BAB II
TINJAUAN UMUM INDUSTRI
2.1 Industri Farmasi
2.1.1 Pengertian Industri Farmasi
Industri Farmasi menurut Peraturan Menteri Kesehatan RI Nomor 1799/Menkes/Per/XII/2010 adalah badan usaha yang memiliki izin dari menteri kesehatan untuk melakukan kegiatan pembuatan obat atau bahan obat. Industri Farmasi harus membuat obat sesuai aturan CPOB agar sesuai dengan tujuan penggunaannya, memenuhi persyaratan yang tercantum dalam dokumen izin edar (registrasi) dan tidak menimbulkan risiko yang membahayakan konsumen, baik karena ketidakamanan, ketidakefektifan, maupun mutu obat yang substandar (Depkes RI, 2010).
2.1.2 Persyaratan Industri Farmasi
Proses pembuatan obat atau bahan obat hanya dapat dilakukan oleh Industri Farmasi. Setiap pendirian Industri Farmasi wajib memperoleh Izin Industri Farmasi dari Direktur Jenderal. Direktur Jenderal yang dimaksud adalah Direktur Jenderal pada Kementerian Kesehatan yang bertugas dan bertanggung jawab dalam pembinaan kefarmasian dan alat kesehatan (Depkes RI, 2010).
Persyaratan untuk memperoleh Izin Industri Farmasi tercantum dalam Permenkes RI Nomor 1799/Menkes/Per/XII/2010 adalah sebagai berikut:
1. Berbadan usaha berupa perseroan terbatas
4. Memiliki secara tetap paling sedikit 3 (tiga) orang Apoteker Warga Negara Indonesia masing-masing sebagai penanggung jawab pemastian mutu, produksi, dan pengawasan mutu
5. Komisaris dan direksi tidak pernah terlibat, baik langsung ataupun tidak langsung dalam pelanggaran peraturan perundang-undangan di bidang kefarmasian (Depkes RI, 2010).
2.1.3 Izin Usaha Industri Farmasi
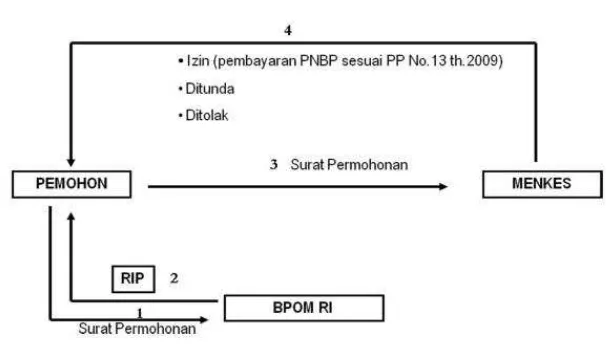
Berdasarkan Permenkes RI Nomor 1799/Menkes/Per/XII/2010, untuk memperoleh Izin Usaha Industri Farmasi diperlukan persetujuan prinsip. Tata cara permohonan persetujuan prinsip Industri Farmasi sebagai berikut:
a. Permohonan persetujuan prinsip diajukan kepada Direktur Jenderal dengan tembusan kepada Kepala Badan dan kepala dinas kesehatan provinsi.
b. Sebelum pengajuan permohonan persetujuan prinsip, pemohon wajib mengajukan permohonan persetujuan Rencana Induk Pembangunan (RIP) kepada Kepala Badan.
c. Persetujuan Rencana Induk Pembangunan (RIP) diberikan oleh Kepala Badan dalam bentuk rekomendasi hasil analisis Rencana Induk Pembangunan (RIP) paling lama dalam jangka waktu 14 (empat belas) hari kerja sejak permohonan diterima.
d. Permohonan persetujuan prinsip diajukan dengan kelengkapannya.
e. Persetujuan prinsip diberikan oleh Direktur Jenderal paling lama dalam waktu 14 (empat belas) hari kerja setelah permohonan diterima atau menolaknya. f. Pemohon izin industri farmasi dengan status Penanaman Modal Asing atau
Penanaman Modal dari instansi yang menyelenggarakan urusan penanaman modal, wajib mengajukan permohonan persetujuan prinsip sesuai dengan ketentuan sebagaimana dimaksud dalam Pasal ini (Ditjen Binfar dan Alkes RI, 2011)
Gambar 2.1 Tata cara pemberian persetujuan prinsip (Ditjen Binfar dan Alkes RI, 2011).
Setelah memperoleh persetujuan prinsip, Industri Farmasi dapat mengurus Izin Industri Farmasi dengan tata cara sebagai berikut:
a. Pemohon yang telah selesai melaksanakan tahap persetujuan prinsip dapat mengajukan permohonan izin industri farmasi.
b. Surat permohonan izin industri farmasi harus ditandatangani oleh Direktur Utama dan Apoteker penanggung jawab pemastian mutu diajukan ke Kementerian Kesehatan beserta kelengkapannya.
c. Permohonan izin industri diajukan kepada Direktur Jenderal dengan tembusan kepada Kepala Badan dan Kepala Dinas Kesehatan Provinsi setempat.
e. Paling lama dalam waktu 20 (dua puluh) hari kerja sejak diterimanya tembusan permohonan, Kepala Dinas Kesehatan Provinsi melakukan verifikasi kelengkapan persyaratan administratif.
f. Paling lama dalam waktu 10 (sepuluh) hari kerja sejak dinyatakan memenuhi persyaratan CPOB, Kepala Badan mengeluarkan rekomendasi pemenuhan persyaratan CPOB kepada Direktur Jenderal dengan tembusan kepada Kepala Dinas Kesehatan Provinsi dan pemohon.
g. Paling lama dalam waktu 10 (sepuluh) hari sejak dinyatakan memenuhi kelengkapan persyaratan administratif, Kepala Dinas Kesehatan Provinsi mengeluarkan rekomendasi pemenuhan persyaratan administratif kepada Direktur Jenderal dengan tembusan kepada Kepala Badan dan pemohon. h. Paling lama dalam waktu 10 (sepuluh) hari kerja setelah menerima
rekomendasi serta persyaratan lainnya, Direktur Jenderal menerbitkan izin industri farmasi (Ditjen Binfar dan Alkes RI, 2011).
2.1.4 Pembinaan dan Pengawasan Industri Farmasi
Pembinaan terhadap pengembangan Industri Farmasi dilakukan oleh Direktur Jenderal, sedangkan pengawasan dilakukan oleh Kepala Badan. Pelanggaran terhadap ketentuan dalam Permenkes RI Nomor 1799/Menkes/Per/XII/2010 dapat dikenakan sanksi administratif berupa:
1. Peringatan secara tertulis
2. Larangan mengedarkan untuk sementara waktu dan/atau perintah untuk penarikan kembali obat atau bahan obat dari peredaran bagi obat atau bahan obat yang tidak memenuhi standar dan persyaratan keamanan, khasiat/kemanfaatan, atau mutu
3. Perintah pemusnahan obat atau bahan obat, jika terbukti tidak memenuhi persyaratan keamanan, khasiat/kemanfaatan, atau mutu
4. Penghentian sementara kegiatan 5. Pembekuan Izin Industri Farmasi 6. Pencabutan Izin Industri Farmasi
2.1.5 Pencabutan Izin Usaha Industri Farmasi a. Persetujuan Prinsip
Persetujuan prinsip batal apabila setelah jangka waktu 3 (tiga) tahun dan/atau setelah jangka waktu 1 (satu) tahun perpanjangan, pemohon belum menyelesaikan pembangunan fisik (Ditjen Binfar dan Alkes RI, 2011).
b. Izin Industri Farmasi
2.2 Peran, Fungsi dan Tugas Apoteker di Industri Farmasi 2.2.1. Apoteker Sebagai Penanggung Jawab Produksi
Penanggung jawab produksi (kepala bagian produksi/manajer produksi) hendaklah seorang apoteker yang terdaftar dan terkualifikasi, memperoleh pelatihan yang sesuai, memiliki pengalaman praktis paling sedikit 5 tahun bekerja di bagian produksi pabrik farmasi, memiliki pengalaman dan pengetahuan di bagian pembuatan obat dan perencanaan produksi, pengetahuan mengenai peralatan yang digunakan dalam pembuatan obat, CPOB, penguasaan bahasa asing yang baik, serta keterampilan dalam kepemimpinan yang dibuktikan dengan sertifikasi lembaga yang ditunjuk.
Secara rinci, ruang lingkup tugas dan tanggung jawab seorang penanggung jawab produksi adalah sebagai berikut:
1. Bertanggung jawab dalam memastikan bahwa obat diproduksi dan disimpan sesuai prosedur sehingga memenuhi persyaratan mutu yang ditetapkan. 2. Bertanggung jawab atas terlaksananya pembuatan obat dari perolehan bahan,
pengolahan, pengemasan, sampai pengiriman obat ke gudang jadi.
3. Memberikan pengarahan teknis dan administratif untuk semua pelaksanaan operasi di gudang, penimbangan, pengolahan, dan pengemasan.
4. Bersama-sama dengan manajer perencanaan dan pengadaan bahan menyusun rencana produksi.
5. Bertanggung jawab memeriksa catatan pengolahan bets dan catatan pengemasan bets serta menjamin bahwa produksi dilaksanakan sesuai dengan prosedur pengolahan bets dan prosedur pengemasan bets.
7. Bertanggung jawab atas peralatan yang digunakan dalam proses produksi, peralatan yang digunakan harus selalu dikualifikasi dan divalidasi dengan benar.
8. Ikut membantu pelaksanaan inspeksi CPOB dan menjaga pelaksanaan serta pematuhan terhadap peraturan CPOB.
9. Bertanggung jawab atas kebersihan di daerah produksi.
10. Bertanggung jawab untuk menjaga moral kerja yang tinggi, kemampuan pengembangan, dan pelatihan serta melakukan evaluasi tahunan atas semua karyawan yang dibawahinya.
11. Membuat laporan bulanan.
12. Membuat anggaran tahunan untuk bagian produksi. 13. Mengusahakan perbaikan biaya produksi.
14. Menjaga hubungan kerja yang baik dengan Penanggung jawab Pengawasan Mutu, Teknik dan Perencanaan dan Pengadaan Bahan serta Pemasaran. 15. Berhubungan dengan pemerintah, dalam hal ini Pengawas Obat dan Makanan
berkaitan dengan kualitas obat.
2.2.2 Apoteker Sebagai Penanggung Jawab Pengawasan Mutu (Quality Control)
Pengawasan mutu merupakan bagian yang penting dari CPOB untuk memberikan kepastian bahwa produk secara konsisten mempunyai mutu yang sesuai dengan tujuan pemakaiannya. Pengawasan mutu hendaklah mencakup semua kegiatan analitik yang dilakukan di laboratorium, termasuk pengambilan sampel, pemeriksaan dan pengujian bahan awal, produk antara, produk ruahan dan produk jadi. Kegiatan ini juga mencakup uji stabilitas, program pemantauan lingkungan, pengujian yang dilakukan dalam rangka validasi, penanganan sampel pertinggal, menyusun dan memperbaharui spesifikasi bahan, produk serta metode pengujiaannya.
Dokumentasi dan prosedur pelulusan yang diterapkan bagian pengawasan mutu hendaklah menjamin bahwa pengujian yang diperlukan telah dilakukan sebelum bahan digunakan dalam produksi dan produk disetujui sebelum didistribusikan. Personil pengawasan mutu hendaklah memiliki akses ke area produksi untuk melakukan pengambilan sampel dan penyelidikan bila diperlukan Seorang penanggung jawab pengawasan mutu (Kepala Bagian Pengawasan Mutu/ Manajer Pengawasan Mutu) adalah seorang apoteker yang terkualifikasi, memperoleh pelatihan yang sesuai, memiliki pengalaman praktis yang memadai dalam bidang pembuatan obat dan keterampilan manajerial sehingga memungkinkan untuk melaksanakan tugas secara profesional.
mikrobiologi, pemeriksaan bahan pengemas, CPOB dan keterampilan dalam kepemimpinan. Seorang penanggung jawab pengawasan mutu memiliki kewenangan dan tanggung jawab penuh dalam pengawasan mutu, termasuk: 1. Menyetujui atau menolak bahan awal, bahan pengemas, produk
2. Memastikan bahwa seluruh pengujian yang diperlukan telah dilaksanakan. 3. Memberi persetujuan terhadap spesifikasi, petunjuk kerja pengambilan contoh,
metode pengujian dan prosedur pengawasan mutu lain.
4. Memberikan persetujuan dan memantau semua kontrak analisis.
5. Memeriksa pemeliharaan bangunan dan fasilitas serta peralatan di bagian pengawasan mutu.
6. Memastikan bahwa validasi yang sesuai telah dilaksanakan.
7. Memastikan bahwa pelatihan awal dan berkesinambungan bagi personil 8. Di departemennya dilaksanakan dan diterapkan sesuai kebutuhan.
2.2.3. Apoteker Sebagai Penanggung Jawab Pemastian Mutu (Quality Assurance)
Seorang penanggung jawab Pemastian Mutu/Manajemen Mutu (Quality Assurance) adalah seorang apoteker yang terdaftar dan terkualifikasi, memperoleh
pelatihan yang sesuai, memiliki pengalaman praktis yang memadai dalam bidang pembuatan obat dan keterampilan manajerial sehingga memungkinkan untuk melaksanakan tugas secara profesional.
dan peralatan laboratorium modern, kemampuan untuk menguraikan metode analisis serta fasih berbahasa inggris, kesanggupan dalam manajemen dan motivasi personalia serta memiliki pengetahuan yang baik dalam proses pembuatan obat dan CPOB baik nasional maupun internasional.
Penanggung jawab Pemastian Mutu memiliki kewenangan dan tanggung jawab penuh dalam sistem mutu, termasuk:
1. Memastikan penerapan (bila diperlukan, membentuk) sistem mutu.
2. Ikut serta dalam atau memprakarsai pembentukan acuan mutu perusahaan. 3. Memprakarsai dan mengawasi audit internal atau inspeksi diri berkala. 4. Melakukan pengawasan terhadap fungsi bagian pengawasan mutu. 5. Memprakarsai dan mengawasi audit eksternal (audit terhadap pemasok). 6. Memprakarsai dan berpartisipasi dalam program validasi.
7. Memastikan pemenuhan persyaratan teknik atau peraturan Otoritas Pengawasan Obat (OPO) yang berkaitan dengan mutu produk jadi.
8. Mengevaluasi/mengkaji catatan bets.
9. Meluluskan atau menolak produk jadi untuk penjualan dengan mempertimbangkan semua faktor terkait.
10. Memantau kinerja sistem mutu dan prosedur serta menilai efektifitasnya. Penekanan difokuskan pada pencegahan kerugian/cacat dan realisasi peluang perbaikan yang berkesinambungan.
11. Menyiapkan prosedur dalam penerapan CPOB dalam pembuatan obat, pengemasan, penyimpanan dan pengawasan mutu.
14. Menyusun prosedur tetap (Protap) dan mengelola sistem protap.
15. Melakukan penilaian terhadap keluhan teknik farmasi dan mengambil keputusan serta tindakan atas hasil penilaian, bila perlu bekerja sama dengan bagian lain.
16. Memastikan penyelanggaraan validasi proses pembuatan dan sistem pelayanan.
17. Memantau penyimpangan bets.
18. Mengawasi sistem pengendalian perubahan dan menyetujui perubahan. 19. Menyetujui prosedur pengolahan induk dan prosedur pengemasan induk. 20. Menyetujui atau menolak pasokan bahan baku.
21. Bertanggung jawab dalam pelulusan atau penolakan obat jadi sesuai Protap terkait.
2.3 Cara Pembuatan Obat yang Baik (CPOB)
Cara Pembuatan Obat yang Baik adalah pedoman pembuatan obat bagi industri farmasi di Indonesia yang bertujuan untuk memastikan agar sifat dan mutu obat yang dihasilkan senantiasa memenuhi persyaratan mutu yang telah ditentukan dan sesuai dengan tujuan penggunaannya (Badan POM RI, 2012).
Berikut adalah aspek-aspek yang diatur dalam CPOB 2012: 2.3.1 Manajemen Mutu
dengan tujuan pemakaiannya. Sedangkan pengawasan mutu (QC) adalah bagian dari CPOB yang berhubungan dengan pengambilan sampel, spesifikasi, pengujian serta organisasi, dokumentasi, prosedur pelulusan (Badan POM RI, 2012).
Konsep dasar Pemastian Mutu, Cara Pembuatan Obat yang Baik (CPOB), Pengawasan Mutu dan Manajemen Risiko Mutu adalah aspek manajemen mutu yang saling terkait (Badan POM RI, 2012).
Pemastian mutu mencakup Cara Pembuatan Obat yang Baik (CPOB) ditambah dengan faktor lain di luar pedoman ini seperti desain dan pengembangan produk. Sistem pemastian mutu yang benar dan tepat bagi industri farmasi hendaklah memastikan bahwa:
- Desain dan pengembangan obat dilakukan dengan memperhatikan persyaratan Cara Pembuatan Obat yang Baik (CPOB) dan semua langkah produksi dan pengawasan diuraikan secara jelas.
- Tanggung jawab manajerial diuraikan dengan jelas dalam uraian jabatan. - Pengaturan disiapkan untuk pembuatan pasokan dan penggunaan bahan awal
dan pengemas yang benar.
- Semua pengawasan terhadap produk antara dan pengawasan selama - proses (In Process Control/IPC) lain memenuhi persyaratan yang ditetapkan.
- Pengkajian terhadap semua dokumen yang terkait dengan proses pengemasan dan pengujian Bets (Batch) dilakukan sebelum memberikan pengesahan pelulusan untuk distribusi.Penilaian hendaklah meliputi semua faktor yang relevan termasuk kondisi pembuatan, hasil dan PengawasanSelamaProses (In Process Control/IPC), pengkajian dokumen produksi termasuk pengemasan,
persyaratan dari spesifikasi produk jadi dan pemeriksaan produk dalam kemasan akhir.
- Obat tidak dijual atau dipasok sebelum Kepala Bagian Manajemen Mutu (pemastian mutu) menyatakan bahwa tiap bets (Batch) produksi dibuat dan dikendalikan sesuai dengan persyaratan yang tercantum dalam izin edar dan peraturan lain yang berkaitan dengan aspek produksi, pengawasan mutu dan pelulusan produk.
- Tersedia pengaturan yang memadai untuk memastikan bahwa sedapat mungkin produk disimpan, didistribusikan dan selanjutnya ditangani sedemikian rupa agar mutu tetap dijaga selama masa edar atau masa simpan obat.
- Tersedia prosedur inspeksi diri dan audit mutu yang secara berkala mengevaluasi efektivitas dan penerapan sistem pemastian mutu.
- Pemasok bahan awal dan bahan pengemas dievaluasi dan disetujui untuk memenuhi spesifikasi mutu yang telah ditentukan oleh perusahaan.
- Penyimpangan dilaporkan, diselidiki dan dicatat.
- Tersedia sistem persetujuan terhadap perubahan yang berdampak pada mutu produk.
- Prosedur pengolahan ulang dievaluasi dan disetujui.
Manajemen resiko mutu adalah suatu proses sistematis untuk melakukan penilaian, pengendalian dan pengkajian resiko terhadap mutu suatu produk. Manajemen risiko mutu hendaklah memastikan bahwa:
- Evaluasi risiko terhadap mutu dilakukan berdasarkan pengetahuan secara ilmiah, pengalaman dengan proses dan pada akhirnya terkait pada perlindungan pasien
- Tingkat usaha, formalitas dan dokumentasi dari proses manajemen risiko mutu sepadan dengan tingkat risiko (Badan POM RI, 2012).
2.3.2 Personalia
Suatu industri farmasi bertanggung jawab menyediakan personil yang sehat, terkualifikasi dan dalam jumlah yang memadai agar proses produksi dapat berjalan dengan baik. Semua personil harus memahami prinsip CPOB agar produk yang dihasilkan bermutu (Badan POM RI, 2012).
Dalam kualifikasi dan pengalaman personil yang diperlukan untuk tiap posisi hendaklah ditetapkan secara tertulis yang disimpan oleh bagian SDM, juga dapat ditampilkan pada uraian tugas masing-masing (Badan POM RI, 2012).
Jumlah personil yang memadai sangat mempengaruhi proses produksi. Kekurangan jumlah personil cenderung mempengaruhi kualitas obat karena jumlah karyawan yang sedikit biasanya mengakibatkan kerja lembur sehingga dapat menimbulkan kelelahan fisik dan mental baik bagi operator ataupun supervisor yang melakukan evaluasi atau mengambil keputusan (Badan POM RI, 2012).
Berdasarkan Peraturan Pemerintah RI Nomor 51 tahun 2009 pasal 9, Industri Farmasi minimal harus memiliki 3 (tiga) orang Apoteker sebagai penanggung jawab masing-masing pada bidang Pemastian Mutu, Produksi dan Pengawasan Mutu setiap produksi sediaan farmasi.
2.3.3 Bangunan dan Fasilitas
Tingkat kebersihan ruang/area untuk pembuatan obat hendaklah diklasifikasikan sesuai dengan jumlah maksimum partikulat udara yang diperbolehkan untuk tiap kelas kebersihan sesuai tabel di bawah ini:
Kelas A, B, C dan D adalah kelas kebersihan ruang untuk pembuatanproduk steril. Kelas E adalah kelas kebersihan ruang untuk pembuatan produknonsteril (Badan POM RI, 2012).
Ruangan produksi hendaklah dilengkapi dengan sistem ventilasi dengan pengontrol udara yang sesuai bagi produk dan aktifitas yang dilakukan, baik terhadap ruangan lain maupun terhadap udara luar (Badan POM RI, 2013).
Rancang bangunan hendaklah dibuat sehingga untuk kegiatan yang berhubungan langsung dengan daerah luar sarananya dikelompokkan. Kegiatan yang berhubungan langsung dengan daerah luar antara lain:
- Penyerahan produk jadi untuk distribusi.
Rancangan diatas perlu ditekankan agar tidak berdampak negatif terhadap kegiatan produksi yang dilakukan di area dengan kelas kebersihan lebih tinggi (Badan POM RI, 2013).
Tata letak ruang hendaklah dikaji sejak tahap perencanaan kontruksi bangunan demi keefektifan semua kegiatan, kelancaran arus kerja, komunikasi, dan pengawasan serta untuk menghindari ketidakteraturan (Badan POM RI, 2013).
Tata letak ruang dalam area produksi yang harus dipenuhi antara lain sebagai berikut:
- Untuk memperkecil risiko bahaya medis yang serius akibat terjadi pencemaran silang, suatu sarana khusus dan self-contained harus disediakan untuk produksi obat tertentu.
- Luas area kerja produksi minimal 2 kali luas yang diperlukan untuk penempatan peralatan (termasuk wadah yang diperlukan untuk suatu kegiatan) ditambah luas area untuk keperluan pembersihan dan perawatan mesin oleh operator produksi atau teknisi.
- Permukaan lantai, dinding, langit-langit dan pintu hendaklah: Kedap air.
Tidak terdapat sambungan untuk mengurangi pelepasan atau pengumpulan
partikel.
Mudah dibersihkan, serta tahan terhadap proses pembersihan, bahan
memperhatikan faktor kepadatan, porositas, tekstur, dan sifat elektrostatis (Badan POM RI, 2013).
2.3.4 Peralatan
Peralatan yang digunakan dalam pembuatan obat hendaklah memiliki rancang bangun dan konstruksi yang tepat, ukuran yang memadai, dan ditempatkan dengan tepat sehingga mutu dari setiap produk obat terjamin secara seragam dari bets ke bets, serta untuk memudahkan pembersihan dan perawatannya (Badan POM RI, 2012).
Rancangan bangunan dan kontruksi peralatan hendaklah memenuhi persyaratan sebagai berikut:
1. Permukaan peralatan yang bersentuhan dengan bahan baku, produk antara, produk jadi tidak boleh bereaksi, mengadisi atau mengasorbsi, yang dapat mengubah identitas, mutu atau kemurniannya di luar batas yang ditentukan. 2. Peralatan tidak boleh menimbulkan akibat yang merugikan terhadap produk 3. Bahan-bahan yang diperlukan untuk suatu tujuan khusus, seperti pelumas
atau pendingin tidak boleh bersentuhan langsung dengan bahan yang diolah 4. Peralatan hendaknya dapat dibersihkan dengan mudah, baik bagian dalam
maupun bagian luar
5. Peralatan yang digunakan untuk menimbang, mengukur, menguji, dan mencatat hendaklah diperiksa ketelitiannya secara teratur serta dikalibrasi menurut suatu program dan prosedur yang tepat
6. Peralatan hendaknya dirawat sesuai jadwal yang tepat
8. Daerah yang digunakan sebagai tempat penyimpanan bahan yang mudah terbakar hendaklah dilengkapi dengan perlengkapan elektris yang kedap eksplosi serta dibumikan dengan sempurna (Badan POM RI, 2012).
2.3.5 Sanitasi dan Higiene
Tingkat sanitasi dan higiene yang tinggi hendaklah diterapkan pada setiap aspek pembuatan obat. Ruang lingkup meliputi personalia, bangunan, peralatan dan kelengkapan, bahan produksi serta wadahnya, dan setiap hal yang dapat merupakan sumber pencemaran produk. Sumber pencemaran hendaklah dihilangkan melalui suatu program sanitasi dan higiene yang menyeluruh serta terpadu (Badan POM RI, 2012).
Sanitasi dan higiene yang diatur dalam pedoman CPOB 2012 adalah terhadap personalia, bangunan, dan peralatan. Prosedur pembersihan, sanitasi dan higiene hendaklah divalidasi serta dievaluasi secara berkala untuk memastikan efektivitas prosedur dan selalu memenuhi persyaratan (Badan POM RI, 2012). 2.3.6 Produksi
Produksi hendaklah dilaksanakan dengan mengikuti prosedur yang telah ditetapkan dan memenuhi ketentuan CPOB yang menjamin senantiasa menghasilkan produk yang memenuhi persyaratan mutu serta memenuhi ketentuan izin pembuatan dan izin edar (Badan POM RI, 2012).
bangunan, peralatan, kebersihan dan higiene sampai dengan pengemasan. Prinsip utama produksi adalah:
a. Adanya keseragaman atau homogenitas dari bets ke bets.
b. Proses produksi dan pengemasan senantiasa menghasilkan produk yang seidentik mungkin (dalam batas syarat mutu) baik bagi bets yang sudah diproduksi maupun yang akan diproduksi.
Sedangkan hakikat produksi adalah:
a. Mutu produk obat tidak ditentukan oleh hasil akhir analisa saja, tetapi ditentukan oleh keseluruhan proses produksi (built in process).
b. Adanya prosedur baku (standar) untuk setiap langkah (tahapan) proses produksi dengan persyaratan yang harus diikuti dengan konsisten (Badan POM RI, 2012).
Hal-hal yang perlu diperhatikan dalam produksi antara lain: a. Pembelian Bahan Awal
Pembelian bahan awal hendaklah hanya dari pemasok yang telah disetujui dan memenuhi spesifikasi yang relevan.Semua penerimaan, pengeluaran dan jumlah bahan tersisa hendaklah dicatat. Catatan hendaklah berisi keterangan mengenai pasokan, nomor bets/lot, tanggal penerimaan, tanggal pelulusan, dan tanggal daluarsa (Badan POM RI, 2012).
b. Pencegahan Pencemaran Silang
pakaian kerja operator. Tingkat resiko pencemaran ini tergantung dari jenis pencemaran dan produk yang tercemar. Pencemaran silang hendaklah dihindari dengan tindakan teknis atau pengaturan yang tepat, antara lain:
- Produksi di dalam gedung yang terpisah (diperlukan untuk produk seperti
penisilin, hormon, sitotoksik, dan produk biologi). - Tersedia ruang penyangga udara dan penghisap udara.
- Memakai pakaian pelindung yang sesuai di area dimana produk yang
beresiko tinggi terhadap pencemaran silang diproses.
- Melaksanakan prosedur pembersihan dan dekontaminasi yang terbukti
efektif (Badan POM RI, 2012). c. Penimbangan dan Penyerahan
Penimbangan dan penyerahan bahan awal, bahan pengemas, produk antara dan produk ruahan dianggap sebagai bagian dari siklus produksi dan memerlukan dokumentasi yang lengkap. Hanya bahan awal, bahan pengemas, produk antara dan produk ruahan yang telah diluluskan oleh pengawasan mutu dan masih belum daluarsa yang boleh diserahkan (Badan POM RI, 2012).
d. Pengembalian
Semua bahan awal dan bahan pengemas yang dikembalikan ke gudang penyimpanan hendaklah didokumentasikan dengan benar (Badan POM RI, 2012).
e. Pengolahan produk antara dan produk ruahan
prosedur yang tertulis. Tiap penyimpangan hendaklah dilaporkan. Semua produk antara dan ruahan diberi label (Badan POM RI, 2012).
f. Kegiatan Pengemasan
Kegiatan pengemasan berfungsi mengemas produk ruahan menjadi produk jadi. Pengemasan hendaklah dilaksanakan di bawah pengendalian yang ketat untuk menjaga identitas, keutuhan dan mutu produk akhir yang dikemas. Semua kegiatan pengemasan hendaklah dilaksanakan sesuai dengan instruksi yang diberikan dan menggunakan bahan pengemasan yang tercantum dalam prosedur pengemasan induk. Rincian pelaksanaan hendaklah dicatat dalam catatan pengemasan bets (Badan POM RI, 2012).
g. Pengawasan Selama Proses
Pengawasan selama proses hendaklah mencakup:
- Semua parameter produk, volume atau jumlah isi produk diperiksa pada saat awal dan selama proses pengolahan atau pengemasan.
- Kemasan akhir diperiksa selama proses pengemasan dengan selang waktu yang teratur untuk memastikan kesesuaiannya dengan spesifikasi dan memastikan semua komponen sesuai dengan yang ditetapkan dalam prosedur pengemasan induk (Badan POM RI, 2012).
h. Karantina Produk Jadi
2.3.7 Pengawasan Mutu
Pengawasan Mutu merupakan bagian yang esensial dari CPOB untuk memberikan kepastian bahwa produk secara konsisten mempunyai mutu yang sesuai dengan tujuan pemakaiannya. Keterlibatan dan komitmen semua pihak yang berkepentingan pada semua tahap merupakan keharusan untuk mencapai sasaran mutu mulai dari awal pembuatan sampai kepada distribusi produk jadi. Pengawasan mutu tidak terbatas pada kegiatan laboratorium, tapi juga harus terlibat dalam semua keputusan yang terkait dengan mutu produk (Badan POM RI, 2012).
Bagian Pengawasan Mutu secara keseluruhan mempunyai tanggung jawab, antara lain adalah:
- Membuat, memvalidasi dan menerapkan semua prosedur pengawasan mutu
- Menyimpan sampel pembanding dari bahan dan produk
- Memastikan pelabelan yang benar pada wadah bahan dan produk - Memastikan pelaksanaan pemantauan stabilitas dari produk
- Ikut serta pada investigasi dari keluhan yang terkait dengan mutu produk (Badan POM RI, 2012).
Personil, bangunan dan fasilitas serta peralatan laboratorium hendaklah sesuai untuk jenis tugas yang ditentukan dan skala kegiatan pembuatan obat. Kegiatan bagian Pengawasan Mutu yang dipersyaratkan dalam CPOB adalah sebagai berikut:
a. Penanganan baku pembanding
c. Penanganan contoh pertinggal d. Validasi
e. Pengawasan terhadap bahan awal, produk antara, produk ruahan, dan obat jadi meliputi spesifikasi, pengambilan contoh, pengujian untuk bahan-bahan tersebut, serta in process control
f. Pengujian ulang bahan yang diluluskan g. Pengujian stabilitas
h. Penanganan terhadap keluhan produk dan produk kembalian.
Bagian Pengawasan Mutu memiliki wewenang khusus untuk memberikan keputusan akhir meluluskan atau menolak atas mutu bahan baku, produk obat ataupun hal lain yang mempengaruhi mutu obat (Badan POM RI, 2012).
Dokumentasi dan prosedur pelulusan yang diterapkan bagian Pengawasan Mutu hendaklah menjamin bahwa pengujian yang diperlukan telah dilakukan sebelum bahan digunakan dalam produksi dan produk disetujui sebelum didistribusikan (Badan POM RI, 2012).
2.3.8 Inspeksi Diri, Audit Mutu dan Audit & Persetujuan Pemasok
Tujuan inspeksi diri adalah untuk mengevaluasi apakah semua aspek produksi dan pengawasan mutu industri farmasi memenuhi ketentuan CPOB (Badan POM RI, 2012).
Aspek-aspek dalam inspeksi diri antara lain: - Personalia
- Penyimpanan bahan awal, bahan pengemas dan obat jadi - Peralatan
- Pengolahan dan pengawasan selama proses - Pengawasan mutu
- Dokumentasi - Sanitasi dan higiene
- Program validasi dan re-validasi - Kalibrasi alat dan sistem pengukuran - Penanganan keluhan
- Pengawasan label
- Hasil inspeksi sebelumnya dan tindakan perbaikan
Inspeksi diri hendaklah dilakukan oleh tim yang anggotanya ditunjuk secara tertulis atau ditetapkan dalam system inspeksi diri. Anggota tim inspeksi diri hendaklah mempunyai pengetahuan tentang CPOB dan penerapannya, terkualifikasi dan mempunyai pengalaman yang memadai dalam melakukan inspeksi diri (Badan POM RI, 2013).
Hendaklah dibuat daftar pemasok yang disetujui untuk bahan awal dan bahan pengemas. Daftar pemasok hendaklah disiapkan dan ditinjau ulang. Hendaklah dilakukan evaluasi sebelum pemasok disetujui dan dimasukkan ke dalam daftar pemasok atau spesifikasi. Evaluasi hendaklah mempertimbangkan riwayat pemasok dan sifat bahan yang dipasok. Semua pemasok yang telah ditetapkan hendaklah dievaluasi secara teratur (Badan POM RI, 2012).
2.3.9 Penanganan Keluhan Terhadap Produk dan Penarikan Kembali Produk
Semua keluhan dan informasi lain yang berkaitan dengan kemungkinan terjadi kerusakan obat harus dikaji dengan teliti sesuai dengan prosedur tertulis. Untuk menangani semua kasus yang mendesak, hendaklah disusun suatu sistem, bila perlu mencakup penarikan kembali produk yang diketahui atau diduga cacat dari peredaran secara cepat dan efektif (Badan POM RI, 2012).
Keluhan dapat ditangani dengan:
- Menunjuk personil yang bertanggung jawab untuk menangani keluhan dan memutuskan tindakan yang hendak dilakukan bersama staf yang memadai untuk membantunya.
- Tersedia prosedur tertulis yang merinci penyelidikan, evaluasi, tindak lanjut yang sesuai, termasuk pertimbangan untuk penarikan kembali produk, dalam menanggapi keluhan terhadap obat yang diduga cacat. - Memberikan perhatian khusus untuk menetapkan apakah keluhan
- Mencatat tiap keluhan yang menyangkut kerusakan produk yang mencakup rincian mengenai asal-usul keluhan dan diselidiki secara menyeluruh dan mendalam.
Pelaksanaan Penarikan Kembali Produk:
- Tindakan penarikan kembali produk hendaklah dilakukan segera setelah diketahui ada produk yang cacat mutu atau diterima laporan mengenai reaksi yang merugikan.
- Pemakaian produk yang berisiko tinggi terhadap kesehatan, hendaklah dihentikan dengan cara embargo yang dilanjutkan dengan penarikan kembali dengan segera.
- Sistem dokumentasi penarikan kembali produk di industri farmasi, hendaklah menjamin bahwa embargo dan penarikan kembali dilaksanakan secara cepat, efektif dan tuntas.
- Pedoman dan prosedur penarikan kembali terhadap produk hendaklah dibuat untuk memungkinkan embargo dan penarikan kembali dapat dilakukan dengan cepat dan efektif dari seluruh mata rantai distribusi. Produk yang ditarik kembali diberi identifikasi dan disimpan terpisah di area yang aman sementara menunggu keputusan terhadap produk tersebut. Efektivitas penyelenggaraan penarikan kembali hendaklah dievaluasi dari waktu ke waktu (Badan POM RI, 2012).
2.3.10 Dokumentasi
mengutamakan tujuannya, yaitu menentukan, memantau dan mencatat seluruh aspek produksi serta pengendalian dan pengawasan mutu. Dokumentasi sangat penting untuk memastikan bahwa tiap personil menerima uraian tugas secara jelas dan rinci sehingga memperkecil resiko terjadinya kekeliruan yang biasanya timbul karena hanya mengandalkan komunikasi lisan. Dokumentasi meliputi: - Spesifikasi
Spesifikasi menguraikan secara rinci persyaratan yang harus dipenuhi produk atau bahan yang digunakan atau diperoleh selama pembuatan. Dokumen ini merupakan dasar untuk mengevaluasi mutu. Spesifikasi meliputi spesifikasi bahan awal, spesifikasi bahan pengemas, spesifikasi produk antara dan produk ruahan, dan spesifikasi produk jadi.
- Dokumen Produksi
Dokumen produksi meliputi dokumen produksi induk, prosedur pengolahan induk, dan prosedur pengemasan induk (formula pembuatan, instruksi pengolahan, dan instruksi pengemasan) yang menyatakan seluruh bahan awal dan bahan pengemas yang digunakan serta menguraikan semua operasi pengolahan dan pengemasan.
- Prosedur
Prosedur berisi cara untuk melaksanakan operasi tertentu, misalnya pembersihan, berpakaian, pengendalian lingkungan, pengambilan sample, pengujian dan pengoperasian peralatan.
- Laporan dan Catatan
2.3.11 Pembuatan dan Analisis Berdasarkan Kontrak
Kontrak hendaklah dibuat antara pemberi kontrak dan penerima kontrak dengan menetapkan tanggung jawab masing-masing pihak yang berhubungan dengan produksi dan pengendalian mutu produk. Semua pengaturan pembuatan dan analisis harus sesuai dengan izin edar dan disetujui oleh kedua belah pihak. Pemberi kontrak hendaklah:
Bertanggung jawab untuk menilai kompetensi penerima kontrak dalam
melaksanakan pekerjaan atau pengujian yang diperlukan.
Menyediakan semua informasi yang diperlukan penerima kontrak untuk
melaksanakan pekerjaan kontrak secara benar sesuai izin edar dan persyaratan legal lain.
Memastikan semua produk yang diproses dan bahan yang dikirimkan oleh
penerima kontrak memenuhi spesifikasi yang ditetapkan atau telah diluluskan. Penerima kontrak hendaklah:
Mempunyai gedung dan peralatan yang cukup, pengetahuan dan pengalaman,
dan personil yang kompeten
Memastikan bahwa semua produk dan bahan yang diterima sesuai dengan
tujuan penggunaannya.
Tidak mengalihkan pekerjaan atau pengujian kepada pihak ketiga tanpa
persetujuan pihak pemberi kontrak
Membatasi diri dari segala aktifitas yang berpengaruh buruk pada mutu (Badan
2.3.12 Kualifikasi dan Validasi A. Kualifikasi
Validasi untuk mesin, peralatan produksi dan sarana penunjang disebut dengan kualifikasi. Jadi, kualifikasi adalah kegiatan pembuktian (dokumentasi) bahwa perlengkapan, fasilitas atau sistem yang digunakan dalam proses/sistem akan bekerja dengan kriteria yang diinginkan secara konsisten. Kualifikasi merupakan langkah awal (first step) dari keseluruhan pelaksanakan (Priyambodo, 2007).
Validasi atau kualifikasi mesin, peralatan produksi dan sarana penunjang terdiri dari 4 tingkatan, yaitu:
1. Kualifikasi Desain
Untuk menjamin dan mendokumentasikan bahwa sistem atau peralatan atau bangunan yang akan dipasang atau dibangun (rancang bangunan) sesuai dengan ketentuan atau spesifikasi yang diatur dalam ketentuan Cara Pembuatan Obat yang Baik (CPOB) yang berlaku. Jadi kualifikasi desain dilaksanakan sebelum mesin, peralatan produksi atau sarana penunjang (termasuk bangunan untuk industri farmasi) tersebut dibeli atau dipasang atau dibangun.
2. Kualifikasi Instalasi
instalasi dilaksanakan pada saat pemasangan atau instalasi peralatan produksi atau sarana penunjang.
3. Kualifikasi Operasional
Untuk menjamin dan mendokumentasikan bahwa sistem atau peralatan yang telah diinstalasi bekerja (beroperasi) sesuai dengan spesifikasi yang diinginkan. Jadi kualifikasi operasional dilaksanakan setelah pemasangan atau instalasi mesin atau peralatan produksi atau sarana penunjang dan digunakan sebagai mesin atau peralatan percobaan.
4. Kualifikasi Kinerja
Untuk menjamin dan mendokumentasikan bahwa sistem atau peralatan yang telah diinstalasi bekerja (beroperasi) sesuai dengan spesifikasi yang diinginkan dengan cara menjalankan sistem sesuai dengan tujuan penggunaan (Priyambodo, 2007).
Pelaksanaan kualifikasi harus dilakukan secara berurutan dan berkesinambungan. Maka, pelaksanaan kualifikasi dimulai dari kualifikasi desain, kemudian kualifikasi instalasi, kualifikasi operasional dan yang terakhir kualifikasi kinerja, tidak bisa dibolak-balik (Priyambodo, 2007).
B. Validasi
1. Validasi Proses
Validasi Proses diartikan sebagai tindakan pembuktian yang didokumentasikan bahwa proses yang dilakukan dalam batas parameter yang ditetapkan dapat bekerja secara efektif dan memberi hasil yang dapat terulang untuk menghasilkan produk jadi yang memenuhi spesifikasi dan atribut mutu yang ditetapkan sebelumnya (Priyambodo, 2007).
Tujuannya adalah memberikan dokumentasi secara tertulis bahwa prosedur produksi yang berlaku dan digunakan dalam proses produksi (Batch Processing Record), senantiasa mencapai hasil yang diinginkan secara
terus-menerus, mengurangi problem yang terjadi selama proses produksi serta memperkecil kemungkinan terjadinya proses ulang (Priyambodo, 2007).
Secara sederhana, pada umumnya validasi proses dilakukan dengan pendekatan sebagai berikut:
a. Validasi Prospektif
Validasi Prospektif adalah Validasi yang dilakukan sebelum pelaksanaan produksi rutin dari produk yang akan dipasarkan dan dilaksanakan sebelum produk diedarkan yang berlaku untuk:
Produk baru
Modifikasi pada proses produksi yang dapat berdampak pada karakteristik
produk tersebut. Prasyarat lain adalah Laporan produk transfer dari bagian R&D ke bagian Produksi.
b. Validasi konkuren
Produk yang telah divalidasi secara prospektif, karena hal tertentu seperti: Perubahan parameter proses sebagai tindak lanjut dari adanya
penyimpangan atau rekomendasi dari Pengkajian Mutu Produk Perubahan pabrik pembuat eksipien dengan spesifikasi yang sama Perubahan mesin dengan spesifikasi yang sama
Transfer pembuatan produk ke pabrik lain
Dapat dilakukan validasi konkuren (Badan POM RI, 2013).
c. Validasi retrospektif
Validasi retrospektif adalah validasi pembuatan produk yang telah dipasarkan yang dilaksanakan berdasarkan data pembuatan, pengujian dan pengawasan bets yang dikumpulkan sesuai dengan protocol yang telah disiapkan dan disetujui (Badan POM RI, 2013).
2. Validasi Pembersihan
Tujuan dari pelaksanaan Validasi Pembersihan (Cleaning Validation) adalah untuk membuktikan bahwa prosedur yang ditetapkan untuk membersihkan suatu peralatan pengolahan, hingga pengemasan primer mampu membersihkan sisa bahan aktif obat dan deterjen yang digunakan untuk proses pencucian dan juga dapat mengendalikan cemaran mikroba pada tingkat yang dapat diterima (Priyambodo, 2007).
3. Validasi Metode Analisis
Tujuan validasi metode analisis adalah untuk menunjukkan bahwa metode analisis sesuai dengan tujuan penggunaannya.
Uji kuantitatif kandungan impuritas (impurity)
Uji batas impuritas
Uji kuantitatif zat aktif dalam sampel bahan aktif obat atau obat atau komponen
tertentu dalam obat
Metode analisis lain, seperti uji disolusi untuk obat atau penentuan ukuran partikel untuk bahan aktif obat, hendaklah juga divalidasi (Badan POM RI, 2012).
4. Validasi Ulang
Fasilitas, sistem, peralatan, dan proses termasuk proses pembersihan hendaklah dievaluasi secara berkala untuk konfirmasi keabsahannya (Badan POM RI, 2012). Validasi ulang juga diperlukan pada kondisi sebagai berikut:
Melibatkan bahan aktif obat baru/pemasok baru
Melibatkan formulasi baru Perubahan prosedur analisis
Prosedur pembersihan diperbaharui melalui mekanisme perubahan
Melewati jangka waktu yang ditetapkan untuk melakukan validasi ulang