0
[Untuk ka angan send r l i i ] Gun, Denny & Nuz yl
BASIC CLINICAL
PHARMACOKINETIC
S
EDISI 4
(Terjemahan Oleh Gunawan, Denny & Nuzly)
1
[Untuk ka angan send r l i i ] Gun, Denny & Nuz yl
Wajib baca
Kami tak ada maksud mencari untung untuk diri sendiri dari pembuatan buku terjemahan ini. Tujuan kami menterjemahkan dan membuatnya dalam bentuk seperti ini hanyalah untuk memudahkan dalam memahami tentang isi materi dari buku ini, dalam proses perkuliahan, yang mana buku aslinya dalam Bahasa Inggris.
Terima kasih kepada Michael E. Winter dan penerbitnya, yang telah menyediakan kami sumber bacaan sebagai referensi dalam memahami Farmakokinetik, terima kasih juga kepada rekan-rekan seangkatan Apoteker X Unjani (yang tak dapat kami sebutkan satu-persatu), yang mana telah turut berpartisipasi dalam penterjemahan materi ini, kami tidak mungkin bekerja dengan sendiri.
Akhirnya kepada semua pihak yang telah membantu dalam penyusunan terjemahan ini kami sampaikan terima kasih.
Harapan kami kiranya terjemahan ini dapat membantu kita semua dalam memahami Farmakokinetik. (Penggunaan buku ini tidak bisa tepisahkan dari buku aslinya : Basic Clinical Pharmacokinetics (Ed. 4) oleh Michael E. Winter).
Kiranya apa yang telah kami susun bisa disempurnakan oleh angkatan-angkatan setelah kami.
“Tak ada gading yang tak retak”
Medio, Januari 2011
“Salam Jablay”
Gunawan, Denny & Nuzly
[Untuk kalangan sendiri]
Gun, Denny & Nuzly 2 BAGIAN PERTAMA
Prinsip Dasar
I.1 Bioavailabilyty (F)
DEFINISIBioavailabilitas/ketersediaan hayati adalah persentase atau fraksi dari dosis yang mencapai sirkulasi sistemik pasien. Contoh faktor-faktor yang dapat mengubah ketersediaan hayati termasuk pembubaran karakteristik yang melekat absorption suatu bentuk kimia diberikan (misalnya, garam, ester), bentuk sediaan (misalnya, tablet, kapsul), rute pemberian, stabilitas bahan aktif dalam saluran gastrointestinal (GI), dan tingkat metabolisme obat sebelum mencapai sirkulasi sistemik. Obat dapat dimetabolisme oleh Bakteri saluran GI, lapisan permukaan/mukosa GI, dan oleh hati sebelum masuk dalam sistem sirkulasi.
Untuk menghitung jumlah obat yang terabsorpsi, dosis harus dikalikan dengan faktor ketersediaan hayati, yang biasanya ditandai oleh huruf "F". Sebagai contoh, ketersediaan hayati Digoxin (Lanoxin) diperkirakan 0,7 untuk pemberian tablet secara oral.1-3 Ini berarti
bahwa jika 250 µg (0,25 mg) dari digoksin diberikan secara oral, efektifitas atau absorpsi dapat dihitung dengan mengalikan dosis yang diberikan oleh F:
Jumlah Obat yang di serap atau
Yang mencapai sirkulasi sistemik = (F)(Dose) [Eq. 1] Jumlah Obat yang di serap atau
Yang mencapai sirkulasi sistemik = (F)(Dose) = (0,7)(250 µg) = 175 µg
Hal yang harus diperhatikan bahwa faktor ini tidak mempertimbangkan tingkat penyerapan obat, hanya perkiraan tingkat penyerapan. Meskipun tingkat penyerapan dapat menjadi penting ketika mulai cepat efek farmakologis diperlukan, tidak biasanya penting bila obat yang diberikan kronis. Tingkat penyerapan penting hanya ketika sangat lambat sehingga membatasi ketersediaan hayati absolut dari obat, atau ketika begitu cepat sehingga terlalu banyak obat diserap persiapan. "Dosis dumping" dapat terjadi tertentu dalam kondisi dengan berkelanjutan-release beberapa .4,5 Selain penyerapan tidak lengkap, dari bentuk-realease
[Untuk kalangan sendiri]
Gun, Denny & Nuzly
3 dosis berkelanjutan harus dipertimbangkan n pasien yang memiliki waktu transit singkat
4 [Untuk kalangan
sendiri]
Gun, Denny & Nuzly
GI. GI transit waktu dari 24 sampai 48 jam mungkin rata-rata, tapi pasien dengan penyakit usus mungkin memiliki waktu transit hanya beberapa jam. A-dari-averege bioavailabilitas yang lebih rendah harus dipertimbangkan dalam hal ini pasien, khususnya bila penyerapan durasi diperpanjang. BENTUK
DOSIS
Seperti dijelaskan sebelumnya, ketersediaan hayati dapat bervariasi antara bentuk formulasi yang berbeda dan dosis obat, misalnya, obat dogoxin elixir memiliki bioavailabilitas sekitar 80% (F=0,8), sedangkan kapsul gelatin lunak memiliki bioavailabilitas sebesar 100% (F=1,0). Hal ini berbeda dengan tablet, yang mana memiliki bioavailabilitas sebesar 70% (F=0,7).2,6,7 Ketika obat diberikan/dikelola secara parenteral, bioavailabilitas biasanya
dianggap 100% (F = 1.0). Dengan menata ulang Persamaan 1, prinsip ini dapat digunakan untuk menghitung dosis setara dengan obat ketika pasien akan menerima bentuk sediaan yang berbeda dari obat yang sama.
Dosis dari bentuk sediaan obat baru =
Jumlah dosis yang diserap dari obat yang tersedia F dari bentuk sediaan obat baru
[Eq. 2]
Misalnya, jika pasien yang telah menerima digoxin 250 µg (0,25 mg) dalam bentuk tablet dosis, dengan bioavailabilitas sebesar 0,7, perlu menerima eliksir digoksin, dosis setara dengan obat mujarab akan dihitung sebagai berikut:
Dosis dari elixir = (0,7)(250 μg) 0,8
=
(K,L)(MNK OP) K,Q= 219 µg
Jika kapsul gelatin lunak digoxin itu harus diberikan, ketersediaan hayati atau F dari bentuk sediaan baru akan heve menjadi 1,0 dan dosis setara pasti 175µg.
Ketersediaan hayati obat diberikan parenteral biasanya diasumsikan 1.0. Obat yang diberikan sebagai prekursor tidak aktif yang kemudian harus dikonversi ke produk aktif
merupakan pengecualian terhadap peraturan ini. Jika beberapa aktif prekursor dieliminasi dari tubuh (ekskresikan ginjal atau dimetabolisme untuk senyawa tidak aktif) sebelum dapat dikonversi menjadi senyawa aktif, ketersediaan hayati akan <1,0. Sebagai contoh,
kloramfenikol parenteral diberikan sebagai ester suksinat, dan kloramfenikol ester ini harus dihidrolisis menjadi senyawa aktif. Ketersediaan hayati dari kloramfenikol suksinat yang diberikan parenteral berkisar dari 55% sampai 95%, karena 5% menjadi 45% dari kloramfenikol ester tereliminasi di ginjal sebelum dapat dikonversi menjadi senyawa aktif. 8 Secara umum, untuk obat-obatan dengan penyerapan hampir lengkap ( F> 0,8)
bioavailabilitas biasanya konsisten. Untuk obat-obatan dengan bioavailabilitas oral rendah (F <0,5) sering kali ada variasi besar di tingkat penyerapan. Ini bukan aturan keras dan cepat sebagai obat apapun di bawah kondisi yang tepat dapat memiliki ketersediaan hayati diubah. BENTUK KIMIA (S)
Bentuk kimia obat juga harus dipertimbangkan ketika mengevaluasi ketersediaan hayati. Sebagai contoh, ketika garam atau ester suatu obat diberikan, faktor ketersediaan hayati (F) harus dikalikan dengan fraksi berat molekul total bahwa obat aktif merupakan . Jika "S" merupakan bagian dari dosis utamanya adalah obat aktif, maka jumlah obat yang diserap dari bentuk garam atau ester dapat dihitung sebagai berikut:
Jumlah obat yang diserap atau
Yang Mencapai Sirkulasi Sistemik = (S)(F)(dosis) [Eq. 3] Faktor "S" harus disertakan dalam semua persamaan bioavailabilitas sebagai pengingat penting dalam menilai ketersediaan hayati berupa obat aktif. Bila obat diberikan dalam induknya atau bentuk aktif, "S" untuk obat yang 1.0.
Persamaan 2 sekarang dapat diperluas untuk mempertimbangkan faktor garam dan ketersediaan hayati ketika menghitung dosis bentuk sediaan baru:
Dosis dari bentuk Sediaan obat baru =
Jumlah dosis yang di se rap
(S)(F) dosis obat
[eq 4]
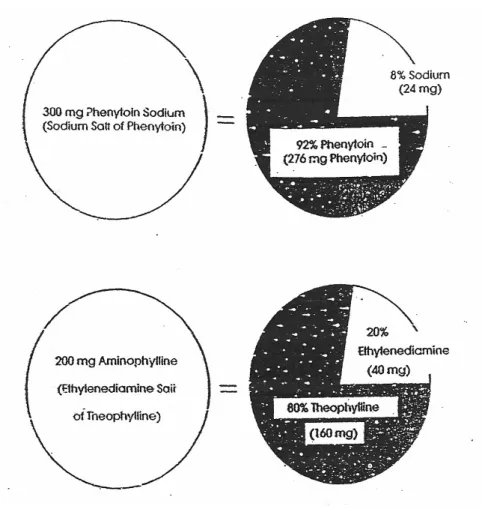
Aminofilin dan fenitoin adalah contoh dari prinsip ini (Gbr. 1). Aminofilin adalah garam etilendiamina dari bagian teofilin yang aktif secara farmakologis. Delapan puluh sampai delapan puluh lima persen (berat) dari garam ini adalah teofilin, sehingga "S" untuk aminofilin adalah sekitar 0,8. Aminofilin tablet tak bersalut dianggap sepenuhnya (100%) bioavailable; factor ketersediaan hayati (F) untuk bentuk sediaan ini, oleh karena itu,
1.0. Penting untuk mempertimbangkan bentuk garam dalam menentukan jumlah teofilin diserap dari tablet aminofilin. Ketika Persamaan 3 diterapkan untuk situasi ini, dapat menunjukkan bahwa 160 mg dari teofilin diserap dari aminofilin tablet 200 mg:
Jumlah obat yang diserap atau Yang Mencapai Sirkulasi Sistemik
= (S) (F) (dosis) = (0.8) (1) (200mg = 160 mg Theophylline
GAMBAR 1. Pengaruh obat kimia dari pada bioavailabilitas, Contoh diatas menekankan pentingnya mempertimbangkan bentuk kimia saat menghitung jumlah obat aktif benar-benar diberikan. Jumlah obat aktif dapat diberikan hanya mewakili sebagian kecil (S) dari garam, ester, atau bentuk kimia lainnya itu sendiri juga harus dipertimbangkan ketika obat yang diberikan oleh rute oral.
Demikian pula, dari 300 mg Natrium Fenitoin dengan S hanya mewakili 0,92 dari 276 mg fenitoin dalam sirkulasi sistemik, dengan asumsi penyerapan lengkap (F = 1).
Jumlah obat yang diserap atau Yang Mencapai Sirkulasi Sistemik
= (S) (F) (dosis)
= (0.92)(1)(300mg Phenytoin Sodium) = 276 mg Phenytoin
Dalam beberapa kasus jumlah obat yang berlabel sudah diperhitungkan jumlah obat aktif. Natrium valporat, garam natrium dari asam valproik, yang berlabel diproduksi dengan jumlah asam valproik dan oleh karena itu, nilai 1 akan sesuai untuk S. Natrium Fosphenytoin adalah garam natrium dari ester fosfat phynytoin. Mium fenitoin setara sama dengan P.E. Oleh karena itu, untuk menghitung jumlah fenitoin dalam 100 mg Fosphenytoin PE, nilai S yang akan digunakan adalah 0,92 .
Konsep penting adalah memahami dan dapat menghitung jumlah obat yang berlabel akan tersedia kepada pasien sebagai obat aktif. Untuk melakukan hal ini, kedua fraksi dari ltu
dosis obat aktif (S) dan ketersediaan hayati atau fraksi dari dosis yang akan mencapai sirkulasi sistemik (F) perlu dipertimbangkan ketika menghitung dosis dan dosis regimen.
FIRST PASS
EFFECT
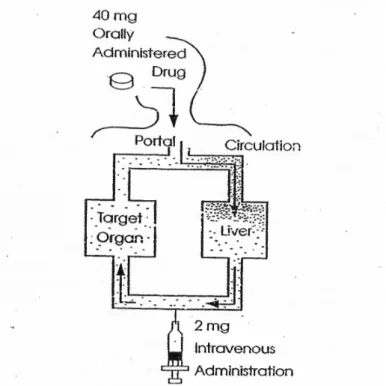
Karena obat-obatan diberikan secara oral diserap dari saluran pencernaan ke dalam sirkulasi portal, beberapa obat dapat dimetabolisme secara ekstensif oleh hati sebelum mencapai sirkulasi sistemik. Istilah "lulus pertama" mengacu pada metabolisme oleh hati sebagai obat melewati hati melalui vena portal berikut penyerapan. Efek pertama ini berlalu "" secara substansial dapat menurunkan jumlah obat aktif yang mencapai sirkulasi sistemik dan ini bioavailabilitas nya (gbr. 2).
Propanolol adalah contoh obat yang memiliki porsi signifikan dari dosis diberikan secara oral yang tidak mencapai sirkulasi sistemik ASN karena dimetabolisme saat melewati hati berikut penyerapan dari saluran pencernaan. Karena ini berlalu Efek pertama "", bioavailabilitas oral dosis rendah dan dikelola secara lisan jauh lebih besar daripada dosis diberikan secara intravena. Namun, isu propanolol lebih rumit oleh kenyataan bahwa salah satu metabolit, 4-hydroxy-propanolol, adalah obat aktif. 9 Lidocain adalah contoh dari obat
dengan efek yang lulus pertama yang begitu besar bahwa oral tidak praktis sebagai rute administrasi jika efek sistemik adalah dinding yang diinginkan. 10 Sebagai tambahan,
beberapa obat yang secara ekstensif dimetabolisme oleh enzim sitokrom, terutama CYP 3A4, yang terletak di dalam usus. Sebagai contoh, rendah dan variabel ketersediaan hayati (F ≈ 0,3) dari siklosporin adalah sebagian karena metabolisme oleh CYP 3A4 di dinding usus. 11
GAMBAR 2. Pertama lewat efek. Ketika obat dengan tinggi "efek pertama lulus" dikelola arally, sejumlah besar obat dimetabolisme diserap sebelum mencapai sirkulasi sistemik. Jika obat ini diberikan intravena, hati dilewati dan fraksi dari dosis yang mencapai sirkulasi meningkat. parenteral dosis obat dengan pertama "lulus tinggi" jauh lebih kecil daripada dosis oral diperlukan untuk menghasilkan efek farmakologis setara.
I.2 Administrasi Rate (R
A)
Kecapatan pemberian adalah nilai rata-rata di mana obat diabsorpsi sehingga mencapai sirkulasi sistemik. Hal ini biasanya dihitung dengan membagi jumlah obat yang diserap pada saat di mana obat itu diberikan (interval dosis) (lihat Persamaan 3). Interval dosis biasanya diwakili oleh simbol, tau (τ).
Administration rate RA = (S)(F)(Dose)
τ [Eq. 5]
Ketika obat-obatan yang diberikan sebagai infus kontinyu, interval dosis dapat dinyatakan dalam unit waktu yang tepat. Sebagai contoh, tingkat administrasi aminofilin teofilin akibat infus dengan kecepatan 40 mg/jam dihitung dari Persamaan 5 sebagai berikut:
Administration rate RA = (S)(F)(Dose)
τ Administration rate RA = (0,8)(1)(40mg)
1 hr = 32 mg/jam
Atau
Administration rate RA = (Y)(Z)([\]^)
τ
Administration rate RA = (K,Q)(_)(`K aP) 60 min
= 0.53 mg/jam
Ketika obat-obatan diberikan pada interval dosis tetap, tingkat administrasi yang diperhitungkan adalah nilai rata-rata. Misalnya, rata-rata tingkat administrasi dogoxin akibat dari dosis oral 250 µg dari digoksin diberikan secara oral dalam bentuk tablet setiap hari akan dihitung dengan menggunakan Persamaan 5 sebagai berikut:
Administration rate RA = (S)(F)(Dose) τ Administration rate RA = (1)(0.7)(250 μg) 1 day = 175 µg/day atau
Administration rate RA = (S)(F)(Dose)
τ
Administration rate RA = (1)(0,7)(250 μg)
24 hr = 7,29 µg/hr
Meskipun setiap tablet digoksin sebenarnya diserap selama 1 sampai 2 jam, tingkat "administrasi rate" dihitung atas seluruh interval pemberian dosis. Meskipun tingkat administrasi rate setara dengan 7,29 µg/jam dan 175 µg/hari, kebanyakan dokter paling memikirkan dosis tingkat yang konsisten dengan bagaimana obat diberikan. Dalam hal ini, interval biasa akan 1 hari karena digoksin ini paling sering diberikan satu kali setiap hari. Pada bagian pada bersihan, dan melaporkan unit biasanya untuk konsentrasi obat semua harus konsisten untuk kepentingan melakukan perhitungan farmakokinetik.
I.3 Konsentrasi Plasma yang Diinginkan (C)
IKATAN PROTEIN
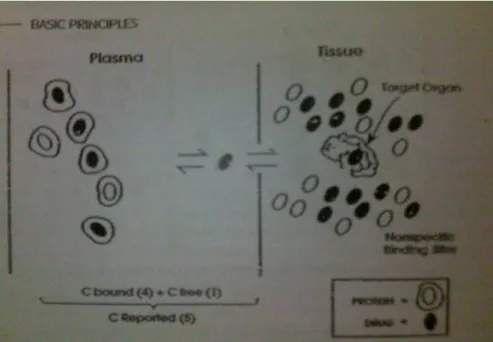
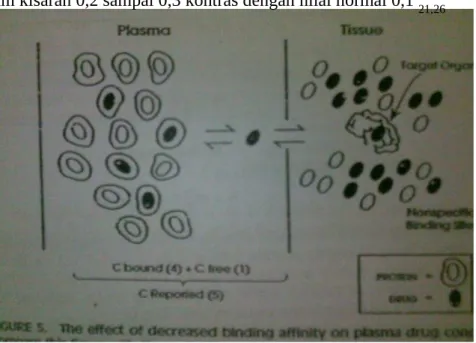
Banyak laboratorium klinik yang melaporkan konsentrasi obat dalam plasma mewakili obat yang terikat oleh protein plasma ditambah dengan obat yang tidak terikat atau bebas. Obat yang terikat bebas harus seimbang dengan reseptor satu sama lainnya, oleh karena itu, farmakologi yang aktif hanya setengahnya. Dengan demikian, dalam kasus obat dengan ikatan plasma yang signifikan, dilaporkan konsentrasi plasma obat secara tidak langsung mencerminkan konsentrasi obat bebas atau aktif. (Gambar 3)
Beberapa negara yang bermasalah dengan penyakit dihubungkan dengan penurunan protein plasma atau dengan penurunan ikatan obat untuk protein plasma. Pada situasi ini, obat-obatan yang biasanya sangat terikat protein memiliki persentase lebih besar dari obat bebas atau tidak terikat dalam plasma. Oleh karena itu, efek farmakologis yang lebih besar dapat diharapkan pada setiap pemberian konsentrasi obat di dalam plasma (C). Klinikal harus selalu mempertimbangkan perubahan ikatan protein dan apakah fraksi konsentrasi obat bebas atau fraksi terikat (fu) adalah perubahan ketika menginterpretasikan atau mendirikan konsentrasi plasma obat yang diinginkan.
fu =konsentrasi obat bebas .
konsentrasi obat total
fu = C bebas
C terikat + C bebas [Eq. 6]
Fraksi obat yang tidak terikat (fu) tidak bervariasi dengan konsentrasi obat untuk sebagian besar obat yang terikat utama pada albumin. ini adalah karena jumlah ikatan protein satu sama lainnya jauh melebihi jumlah molekul obat yang tersedia untuk diikat. ketika konsentrasi plasma obat lebih dari 25 albumin 50 mg/L, namun ikatan albumin satu sama lainnya dapat mulai menjadi jenuh. Hasilnya, fu, atau fraksi obat yang bebas, akan berubah dengan konsentrasi plasma obat. Sebagai contoh, salisilat dan asam valporat dapat menyerap ikatan protein plasma satu dengan yang lainnya, dan kedua obat ini biasanya memiliki konsentrasi plasma antara 25-50 mg/L. Untuk obat-obatan yang tidak mencapai konsentrasi serum dapat mengikat protein jenuh. Konsentrasi protein plasma (dalam banyak kasus, seperti albumin) dan ikatan afinitas obat untuk protein plasma adalah dua faktor utama yang mengontrol fraksi terikat obat (fu).
10 Gambar 3. Konsentrasi plasma obat-protein yang sangat terikat: konsentrasi protein plasma normal. Konsentrasi plasma obat yang dilaporkan oleh laboratorium merupakan total dari keduanya obat "terikat" dan "bebas". Itu adalah obat "bebas" yang berada dalam kesetimbangan dengan organ target dan terikat pada molekul obat aktif. dalam ilustrasi, fu (atau fraksi obat bebas untuk konsentrasi obat total) adalah 0,1.
KONSENTRASI PROTEIN PLASMA
RENDAH
Konsentrasi protein plasma rendah menurunkan konsentrasi plasma obat terikat (Cterikat),
namun konsentrasi obat bebas (Cbebas) umumnya tidak berefek. Oleh karena itu, fraksi obat
yang bebas (fu) meningkat sebagai penurunan konsentrasi protein plasma. Konsentrasi obat bebas atau tidak terikat meningkat secara signifikan, karena obat bebas yang dilepaskan ke dalam plasma sekunder sampai pada konsentrasi plasma protein rendah yang seimbang dengan kompartemen jaringan (bandingkan Gambar. 4 dengan Gambar. 3)
Oleh karena itu, jika volume disribution (V) relatif lebih besar (misalnya, fenitoin 0,65 L/kg), hanya terjadi peningkatan kecil pada C bebas (juga lihat Volume Distribusi).
Hubungan antara konsentrasi plasma obat dan konsentrasi protein plasma dapat dinyatakan sebagai berikut :
C′ Pg
= (1 − fu) f h + fu [Ed. 7]
C Ikatan Normal PNL
Persamaan ini dapat digunakan untuk memperkirakan perubahan kadar konsentrasi plasma protein yang akan mempengaruhi konsentrasi theraupetic obat yang diinginkan. C’ merupakan konsentrasi plasma obat pasien, dan P' merupakan konsentrasi plasma protein pasien. CIkatan Normal
adalah konsentrasi plasma obat yang akan diharapkan jika konsentrasi protein plasma pasien normal (PNL). Dari catatan fu yang merupakan fraksi bebas terkait dengan "ikatan protein plasma
=
Gambar 4. Pengaruh konsentrasi protein plasma menurun pada konsentrasi plasma obat. Bandingkan angka ini dengan Gambar 3. Penurunan konsentrasi protein menurunkan konsentrasi plasma obat yang dilaporkan oleh laboratorium. Dalam situasi ini, konsentrasi bebas, atau aktif, obat tetap sama karena obat bebas yang dilepaskan sebagai hasil penurunan konsentrasi protein plasma yang dilakukan oleh jaringan nonspesifik mengikat dan/atau dibersihkan dari tubuh. Sebagai alasan, efek farmakologis, yang dapat diharapkan dari C melaporkan dari 5, akan sama seperti yang dihasilkan oleh C melaporkan dari 10 pada Gambar 3. Dalam ilustrasi ini, fu (atau fraksi obat bebas untuk konsentrasi obat total) meningkat menjadi 0,2.
C Ikatan Normal = C′
(1 − fu) k PPg l + fu
NL
[Ed. 8]
Sebagai contoh, pasien dengan serum albumin rendah 2,2 gram/dL (normal albumin 4,4 mg/dL) dan konsentrasi plasma fenitoin tampaknya rendah dari 5,5 mg/L masih memiliki konsentrasi teraupetik plasma obat yang diterima bila disesuaikan untuk serum albumin rendah. Ketika fraksi bebas normal (fu) untuk fenitoin dari 0,1 disubstitusikan ke Persamaan 8, konsentrasi plasma fenitoin disesuaikan mencapai 10 mg/L dari hasil perhitungan.
CIkatan Normal = C′ (1 − fu) k PPg l + fu NL 5,5 mg/L (1 − 0,1) m2,2 gm/dL4,4 gm/dLn + 0,1 5,5 mg/L = (0,9)[0,5] + 0,1 = 10 mg/L
Konsentrasi fenitoin yang telah dilaporkan dari laboratorium adalah konsentrasi albumin pasien normal sekitar 10 mg/L. Ini dihitung berdasarkan pada asumsi bahwa fenitoin utama terikat pada albumin dan bahwa rata-rata konsentrasi albumin normal 4,4 mg/dL (kisaran 3,5 ke 5,5 gram/dL). Sedangkan persamaan 8 dapat digunakan untuk menyesuaikan secara signifikan setiap obat yang terikat pada albumin, sejauh mana konsentrasi obat akan disesuaikan atau "normal" untuk perubahan dalam serum albumin antara 3,5 sampai 5,5 gram/dL akan menjadi minimal dan umumnya tidak dibenarkan.
Banyak obat lain yang terikat utama pada globulin dibandingkan dengan albumin. Penyesuaian konsentrasi plasma obat untuk obat-obatan ini didasarkan pada konsentrasi serum albumin yang akan ditentukan, oleh karena itu, menjadi tidak tepat. Sayangnya, penyesuaian atas perubahan ikatan globulin sulit karena obat biasanya diikat oleh globulin spesifik yang hanya sebagian kecil dari konsentrasi globulin total. Secara umum, obat-obatan asam (misalnya, fenitoin, sebagian besar obat anti-epilepsi, dan beberapa senyawa netral) terikat utama pada albumin, obat-obatan dasar (misalnya, lidokain dan kinidin) terikat lebih luas pada globulin. 13,18-21
PENINGKATAN KONSENTRASI PROTEIN PLASMA
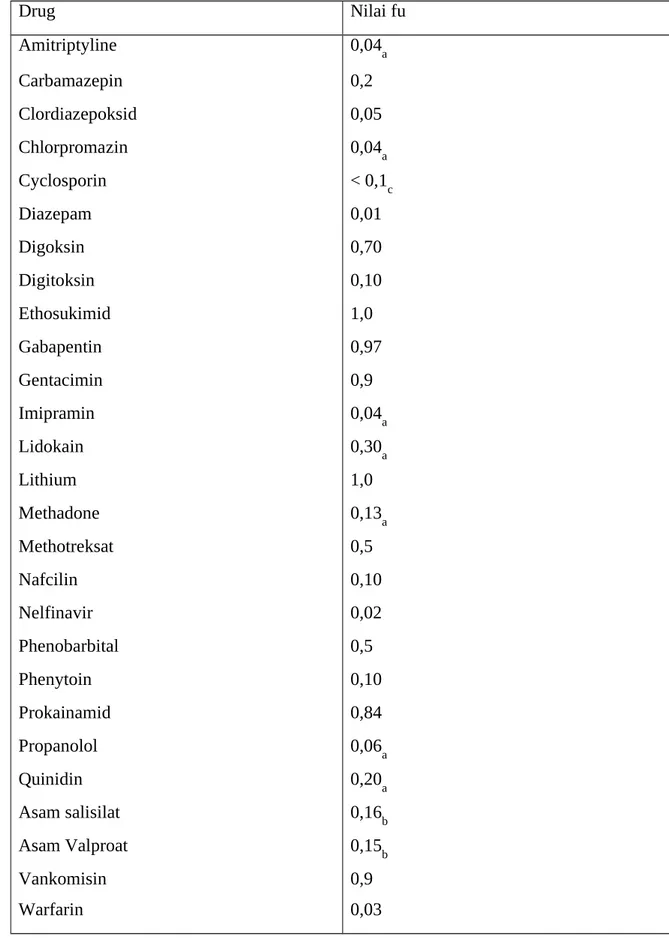
Nilai fu (fraksi konsentrasi total obat bebas atau tidak terikat) untuk obat yang dipilih tertera pada Table.1. Karena peningkatan serum albumin jarang diatur secara klinis, digunakan Persamaan 8 untuk serum albumin tinggi yang jarang terjadi. Banyak obat-obatan umum, yang bagaimanapun, terikat pada fase akut reactif protein, 22,23 alpha1-asam glikoprotein
(AAG). Protein plasma ini telah dikenal secara signifikan akan menurun dan meningkat dalam kondisi klinis tertentu. Sebagai contoh, peningkatan konsentrasi plasma kinidin telah diamati setelah operasi, atau trauma.18 24 Perubahan konsentrasi kinidin adalah hasil dari peningkatan
konsentrasi ikatan protein plasma (alpha1-asam glikoprotein) dan peningkatan konsentrasi
terikat kinidin. Tampaknya ada sedikit atau tidak ada perubahan di tingkat kinidin bebas karena kembali seimbang dengan jaringan perbekalan yang lebih besar. Dalam situasi ini, akan ada penurunan ikatan fraksi bebas, dan tingkat terapetik obat bebas atau tidak terikat harus berhubungan dengan konsentrasi obat lebih tinggi dari biasanya (terikat ditambah bebas). Lainnya senyawa dasar dengan signifikan mengikat alpha1-asam glikoprotein akan diharapkan akan sama terpengaruh. Sayangnya, konsentrasi alpha1-asam glikoprotein hampir
tidak pernah diuji dalam pengaturan klinis, sehingga sulit untuk mengevaluasi hubungan antara konsentrasi total obat dan fraksi tidak terikat atau bebas. Sebat itu, evaluasi tingkat plasma untuk obat dasar yang terikat protein secara signifikan seringkali sulit. Sebuah evaluasi yang teliti terhadap respon klinis pasien ke tingkat obat diukur, serta evaluasi dari setiap masalah medis secara bersamaan (seperti pembedahan, trauma, atau penyakit radang) yang dapat mempengaruhi konsentrasi protein plasma dan ikatan obat, diperlukan.
Pasien dengan sirosis sangat bervariasi dalam karakteristik ikatan protein plasmanya. Beberapa pasien memiliki kemampuan mengikat yang meningkat secara signifikan, sementara yang lain memiliki kemampuan mengikat yang menurun secara signifikan. Variasi ini mungkin mencerminkan kenyataan bahwa beberapa pasien sirosis memiliki stimulus yang
Tabel 1. Obat dan Nilai fu untuk Ikatan Plasma Protein Drug Nilai fu Amitriptyline Carbamazepin Clordiazepoksid Chlorpromazin Cyclosporin Diazepam Digoksin Digitoksin Ethosukimid Gabapentin Gentacimin Imipramin Lidokain Lithium Methadone Methotreksat Nafcilin Nelfinavir Phenobarbital Phenytoin Prokainamid Propanolol Quinidin Asam salisilat Asam Valproat Vankomisin Warfarin 0,04a 0,2 0,05 0,04a < 0,1c 0,01 0,70 0,10 1,0 0,97 0,9 0,04a 0,30a 1,0 0,13a 0,5 0,10 0,02 0,5 0,10 0,84 0,06a 0,20a 0,16b 0,15b 0,9 0,03
aObat dasar yang terikat pada protein plasma secara signifikan selain albumin. 12,18,19,33 bTergantung pada konsentrasi ikatan protein plasma (lihat bab salisilat dan asam valproik) cTerikat pada lipoprotein dan unsur darah lainnya. 34,35
= Cfre e C total C bebas = (fu)(C total)
kuat untuk memproduksi alpha1-asam glikoprotein, sedangkan penyakit hati lain yang lebih serius tidak mampu memproduksi ikatan protein ini. 22,24,25
IKATAN
AFINITAS
Ikatan afinitas protein plasma untuk obat juga dapat mengubah fraksi obat yang bebas (fu) (bandingkan Gbr.5 dengan Gbr.3). Misalnya, protein plasma pada pasien dengan uremia (akhir tingkat-keparahan gagal ginjal) kehilangan afinitas untuk fenitoin dibandingkan protein yang ada dalam individu nonuremic.Hhasilnya, fu untuk fenitoin pada pasien uremic diperkirakan dalam kisaran 0,2 sampai 0,3 kontras dengan nilai normal 0,1 21,26
Gambar 5. Penurunan efektifitas ikatan afinitas konsentrasi protein plasma. Bandinngkan dengan gambar.3. Meskipun konsentrasi protein normal, penurunan ikatan afinitas obat untuk protein dilaporkan terjadi penurunan konsentrasi obat. Konsentrasi bebas, atau aktif, obat tetap sama karena obat bebas yang dilepaskan sebagai hasil penurunan konsentrasi protein plasma yang dilakukan oleh jaringan nonspesifik mengikat dan/atau dibersihkan dari tubuh. Dengan demikian, efek farmakologi yang dapat diharapkan dari yang dilaporkan yaitu C dengan nilai 5 akan sama hasilnya dengan C yang dilaporkan yaitu 10 pada gambar.3. Pada ilustrasi ini, nilai fu (atau fraksi obat bebas dari konsentrasi obat total) menurun sampai nilai 0,2.
Efektivitas atau konsentrasi obat bebas dapat dihitung ulang dari persaamaan 6 : fu = C beb as
C terikat + C bebas
[Eq. 9]
Menurut Persamaan 9, konsentrasi fenitoin bebas pada pasien uremic adalah sebanding dengan yang pasien nonuremia, walaupun konsentrasi plasma fenitoin rendah
(C-total) karena fu untuk fenitoin meningkat pada pasien uremic. Pasien uremic dengan nilai fu 0,2 dan konsentrasi fenitoin sebesar 5 mg/L akan memiliki konsentrasi obat bebas yang sama (dan efek farmakologis yang sama) sebagai pasien dengan fungsi ginjal normal yang memiliki konsentrasi fenitoin sebesar 10 mg/L (menggunakan Persamaan 9):
C bebas = (fu) (C total)
C bebas = (0,2)(5 mg/L)
(pada pasien uremia) = 1 mg/L
C bebas = (0,1)(10 mg/L)
(pada pasien dengan
fungsi ginjal normal) = 1 mg/L
Kesimpulannya, beberapa faktor yang mengubah ikatan protein menjadi penting secara klinis ketika ikatan obat protein sedang tinggi (yaitu, jika fu adalah <0,1 atau terikat 10%).
C bebas = (fu) (C total) = (0,1)(10 mg/L) = 1 mg/L
Vs.
= (0,2)(10 mg/L) = 2 mg/L
Bila di sisi lain, nilai fu untuk obat adalah ≥ 0,5 (50% bebas), kemungkinan bahwa perubahan dalam ikatan protein plasma akan menjadi klinis yang tetap. Sebagai ilustrasi, jika fraksi terikat untuk obat adalah peningkatan dari nilai normal 0,5 (50% gratis) ke 0,6 (60% gratis) karena konsentrasi protein menurun, konsentrasi obat aktif bebas (asumsi yang sama dari konsentrasi total) benar-benar akan meningkat hanya 20%.C bebas = (fu) (C total)
= (0,5)(10 mg/L) = 5 mg/L
Vs.
= (0,6)(10 mg/L) = 6 mg/L
Umumnya, jika fraksi tidak terikat meningkat dalam situasi tertentu, klinisi harus mengurangi C yang diinginkan sampai proporsinya sama. 27Artinya, jika fu meningkat dua kali lipat,
C diinginkan atau "jangkauan terapeutik" harus dikurangi menjadi satu-setengah dari nilai yang lazim.
Apa yang sering disalahartikan adalah bahwa obat signifikan dengan ikatan protein plasma, perubahan dalam ikatan plasma akan memiliki efek besar pada konsentrasi plasma obat, karena konsentrasi terikat telah diubah. Akibatnya, fraksi bebas (fu) obat dalam plasma diubah. Namun, konsentrasi obat terikat adalah, dalam banyak kasus, relatif tidak terpengaruh. 28 Ketika
mempertimbangkan perubahan pada ikatan, harus diingat bahwa fraksi bebas (fu) adalah rasio konsentrasi obat terikat terhadap konsentrasi obat total seperti yang dijelaskan dalam Persamaan 6.
fu = C beb a s .
Cterikat+ Cbebas
Seperti digambarkan dalam Persamaan 6, fu tergantung pada karakteristik ikatan dan bukan disebabkan oleh konsentrasi obat bebas atau terikat yang dapat dilihat dalam Persamaan 9.
Cbebas = (fu)(Ctotal)
Sebagai contoh, kita mempertimbangkan empat pasien, dua pertama dengan konsentrasi fenitoin 10 dan 20 mg/L, masing-masing. Jika kedua pasien memiliki ikatan plasma normal (fu = 0,1), masing-masing C konsentrasi fenitoin bebas adalah 1 dan 2 mg/L. Pengaruh kenaikan potensi konsentrasi C fenitoin total 20 mg/L dengan C bebas dari 2 mg/L tampak jelas secara intuitif. Fakta bahwa konsentrasi obat (Cterikat dan Cbebas) kemungkinan lebih tinggi
pada pasien kedua baik dari hasil dosis lebih tinggi dari rata-rata atau penurunan eliminasi.
Sekarang kita mempertimbangkan dua pasien lain masing-masing dengan konsentrasi fenitoin 10 mg/L. Namun, dalam hal ini pasien pertama memiliki ikatan plasma normal dan fu sebesar 0,1. Pasien kedua telah menurun ikatan plasma dan hasil dari nilai fu sebesar 0,2. Dalam situasi ini pasien pertama dengan ikatan normal fu 0,1 dan C total 10 mg/L akan memiliki C bebas dari 1 mg/L. Pasien kedua dengan ikatan fu berubah dari 0,2 dan total C 10 mg/L akan memiliki Cbebas dari
2 mg/L. Adalah penting untuk menyadari bahwa meskipun kedua pasien memiliki konsentrasi fenitoin 10 mg/L, kedua pasien diharapkan memiliki efek obat meningkat karena Cbebas atau
konsentrasi obat tidak terikat tinggi. Alasan bahwa pasien kedua memiliki Cbebas meningkat bukan
karena ikatannya berubah, tapi mungkin karena pasien telah diberikan dosis lebih tinggi dari rata-rata atau metabolismenya kurang dari rata-rata- rata-rata.
PEMANTAUAN KONSENTRASI PLASMA BEBAS ATAU TIDAK TERIKAT
Meskipun banyak klinikal percaya bahwa pemantauan konsentrasi plasma bebas atau terikat yang diinginkan, tidak umum dalam praktek klinis umum. Alasan umumnya dan mencakup beberapa fakta bahwa uji prosedur untuk obat bebas atau terikat tidak tersedia komersial untuk banyak senyawa. Selanjutnya, prosedur assay tersedia untuk konsentrasi obat bebas yang lebih mahal dan meningkatkan biaya untuk penyediaan perawatan pasien. Juga, kebanyakan pasien cukup menunjukkan karakteristik ikatan normal, oleh karena itu; monitoring secara klinis konsentrasi obat terikat harus unggul, ada sedikit bukti yang menunjukkan bahwa pemantauan tingkat obat terikat meningkatkan hubungan antara konsentrasi plasma dan efek farmakologis atau hasil terapi.
Jika konsentrasi obat terikat harus digunakan dalam praktek klinis, klinisi harus menyadari factor-faktor yang dapat mengubah hubungan antara karakteristik ikatan plasma in vitro in vivo. Sebagai contoh, metode yang digunakan untuk menentukan tingkat obat bebas (dialisis ekuilibrium, ultrafiltrasi, sampling air liur, dll) dan kondisi dimana sampel yang diperoleh dapat mengubah hasil in vitro assay. Hal ini pada gilirannya dapat menghasilkan ketidakakuratan perkiraan pada karakteristik ikatan in vivo.15,29-32 Ini
sebabnya, penggunaan tingkat plasma tidak terikat atau bebas dipantau bukan berdasarkan standar praktek dan digunakan hanya dalam jumlah terbatas pada pengaturan klinis. Jika konsentrasi serum obat tidak terikat digunakan tidak beraturan, hasil harus dievaluasi secara cermat dan dibandingkan keduanya pada tingkat obat bebas dan respon klinis pasien.
I.4 Volume Distribusi (V)
Volume distribusi obat atau “volume distribusi nyata” tidak membutuhkan banyak kompartemen fisiologi di dalam tubuh. Kompartemen dengan bentuk sederhana diperlukan untuk menghitung jumlah total obat dalam tubuh apabila seluruh obat berada di dalam tubuh
pada konsentrasi yang sama
distribusi seperti di bawah ini : ditemukan di dalam plasma (Gambar 6A). Rumus volume
V = qr
s [Eq. 10]
Dimana V adalah volume distribusi nyata, Ab adalah jumlah total obat dalam tubuh, dan C adalah konsentrasi obat dalam plasma.
Gambar 6. Volume distribusi. (A) pemberian obat ke dalam tubuh menghasilkan konsentrasi plasma spesifik. Volume distribusi nyata (V) adalah volume yang terhitung dari jumlah dosis yang diberikan berdasarkan pada pengamatan konsentrasi plasma. (B) banyak faktor yang menurunkan konsentrasi obat dalam plasma (contoh : menurunnya ikatan protein plasma) akan meningkatkan volume distribusi nyata. (C) sebaliknya, banyak faktor yang meningkatkan konsentrasi plasma (contoh : menurunnya ikatan jaringan) akan menurunkan volume distribusi.
Volume plasma rata-rata dari orang dewasa diperkirakan 3 L. Oleh karena itu, volume distribusi nyata yang lebih besar daripada kompartemen plasma (> 3 L) hanya menunjukkan bahwa obat juga berada dalam jaringan atau cairan di luar kompartemen plasma. Tempat sebenarnya dari distribusi tidak dapat ditentukan dari nilai V. Contoh, suatu obat dengan
menunjukkan bahwa obat sama dan seimbang pada seluruh cairan tubuh. Obat mungkin ditemukan dan mungkin juga tidak ditemukan dalam jaringan tertentu. Bagaimanapun, hasil rata-rata ikatan dalam volume distribusi nyata diperkirakan sama dengan jumlah cairan tubuh. Tanpa tambahan informasi, tempat sebenarnya dari distribusi obat hanya spekulasi atau perkiraan saja.
Volume distribusi nyata merupakan suatu fungsi dari kelarutan air vs lemak dan ikatan protein obat dalam plasma dan jaringan. Faktor-faktor yang menjaga obat dalam plasma atau meningkatkan C (seperti kelarutan dalam lemak rendah, meningkatnya ikatan protein plasma, atau menurunnya ikatan jaringan) mengurangi volume distribusi nyata. Faktor-faktor yang menurunkan C (seperti menurunnya ikatan protein plasma, meningkatnya ikatan jaringan, dan meningkatnya kelarutan dalam lemak) meningkatkan volume distribusi nyata.
LOADING DOSE
Dikarenakan volume distribusi adalah faktor yang menghitung semua obat dalam tubuh, ini merupakan variabel yang penting dalam memperkirakan loading dose yang diperlukan untuk mempercepat pencapaian konsentrasi plasma, rumus loading dose seperti di bawah ini :
Loading dose = (t)(s) (Y)(Z) [Eq. 11]
Dimana, V adalah volume distribusi, C adalah tingkat plasma yang diinginkan, (S)(F) mewakili obat yang diberikan yang akan sampai ke sirkulasi sistemik (Sesuai gambar 7 di bawah ini):
Gambar 7. Loading dose. Volume distribusi merupakan penentu utama dari loading dose. Jika V suatu obat diketahui, loading dose akan menghasilkan konsentrasi tertentu yang dapat dihitung dengan mudah (lihat persamaan 11).
Sebagai contoh, jika seseorang ingin menghitung loading dose obat digoxin oral (tablet digoxin) untuk seorang laki-laki dengan berat badan 70 kg akan menghasilkan konsentrasi plasma 1,5 µg/L, dapat menggunakan persamaan 11. Jika diasumsikan S = 1,0; F = 0,7 dan V
20 Loading dose = (t)(s) (Y)(Z) (L,uv/wP)(LK wP)(_,N OP/v) = (_)(K,L) = 1095 µg atau 1,095 mg
Perkiraan yang rasional dari dosis ini akan diberikan tablet oral 1 mg. Pendekatan yang biasa digunakan secara klinik adalah dengan memberikan loading dose dalam dosis terbagi (0,25 - 0,5 mg tiap dosis setiap 6 jam). Pasien diamati dan dievaluasi respon terapinya dan toksisitas digoxin sebelum diberikan dosis berturut-turut. Sebagai tambahan, beberapa dokter menggunakan faktor bioavailabilitas > 0,7 (0,75 atau 0,8), akan mengurangi kemungkinan melampaui dari konsentrasi obat yang diinginkan.
Persamaan 11 dapat juga digunakan untuk memperkirakan loading dose yang akan diberikan untuk mencapai konsentrasi plasma lebih tinggi daripada konsentrasi yang ada (gambar 8). Rumus baru ini diturunkan dengan mengganti C dalam persamaan 11 dengan tanda yang mewakili peningkatan konsentrasi plasma yang diinginkan (persamaan 12).
Peningkatan loading dose = (t)(sxyz{|yx (Y)(Z)}s{~{{€ ) [Eq. 12]
Contoh, jika pasien sebelumnya mendapatkan digoxin 0,5 µg/L dan konsentrasi yang diinginkan adalah 1,5 µg/L, loading dose yang diberikan :
Peningkatan loading dose = (t)(sxyz{|yx }s{~{{€ )
(Y)(Z)
(L,u‚/wP)(LK wP)(_,N OP/v – K,N OP/v)
=
(_)(K,L)
= 730 µg atau 0,73 mg
Gambar 8. Loading dose untuk menghasilkan kenaikan dalam plasma. Jika V dan konsentrasi plasma awal diketahui, kenaikan loading dose akan menghasilkan konsentrasi plasma yang diinginkan lebih tinggi dan dapat dihitung.
FAKTOR YANG DAPAT MENGUBAH VOLUME DISTRIBUSI (V) DAN
LOADING DOSE
Dengan menganalisis Persamaan 11, ini menjadi jelas bahwa banyak faktor yang dapat mengubah volume distribusi yang secara teori juga dapat mempengaruhi loading dose.
Penurunan ikatan jaringan pada pasien uremik biasanya menyebabkan penurunan volume distribusi nyata pada beberapa agen (Gambar 6C). Penurunan ikatan jaringan akan meningkatkan C dengan menyediakan obat untuk tetap dalam plasma. Untuk itu jika nilai plasma yang diinginkan tetap tidak berubah, dibutuhkan loading dose yang lebih kecil. Contoh digoxin dimana loading dose sebaiknya diubah pada pasien uremik. Hal ini dibahas lebih lanjut pada Bagian 2 : Digoksin.
Penurunan ikatan protein plasma. Pada kasus lain perlu untuk meningkatkan volume distribusi nyata karena banyak obat yang tersedia dalam plasma seimbang dengan jaringan dan sisi yang berikatan dengan jaringan (Gambar 6B). Penurunan ikatan protein plasma, bagaimanapun juga meningkatkan fraksi obat bebas atau obat aktif sehingga C yang diinginkan dimana menghasilkan respon terapetik menurun. Penurunan ikatan protein plasma meningkatkan V dan menurunkan C di Persamaan 11, menghasilkan efek yang tidak menguntungkan pada loading dose.
loading dose = (↑t)(↓s) (Y)(Z)
Ini berdasarkan asumsi bahwa mayoritas obat dalam tubuh sebenarnya terdapat di bagian luar kompartemen plasma dan jumlah obat yang berikatan dengan ptotein plasma terdiri dari presentase kecil total obat dalam tubuh.
Prinsip ini digambarkan pada profil farmakokinetika fenitoin pada pasien uremik. Konsentrasi fenitoin dalam plasma pada pasien uremik 1½ daripada pasien normal dengan diberikan dosis yang sama. Tingkat plasma yang rendah, bagaimanapun, memproduksi konsentrasi fenitoin aktif 2 x lebih tinggi daripada pasien non uremik karena fraksi bebas (fu)
(
ditingkatkan dari 0,1 menjadi 0,2 pada individu tersebut, menunjukkan konsentrasi plasma target (terikat + bebas) pada pasien uremik sebaiknya ½ konsentrasi biasanya. Lebih jauh, loading dose fenitoin dimana menghasilkan efek terapetik normal untuk pasien uremik dan non uremik karena volume distribusi meningkat (kira-kira 0,65 L/kg – 14,4 L/kg) pada pasien uremik. Persamaan 11 menunjukkan bahwa tidak ada perubahan pada loading dose jika volume distribusi ditingkatkan oleh faktor 2 dan konsentrasi obat yang diinginkan diturunkan oleh faktor ½.
(M ‡ t) ˆ‡ s)
Loading dose = ‰
(Y)(Z)
MODEL 2 KOMPARTEMEN Parameter farmakokinetika
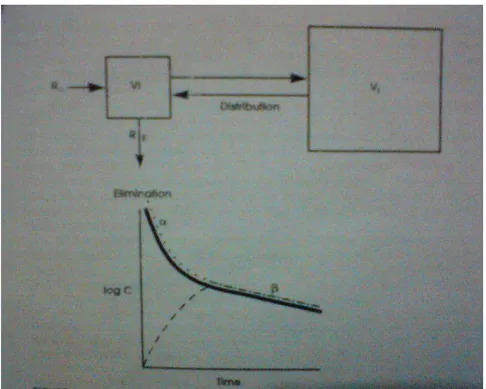
Jika membayangkan tubuh merupakan kompartemen tunggal, perhitungan farmakokinetika menjadi mudah. Bagaimanapun, terdapat beberapa situasi yang lebih cocok membayangkan tubuh meiliki 2 kompartemen dan kadang-kadang lebih dari 2 kompartemen ketika membahas tentang distribusi obat, eliminasi, dan efek farmakologi. Kompartemen pertama dibayangkan lebih kecil, volume kesetimbangan cepat, biasanya terdiri dari plasma atau darah dan organ atau jaringan yang memiliki kecepatan aliran darah tinggi dan kesetimbangan cepat dengan darah atau konsentrasi obat dalam plasma. Kompartemen pertama memiliki volume yang disebut Vi (initial volume of distribution). Kompartemen kedua menyeimbangkan obat dengan waktu lebih lama. Volume ini disebut Vt (tissue volume
of distribution). Waktu paruh untuk fase distribusi disebut alpha (α) half life dan waktu untuk eliminasi obat dari tubuh disebut beta (β) half life. Jumlah Vi dan Vt adalah volume distribusi nyata. Obat diasumsikan masuk ke dalam tubuh dan dieliminasi dari Vi. Banyak obat yang didistribusikan ke dalam kompartemen jaringan harus diseimbangkan ulang ke dalam Vi sebelum dieliminasi (Gambar 9).
Efek model 2 Kompartemen terhadap Loading dose dan konsentrasi plasma (C)
Karena terkadang dibutuhkan obat didistribusikan ke dalam Vt, penggunaan perhitungan loading dose yang cepat berdasarkan V (Vi + Vt) akan menghasilkan C awal yang lebih tinggi daripada yang diprediksi karena volume distribusi awal (Vi) selalu lebih kecil dari V. Konsekuensi dari prediksi yang kurang tepat tergantung profil organ target seolah-olah berlokasi di Vi atau Vt.
Obat seperti lidokain, fenobarbital, prokainamida, dan teofilin menggunakan efek terapetik dan toksik pada pada organ target yang berkelakuan seolah-olah berlokasi di Vi. Dalam hal ini, ketika loading dose dihitung berdasarkan total volume distribusi, konsentrasi obat yang dihantarkan ke organ target harus lebih tinggi daripada yang diinginkan dan menghasilkan toksisitas jika loading dose tidak digunakan dengan tepat. Masalah ini dapat dicegah dengan menghitung terlebih dahulu loading dose berdasarkan total volume distribusi
(V) kemudian penggunaan loading dose pada kecepatan rendah untuk mengijinkan obat terdistribusi ke dalam Vt. Pendekatan ini umum dalam praktek klinis, dan pedoman untuk tingkatadministrasi obat seringkali didasarkan pada prinsipnya pemodelan dua- compatrtement dengan reseptor untuk respon clinicals (beracun atau terapeutik) menanggapi seolah-olah mereka ware terletak di Vi. Pendekatan kedua adalah addminister dosis pembebanan pada dosis bolus cukup kecil perorangan tersebut bahwa C di Vi tidak melebihi beberapa yang telah ditentukan concentration kritis. 41, 42
Gambar 9. Model 2 kompartemen. Volume distribusi untuk model 2 kompartemen. Vi adalah volume distribusi awal. Pemberian obat (RA) dan eliminasi (RE) diasumsikan terjadi di Vi. Pada grafik bawah menunjukkan bahwa pemberian obat yang cepat ke dalam Vi, konsentrasi plasma ( ) mengikuti pola penurunan bifase. Waktu paruh penurunan awal (t½) biasanya dikarenakan distribusi obat ke dalam Vt. Waktu paruh penurunan kedua (t½) biasanya dikarenakan eliminasi obat dari tubuh. Garis titik-titik ( ... ) menunjukkan efek obat ketika organ terakhir yang berefek berada dalam Vi. Dengan catatan bahwa efek obat setara dengan konsentrasi obat dalam plasma sepanjang waktu. Garis putus-putus ( --- ) menunjukkan efek obat ketika organ terakhir yang berefek berada dalam Vt. Dengan catatan ketika semua obat berada dalam Vi, disana tidak ada efek obat. Bagaimanapun, ketika terjadi distribusi, efek obat meningkat dan mulai setara dengan konsentrasi plasma hanya pada fase eliminasi setelah fase distribusi selesai.
Walaupun jarang dibicarakan dalam farmakokinetika, potasium merupakan contoh obat yang baik, yang mengikuti prinsip model 2 kompartemen dengan organ terakhir berada dalam Vi. Potasium merupakan elektrolit intrasel utama tetapi efek terhadap jantung setara dengan konsentrasi plasma. Dengan tambahan, terjadi keseimbangan secara perlahan konsentrasi potasium antara plasma dan jaringan. Ketika potasium diberikan intravena, kecepatan administrasi harus hati-hati karena jika pasien kelebihan konsentrasi plasma (Vi) dapat terjadi toksisitas pada jantung yang serius dan kematian.
Konsep model 2 kompartemen juga penting untuk mengevaluasi efek lain dari obat. Untuk obat dengan organ terakhiryang memberikan respon klinik berada dalamVi, penerimaan yang cepat respon terapetik diikuti dengan cepat hilangnya respon terapetik memungkinkan hasil dari obat didistribusikan ke dalam volume distribusi yang lebih besar daripada obat yang dieliminasi dari tubuh (lihat Bagian Dua : Lidokain).
Ketika organ target adalah kompartemen kedua atau jaringan, Vt (contoh digoxin, litium), C cukup tinggi, dimana mungkin diamati sebelum distribusi terjadi, ini tidak berbahaya. Bagaimanapun, konsentrasi plasma yang ditetapkan sebelum distribusi selesai tidak dapat merefleksikan konsentrasi jaringan saat keseimbangan. Untuk itu, sampel plasma tidak dapat digunakan untuk memprediksi kemampuan terapetik atau toksik dari obat. Contoh ahli klinik selalu menunggu 1-3 jam setelah pemberian digoksin secara bolus intravena sebelummengevaluasi efeknya. Penundaan mengijinkan digoksin untuk terdistribusi ke sisi aksinya (miokardium) sehingga efek terapetik atau toksiknya dapat diamati (lihat Bagian Dua : Digoksin dan Gambar 4.1).
Distribusi obat yang lambat ke dalam kompartemen jaringan dapat mengemukakan masalah pada interpretasi konsentrasi obat yang tidak akurat ketika obat diberikan secara intravena. Masalah yang tidak umum ketika obat diberikan per oral, karena laju absorpsi selalu lebih lambat daripada laju distribusi dari Vi ke Vt. Sekalipun demikian, digoksin dan litium pengecualian pada kasus ini. Walaupun obat ini diberikan per oral, dibutuhkan beberapa jam untuk menyelesaikan absorpsi dan distribusi.
Sampel/contoh plasma yang diperoleh kurang dari 6 jam setelah pemberian digoksin per oral atau kurang dari 12 jam setelah pemberian litium per oral hasilnya dipertanyakn. Untuk 2 obat tersebut, reseptor pada organ terakhir berperilaku seolah-olah mereka berada pada kompartemen jaringan dengan keseimbangan yang lambat. Konsentrasi plasma ditetapkan selama fase distribusi (sebelum keseimbangan dengan kompartemen jaringan selesai) akan meningkat, dan respon farmakologi akan jauh lebih kurang daripada konsentrasi plasma yang diperkirakan.
Obat dengan model 2 kompartemen signifikan dan tidak signifikan
Seperti pada Gambar 9, fase alfa untuk sebagian besar obat menunjukkan distribusi obat dari Vi ke Vt, dan sebagian kecil obat dieliminasi selama fase distribusi. Obat dengan karakteristik seperti itu disebut model 2 kompartemen “non signifikan”. Non signifikan berarti jika pasien tidak dirugikan oleh elevasi awal konsentrasi obat pada fase alfa dan tidak ada sampel obat yang diambil dari fase alfa, obat dianggap sebagai obat kompartemen satu (hanya eliminasi atau fase beta yang dipertimbangkan). Ini penting untuk mengenali obat, peningkatan konsentrasi obat dalam plasma selama fase alfa secara klinik signifikan karena pasien dapat mengalami toksisitas yang serius jika organ terakhir memiliki karakteristik seolah-olah ini terdapat pada volume distribusi awal. Obat ini menunjukkan non signifikan terhadap dua model kompartemen setelah fase alfa atau setelah distribusi selesai. Dari sampel plasma didapatkan model farmakokinetik selama fase eliminasi atau beta.
Obat dengan signifikan terhadap model dua kompartemen yang dieliminasi menjadi tingkat penting selama awal fase alfa. Obat ini (metotrexate), fase alfa tidak dapat dipikirkan secara sederhana sebagai distribusi, karena eliminasi terjadi dengan baik. Dua obat yang berdekatan yang signifikan dengan model dua kompartemen adalah lithium dan lidokain. Ketika model satu kompartemen digunakan untuk obat-obat yang menunjukkan eliminasi obat dalam fase alfa, konsentrasi sebenarnya akan lebih rendah daripada yang diprediksikan dengan model satu kompartemen.
Beberapa dokter sudah menyarankan bahwa obat ini dapat lebih mudah dimonitor dengan menggunakan model dua kompartemen farmakokinetik. Kesulitan dari model ini, sejumlah sampel plasma pada pasien khusus membutuhkan penyesuaian dosis, biasanya terbatas pada penggunaan teknik model dua kompartemen.
Model komputer dua kompartemen tersedia untuk memonitor obat. Biasanya, nilai dari model komputer dua kompartemen adalah mereka bisa mengganti atau menyesuaikan agar sampel obat yang sudah dihasilkan dalam fase distribusi. Jika perawatan di ambil untuk menghindari perolehan sampel dari fase distribusi, interpretasi farmakokinetik hampir sama biaasanya dengan menggunakan model satu kompartemen yang lebih sederhana.
I.5 Clearance (Cl)
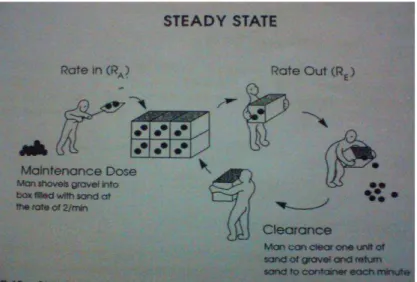
Clearance dapat diartikan sebagai kemampuan intrinsik dari tubuh atau organ-organ eliminasi (biasanya ginjal dan hati) untuk menghilangkan obat dari darah atau plasma. Clearance dinyatakan sebagai volume per waktu. Ini penting untuk memperhatikan bahwa clearance bukan suatu indikator dari berapa banyak obat telah dibuang, itu hanya mewakili secara teori volume darah atau plasma yang sudah di bersihkan dari obat Selama periode pemberian obat. Jumlah dari obat yang terbuang tergantung pada konsentrasi plasma obat dan clearance. Seperti pada gambar 10.
Gambar 10. Keadaan tunak, dosis pemeliharaan, clearance, konstanta laju eliminasi. Pada keadaan tunak, kecepatan pemberian obat (RA) sama dengan kecepatan eliminasi obat (RE), dan konsentrasi sisa obat adalah tetap.
Pada contoh ini, laki-laki yang di kiri bisa menyekop batu atau “obat” ke dalam wadah pasir dengan kecepatan rata-rata 2/menit. Laki-laki di sebelah kanan bisa menghilangkan satu unit pasir yang mengandung batu atau ‘obat” dari wadah, mengeluarkan batu, dan mengembalikan pasir ke dalam wadah tipa menit. Jumlah batu atau “obat” yang dibuang per waktu (laju eliminasi) akan ditentukan oleh konsentrasi batu per unit pasir sebanding dengan clearance (volume pasir yang dibersihkan dari batu). Konstanta laju eliminasi (K) dapat diartikan sebagai fraksi dari volume total yang dibersihkan per waktu. Pada kasus ini, K sama dengan 1/6 atau
0,17/menit-1.
Pada keadaan tunak, kecepatan pemberian obat (RA) dan kecepatan eliminasi obat
(RE) harus sama. Rumus:
RA = RE [Eq. 13]
Clearance (CL) bisa menjadi pikiran baik sebagai perbandingan konstan. Itu membuat rata-rata tetap-negara level plasma obat sama untuk tingkat administrasi obat (RA).
dimana RA adalah (S)(D)(Dose)/τ (lihat persamaan 5), dan Css ave adalah nilai rata-rata
steady-state konsentrasi obat.
Jika sebuah nilai rata-rata konsentrasi steady-state plasma dan tingkatan dari pemberian obat diketahui, clearance bias dihitung dengan pengulangan kembali persamaan 14 :
CL = ( S ) ( FCss ave ) ( Dos e / T ) [Eq. 15]
Misalnya, jika lidocaine kedalam pembuluh darah diinfuskan secara berlanjut pada tingkat 2mg/min dan jika konsentrasi lidocaine pada tetap-negara adalah 3mg/L, nilai kebersihan lidocaine dihitung menggunakan persamaan 15 akan menjadi 0667 L/min :
CL = ( S ) ( Fsave ) ( Dos e / T ) Cs = ( 1) ( 1) ( 2mg/min )
3 mg/L = 0,667 L/min
atau nilai kebersihan 40 L/hr jika tingkat administrasi dari lidocaine ditulis sebagi mg/hr. CL = ( S ) ( Fsave ) ( Dos e / T ) Cs
= ( 1) ( 1) ( 120mg/h r) 3 mg/L
= 40L/hr
F dipertimbangkan menjadi 1,0 karena obat itu telah diadminstrasi di dalam pembuluh darah. S juga bias diasumsikan menjadi 1,0 karena garam hidroklorida menampilkan hanya sebagian kecil dari berat total molekul untuk lidocaine dan perbaikan untuk bentuk garam adalah tidak perlu.
PEMELIHARAAN DOSIS
Jika perkiraan clearance diperoleh dari literatur, rumus clarance (Persamaan 15) dapat diatur kembali sedikit dan digunakan untuk menghitung tingkat atau administrasi atau dosis pemeliharaan yang akan menghasilkan konsentrasi plasma yang diinginkan rata-rata pada steady state:
Dosis Pemeliharaan = ( C L ) ( Cs sa(S)(F) v e) / T ) [Eq.16]
Misalnya, dengan menggunakan perkiraan literatur untuk clearance theophyllne dari 2,8 L/Jam, tingkat pemberian intravena untuk teofilin yang akan menghasilkan plasma steady-state teofilin konsentrasi 10 mg/L digambarkan di bawah ini:
Dosis pemeliharan = ( C L ) ( Cs sa(F) v e) / τ ) (S)
= ( 2 , 8 L / h r ) ( 10(1) m g / L ) ( 1 h r ) (1) = 28 mg diberikan setiap jam
Karena τ adalah 1 jam, tingkat administrasi adalah 28 mg/jam. jika teofilin itu diberikan setiap 12 jam, dosis akan 336 mg atau 12 kali tingkat administrasi per jam untuk menjaga konsentrasi rata-rata sama kondisi mapan.
Dosis pemeliharaan = ( C L ) ( Css a v e)(F) / τ ) (S)
= ( 2 , 8 L / h r ) ( 10(1) m g / L ) ( 12r ) (1) = 336 mg diberikan setiap 12 jam
Satuan untuk volume dan waktu pada clearance agak sewenang-wenang, tetapi harus konsisten dengan dua unit untuk tingkat pemberian obat dan konsentrasi obat.
Laju Administrasi Massa/waktu Konsentrasi obat Massa/volume
[Untuk kalangan sendiri]
Gun, Denny & Nuzly
–
Sebagai contoh, jika tingkat pemberian obat adalah dalam mg/jam dan konsentrasi dalam mg/L, maka izin harus dalam L/jam. Sebaliknya, jika tingkat administrasi adalah mg/hari suatu konsentrasi dalam mg/L, maka izin harus dalam L/hari. Sekali lagi, unit ini agak sewenang-wenang, tetapi dokter biasanya menggunakan nilai yang konsisten dengan bagaimana obat digunakan dalam praktek klinis. Dalam beberapa kasus konversi perlu dibuat. Methotrexateisbiasanya diberikan sebagai gram atau miligram, tetapi konsentrasi metotreksat dilaporkan dalam satuan micromolar atau micromoles/L. Perawatan harus diambil untuk memastikan unit yang tepat digunakan (lihat Bagian dua: Methotrexate).
FAKTOR YANG MENGUBAH CLEARANE (Cl) Luas Permukaan Tubuh (BSA)
Kebanyakan nilai-nilai literatur untuk clearence dinyatakan sebagai volume/kg/waktu atau sebagai volume/70kg/waktu. Ada beberapa bukti-bukti, bagaimanapun, clearane obat terbaik jika disesuaikan atas dasar luas permukaan tubuh daripada berdasarkan pada berat badan. Luas permukaan tubuh dapat dihitung menggunakan persamaan 17 atau dapat diperoleh dari berbagai nomogram dan tabel.
BSA dalam m2 = Žr^‘ r’“ ”]•^“ ’‚a wP K.L(1,73 m2) [Eq. 17] LK wP
Nilai dari suatu berat badan pasien yang dibagi 70 dipangkatkan 0,7 adalah untuk mengukur pasien sebagai rata-rata 1,73 m2 atau 70 kg individu. Berat badan dibagi 70 dipangkatkan 0.7 tidak
punya unit dan harus diingat sebagai pecahan menyangkut ukuran rata-rata seseorang.
Sebagai contoh, pasien dengan berat badan 7 kg mempunyai suatu perbandingan-bobot 70 kg dalam 0,1 oleh karena itu, mempunyai ukuran kapasitas ginjal dan metabolisme sebesar sepersepuluh dari rata-rata orang dengan bobot 70 kg.
( LK wPL wP ) = 0.1
Tabel 2. Faktor Yang Mengubah Clearane ( Cl) Berat badan
Luas permukaan tubuh Keluaran jantung Interaksi obat
Perbandingan ekstraksi Genetika
Fungsi hati
Ikatan Protein plasma
30
— —
)
Jika berat individu sama dengan standar 70 kg yang menggunakan berat 0,7, perbandingan menjadi 0,2 atau 20% kapasitas clearane dalam standar 70 kg atau 1,73 m2 individu.
7 kg (
70 kg)
K.L = 0.2
Dalam contoh di atas, perbedaan antara 0,1 dan 0,2 adalah besar. Bagaimanapun, ketika pasien tidak berbeda secara signifikan dari 70 kg, Perbedaan antara menggunakan berat badan dengan luas permukaan tubuh menjadi kurang penting.
Penting juga untuk mengingat bahwa 0,2 tidak punya unit dan menggunakan ukuran rata-rata (1,73 m2 atau 70 kg) individu. Adakalanya, nilai 0,2 merupakan kesalahan untuk ukuran atau luas permukaan tubuh pasien dalam m2. Ini tidak benar dan dapat memicu kesalahan dalam
pemberian dosis.
Rumusan berikut dapat digunakan untuk melakukan penyesuaian nilai-nilai clearane yang dilaporkan dalam literatur untuk pasien yang spesifik. Ada beberapa persamaan yang salah satunya dapat digunakan tergantung pada unit yang yang digunakan dalam literatur untuk clearane.
Patent’s Cl = (literature Cl per m2)(patient’s BSA) [Eq. 18]
Patient’s Cl = (literature Cl per 70 kg)( ”‘•^“‘ ] ˜Yq_.Lu a‰ ) [Eq. 19] Patient’s Cl = (literature Cl per 70 kg)( ”‘•^“‘ ] ™^•Pš‘ •“ wPLK wP [Eq. 20]
Patient’s Cl = (literature Cl per kg)(patientgs weight in kg) [Eq. 21]
Persamaan 20 dan 21 melakukan penyesuaian clearane dibandingkan dengan berat badan sedangkan penyamaan 18 dan 19 melakukan penyesuaian clearane dibandingkan dengan luas permukaan tubuh.
Asumsi dasar dalam penggunaan berat badan atau luas permukaan tubuh untuk melakukan penyesuaian clearane adalah bahwa ukuran ginjal dan hati pasien sangat bergantung pada pengukuran fisik ini. Tidak selalu terjadi kasus; oleh karena itu, nilai clearane diperoleh dari populasi pasien yang mempunyai suatu ukuran dan umur serupa harus digunakan pada kemungkinan kapanpun. Jika berat pasien mendekati 70 kg ( BSA=
1.73 m2), clearane pasien dihitung akan serupa apakah berat badan atau luas permukaan tubuh yang digunakan untuk perhitungan clearane. Jika, berat badan pasien berbeda signifikan dari 70 kg, penggunaan luas permukaan tubuh atau berat badan dapat dilakukan
untuk mengetahui perbedaan yang substansial pada clearane pasien itu. Ketika ukuran pasien lebih besar atau kurang dari standar 70 kg, atau 1,73 m2, maka seharusnya dilakukan penaksiran yang teliti
untuk menjelaskan jika tinggi badan pasien normal, gemuk, atau kurus. Pada pasien gemuk dan kurus, baik berat badan maupun luas permukaan kemungkinan besar membantu dalam memprediksi clearance, karena ukuran badan pasien tidak merefleksikan ukuran atau fungsi hati dan ginjal.
Ikatan Protein Plasma
Untuk obat yang terikat kuat dengan protein, pengurangan ikatan protein plasma berhubungan dengan pengurangan pada konsentrasi steady state obat pada plasma yang dilaporkan (total dari obat yang tidak terikat dan obat bebas) untuk beberapa dosis yang diadministrasikan [lihat Gambar 5 dan Konsentrasi Plasma yang Diinginkan (C)]. Berdasarkan persamaan 15, penurunan denominator, Css ave, meningkatkan calculated clearance yang dihitung.
Cl = (S)(F)(Dose⁄τ) Css ave
Akan tetapi, nilai mungkin berbeda, hal ini diasumsikan karena calculated clearance meningkat, jumlah yang dieliminasi per satuan waktu meningkat juga. Persamaan 15 mengasumsikan bahwa ketika Css ave (total obat yang berikatan dan obat bebas) berubah, konsentrasi obat bebas yang tersedia untuk metabolisme dan eliminasi renal berubah secara seimbang. Kenyataannya, fraksi bebas atau yang tidak terikat pada plasma pada umumnya meningkat (walaupun Css ave menurun) dengan pengurangan ikatan protein plasma. Sebagai hasilnya, sejumlah obat bebas dieliminasi per satuan waktu harusnya tidak berubah. Hal ini dapat terjadi jika dipertimbangkan pada steady state, jumlah obat yang diadministrasikan per satuan waktu (RA) harus sama dengan jumlah yang
dieliminasi per satuan waktu (RE). Jika RA tidak berubah, RE harusnya sama.
Ringkasnya, ketika dosis harian yang sama dari suatu obat diberikan, dengan adanya pengurangan ikatan protein, jumlah yang sama dari dosis akan dieliminasi dari tubuh setiap hari pada steady state walaupun ada pengurangan konsentrasi plasma steady state dan peningkatan pada calculated clearance. Konsentrasi plasma yang lebih rendah (C bound + C free) berhubungan dengan penurunan C bound, tidak berubah pada C free, dan hasilnya ada peningkatan pada fraksi obat yang tidak terikat (fu).
↑ fu = s œ^^
↓s r\“’ ž s œ^^ [Eq. 22]
Oleh karena itu, efek farmakologis yang diperoleh akan sama dengan yang diproduksi pada konsentrasi serum yang lebih besar yang diamati di bawah kondisi ikatan protein
normal. Contoh ini menekankan kembali prinsip bahwa clearance sendiri bukan indikator yang baik jika jumlah obat yang dieliminasi per satuan waktu (Gambar 11 dan 12).
Gambar 11. Clearance (Cl) pada obat dengan ikatan protein kuat dan rasio ektraksi rendah. Obat bebas atau tidak terikat dapat dilakukan clearance. Obat dengan ikatan protein dapat kembali sehingga volume yang dibersihkan adalah ¼ dari total volume yang dibersihkan oleh organ pembersih (seperti hati dan ginjal).
Gambar 12. Efek penurunan ikatan protein pada clearance (Cl) dari obat dengan ikatan protein tinggi dan rasio ekstraksi rendah. Bandingkan dengan Gambar 11. Konsentrasi obat dalam plasma menurun, tetapi konsentrasi obat yang bebas tetap sama (fu meningkat) (lihat Gambar 4). Volume yang dibersihkan meningkat (1/2) dibandingkan Gambar 11, walaupun konsentrasi obat yang tidak terikat dan jumlah obat yang dibersihkan per satuan waktu tetap tidak berubah. Ini menggambarkan prinsip bahwa jumlah obat terikat protein kuat yang dibersihkan per satuan waktu atau Rate of Elimination (RE) tetap sama jika peningkatan clearance bertujuan untuk menurunkan ikatan protein plasma
dan metabolisme intrinsik atau eliminasi oleh ginjal tetap tidak berubah.
Prinsip ini dijelaskan dengan membandingkan fenitoin pada pasien uremik dan non- uremik pada kondisi steady state. Seperti yang telah dibahas sebelumnya pada konsentrasi plasma yang diinginkan, konsentrasi steady state fenitoin yang tidak terikat pada plasma (C free) akan sama dengan individu uremik dan non-uremik yang menerima dosis yang sama dan memiliki kemampuan metabolisme yang sama. Akan tetapi, karena penurunan ikatan
protein, konsentrasi yang terikat (Cbound) dan Ctotal akan lebih rendah pada pasien uremik
dibandingkan pasien non-uremik.Sebagai contoh, dua pasien dengan kemampuan metabolisme yang sama menerima 300 mg/hari. Pasien pertama yang non-uremik dengan konsentrasi fenitoin 10 mg/L dan ikatan plasma normal (fu = 0,1). Pasien kedua yang uremik dengan konsentrasi fenitoin 5 mg/L dan penurunan ikatan plasma (fu = 0,2). Jika kedua pasien dihitung clearance-nya dengan Persamaan 15, maka akan diketahui pasien uremik memiliki clearance yang lebih besar. Cl = (Y)(Z)([\]^⁄ ) s]] ¡^ Non-Uremik Cl = (Y)(Z)([\]^⁄ )s]] ¡^
=
(_)(_)(uKKaP⁄š•) _K aP⁄v = 30 L⁄hari Uremik Cl = (S)(F)(Dose⁄τ) Css ave = (_)(_)(uKKaP⁄š•)N aP⁄v = 60 L⁄hariWalaupun calculated clearance untuk pasien uremik lebih tinggi dibandingkan dengan pasien non-uremik (60 L/hari vs 30 L/hari), jumlah obat yang dibuang per hari (300 mg) adalah sama, karena pada steady state laju pemberian obat (RA) sama dengan laju eliminasi obat (RE) baik pada
pasien uremik maupun non-uremik.
Rq = R¢
300 mg⁄hari = 300 mg⁄hari
Ketika ikatan protein menurun, peningkatan pada calculated clearance umumnya seimbang dengan perubahan pada fu. Walaupun calculated clearance mungkin bisa digunakan untuk memperkirakan maintenance dose, seleksi level plasma yang teliti akan menghasilkan
level obat yang tidak terikat atau bebas plasma yang diinginkan dan efek farmakologis kritikal menjelaskan maintenance dose yang tepat secara terapeutis.
Rasio Ekstraksi
Perbandingan langsung antara calculated clearance dan fraksi yang tidak terikat (fu) tidak diterapkan pada obat yang secara efisien termetabolisme atau beberapa terekskresi (kemungkinan semua) dari obat yang terikat pada protein plasma dibuang ketika melalui organ pengeliminasi. Pada saat protein plasma berperan sebagai suatu “sistem transportasi” suatu obat, membawanya ke organ pengeliminasi, dan clearance jadi tergantung pada aliran darah atau plasma ke organ pengeliminasi. Untuk menjelaskan apakah clearance obat dengan ikatan plasma signifikan akan dipengaruhi terutama oleh aliran darah atau ikatan protein plasma, rasio ekstraksinya diperkirakan dan dibandingkan dengan nilai fu-nya.
Rasio ekstraksi adalah fraksi obat di organ pengeliminasi yang dibuang setelah melewati organ tersebut. Rasio ini dapat diperkirakan dengan membagi clearance darah atau plasma suatu obat dengan fraksi bebas yang dikeluarkan (fu), lalu protein plasma berperan sebagai sistem transportasi dan clearance tidak akan berubah dengan proporsi fu. Begitupun jika rasio ekstraksi lebih rendah dibandingkan fu, clearance kemungkinan besar akan meningkat dengan proporsi yang sama dengan perubahan fu. Pendekatan ini tidak digunakan dengan faktor lainnya yang dapat mempengaruhi clearance seperti ikatan sel darah merah, eliminasi sel darah merah, atau perubahan fungsi metabolism.
Fungsi Renal dan Hepatik
Obat dapat dieliminasi atau dibuang sebagai obat yang tidak berubah setelah melewati ginjal (renal clearance) dan oleh metabolism pada hati (metabolic clearance). Kedua rute clearance diasumsikan independen satu sama lain dan aditif.
Cl‘ = Cla + Cl [Eq. 23]
Dimana Clt adalah total clearance, Clm adalah metabolic clearance atau fraksi yang terbuang oleh
metabolisme, dan Clr adalah renal clearance atau fraksi yang dibuang lewat rute renal. Karena ginjal
dan hati memiliki fungsi yang independen, diasumsikan bahwa perubahan pada salah satu tidak mempengaruhi yang lainnya. Maka, Clt dapat diperkirakan ada adanya kegagalan ginjal atau
hati atau keduanya. Karena fungsi metabolism susah dihitung, Clt biasanya disesuaikan ketika
ada penurunan fungsi renal:
Cl Adjusted = (Cla ) + m(Cl) £
Fraction of Normal Renal
µ
µ
Clearance dapat digunakan untuk mengatur fungsi ginjal dan memperkirakan dosis pemeliharaan untuk pasien dengan penurunan fungsi ginjal. Bagaimanapun juga, persamaan pengaturan clearance hanya dapat valid jika metabolit obat dalam keadaan tidak aktif dan metabolic clearance tidak berefek pada disfungsi ginjal. Penurunan fungsi dari organ eliminasi terjadi secara signifikan ketika organ tersebut melakukan rute eliminasi primer obat. Bagaimanapun juga, ketika jalur eliminasi mayor meningkat, jalur minor menjadi lebih signifikan karena ini mengambil proporsi yang terbesar dari total clearance. Contohnya, obat biasanya 67% dieliminasi dengan rute ginjal dan 33% dengan rute metabolisme akan menjadi
100% dimetabolisme pada saat gagal ginjal komplit, total clearance, bagaimanapun hanya 1/3 dari hasil normal.
Pengaturan Clt dapat digunakan sebagai alternatif penghitungan besarnya dosis, sebagai
salah satu pengganti fraksi dari total clearance adalah metabolic dan ginjal untuk Clm dan Clr. Dengan
menggunakan teori tersebut dapat diperoleh persamaan sebagai berikut :
Faktor penyesuaian besarnya dosis : [Eq. 25]
Ž¥¦§¨©ª «¬ªª®§©ª«¯§°±¬ª¨ – + m£¥¦§¨©ª «¬ªª®§©ª²ª®³§¬ ¤ £ ¶®²©ª ²ª®³§¬ ®±¦§¬¥¦§¨©ª ´§¦ª ¤n
Faktor penyesuaian dosis dapat digunakan untuk menentukan dosis pemeliharaan untuk pasien dengan fungsi ginjal yang berubah.
Sebagai contoh, suatu obat 25% termetabolisme dan 75% mengalami clearance ginjal dan normalnya teradministrasi 100 mg setiap 12 jam. Jika obat ini diberikan pada pasien yang hanya memiliki 33% fungsi ginjal normal, besarnya dosis dengan faktor penyesuaian akan menjadi 0,5 Besarnya dosis dengan faktor penyesuaian:
Ž¥¦§¨©ª «¬ªª®§©ª«¯§°±¬ª¨ – + m£¥¦§¨©ª «¬ªª®§©ª²ª®³§¬ ¤ £ ¶®²©ª ²ª®³§¬ ®±¦§¬¥¦§¨©ª ´§¦ª ¤n
= (0,25) + [(0,75)(0,33)] = 0,25 + [(0,25)]
Faktor penyesuaian dosis yang memiliki nilai 0,5 menunjukkan bahwa obat tersebut akan teradministrasi setengah dari dosis yang sebenarnya. Hal ini dapat diselesaikan dengan penurunan dosis dan pemeliharaan dengan interval yang sama (misalnya 50 mg setiap 12 jam) atau dengan pemeliharaan yang sama tapi intervalnya ditingkatkan (misalnya 100 mg setiap 24 jam). Berdasarkan pada situasi dan tujuan terapi, tiap metode (kombinasi dari pengaturan dosis dan interval dosis) mungkin dapat dipergunakan.
Banyak penetapan farmakokinetik untuk eliminasi obat adalah berdasarkan pada fungsi ginjal karena fungsi hati biasanya lebih sulit untuk dikuantifikasi. Peningkatan enzim hati dapat menggambarkan kerusakan hati tapi bukan tindakan yang baik untuk fungsi hati. Fungsi hati sering dievaluasi dengan waktu protombin, konsentrasi serum albumin, dan konsentrasi serum bilirubin. Namun, setiap tes laboratorium dipengaruhi oleh variabel lain selain perubahan fungsi hati. Contohnya, serum albumin rendah mungkin karena penurunan pemasukkan protein atau peningkatan ginjal atau penurunan GI, akan menurunkan fungsi hati. Meskipun tes fungsi hati tidak memberikan data kuantitatif, penetuan farmakokinetik harus tetap mempertimbangkan fungsi hati karena rute eliminasi ini penting untuk beberapa obat.
Keluaran Jantung
Hasil keluaran jantung merupakan efek dari metabolisme obat. Aliran hati atau clearance metabolik untuk beberapa obat dapat mengalami penurunan 25% - 50% pada pasien dengan CHF. Contohnya, clearance metabolic dari teofilin dan digoksin pada pasien CHF mengalami pengurangan kira-kira 1,5. Karena clearance metabolic untuk kedua obat ini lebih rendah dari pada darah ke hati atau aliran plasma (rasio ekstraksi hati rendah), ini akan sulit untuk ditentukan apakah clearancenya dipengaruhi oleh keluaran jantung atau aliran darah hati untuk tingkat ini. Penurunan keluaran jantung dan hasil penyumbatan hati pada suatu aliran akan menurunkan kapasitas metabolisme intrinsik dari hati. Efek penurunan clearance pada konsentrasi obat plasma digambarkan pada Gambar 13.