nol untuk asetosal dan kofein (titik zero crossing), serta nilai absorbansi parasetamol pada λ sama atau hampir sama dengan nilai absorbansi dari spektrum campurannya.
3.3.18 Penentuan Panjang Gelombang (λ) Analisis untuk Metode Zero Crossing Derivatif pada Asetosal
Spektrum asetosal dan spektrum campuran derivat ke-1, 2, 3, dan 4 dengan
Δλ 2, 4, 8, dan 16 yang saling ditumpangtindihkan, dicari λ dimana pada λ tersebut memberikan absorbansi yang bernilai untuk asetosal dan memberikan
absorbansi yang bernilai nol untuk parasetamol dan kofein (titik zero crossing), serta nilai absorbansi asetosal pada λ sama atau hampir sama dengan nilai absorbansi dari spektrum campurannya.
3.3.19 Penentuan Panjang Gelombang (λ) Analisis untuk Metode Zero Crossing Derivatif pada Kofein
Spektrum kofein dan spektrum campuran derivat ke-1, 2, 3, dan 4 dengan
Δλ 2, 4, 8, dan 16 yang saling ditumpangtindihkan, dicari λ dimana pada λ tersebut memberikan absorbansi yang bernilai untuk kofein dan memberikan
absorbansi yang bernilai nol untuk parasetamol dan asetosal (titik zero crossing), serta nilai absorbansi kofein pada λ sama atau hampir sama dengan nilai absorbansi dari spektrum campurannya.
3.3.20 Pembuatan Kurva Kalibrasi Metode Zero Crossing Parasetamol
Dibuat larutan standar parasetamol dengan konsentrasi 2,5 μg/mL;
3,5 µg/mL; 4,5 μg/mL; 5,5 µg/mL; dan 6,5 μg/mL, kemudian diukur absorbansi pada derivat ketiga (Δλ 2 nm) pada panjang gelombang analisis yang telah ditentukan dan dilakukan analisis hubungan antara konsentrasi dan nilai serapan dihitung untuk mendapatkan persamaan regresi linear.
3.3.21 Pembuatan Kurva Kalibrasi Metode Zero Crossing Asetosal
Dibuat larutan standar asetosal dengan konsentrasi 5 μg/mL; 7 µg/ mL;
9 μg/mL; 11 µg/ mL; dan 13 μg/mL, kemudian diukur absorbansi pada derivat ketiga (Δλ 2 nm) pada panjang gelombang analisis yang telah ditentukan dan dilakukan analisis hubungan antara konsentrasi dan nilai serapan dihitung untuk mendapatkan persamaan regresi linear.
3.3.22 Pembuatan Kurva Kalibrasi Metode Zero Crossing Kofein
Dibuat larutan standar kofein dengan konsentrasi 5 μg/mL; 7,5 μg/mL; 10 μg/mL; 12,5 μg/mL dan 15 μg/mL, kemudian diukur absorbansi pada derivat kedua (Δλ 16 nm) pada panjang gelombang analisis yang telah ditentukan dan dilakukan analisis hubungan antara konsentrasi dan nilai serapan dihitung untuk mendapatkan persamaan regresi linear.
3.4 Validasi metode
3.4.1 Linearitas, batas deteksi (Limit of Detection, LOD) dan batas kuantitasi (Limit of Quantification, LOQ)
Diambil sebanyak 0,5 mL; 0,7 mL; 0,9 mL; 1,1 mL; dan 1,3 mL dari LIB II parasetamol ke dalam labu tentukur 10 mL, kemudian dicukupkan dengan menggunakan pelarut etanol sampai garis tanda untuk membuat larutan
parasetamol dengan konsentrasi 2,5 μg/mL; 3,5 µg/mL; 4,5 μg/mL; 5,5 µg/mL;
dan 6,5 μg/mL.
Diambil sebanyak 1 mL; 1,4 mL; 1,8 mL; 2,2 mL; dan 2,6 mL dari LIB II asetosal ke dalam labu tentukur 10 mL, kemudian dicukupkan dengan menggunakan pelarut etanol sampai garis tanda untuk membuat larutan asetosal dengan konsentrasi 5 μg/mL; 7 µg/mL; 9 μg/mL; 11 µg/mL; dan 13 μg/mL.
Diambil sebanyak 1 mL; 1,5 mL; 2 mL; 2,5 mL; dan 3 mL dari LIB II kofein ke dalam labu tentukur 10 mL, kemudian dicukupkan dengan menggunakan pelarut etanol sampai garis tanda untuk membuat larutan kofein dengan konsentrasi 5 μg/mL; 7,5 µg/mL; 10 μg/mL; 12,5 µg/mL; dan 15 μg/mL.
Larutan-larutan diatas dilihat serapannya pada λ analisis masing-masing zat yang telah ditentukan. Kemudian dilakukan analisis hubungan antara konsentrasi dengan serapan untuk masing-masing zat, sehingga didapat persamaan regresi linear dan juga nilai korelasinya.
y = a + bx.
Dan berdasarkan nilai serapan pada panjang gelombang analisis, dilakukan pula perhitungan LOD dan LOQ.
Rumus yang digunakan adalah :
SD =
2 - n
Yi -
Y 2
LOD =
slope 3SD
LOQ =
slope 10 SD
Keterangan :
SD = Standard Deviation / simpangan baku LOD = Limit of Detection / batas deteksi LOQ = Limit of Quantitation / batas kuantitasi 3.4.2 Uji Akurasi
Uji akurasi dilakukan dengan metode penambahan bahan baku yaitu dengan membuat 3 konsentrasi sampel dengan rentang spesifik 80%, 100%, 120%. Dimana pada masing-masing rentang spesifik digunakan 70% sampel dan 30% baku yang akan ditambahkan (Harmita, 2004).
Kemudian campuran sampel dan baku diukur serapannya pada panjang gelombang 200–400 nm, selanjutnya spektrum serapan ditransformasikan menjadi spektrum serapan derivat kedua dengan Δλ 2 nm pada panjang gelombang analisis parasetamol, asetosal dan kofein yang akan ditentukan. Persen perolehan kembali dapat dihitung dengan rumus:
Y = 100 % Keterangan:
Y = Persentase perolehan kembali CF = Jumlah analit yang terukur
CA = Jumlah analit dalam sampel (70% berasal dari sampel) = Jumlah baku yang ditambahkan (30% berasal dari baku) 3.4.3 Uji Presisi
Presisi diukur sebagai simpangan baku relatif atau koefisien variasi.
Presisi merupakan ukuran yang menunjukkan derajat kesesuaian antara hasil uji individual ketika suatu metode dilakukan secara berulang untuk sampel yang
homogen. Nilai simpangan baku relatif yang memenuhi persyaratan menunjukkan adanya keseksamaan metode yang dilakukan (Harmita, 2004). Simpangan baku relatif dapat dihitung dengan rumus berikut ini :
RSD =
̅
x 100%
Keterangan : = Kadar zat rata-rata dalam sampel SD = Standard Deviation
RSD = Relative Standar Deviation
3.5 Penentuan Kadar Campuran Parasetamol, Asetosal dan Kofein dalam Sediaan Tablet
Dua puluh tablet ditimbang dan digerus homogen. Serbuk ditimbang setara dengan 100 mg parasetamol dan dihitung kesetaraan asetosal dan kofein yang terkandung didalamnya, penimbangan dilakukan sebanyak enam kali pengulangan. Dimasukkan ke dalam labu tentukur 100 mL dan dicukupkan dengan pelarut etanol sampai garis tanda. Dibantu pelarutannya menggunakan sonikator selama 15 menit, lalu disaring (± 25 ml filtrat pertama dibuang, filtrat selanjutnya ditampung). Kemudian dipipet 0,1 mL larutan filtrat, dimasukkan kedalam labu tentukur 25 mL, lalu dicukupkan dengan etanol hingga garis tanda.
Selanjutnya diukur absorbansinya pada panjang gelombang yang telah ditetapkan dengan menggunakan metode zero crossing.
3.5.1 Perhitungan Kadar Parasetamol, Asetosal dan Kofein dalam Sediaan Tablet
Nilai absorbansi yang didapatkan dari analisis disubstitusikan kedalam persamaan garis regresi yang didapatkan sehingga akan didapatkan kadar analit (parasetamol, asetosal, dan kofein) dalam sampel (Wulandari, 2007). Kadar analit
(parasetamol, asetosal, dan kofein) dalam satu tablet dapat dihitung dengan menggunakan rumus:
Keterangan:
X = Kadar analit dalam satu tablet C = Kadar analit dalam sampel Fp = Faktor pengenceran (10000)
A = Berat sampel setara dengan satu tablet (berat 20 tablet dibagi 20) S = Berat sampel yang dilarutkan (berat sampel setara 100 mg parasetamol) B = Kadar yang tertera dalam sertifikat analisis
3.6 Analisis Data Penetapan Kadar Secara Statistik
Data perhitungan kadar parasetamol, asetosal, dan kofein dianalisis secara statistik dengan menggunakan uji T.
Rumus yang digunakan adalah :
SD = √
1 - n
X - Xi
2
Untuk mencari t hitung digunakan rumus:
t hitung =
|
̅√
|
Data diterima jika -ttabel < thitung < ttabel pada interval kepercayaan 99%
dengan nilai α = 0,01.
Keterangan :
SD = Standard deviation / simpangan baku Xi = Kadar zat dalam satu perlakuan
X = Kadar zat rata-rata dalam sampel n = jumlah pengulangan
α = tingkat kepercayaan
Untuk menghitung kadar yang sebenarnya dalam sampel secara statistik dapat digunakan rumus (Sudjana, 2005):
µ = X ± (t(α/2, dk) x SD / √n )
Keterangan :
SD = standard deviation / simpangan baku
X = Kadar zat rata-rata dalam sampel n = jumlah pengulangan
t = harga ttabel sesuai dengan derajat kepercayaan
BAB IV
HASIL DAN PEMBAHASAN
4.1 Hasil Penentuan Spektrum Serapan Maksimum
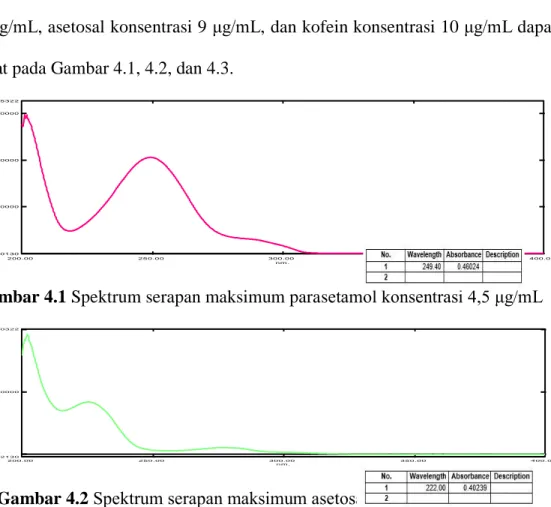
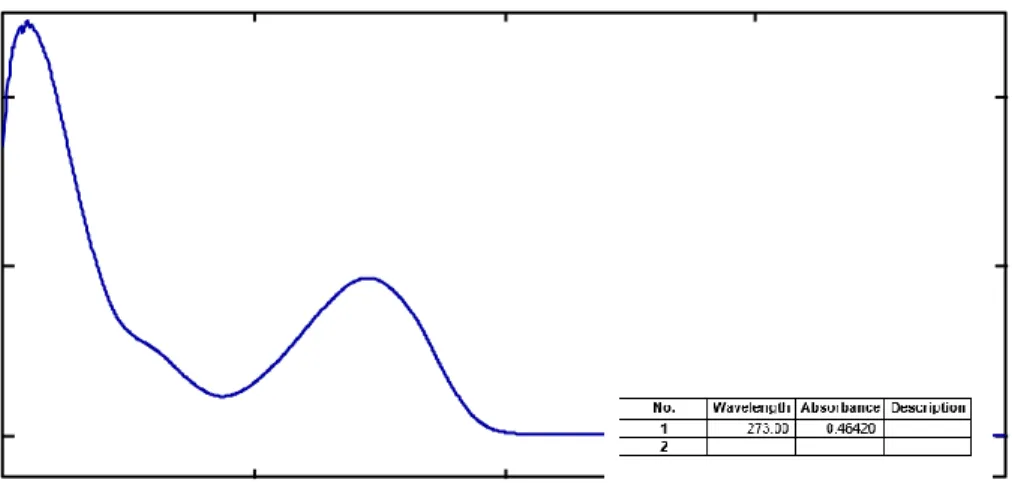
Penentuan spektrum serapan maksimum dilakukan pada panjang gelombang 200-400 nm. Spektrum serapan parasetamol (2,5-6,5 μg/mL), asetosal (5-13 μg/mL), dan kofein (5-15 μg/mL) menggunakan pelarut etanol dalam berbagai konsentrasi. Hasil penentuan spektrum serapan maksimum parasetamol diperoleh pada konsentrasi 4,5 μg/mL, asetosal pada konsentrasi 9 μg/mL, dan untuk kofein pada konsentrasi 10 μg/mL. Berdasarkan hasil penelitian, diperoleh panjang gelombang maksimum parasetamol pada 249,4 nm, asetosal pada 222 nm dan kofein pada 273 nm. Spektrum serapan maksimum parasetamol konsentrasi 4,5 μg/mL, asetosal konsentrasi 9 μg/mL, dan kofein konsentrasi 10 μg/mL dapat dilihat pada Gambar 4.1, 4.2, dan 4.3.
Gambar 4.1 Spektrum serapan maksimum parasetamol konsentrasi 4,5 μg/mL
Gambar 4.2 Spektrum serapan maksimum asetosal konsentrasi 9 μg/mL
nm .
200.00 250.00 300.00 350.00 400.00
Abs.
0.65322
0.60000
0.40000
0.20000
-0.00130
n m .
2 0 0 .0 0 2 5 0 .0 0 3 0 0 .0 0 3 5 0 .0 0 4 0 0 .0 0
Abs.
1 .0 0 3 2 2
0 .5 0 0 0 0
-0 .0 2 1 3 0
Gambar 4.3 Spektrum serapan maksimum kofein konsentrasi 10 μg/mL
4.2 Hasil Penentuan Spektrum Serapan
Hasil penentuan spektrum serapan dibuat terhadap larutan parasetamol dengan konsentrasi 4,5 µg/mL, larutan asetosal dengan konsentrasi 9 µg/mL dan larutan kofein dengan konsentrasi 10 µg/mL, kemudian dibuat spektrum serapan pada panjang gelombang 200-400 nm. Ketika dilakukan orientasi dengan berbagai konsentrasi yaitu 2,5 µg/mL : 5 µg/mL : 5 µg/mL, 3,5 µg/mL : 7 µg/mL : 7,5 µg/mL, 4,5 µg/mL : 9 µg/mL : 10 µg/mL, 6,5 µg/mL : 13 µg/mL : 15 µg/mL ternyata konsentrasi yang terbaik adalah 4,5 µg/mL : 9 µg/mL : 10 µg/mL. Spektrum campuran parasetamol, asetosal, dan kofein akan menghasilkan spektrum yang berbeda dengan spektrum masing-masing dari parasetamol, asetosal, dan kofein, hal ini dikarenakan spektrum campuran merupakan kombinasi dari spektrum zat yang menyusunnya. Spektrum campuran parasetamol, asetosal, dan kofein dapat dilihat pada Gambar 4.4. Metode spektrofotometri biasa tidak dapat dilakukan untuk menetapkan kadar parasetamol, asetosal maupun kofein dalam campuran parasetamol, asetosal, dan
nm .
200.00 250.00 300.00 350.00 400.00
Abs.
1.24824
1.00000
0.50000
0.00000 -0.12305