OPTIMASI DAN VALIDASI METODE
KROMATOGRAFI CAIR KINERJA TINGGI (KCKT)
PADA PENETAPAN KADAR FLUKONAZOL DALAM
SEDIAAN KAPSUL
SKRIPSI
Diajukan untuk Melengkapi Salah Satu Syarat Untuk Memperoleh Gelar Sarjana Farmasi pada Fakultas Farmasi
Universitas Sumatera Utara
OLEH:
SAFRINA
NIM 081501033
PROGRAM STUDI SARJANA FARMASI
FAKULTAS FARMASI
UNIVERSITAS SUMATERA UTARA
MEDAN
OPTIMASI DAN VALIDASI METODE
KROMATOGRAFI CAIR KINERJA TINGGI (KCKT)
PADA PENETAPAN KADAR FLUKONAZOL DALAM
SEDIAAN KAPSUL
SKRIPSI
Diajukan untuk Melengkapi Salah Satu Syarat Untuk Memperoleh Gelar Sarjana Farmasi pada Fakultas Farmasi
Universitas Sumatera Utara
OLEH:
SAFRINA
NIM 081501033
PROGRAM STUDI SARJANA FARMASI
FAKULTAS FARMASI
UNIVERSITAS SUMATERA UTARA
MEDAN
PENGESAHAN SKRIPSI
OPTIMASI DAN VALIDASI METODE KROMATOGRAFI
CAIR KINERJA TINGGI (KCKT) PADA PENETAPAN
KADAR FLUKONAZOL DALAM SEDIAAN KAPSUL
OLEH: SAFRINA NIM: 081501033
Dipertahankan di Hadapan Panitia Penguji Skripsi Fakultas Farmasi
Universitas Sumatera Utara Pada Tanggal: 15 Juni 2013
Pembimbing I, Panitia Penguji:
Drs. Fathur Rahman Harun, M.Si., Apt. Prof. Dr. Siti Morin Sinaga, M.Sc., Apt.
NIP 195201041980031002 NIP 195008281976032002
Pembimbing II, Drs. Fathur Rahman Harun, M.Si., Apt. NIP 195201041980031002
Prof. Dr. rer. nat. E. De Lux Putra, S.U., Apt. Dra. Salbiah, M.Si., Apt. NIP 195306191983031001 NIP 194810031987012001
Drs. Syafruddin, M.Si., Apt. NIP 194811111976031003
Medan, Juni 2013 Fakultas Farmasi
Universitas Sumatera Utara Dekan
KATA PENGANTAR
Alhamdulillah, segala Puji dan syukur kehadirat Allah SWT yang telah
melimpahkan rahmat, hidayah, dan kemudahan kepada penulis sehingga dapat
menyelesaikan penelitian dan penyusunan skripsi dengan judul “Optimasi dan
Validasi Metode Kromatografi Cair Kinerja Tinggi (KCKT) pada Penetapan
Kadar Flukonazol dalam Sediaan Kapsul”. Skripsi ini diajukan sebagai salah satu
syarat guna memperoleh gelar Sarjana Farmasi dari Fakultas Farmasi Universitas
Sumatera Utara.
Pada kesempatan ini penulis menyampaikan terima kasih yang
sebesar-besarnya kepada Bapak Prof. Dr. Sumadio Hadisahputra, Apt., selaku Dekan
Fakultas Farmasi USU Medan yang telah memberikan fasilitas sehingga penulis
dapat menyelesaikan pendidikan . Bapak Drs. Fathur Rahman Harun, M.Si., Apt.,
dan Prof. Dr. rer. nat. Effendy De Lux Putra, S.U., Apt., selaku dosen pembibing
yang telah banyak memberikan bimbingan dengan penuh kesabaran, tulus dan
ikhlas selama penelitian dan penulisan skripsi ini.
Ucapan terima kasih juga penulis sampaikan kepada Ibu Prof. Dr. Siti
Morin Sinaga, M.Sc., Apt., Bapak Drs. Syafruddin, M.Si., Apt., dan Ibu Dra.
Salbiah, M.Si., Apt., selaku dosen penguji yang telah memberikan kritik dan saran
demi kesempurnaan skripsi ini. Serta kepada Bapak Drs. Nahitma Ginting, M.Si.,
Apt., sebagai dosen penasehat akademik yang telah membimbing penulis selama
masa pendidikan.
Penulis juga mengucapkan terima kasih dan penghargaan yang tiada
Ibunda tercinta Hj. Iriany, S.Pd., untuk Saudara tersayang Maulana Saputra, SH.
dan Ramadhan, atas do’a, dukungan, motivasi dan perhatian yang tiada hentinya
kepada penulis, serta teman-teman mahasiswa Fakultas Farmasi USU yang
memberikan saran, arahan, dan masukan kepada penulis dalam penyelesaian
skripsi ini.
Penulis menyadari bahwa tulisan ini masih memiliki banyak kekurangan,
oleh karena itu dengan segala kerendahan hati penulis bersedia menerima kritik
dan saran yang membangun pada skripsi ini. Semoga skripsi ini bermanfaat bagi
kita semua.
Medan,15 Juni 2013 Penulis,
OPTIMASI DAN VALIDASI METODE KROMATOGRAFI CAIR KINERJA TINGGI (KCKT) PADA PENETAPAN KADAR FLUKONAZOL
DALAM SEDIAAN KAPSUL ABSTRAK
Kapsul flukonazol merupakan salah satu antifungi golongan triazol yang bekerja menghambat sintesis ergosterol sehingga menghambat pertumbuhan jamur. Obat ini berspektrum antifungal luas dan efektif pada pemberian per oral. Obat ini digunakan untuk mengobati candidiasis vagina akut dan kronis, candidiasis mulut, candidiasis sistemik dan infeksi kriptokokus. Penetapan kadar flukonazol dalam United States Pharmacopeia (USP) Edisi XXIX tahun 2006
ditentukan secara KCKT menggunakan fase gerak campuran air dan asetonitril (80:20). Tujuan penelitian ini adalah untuk melakukan optimasi dan validasi metode KCKT pada penetapan kadar flukonazol dalam sediaan kapsul. Selanjutnya metode yang tervalidasi ini diaplikasikan pada penetapan kadar kapsul flukonazol dengan nama generik dan dagang
Penetapan kadar flukonazol dalam kapsul dilakukan dengan metode kroma tografi cair kinerja tinggi (KCKT) menggunakan kolom VP-ODS (250 x 4,6 mm) (Shimadzu) dengan perbandingan fase gerak asetonitrill:air (45:55), laju air 1,0 ml/menit dan dideteksi pada panjang gelombang 260 nm.
Hasil identifikasi flukonazol diperoleh waktu retensi flukonazol dalam sediaan kapsul sama dengan waktu retensi flukonazol baku yaitu pada 3,3 menit. Hasil penelitian diperoleh kadar Flukonazol (PT Kimia Farma) = 100,70% ± 1,57%, kapsul Zemyc® (PT Pharos) = 101,77% ± 3,21%. kapsul Cancid® (PT Sunthi Sepuri) = 102,72% ± 1,00%, dan kapsul Govazol® (PT Guardian Pharmatama) = 103,98 ± 2,53%. Hasil yang diperoleh ini memenuhi persyaratan yang tercantum dalam USP Revision Bulletin 33th Edition tahun 2010, yaitu
mengandung flukonazol tidak kurang dari 90% dan tidak lebih dari 110% dari jumlah yang tertera pada etiket. Uji validasi metode yang dilakukan terhadap kapsul Flukonazol (PT Kimia Farma) diperoleh persen perolehan kembali 100,34%, Relatif Standar Deviasi (RSD) = 0,75%. Ini berarti metode yang
digunakan memiliki ketepatan dan ketelitian yang baik. Batas Deteksi = 1,4846 µg/ml dan Batas Kuantitasi = 4,9489 µg/ml.
OPTIMATION AND VALIDATION HIGH PERFORMANCE LIQUID CHROMATOGRAPHY (HPLC) METHOD OF ANALYSIS FOR
FLUCONAZOLE IN CAPSULE ABSTRACT
Fluconazole capsule is a triazole antifungal drug which acts by inhibition of the ergosterol component of the fungal cell membrane which inhibits fungal growth. It is active against a broad spectrum fungal pathogens and is available for oral use. It is indicated in the treatment of accute and recurrent vaginal candidiasis, mucosal candidiasis, systemic candidiasis and criptococcal infections. Determination of fluconazole in United States Pharmacopheia (USP) 29th Edition 2006 confirmed by HPLC using mobile phase of water and acetonitrile (80:20). The purpose of this study is to optimate and validate HPLC method in determining fluconazole levels in generic and brand capsule dosage.
Determination of Fluconazole contents in capsules was perfomed on Reversed Phase High Peformance Liquid Chromatography system VP-ODS (250 x 4.6 mm) (Shimadzu) using a mobile phase of acetonitrile and water (45 : 55 v/v) with flow rate 1.0 ml/min at 260 nm detector wave length.
The identification results esthablished in similar retention time between Fluconazole capsule dosage form and Fluconazole reference standard at 3.3 minute. The results showed contents of generic capsule of Fluconazole (PT Kimia Farma) = 100.70% ± 1.57%, and the contents of brand capsule Zemyc® (PT
Pharos) = 101. 77% ± 3.21%, Cancid® (PT Sunthi Sepuri) = 100.72% ± 1.00%, Govazol® (PT Guardian Pharmatama) = 103.98 ± 2.53%. The results showed the contents of generic and brand capsules of Fluconazoles are accepted in requirement of fluconazole pharmacy dosage form levels specified in USP Revission Bulletin 33th Edition which contains not less than 90.0% and not more than 110.0% of the labeled amount. Validation test was held on the Fluconazole capsules (PT Kimia Farma) showed the percent recovery 100.34%, the Relative Standard Deviation (RSD) = 0.75%. It means the method was obtained has good accuracy and precision. The Limit of Detection (LOD) = 1.4846 µg/ml and the Limit of Quantitation (LOQ) = 4.9489 µg/ml.
Keywords : Fluconazole, HPLC, optimation, validation.
2.3.6 Rentang ... 21
3.5.3.1 Penyiapan alat kromatografi cair kinerja tinggi ... 23
3.5.3.2 Penentuan perbandingan fase gerak yang optimum ... 23
3.5.4 Analisis kualitatif menggunakan KCKT ... 24
3.5.4.1 Uji identifikasi flukonazol menggunakan KCKT ... 24
3.5.6 Validasi metode ... 27
3.5.6.1 Akurasi (kecermatan) ... 27
3.5.6.2 Presisi (keseksamaan) ... 27
3.5.6.3 Batas deteksi (LOD) dan batas kuantitasi (LOQ) ... 28
BAB IV HASIL DAN PEMBAHASAN ... 30
4.1 Optimasi Komposisi Fase Gerak ... 30
4.2 Analisis Kualitatif ... 31
4.3 Analisis Kuantitatif ... 35
4.3.1 Penentuan kurva kalibrasi ... 35
4.3.2 Penetapan kadar analit dalam sampel yang dianalisis ... 36
4.4 Hasil Uji Validasi .... ... 36
BAB V KESIMPULAN DAN SARAN ... 39
5.1 Kesimpulan ... 39
5.2 Saran ... 39
DAFTAR PUSTAKA ... 40
DAFTAR TABEL
Halaman Tabel 1. Data Optimasi Perbandingan Fase Gerak Asetonitril:Air... 30
Tabel 2. Data hasil penetapan kadar flukonazol
dalam sediaan kapsul ... 36
Tabel 3. Hasil pengujian validasi, dengan parameter akurasi, dan
presisi flukonazol pada kapsul Flukonazol ( PT Kimia
DAFTAR GAMBAR
Halaman
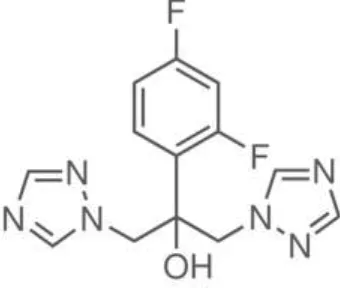
Gambar 1. Struktur Flukonazol . ... 4
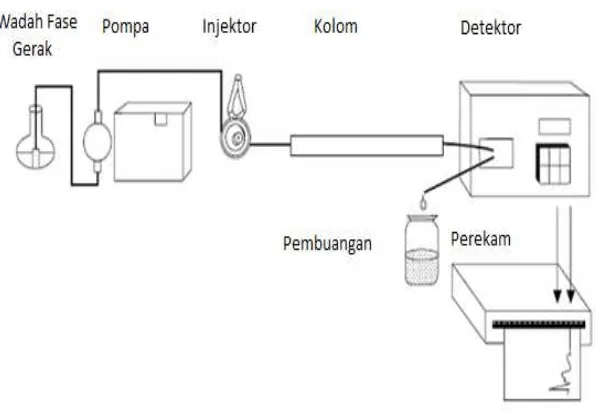
Gambar 2. Diagram Blok KCKT . ... 10
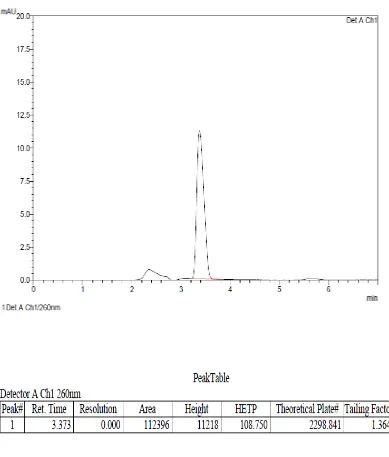
Gambar 3. Kromatogram bahan baku flukonazol secara KCKT
menggunakan kolom Shimadzu VP-ODS (250 x 4,6 mm)
dengan perbandingan fase gerak asetonitril : air (45:55)
dan Laju alir 1 ml/menit, volume penyuntikan 20 µl dan
deteksi pada panjang gelombang 260 nm ... 31
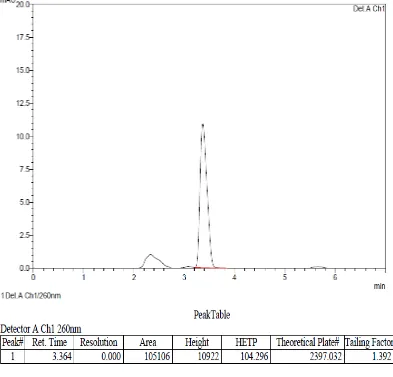
Gambar 4. Kromatogram kapsul flukonazol secara KCKT menggunakan
kolom Shimadzu VP-ODS(250 x 4,6 mm) dengan
perbandingan fase gerak asetonitril : air (45:55) dan
laju alir 1 ml/menit, volume penyuntikan 20 µl dan deteksi
pada panjang gelombang 260 nm ... 32
Gambar 5. Kromatogram kapsul flukonazol menggunakan kolom
Shimadzu VP-ODS (250 x 4,6 mm) dengan perbandingan
fase gerak asetonitril : air (45:55) dan laju alir 1 ml/menit,
volume penyuntikan 20 µl dan deteksi pada
panjang gelombang 260 nm ... 33
Gambar 6. Kromatogram hasil Spike secara KCKT menggunakan
kolom Shimadzu VP-ODS(250 x 4,6 mm) dengan
perbandingan fase gerak asetonitril : air (45:55) dan
laju alir 1 ml/menit, volume penyuntikan 20 µl dan deteksi
Gambar 7. Kurva kalibrasi flukonazol baku menggunakan kolom
Shimadzu VP-ODS (250 x 4,6 mm) dengan perbandingan
fase gerak asetonitril : air (45:55) dan laju alir 1 ml/menit,
volume penyuntikan 20 µl dan deteksi pada
panjang gelombang 260 nm ... 35
Gambar 8. Alat KCKT Shimadzu ... 107
Gambar 9. Alat sonifikator Branson (1510) ... 108
Gambar 10. Pompa vakum dan alat penyaring fase gerak ... 108
Gambar 11. Sonifikator Kudos ... 109
Gambar 12. Neraca analitik ... 109
Gambar 13. Syringe KCKT ... 110
DAFTAR LAMPIRAN
Halaman
Lampiran 1. Kromatografi penyuntikan larutan kapsul flukonazol
untuk mencari komposisi fase gerak asetonitril : air
yang optimum pada analisis ... 42
Lampiran 2. Kromatogram larutan flukonazol baku pada pembuatan
kurva kalibrasi ... 45
Lampiran 3. Perhitungan persamaan regresi dari kurva kalibrasi
flukonazol bakuI yang diperoleh secara KCKT pada
panjang gelombang 260 nm ... 53
Lampiran 4. Perhitungan recovery dengan metode adisi standar ... 55
Lampiran 5. Kromatogram hasil reovery dari sampel Flukonazol
(PT Kimia Farma) ... 59
Lampiran 6. Hasil pengujian validasi dengan parameter akurasi, dan
presisi flukonazol pada kapsul Flukonazol
(PT Kimia Farma) ... 71
Lampiran 7. Contoh perhitungan % recovery dengan metode adisi
standar ... 72
Lampiran 8. Perhitungan batas deteksi (LOD) dan batas kuantitasi
(LOQ) persamaan regresi : Y = ax + b ... 73
Lampiran 9. Kromatogram dari larutan kapsul Flukonazol
(PT Kimia Farma) ... 74
Lampiran 10. Analisis data statistik untuk mencari kadar
(PT Kimia Farma) ... 78
Lampiran 11. Kromatogram dari larutan kapsul Cancid® (PT Sunthi Sepuri) ... 80
Lampiran 12. Analisis data statistik untuk mencari kadar sebenarnya dari penyuntikan larutan kapsul Cancid® (PT Sunthi Sepuri) ... 84
Lampiran 13. Kromatogram dari larutan kapsul Govazol® (PT Guardian Pharmatama) ... 86
Lampiran 14. Analisis data statistik untuk mencari kadar sebenarnya dari penyuntikan larutan kapsul Govazol® (PT Guardian Pharmatama) ... 90
Lampiran 15. Lanjutan analisis data statistik untuk mencari kadar sebenarnya dari larutan kapsul Govazol® (PT Guardian Pharmatama) ... 91
Lampiran 16. Kromatogram dari larutan kapsul Zemyc® (PT Pharos) ... 93
Lampiran 17. Analisis data statistik untuk mencari kadar sebenarnya dari penyuntikan larutan kapsul Zemyc® (PT Pharos) ... 97
Lampiran 18. Perhitungan penimbangan sampel ... 99
Lampiran 19. Hasil analisa kadar flukonazol dalam sampel ... 100
Lampiran 20. Contoh perhitungan untuk mencari kadar flukonazol ... 102
Lampiran 21. Daftar spesifikasi sampel ... 103
Lampiran 23. Sertifikat flukonazol baku ... 106
Lampiran 24. Gambar alat KCKT (Simadzu) ... 107
OPTIMASI DAN VALIDASI METODE KROMATOGRAFI CAIR KINERJA TINGGI (KCKT) PADA PENETAPAN KADAR FLUKONAZOL
DALAM SEDIAAN KAPSUL ABSTRAK
Kapsul flukonazol merupakan salah satu antifungi golongan triazol yang bekerja menghambat sintesis ergosterol sehingga menghambat pertumbuhan jamur. Obat ini berspektrum antifungal luas dan efektif pada pemberian per oral. Obat ini digunakan untuk mengobati candidiasis vagina akut dan kronis, candidiasis mulut, candidiasis sistemik dan infeksi kriptokokus. Penetapan kadar flukonazol dalam United States Pharmacopeia (USP) Edisi XXIX tahun 2006
ditentukan secara KCKT menggunakan fase gerak campuran air dan asetonitril (80:20). Tujuan penelitian ini adalah untuk melakukan optimasi dan validasi metode KCKT pada penetapan kadar flukonazol dalam sediaan kapsul. Selanjutnya metode yang tervalidasi ini diaplikasikan pada penetapan kadar kapsul flukonazol dengan nama generik dan dagang
Penetapan kadar flukonazol dalam kapsul dilakukan dengan metode kroma tografi cair kinerja tinggi (KCKT) menggunakan kolom VP-ODS (250 x 4,6 mm) (Shimadzu) dengan perbandingan fase gerak asetonitrill:air (45:55), laju air 1,0 ml/menit dan dideteksi pada panjang gelombang 260 nm.
Hasil identifikasi flukonazol diperoleh waktu retensi flukonazol dalam sediaan kapsul sama dengan waktu retensi flukonazol baku yaitu pada 3,3 menit. Hasil penelitian diperoleh kadar Flukonazol (PT Kimia Farma) = 100,70% ± 1,57%, kapsul Zemyc® (PT Pharos) = 101,77% ± 3,21%. kapsul Cancid® (PT Sunthi Sepuri) = 102,72% ± 1,00%, dan kapsul Govazol® (PT Guardian Pharmatama) = 103,98 ± 2,53%. Hasil yang diperoleh ini memenuhi persyaratan yang tercantum dalam USP Revision Bulletin 33th Edition tahun 2010, yaitu
mengandung flukonazol tidak kurang dari 90% dan tidak lebih dari 110% dari jumlah yang tertera pada etiket. Uji validasi metode yang dilakukan terhadap kapsul Flukonazol (PT Kimia Farma) diperoleh persen perolehan kembali 100,34%, Relatif Standar Deviasi (RSD) = 0,75%. Ini berarti metode yang
digunakan memiliki ketepatan dan ketelitian yang baik. Batas Deteksi = 1,4846 µg/ml dan Batas Kuantitasi = 4,9489 µg/ml.
OPTIMATION AND VALIDATION HIGH PERFORMANCE LIQUID CHROMATOGRAPHY (HPLC) METHOD OF ANALYSIS FOR
FLUCONAZOLE IN CAPSULE ABSTRACT
Fluconazole capsule is a triazole antifungal drug which acts by inhibition of the ergosterol component of the fungal cell membrane which inhibits fungal growth. It is active against a broad spectrum fungal pathogens and is available for oral use. It is indicated in the treatment of accute and recurrent vaginal candidiasis, mucosal candidiasis, systemic candidiasis and criptococcal infections. Determination of fluconazole in United States Pharmacopheia (USP) 29th Edition 2006 confirmed by HPLC using mobile phase of water and acetonitrile (80:20). The purpose of this study is to optimate and validate HPLC method in determining fluconazole levels in generic and brand capsule dosage.
Determination of Fluconazole contents in capsules was perfomed on Reversed Phase High Peformance Liquid Chromatography system VP-ODS (250 x 4.6 mm) (Shimadzu) using a mobile phase of acetonitrile and water (45 : 55 v/v) with flow rate 1.0 ml/min at 260 nm detector wave length.
The identification results esthablished in similar retention time between Fluconazole capsule dosage form and Fluconazole reference standard at 3.3 minute. The results showed contents of generic capsule of Fluconazole (PT Kimia Farma) = 100.70% ± 1.57%, and the contents of brand capsule Zemyc® (PT
Pharos) = 101. 77% ± 3.21%, Cancid® (PT Sunthi Sepuri) = 100.72% ± 1.00%, Govazol® (PT Guardian Pharmatama) = 103.98 ± 2.53%. The results showed the contents of generic and brand capsules of Fluconazoles are accepted in requirement of fluconazole pharmacy dosage form levels specified in USP Revission Bulletin 33th Edition which contains not less than 90.0% and not more than 110.0% of the labeled amount. Validation test was held on the Fluconazole capsules (PT Kimia Farma) showed the percent recovery 100.34%, the Relative Standard Deviation (RSD) = 0.75%. It means the method was obtained has good accuracy and precision. The Limit of Detection (LOD) = 1.4846 µg/ml and the Limit of Quantitation (LOQ) = 4.9489 µg/ml.
Keywords : Fluconazole, HPLC, optimation, validation.
BAB I PENDAHULUAN
1.1 Latar Belakang
Flukonazol merupakan antifungal golongan triazol yang bekerja
menghambat sintesis ergosterol, yaitu komponen utama pembentukan membran
sel jamur dan efektif terhadap candidiasis mulut, kerongkongan, dan vagina. (Tan
dan Rahardja, 2007).
Optimasi dalam sistem KCKT dilakukan untuk menemukan kondisi yang
optimal guna menghasilkan pemisahan yang baik pada kondisi percobaan tersebut
dilakukan. Keberhasilan suatu pemisahan analit sangat dipengaruhi oleh
pemilihan sistem kromatografi dan komposisi fase gerak yang tepat. Pemilihan
komposisi fase gerak merupakan aspek utama dalam optimasi. Pemilihan fase
gerak juga berhubungan denngan parameter lain seperti laju alir dan suhu kolom
yang menjadi pertimbangan penting dalam pemilihan komposisi fase gerak yang
digunakan (Berridge, 1985).
Menurut United States Pharmacopeia (USP) Edisi XXVIII Tahun 2006
penetapan kadar flukonazol secara KCKT menggunakan kolom ODS (4,6 mm x
15 cm) sedangkan kolom yang digunakan dalam penelitian adalah kolom
VP-ODS (4,6 mm x 25 cm). Perbedaan panjang kolom yang digunakan pada sistem
kromatografi merupakan parameter penting untuk dilakukan optimasi metode.
Menurut beberapa literatur, penetapan kadar flukonazol sebagai bahan
baku dapat ditentukan secara KCKT menggunakan kolom C18, laju alir 1,0
ml/menit dengan fase gerak asetonitril:air (20:80) (USP Conventional Inc., 2006);
(Moffat, et al., 2005). Sedangkan penetapan kadar flukonazol dalam sediaan
kapsul ditentukan secara KCKT menggunakan kolom C 18, dideteksi pada
panjang gelombang 260 nm, laju alir 1,0 ml/menit dengan perbandingan fase
gerak metanol:air (40:60) (Corrêa, et al., 2012); asetonitril:air (35:65)
(Sadasivudu, et al., 2009). Penetapan kadar flukonazol dapat juga ditentukan
secara spektrofotometri UV menggunakan pelarut HCl 0,1 N pada panjang
gelombang maksimum 260 nm (Sadasivudu, et al., 2009).
Berdasarkan hal tersebut di atas, peneliti tertarik melakukan optimasi dan
validasi metode KCKT dengan kolom VP-ODS (4,6 mm x 25 cm) menggunakan
fase gerak asetonitril:air dengan perbandingan tertentu, laju alir 1 ml/menit
dideteksi pada λ= 260 nm pada penetapan kadar flukonazol dalam sediaan kapsul.
Metode yang tervalidasi ini diaplikasikan pada penetapan kadar kapsul flukonazol
dengan nama dagang dan generik yang beredar di pasaran. Adapun parameter
validasi metode yang dilakukan meliputi uji akurasi, presisi, batas deteksi (LOD),
dan batas kuantitasi (LOQ).
1.2 Perumusan Masalah
1. Apakah metode KCKT menggunakan fase gerak asetonitril:air dengan
perbandingan tertentu dapat digunakan pada penetapan kadar flukonazol
dalam sediaan kapsul dan memberikan uji validasi metode yang memenuhi
syarat?
2. Apakah kadar flukonazol dalam sediaan kapsul dengan nama dagang dan
generik yang beredar di pasaran memenuhi persyaratan kadar yang
1.3 Hipotesis
1. Metode KCKT menggunakan fase gerak asetonitril:air dapat digunakan
pada penetapan kadar flukonazol dalam sediaan kapsul dan
memberikan uji validasi metode yang memenuhi syarat.
2. Kadar flukonazol dalam sediaan kapsul dengan nama dagang dan generik
yang beredar di pasaran memenuhi persyaratan kadar yang ditetapkan
dalam USP Convention Inc. (2010).
1.4 Tujuan Penelitian
1. Melakukan optimasi fase gerak dan validasi metode KCKT pada
penetapan kadar flukonazol dalam sediaan kapsul.
2. Menerapkan metode KCKT dengan fase gerak dari hasil optimasi yang
terbaik pada penetapan kadar kapsul flukonazol dengan nama dagang dan
generik yang beredar di pasaran.
1.5 Manfaat Penelitian
Sebagai metode alternatif bagi industri farmasi pada penetapan kadar
flukonazol dalam sediaan kapsul yang mengandung flukonazol dengan metode
BAB II
TINJAUAN PUSTAKA
2.1 Flukonazol
2.1.1 Sifat fisikokimia
Menurut USP Convention Inc. (2006), sifat fisikokimia flukonazol adalah:
Gambar 1 Struktur Flukonazol
Nama Kimia : 2,4-Difluoro-1’,1’-bis(1H-1,2,4-triazol-1-ylmethyl)benzyl alcohol
Rumus Molekul : C13H12F2N6O
Berat Molekul : 306,27
Pemerian : Serbuk hablur putih sampai hampir putih, melebur pada suhu
1380 sampai suhu 1420.
Kelarutan : Mudah larut dalam metanol; larut dalam etanol dan aseton;
agak sukar larut dalam isopropanol dan kloroform; sukar
larut dalam air; sangat sukar larut dalam toluen.
Menurut Moffat, et al., (2005) Spektrum UV dalam larutan asam 261, 266
2.1.2 Farmakologi
Flukonazol termasuk golongan antifungi golongan triazol yang bekerja
menghambat sintesis ergosterol pada membran sel jamur. Flukonazol diberikan
peroral absorbsinya baik dan tidak bergantung pada keasaman lambung. Waktu
paruh obat berkisar pada 30 jam dengan ikatan obat pada protein plasma rendah
dan obat ini terdistribusi merata dalam cairan tubuh. Flukonazol diberikan pada
penderita candidiasis mulut, kerongkongan dan vagina. Flukonazol berguna untuk
mencegah relaps meningitis yang disebabkan oleh Cryptococcus pada pasien
AIDS (Setiabudi dan Bahry, 2007).
2.1.3 Bentuk Sediaan
Kapsul 50 mg, 100 mg, 150 mg, dan 200 mg; tablet 50 mg, 150 mg, dan
200 mg (Anonim, 2010). Flukonazol tersedia untuk pemakaian sistemik (IV)
dalam formula yang mengandung 2 mg/ml dan untuk pemakaian oral dalam
kapsul yang mengandung 50, 100, 150, 200 mg. Di Indonesia, yang tersedia
adalah sediaan 50 dan 150 mg (Setiabudi dan Bahry, 2007).
2.2 Teori Kromatografi Cair Kinerja Tinggi 2.2.1 Sejarah Kromatografi
Kromatografi adalah suatu istilah umum yang digunakan untuk
bermacam-macam teknik pemisahan, yaitu berdasarkan absorbsi sampel diantara suatu fase
gerak dan fase diam. Penemu Kromatografi adalah Tswett yang pada tahun 1903
mencoba memisahkan pigmen-pigmen dari daun dengan menggunakan suatu
kolom yang berisi kapur (CaSO4). Istilah kromatografi diciptakan oleh Tswett
Pada waktu yang hampir bersamaan, D.T. Day juga menggunakan kromatografi
untuk memisahan fraksi-fraksi petroleum, namun Tswett adalah yang pertama
diakui sebagai penemu dan yang menjelaskan tentang proses kromatografi
(Johnson dan Stevenson, 1978).
2.2.2 Pembagian Kromatografi
Kromatografi dapat dibedakan atas berbagai macam, tergantung pada
pengelompokannya. Berdasarkan pada mekanisme pemisahannya, kromatografi
dibedakan menjadi : (a) kromatografi adsorbsi; (b) kromatografi partisi; (c)
kromatografi pasangan ion; (d) kromatografi penukar ion (e) kromatografi
eksklusi ukuran dan (f) kromatografi afinitas (Johnson dan Stevenson, 1978;
Gandjar dan Rohman, 2007).
Berdasarkan pada alat yang digunakan, kromatografi dapat dibagi atas: (a)
kromatografi kertas; (b) kromatografi lapis tipis, yang kedua sering disebut
kromatografi planar; (c) kromatografi cair kinerja tinggi (KCKT) dan (d)
kromatografi gas (KG) (Johnson dan Stevenson, 1978; Gandjar dan Rohman,
2007).
2.2.3 Kromatografi Cair Kinerja Tinggi
Kromatografi cair kinerja tinggi (KCKT) merupakan sistem pemisahan
dengan kecepatan dan efisiensi yang tinggi karena didukung oleh kemajuan dalam
teknologi kolom, sistem pompa tekanan tinggi serta detektor yang sangat sensitif
dan beragam sehingga mampu menganalisis berbagai analit secara kualitatif
maupun kuantitatif, baik dalam komponen tunggal ataupun campuran (Ditjen
Kegunaan umum KCKT adalah untuk pemisahan sejumlah senyawa
organik, anorganik, maupun senyawa biologis, analisis ketidakmurnian
(impurities) dan analisis senyawa-senyawa yang tidak mudah menguap
(nonvolatile). KCKT sering digunakan untuk menetapkan kadar senyawa-senyawa
tertentu seperti asam-asam amino, asam-asam nukleat dan protein-protein dalam
cairan fisiologis, menentukan kadar senyawa-senyawa aktif obat dan lain-lain
(Gandjar dan Rohman, 2007).
2.2.3.1 Jenis-jenis KCKT
Hampir semua jenis campuran solut dapat dipisahkan dengan KCKT
karena banyaknya fase diam yang tersedia dan selektifitas yang dapat ditingkatkan
dengan mengatur fase gerak. Pemisahan dapat dilakukan dengan fase normal atau
fase terbalik tergantung pada polaritas relatif fase diam dan fase gerak (Gandjar
dan Rohman, 2007).
Pada KCKT fase normal, kemampuan elusi meningkat dengan
meningkatnya polaritas pelarut. Fase gerak biasanya non polar, seperti dietil eter,
benzen, hidrokarbon lurus seperti pentana, heksana, heptana maupun iso-oktana.
Halida alifatis seperti diklorometana, dikloroetana, butilklorida dan kloroform
juga digunakan. Umumnya gas terlarut tidak menimbulkan masalah pada fase
normal (Gandjar dan Rohman, 2007).
Pada KCKT fase terbalik paling sering digunakan fase diam berupa
oktadesilsilan (ODS atau C18) dan fase gerak campuran metanol atau asetonitril
dengan air atau dengan larutan buffer. Untuk solut yang bersifat asam lemah
,peranan pH sangat krusial karena bila pH fase gerak tidak diatur maka solut akan
menyebabkan ikatannya dengan fase diam menjadi lebih lemah dibanding jika
solut dalam bentuk yang tidak terionisasi akan terelusi lebih cepat (Gandjar dan
Rohman, 2007).
2.2.3.2 Kriteria Optimasi KCKT
Menurut Berridge (1985), optimasi dalam sistem KCKT disyaratkan untuk
menemukan kondisi yang optimal guna menghasilkan pemisahan yang baik pada
kondisi percobaan tersebut dilakukan. Meskipun demikian, kondisi terbaik sistem
KCKT sulit untuk ditemukan. Adapun tujuan dipersyaratkannya optimasi pada
sistem KCKT antara lain :
- Menghemat biaya penelitian
- Mendapatkan hasil pemisahan yang baik dengan waktu yang singkat
- Menciptakan pemisahan terbaik yang mungkin dihasilkan oleh sampel
- Menyeleksi / memilih komposisi fase gerak dan kolom yang menunjukkan
pemisahan yang baik pada waktu yang singkat
- Memperoleh kombinasi optimum pada kecepatan elusi / laju alir, ukuran
sampel, dan resolusi dari larutan sampel
- Melokasikan kriteria optimasi untuk tempat / daerah percobaan tersebut
dilakukan.
Keberhasilan suatu pemisahan analit sangat dipengaruhi oleh pemilihan
sistem kromatografi dan komposisi fase gerak yang tepat. Meskipun dari segi
instrumennya sering diabaikan. Proses pemisahan dikatakan baik bergantung pada
kondisi kolom, detektor, dan pompa instrumen KCKT. Ditinjau lebih luas lagi,
pemilihan komposisi fase gerak merupakan aspek utama dalam optimasi.
sampel oleh pelarut karena adanya parameter seperti laju alir dan suhu kolom
yang menjadi pertimbangan penting dalam pemilihan komposisi fase gerak yang
digunakan (Berridge, 1985).
2.2.4 Cara Kerja KCKT
Secara teori, pemisahan kromatografi yang paling baik akan diperoleh jika
fase diam mempunyai luas permukaan sebesar-besarnya sehingga memastikan
kesetimbangan yang baik antara fase dan bila fase gerak bergerak dengan cepat
sehingga difusi sekecil-kecilnya (Gritter, dkk., 1985).
Kromatografi merupakan teknik pemisahan dimana analit atau zat-zat
terlarut terpisah oleh perbedaan kecepatan elusi saat melewati suatu kolom
kromatografi, pemisahan tersebut diatur oleh distribusi analit dalam fase gerak
dan fase diam (Rohman, 2009).
Komponen yang telah terpisah akan dibawa oleh fase gerak menuju
detektor dan sinyal yang terekam oleh detektor disebut sebagai puncak, sedangkan
keseluruhan puncak yang direkam oleh detektor selama analisis dinamakan
kromatogram. Puncak yang diperoleh dalam analisis memiliki dua informasi
penting yakni informasi kualitatif dan kuantitatif (Meyer, 2004).
Untuk mendapatkan hasil analisis yang baik, diperlukan penggabungan
secara tepat dari kondisi operasional seperti jenis kolom, fase gerak, panjang dan
diameter kolom, kecepatan alir fase gerak, suhu kolom dan ukuran sampel
(Rohman, 2009).
2.2.5 Migrasi dan Retensi Solut
Kecepatan migrasi solut melalui fase diam ditentukan oleh perbandingan
fase (fase diam dan fase bergerak). Dalam konteks kromatografi, nilai D
didefinisikan sebagai perbandingan konsentrasi solut dalam fase diam (Cs) dan
dalam fase gerak (Cm) (Gandjar dan Rohman, 2007).
Jadi semakin besar nilai D maka migrasi solut semakin lambat dan
semakin kecil nilai D migrasi solut semakin cepat. Solut akan terelusi menurut
perbandingan distribusinya. Jika perbedaan perbandingan distribusi solut cukup
besar maka campuran-campuran solut akan mudah dan cepat dipisahkan (Gandjar
dan Rohman, 2007).
2.2.6 Instrumen KCKT
Instrument KCKT tersusun atas 6 bagian dasar, yaitu wadah fase gerak
(reservoir), pompa (pump), tempat injeksi sampel (injector), kolom(column),
detector (detector) dan perekam (recorder) (Rohman, 2009). Instrument dasar
KCKT dapat dilihat pada gambar
Gambar 2 . Diagram Blok KCKT (McMaster, 2007) m
S
2.2.6.1 Wadah Fase Gerak
Wadah fase gerak harus bersih dan inert. Wadah pelarut kosong ataupun
labu dapat digunakan sebagai wadah fase gerak dan biasanya dapat menampung
fase gerak antara 1 sampai 2 liter pelarut. Fase gerak sebelum digunakan harus
dilakukan degassing (penghilangan gas) pada fase gerak, sebab adanya gas akan
berkumpul dengan komponen lain terutama pompa dan detektor sehingga akan
mengacaukan analisis (Rohman, 2009).
2.2.6.2 Pompa
Pompa yang cocok digunakan untuk KCKT adalah pompa yang
mempunyai syarat sebagaimana syarat wadah pelarut yakni harus inert terhadap
fase gerak. Bahan yang umum dipakai untuk pompa adalah gelas, baja tahan
karat, Teflon, dan batu nilam. Pompa yang dgunakan sebaiknya mampu
memberikan tekanan sampai 5000 psi dan mampu mengalirkan fase gerak dengan
kecepatan alir 3 ml/menit (Rohman, 2009).
Ada dua jenis utama pompa yang digunakan: tekanan-tetap. Pompa
pendesakan tetap dapat dibagi lagi menjadi pompa torak dan pompa semprit.
Pompa torak menghasilkan aliran yang berdenyut, jadi memerlukan peredam
denyut atau peredam elektronik untuk menghasilkan garis alas detektor yang
stabil jika detektor peka terhadap aliran. Kelebihan utamanya ialah tandonnya
tidak terbatas. Pompa semprit menghasilkan aliran yang tak berdenyut, tetapi
2.2.6.3 Injektor
Ada 3 jenis injektor, yakni syringe injector, loop valve dan automatic
injector (autosampler). Syringe injector merupakan bentuk injektor yang paling
sederhana (Dong, 2005).
Pada waktu sampel diinjeksikan ke dalam kolom, diharapkan agar aliran
pelarut tidak mengganggu masuknya keseluruhan sampel ke dalam kolom.
Sampel dapat langsung diinjeksikan ke dalam kolom (on column injection) atau
digunakan katup injeksi (Dong, 2005).
Katup putaran (loop valve), tipe injektor ini umumnya digunakan untuk
menginjeksi volume lebih besar daripada 10 µl dan sekarang digunakan dengan
cara otomatis (dengan adaptor khusus, volume-volume lebih kecil dapat
diinjeksikan secara manual). Bila katup difungsikan, maka cuplikan di dalam
putaran akan bergerak ke dalam kolom (Dong, 2005).
Automatic injector atau disebut juga autosampler memiliki prinsip yang
mirip, hanya saja sistem penyuntikannya bekerja secara otomatis (Meyer, 2004).
2.2.6.4 Kolom
Kolom dapat dibagi menjadi dua kelompok:
a. Kolom analitik: garis tengah-dalam 2-6 mm. Panjang bergantung pada
jenis kemasan, untuk kemasn pelikel biasanya panjang kolom 50-100 cm,
untuk kemasan mikropartikel berpori biasanya 10-30 cm.
b. Kolom preparatif: umumnya bergaris tengah 6 mm atau lebih besar dari
Kolom hampir selalu terbuat dari baja nirkarat. Kolom biasanya dipakai
pada suhu kamar, tetapi pada suhu yang lebih tinggi dapat juga dipakai (Johnson
dan Stevenson, 1978).
2.2.6.5 Detektor
Suatu detektor dibutuhkan untuk mendeteksi adanya komponen cuplikan
dalam aliran yang keluar dari kolom. Detektor-detektor yang baik memiliki
sensitifitas yang tinggi, gangguan (noise) yang rendah, kisar respon linier yang
luas, dan memberi tanggapan/respon untuk semua tipe senyawa. Suatu kepekaan
yang rendah terhadap aliran dan fluktuasi temperatur sangat diinginkan, tetapi
tidak selalu dapat diperoleh (Johnson dan Stevenson, 1978).
Detektor yang paling banyak digunakan dalam kromatografi cair modern
kecepatan tinggi adalah detektor spektrofotometer UV 254 nm. Bermacam-macam
detektor dengan variasi panjang gelombang UV-Vis sekarang menjadi populer
karena mereka dapat digunakan untuk mendeteksi senyawa-senyawa dalam
rentang yang luas. Detektor indeks refraksi juga secara luas digunakan, terutama
dalam kromatografi eksklusi, tetapi umumnya kurang sensitif dari pada detektor
spektrofotometer UV. Detektor lainnya seperti detektor fluometer, detektor
ionisasi nyala, dan detektor elektrokimia juga telah digunakan (Johnson dan
Stevenson, 1978).
2.2.6.6 Perekam
Alat pengumpul data seperti komputer, integrator, rekorder dihubungkan
dengan detektor. Alat ini akan mengukur sinyal elektronik yang dihasilkan oleh
detektor lalu mem-plotkannya sebagai suatu kromatogram yang selanjutnya dapat
2.2.7 Parameter Penting dalam KCKT 2.2.7.1 Tinggi dan Luas Puncak
Tinggi dan luas puncak berkaitan secara proporsional dengan kadar atau
jumlah analit tertentu yang terdapat dalam sampel (memiliki informasi
kuantitatif). Namun demikian, luas puncak lebih umum digunakan dalam
perhitungan kuantitatif karena lebih akurat/cermat daripada perhitungan
menggunakan tinggi puncak (Dong, 2005). Hal ini dikarenakan luas puncak relatif
tidak banyak dipengaruhi oleh kondisi kromatografi, kecuali laju alir. Sementara
itu, tinggi puncak dipengaruhi oleh banyak faktor seperti misalnya faktor tambat,
suhu kolom serta cara injeksi sampel (Miller, 2005). Hal ini akan menyebabkan
tinggi puncak relatif labil selama analisis. Namun demikian tinggi puncak masih
dapat digunakan dalam perhitungan kuantitatif bila puncak analit simetris (Meyer,
2004).
2.2.7.2 Waktu Tambat
Periode waktu antara penyuntikan sampel dan puncak maksimum yang
terekam oleh detektor disebut sebagai waktu tambat. Waktu tambat dari suatu
komponen yang tidak ditahan oleh fase diam disebut sebagai waktu hampa/void
time (t0). Waktu tambat merupakan fungsi dari laju alir fase gerak dan panjang
kolom. Jika fase gerak mengalir lebih lambat atau kolom semakin panjang, waktu
hampa dan waktu tambat akan semakin besar, dan sebaliknya bila fase gerak
mengalir lebih cepat atau kolom semakin pendek, maka waktu hampa dan waktu
2.2.7.3 Faktor Kapasitas
Waktu tambat dipengaruhi oleh laju alir, ukuran kolom dan parameter
yang lain. Oleh karena itu, diperlukan suatu ukuran derajat tambatan dari analit
yang lebih independen yakni faktor kapasitas (Meyer, 2004).
Dalam beberapa literatur lain, faktor kapasitas juga disebut sebagai faktor
tambat (k’). Idealnya, analit yang sama jika diukur pada dua instrumen berbeda
dengan ukuran kolom yang berbeda namun memiliki fase diam dan fase gerak
yang sama, maka faktor tambat dari analit pada kedua sistem KCKT tersebut
secara teoritis adalah sama (Meyer, 2004).
Faktor tambat yang disukai berada di antara nilai 1 hingga 10. Jika nilai k’
terlalu kecil menunjukkan bahwa analit terlalu cepat melewati kolom sehingga
tidak terjadi interaksi dengan fase diam dan oleh karena itu tidak akan muncul
dalam kromatogram. Sebaliknya, nilai k’ yang terlalu besar mengindikasikan
waktu analisis akan panjang. Nilai k’ dari analit yang lebih besar dari 10 akan
menjadi masalah dalam analisis KCKT karena waktu analisis yang terlalu panjang
dan sensitifitas yang buruk sebagai akibat dari pelebaran puncak yang berlebihan
(Meyer, 2004).
2.2.7.4 Selektifitas
Proses pemisahan antara dua komponen dalam KCKT hanya
memungkinkan bila kedua komponen memiliki kecepatan yang berbeda dalam
melewati kolom. Kemampuan sistem kromatografi dalam
memisahkan/membeda-kan analit yang berbeda dikenal sebagai selektifitas (α). Selektifitas umumnya
tergantung pada sifat analit itu sendiri, interaksinya dengan permukaan fase diam
sistem KCKT harus lebih besar dari 1. Selektifitas disebut juga sebagai faktor
pemisahan atau tambatan relatif (Meyer, 2004).
2.2.7.5 Efisiensi Kolom
Salah satu karakteristik sistem kromatografi yang paling penting adalah
efisiensi atau jumlah lempeng teoritis. Bilangan lempeng (N) yang tinggi
disyaratkan untuk pemisahan yang baik yang nilainya semakin kecilnya nilai H.
Istilah H merupakan tinggi ekivalen lempeng teoritis atau HETP (high equivalent
theoretical plate) yang mana merupakan panjang kolom yang dibutuhkan untuk
menghasilkan satu lempeng teoritis. Kolom yang baik akan mempunyai bilangan
lempeng yang tinggi dan nilai H yang rendah, untuk mencapai hal ini ada
beberapa faktor yang mendukung yaitu kolom yang dikemas dengan baik, kolom
yang lebih panjang, partikel fase diam yang lebih kecil, viskositas fase gerak yang
lebih rendah dan suhu yang lebih tinggi, molekul-molekul sampel yang lebih
kecil, dan pengaruh di luar kolom yang minimal (Gandjar dan Rohman, 2007).
2.2.7.6 Resolusi
Tingkat pemisahan komponen dalam suatu campuran dengan metode
kromatografi direfleksikan dalam kromatogram yang dihasilkan, untuk hasil
pemisahan yang baik puncak-puncak dalam kromatogram harus terpisah secara
sempurna dari puncak lainnya. Resolusi adalah perbedaan waktu retensi 2 puncak
yang saling berdekatan, dibagi dengan rata-rata lebar puncak, dengan rumus sbb:
Keterangan:
Nilai Rs mendekati atau lebih dari 1,5 akan memberikan pemisahan yang
baik (Gandjar dan Rohman, 2007).
2.2.7.7 Faktor Asimetri
Adanya puncak yang asimetris dapat disebabkan oleh hal–hal berikut:
• Ukuran sampel yang dianalisis terlalu besar. Jika sampel terlalu besar maka fase
gerak tidak mampu membawa solut dengan sempurna karenanya terjadi
pengekoran atau tailing.
• Interaksi yang kuat antara solut dengan fase diam dapat menyebabkan solut
sukar terelusi sehingga dapat menyebabkan terbentuknya puncak yang mengekor.
• Adanya kontaminan dalam sampel yang dapat muncul terlebih dahulu sehingga
menimbulkan puncak mendahului (fronting) (Gandjar dan Rohman, 2007).
2.3 Validasi Metode
Validasi metoda analisis adalah suatu tindakan penilaian terhadap
parameter tertentu, berdasarkan percobaan laboratorium, untuk membuktikan
bahwa parameter tersebut memenuhi persyaratan untuk penggunaannya (Harmita,
2004). Berikut delapan karakterisitik utama yang digunakan dalam validasi
metode analitik menurut USP:
Karakteristik Pengertian
Akurasi Kedekatan antara nilai hasil uji yang diperoleh lewat metode analitik dengan nilai sebenarnya.
Presisi Ukuran keterulangan metode analitik, termasuk di antaranya kemampuan instrumen dalam memberikan hasil analitik yang reprodusibel.
Spesifisitas Kemampuan untuk mengukur analit yang dituju secara tepat dan spesifik dengan adanya komponen lain dalam matriks sampel seperti ketidakmurnian, produk degradatif dan komponen matriks.
Batas deteksi Konsentrasi analit terendah dalam sampel yang masih dapat dideteksi, meskipun tidak selalu dapat dikuantifikasi.
ditentukan dengan presisi dan akurasi yang dapat diterima pada kondisi operasional metode yang digunakan.
Linieritas
Rentang
Kemampuan suatu metode untuk memperoleh hasil uji yang secara langsung proposional dengan konsentrasi analit pada kisaran yang diberikan.
Konsentrasi terendah dan tertinggi yang mana suatu metode analitik menunjukkan akurasi, presisi dan linieritas yang cukup. Kekasaran Tingkat reprodusibilitas hasil yang diperoleh dibawah berbagai
kondisi yang diekspresikan sebagai % RSD.
Ketahanan Kapasitas metode untuk tidak terpengaruh oleh adanya variasi parameter yang kecil.
(Rohman, 2009).
2.3.1 Akurasi
Akurasi/kecermatan dapat ditentukan dengan dua metode, yakni spiked
placebo recovery dan standard addition method. Pada spiked placebo recovery
atau metode simulasi, analit murni ditambahkan (spiked) ke dalam campuran
bahan pembawa sediaan farmasi, lalu campuran tersebut dianalisis dan jumlah
analit hasil analisis dibandingkan dengan jumlah analit teoritis yang diharapkan
(Harmita, 2004).
Jika plasebo tidak memungkinkan untuk disiapkan, maka sejumlah analit
yang telah diketahui konsentrasinya dapat ditambahkan langsung ke dalam
sediaan farmasi otentik. Metode ini dinamakan metode standard addition method
atau metode penambahan baku (Harmita, 2004).
Jumlah keseluruhan analit kemudian diukur dan dibandingkan dengan
jumlah teoritis, yaitu jumlah analit yang murni berasal dari sediaan farmasi otentik
tersebut, ditambah dengan jumlah analit yg di-spiked ke dalam sediaan. Akurasi
kemudian dinyatakan dalam persen perolehan kembali (%Recovery) (Harmita,
2004).
Persen perolehan kembali ditentukan sebagai rasio antara hasil yang
yang penting untuk diperhatikan adalah metode kuantitasi yang digunakan dalam
penentuan akurasi harus sama dengan metode kuantitasi yang digunakan untuk
menganalisis sampel dalam penelitian (Harmita, 2004).
2.3.2 Presisi
Presisi diekspresikan dengan standar deviasi atau standar deviasi relatif
(RSD) dari serangkaian data. Data untuk menguji presisi seringkali dikumpulkan
sebagai bagian dari kajian-kajian lain yang berkaitan dengan presisi seperti
linearitas atau akurasi. Biasnya replikasi 6-15 dilakukan pada sampel tunggal
untuk tiap-tiap konsentrasi. Pada pengujian dengan KCKT, nilai RSD antara 1-2%
biasanya dipersyaratkan untuk senyawa-senyawa aktif dalam jumlah yang banyak
sedangkan untuk senyawa-senyawa dengan kadar sekelumit RSD berkisar antara
5-15% (Gandjar dan Rohman, 2007).
2.3.3 Spesifitas
Penentuan spesifitas metode dapat diperoleh dengan dua jalan. Cara
pertama adalah dengan melakukan optimasi sehingga diperoleh senyawa yang
dituju terpisah secara sempurna dari senyawa-senyawa lain (resolusi senyawa
yang dituju ≥ 2). Cara kedua untuk memperoleh spesifitas adalah dengan
menggunakan detektor selektif terutama untuk senyawa-senyawa yang terelusi
secara bersama-sama sebagai contoh detektor elektrokimia hanya akan
mendeteksi senyawa tertentu, sementara senyawa yang lainnya tidak terdeteksi.
Penggunaan detektor UV pada panjang gelombang yang spesifik juga merupakan
cara yang efektif untuk melakukan pengukuran selektifitas (Gandjar dan Rohman,
2.3.4 Batas Deteksi dan Batas Kuantitasi
Batas deteksi dan batas kuantitasi dapat ditentukan dengan 2 metode yakni
metode non instrumental visual dan metode perhitungan. Metode non instrumental
visual digunakan pada teknik kromatografi lapis tipis dan metode titrimetri.
Metode perhitungan didasarkan pada simpangan baku respon (SB) dan derajat
kemiringan/slope (b) dengan rumus perhitungan batas deteksi dan batas kuantitasi
sbb:
Simpangan baku respon dapat ditentukan berdasarkan simpangan baku
blanko, simpangan baku residual dari garis regresi atau simpangan baku intersep y
pada garis regresi (Gandjar dan Rohman, 2007).
2.3.5 Linearitas
Lineritas merupakan kemampuan suatu metode untuk memperoleh
hasil-hasil uji yang secara langsung proporsional dengan konsentrasi analit pada kisaran
yang diberikan. Linearitas suatu metode merupakan ukuran seberapa baik kurva
kalibrasi yang menghubungkan antara respon (y) dengan konsentrasi (x).
Linearitas dapat diukur dengan melakukan pengukuran tunggal pada konsentrasi
yang berbeda-beda. Data yang diperoleh selanjutnya diproses dengan metode
kuadrat terkecil, untuk selanjutnya dapat ditentukan nilai kemiringan (slope),
intersep, dan koefisien korelasinya (Gandjar dan Rohman, 2007).
2.3.6 Rentang
Rentang atau kisaran suatu metode didefinisikan sebagai konsentrasi
terendah dan tertinggi yang mana suatu metode analisis menunjukkan akurasi,
presisi, dan linearitas yang mencukupi. Kisaran-kisaran konsentrasi yang diuji
tergantung pada jenis metode dan kegunaannya (Gandjar dan Rohman, 2007).
2.3.7 Kekuatan
Kekuatan/ketahanan dievaluasi dengan melakukan variasi
parameter-parameter metode seperti persentase pelarut organik, pH, kekuatan ionik, suhu,
dan sebagainya. Suatu praktek yang baik untuk mengevaluasi ketahanan suatu
metode adalah dengan memvariasi parameter-parameter penting dalam suatu
metode secara sistematis lalu mengukur pengaruhnya pada pemisahan (Gandjar
BAB III
METODE PENELITIAN
3.1 Tempat dan waktu penelitian
Penelitian ini dilakukan pada Laboratorium Penelitian Fakultas Farmasi
Universitas Sumatera Utara pada bulan Desember 2012 – Januari 2013.
3.2Alat-alat
Alat – alat yang digunakan dalam penelitian ini adalah seperangkat instrumen
KCKT lengkap (Shimadzu) dengan pompa, degasser, penyuntik mikroliter (50µl),
kolom Shimadzu VP-ODS (250 x 4,6 mm), detektor UV-Vis, wadah fase gerak,
vial, Sonifikator (Branson 1510), pompa vakum (Gast DOA – P604 – BN), neraca
analitik (Mettler Toledo), membrane filter PTFE 0,5 µm dan 0,2 µm, cellulose
nitrate membran filter 0,45 µm.
3.3Bahan-bahan
Bahan-bahan yang digunakan adalah asetonitril grade for HPLC (E.Merck®),
akuabides (PT. Ikapharmindo Putramas), flukonazol baku, kapsul Govazol® 150
mg (PT Guardian Pharmatama), kapsul Zemyc® 150 mg (PT Pharos), kapsul Cancid® 150 mg (PT Sunthi Sepuri), dan kapsul Flukonazol 150 mg (PT kimia Farma).
3.4Pengambilan Sampel
Pengambilan sampel dilakukan secara purposif (Sudjana, 2005) yaitu tanpa
membandingkan satu tempat dengan tempat yang lain karena semua sampel
3.5Prosedur Penelitian 3.5.1Pembuatan Fase Gerak
Asetonitril 500 ml disaring dengan menggunakan cellulose nitrate
membrane filter 0,45 µm dan diawaudarakan selama 30 menit. Akuabides 500 ml
disaring dengan menggunakan cellulose nitrate membrane filter 0,45 µm dan
diawaudarakan selama 30 menit.
3.5.2 Pembuatan pelarut
Pelarut dibuat dari asetonitril dan akuabides dengan perbandingan 45:55
dalam labu takar 500 ml. Pelarut lalu disaring dengan penyaring membran
Cellulose Nitrate 0,45 μm dan diawaudarakan selama ± 20 menit menggunakan
sonifikator.
3.5.3 Prosedur Analisis Menggunakan KCKT
3.5.3.1 Penyiapan Kromatografi Cair Kinerja Tinggi
Masing-masing unit diatur, kolom yang digunakan Shimadzu VP-ODS
(250 x 4,6 mm), detektor UV-Vis dan dideteksi pada panjang gelombang 260 nm.
Setelah alat KCKT dihidupkan, maka pompa dijalankan dan fase gerak dibiarkan
mengalir selama 30 menit dengan laju alir 1 ml/menit sampai diperoleh garis alas
yang datar, menandakan sistem tersebut telah stabil.
3.5.3.2 Penentuan Perbandingan Fase Gerak yang Optimum
Pada kondisi kromatografi komposisi fase gerak divariasikan untuk
mendapatkan hasil analisis yang optimum. Perbandingan fase gerak asetonitril:air
yang divariasikan adalah 50:50, 45:55, 40:60, 35:65, dan 30:70 dengan laju alir 1
2,0, theoretical plate lebih besar dari 2000 dan waktu retensi yang singkat yang
akan dipilih dan digunakan dalam penelitian ini.
3.5.4 Analisis Kualitatif Menggunakan KCKT
3.5.4.1 Uji Identifikasi Flukonazol Menggunakan KCKT
Sampel dan bahan baku flukonazol masing-masing dengan konsentrasi 40
µg/ml diinjeksikan sebanyak 20 µl, dianalisis pada kondisi KCKT dengan
perbandingan fase gerak asetonitril:air (45:55) dan laju alir 1 ml/menit serta
panjang gelombang 260 nm. Sampel dinyatakan mengandung flukonazol dengan
membandingkan waktu retensi sampel dan bahan baku flukonazol. Selanjutnya
untuk identifikasi lanjutan, pada larutan sampel flukonazol ditambahkan sedikit
larutan baku flukonazol (spiking) kemudian diinjeksikan dan dianalisa kembali
pada kondisi KCKT yang sama. Diamati luas area dan dibandingkan antara
kromatogram hasil spike dengan kromatogram larutan sampel sebelum spike.
Sampel dinyatakan mengandung flukonazol, jika terjadi peningkatan tinggi
puncak dan luas area pada kromatogram hasil spike.
3.5.5 Analisis Kuantitatif Menggunakan KCKT 3.5.5.1 Pembuatan Larutan Induk Baku Flukonazol
Ditimbang seksama sejumlah 25,0 mg flukonazol baku, dimasukkan ke
dalam labu tentukur 50 ml, dilarutkan dan diencerkan dengan akuabides hingga
garis tanda sehingga diperoleh larutan dengan konsentrasi 500 µg/ml (LIB I).
Dipipet LIB I sebanyak 0,24 ml; 0,44 ml; 0,64 ml; 0,84 ml; dan 1,04 ml,
dimasukkan ke dalam labu tentukur 10 ml, diencerkan dengan pelarut hingga garis
tanda. Kocok sehingga diperoleh konsentrasi 12,0 µg/ml, 22,0 µg/ml, 32,0 µg/ml,
42,0 µg/ml, dan 52,0 µg/ml. Kemudian masing-masing larutan disaring dengan
membran filter PTFE 0,2 µm, dan diinjeksikan ke sistem KCKT sebanyak 20 µl
dan dideteksi pada panjang gelombang 260 nm. Dari luas area yang diperoleh
pada kromatogram dibuat kurva kalibrasi kemudian dihitung persamaan regresi
dan faktor korelasinya
3.5.4.3 Penetapan Kadar Sampel
Ditimbang isi 20 kapsul untuk masing-masing jenis kapsul, kemudian
digerus homogen dan ditimbang seksama sejumlah serbuk setara dengan 25 mg
flukonazol, lalu dimasukkan ke dalam labu tentukur 50 ml, dilarutkan dengan 30
ml akuabides, disonikasi 10 menit dan dicukupkan dengan akuabides hingga garis
tanda sehingga diperoleh larutan dengan konsentrasi 500 µg/ml, dikocok ± 5
menit, kemudian disaring dengan kertas saring, ± 5 ml filtrat pertama dibuang.
Dipipet 0,8 ml filtrat, dimasukkan ke dalam labu tentukur 10 ml, dan dicukupkan
hingga garis tanda dengan pelarut sehingga diperoleh larutan dengan konsentrasi
40 µg/ml. Dikocok ± 5 menit lalu disaring dengan membran filter PTFE 0,2 µm.
Diinjeksikan sebanyak 20 µl ke sistem KCKT dan dideteksi pada panjang
gelombang 260 nm dengan perbandingan fase gerak asetonitril:air (45:55), laju
alir 1 ml/menit. Dilakukan sebanyak 6 kali perlakuan untuk setiap sampel.
Kadar dapat dihitung dengan mensubstitusikan luas area sampel pada Y
dari persamaan regresi: Y = ax + b.
Data perhitungan kadar dianalisis secara statistik menggunakan uji t.
Menurut Harmita (2004), rumus yang digunakan untuk menghitung Standar
Deviasi (SD) adalah:
Kadar dapat dihitung dengan persamaan garis regresi dan untuk
menentukan data diterima atau ditolak digunakan rumus:
t hitung
Dengan dasar penolakan apabila t hitung ≥ t tabel, pada taraf kepercayaan
99% dengan nilai α = 0,01, dk = n – 1.
Keterangan :
SD = Standar deviasi X = Kadar sampel
X = Kadar rata-rata dalam satu sampel
n = Banyaknya data
Menurut Wibisono (2005), untuk mencari kadar sebenarnya dapat
digunakan rumus:
t = Harga ttabel sesuai dengan derajat kepercayaan
dk= Derajat kebebasan
3.5.6 Validasi Metode
3.5.6.1 Akurasi (kecermatan)
Ditimbang 20 kapsul flukonazol yang mengandung kadar zat berkhasiat
Ditimbang serbuk yang mengandung 70% analit dari kadar zat berkhasiat lalu
dilakukan prosedur yang sama seperti pada penetapan kadar sampel. Ditimbang
lagi serbuk yang mengandung 70% analit dari kadar zat berkhasiat dan 30% bahan
baku lalu dilakukan prosedur yang sama seperti pada penetapan kadar sampel.
Dilakukan 3 kali replikasi untuk masing-masing rentang spesifik tersebut.
Menurut Harmita (2004), hasil dinyatakan dalam persen perolehan kembali (%
recovery). Persen perolehan kembali dapat dihitung dengan rumus:
% Perolehan kembali = A
C*A = konsentrasi analit yang ditambahkan (µg/ml)
3.5.6.2 Presisi (Keseksamaan)
Untuk menguji data presisi (RSD), diambil rata-rata dari data % perolehan
kembali (9 kali replikasi) kemudian dihitung standar deviasi. Setelah itu, dihitung
% RSD dengan cara standar deviasi dibagi rata-rata dari % perolehan kembali
kemudian dikali 100%
Menurut Gandjar dan Rohman (2007), nilai RSD dirumuskan dengan:
%
RSD = Standar Deviasi Relatif (%) SD = Standar deviasi
Sementara itu, nilai SD dihitung dengan :
X = nilai dari masing-masing pengukuran
X = rata-rata (mean) dari pengukuran
n = banyaknya data n-1 = derajat kebebasan
3.5.6.3 Batas Deteksi (LOD) dan Batas Kuantitasi (LOQ)
Nilai batas deteksi (LOD) dan batas kuantitasi (LOQ) dihitung dari
persamaan regresi yang diperoleh dari kurva kalibrasi. Menurut Ephstein (2004),
Batas Deteksi (Limit Of Detection/ LOD) dan Batas Kuantitasi (Limit Of
Quantitation/ LOQ) dapat dihitung dengan menggunakan rumus sebagai berikut:
BAB IV
HASIL DAN PEMBAHASAN
4.1 Optimasi komposisi fase gerak asetonitril:air
Pada awal penelitian ini dilakukan optimasi untuk mendapatkan kondisi
kromatografi yang optimal. Adapun fase gerak yang dioptimasi yaitu
perbandingan 50:50, 45:55, 40:60, 35:65, dan 30:70, pada laju alir 1 ml/menit,
deteksi dilakukan pada panjang gelombang 260 nm menggunakan kolom
Shimadzu VP-ODS (250 x 4,6 mm). Dari Tabel 1 di bawah dapat dilihat hasil
optimasi dari perbandingan fase gerak asetonitril:air yang digunakan.
Tabel 1 Data Optimasi Perbandingan Fase Gerak Asetonitril:Air
Perbandingan fase gerak yang dipilih dari hasil optimasi yaitu pada
perbandingan asetonitril:air (45:55). Pemilihan fase gerak ini didasarkan pada
nilai tailing factor lebih kecil dari 2,0 dan nilai theoritical plate lebih besar dari
2000 dengan waktu retensi paling kecil, yaitu 3,364 menit.
4.2 Analisis Kualitatif
Analisis kualitatif flukonazol ditentukan dengan parameter waktu retensi,
yaitu dengan cara membandingkan waktu retensi sampel dengan bahan baku.
Hasil kromarogram dapat dilihat pada Gambar 3 dan Gambar 4 di bawah ini.
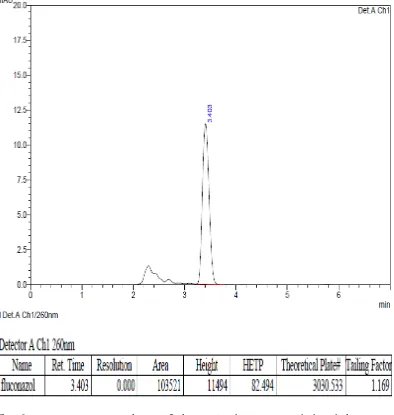
Gambar 4 Kromatogram kapsul flukonazol secara KCKT menggunakan kolom Shimadzu VP-ODS (250 x 4,6 mm) dengan perbandingan fase gerak asetonitiril:air (45:55) dan laju alir 1 ml/menit, volume penyuntikan 20 µl dan deteksi pada panjang gelombang 260 nm.
Dari kromatogram di atas dapat dilihat bahwa waktu retensi flukonazol
dalam sediaan kapsul sama dengan waktu retensi flukonazol baku, yaitu 3,3
menit. Hal ini berarti sampel yang ditentukan mengandung flukonazol.
Selanjutnya untuk dapat memastikan kebenaran analisa sampel
mengandung flukonazol maka dilakukan spiking yaitu menambahkan bahan baku
flukonazol ke dalam sampel dan ditentukan secara KCKT. Hasil kromatogram
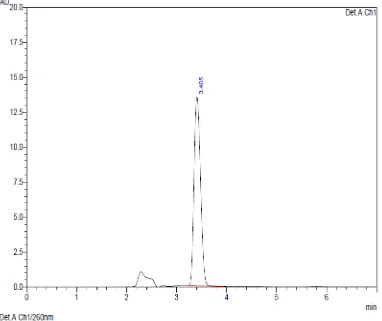
Gambar 6 Kromatogram kapsul flukonazol hasil spike secara KCKT
menggunakan kolom Shimadzu VP-ODS (250 x 4,6 mm) dengan perbandingan fase gerak asetonitril:air (45:55) dan laju alir 1 ml/menit, volume penyuntikan 20 µl dan deteksi pada panjang gelombang 260 nm.
Dari kromatogram diatas dapat dilihat bahwa terjadi peningkatan luas area
dan tinggi puncak pada kromatogram setelah penambahan baku dibandingkan
dengan sebelum penambahan bahan baku maka dapat diambil kesimpulan sampel
4.3 Analisis Kuantitatif
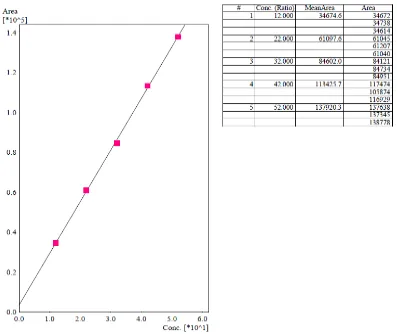
4.3.1 Penentuan Kurva Kalibrasi
Dari hasil penentuan kurva kalibrasi flukonazol baku yang ditentukan
berdasarkan luas area pada konsentrasi 12,0 µg/ml, 22,0 µg/ml, 32,0 µg/ml, 42,0
µg/ml, dan 52,0 µg/ml diperoleh hubungan yang linier dengan koefisien korelasi,
r = 0,9996 dan persamaan regresi Y = 2588,195X + 3521,8. Nilai r mendekati 1
menunjukkan adanya korelasi linier yang menyatakan adanya hubungan antara X
dan Y (Sudjana, 2005). Hasil penentuan kalibrasi dapat dilihat pada Gambar 5di
bawah ini.
4.3.2 Penetapan Kadar Analit dalam Sampel yang dianalisis
Analisis kuantitatif flukonazol dapat ditentukan berdasarkan luas area
kromatogram atau tinggi puncak. Dalam penelitian ini ditentukan berdasarkan
luas area kromatogram karena luas area dianggap merupakan parameter yang
lebih akurat untuk pengukuran kuantitatif (Ditjen POM, 1995). Hasil penetapan
kadar flukonazol dalam sediaan kapsul dengan nama dagang dan generik dapat
dilihat pada Tabel 2di bawah ini.
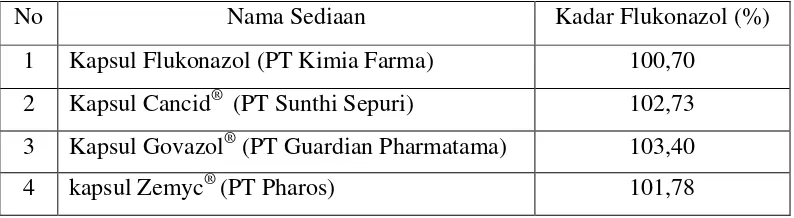
Tabel 2 Hasil penetapan kadar flukonazol dalam sediaan kapsul dengan nama dagang dan generik
Persyaratan kadar sediaan ditetapkan berdasarkan persyaratan tablet
flukonazol menurut USP Revision Bulletin 33th Edition tahun 2010 yaitu
mengandung flukonazol tidak kurang dari 90,0% dan tidak lebih dari 110,0% dari
jumlah yang tertera pada etiket. Hal ini dikarenakan tidak terdapatnya monografi
kapsul flukonazol dalam USP maupun FI edisi IV. Dari tabel diatas diperoleh
kesimpulan kadar kapsul flukonazol memenuhi persyaratan yang ditetapkan.
4.4 Hasil Uji Validasi
Pada penelitian ini dilakukan uji validasi metode dengan metode standar
adisi terhadap sampel kapsul Flukonazol (PT Kimia Farma) yang meliputi uji
akurasi dengan parameter % recovery dan uji presisi dengan parameter RSD
No Nama Sediaan Kadar Flukonazol (%)
1 Kapsul Flukonazol (PT Kimia Farma) 100,70 2 Kapsul Cancid® (PT Sunthi Sepuri) 102,73 3 Kapsul Govazol® (PT Guardian Pharmatama) 103,40
(Relative Standard Deviasi), LOD (Limit of Detection) dan LOQ (Limit of
Quantitation).
Uji akurasi dengan parameter % recovery dilakukan dengan membuat tiga
konsentrasi analit dengan rentang spesifik 80%, 100%, dan 120%, masing-
masing dengan tiga replikasi dan setiap rentang spesifik mengandung 70% analit
dan 30% baku pembanding (Harmita, 2004).
Data hasil ujivalidasi metode yang dilakukan dengan metode adisi standar
dapat dilihat padaTabel 3.
Tabel 3 Hasil Pengujian Validasi, dengan parameter Akurasi, dan Presisi Flukonazol pada Kapsul Flukonazol (PT Kimia Farma) dengan Menggunakan Metode Adisi Standar
Dari tabel di atas diperoleh hasil pengujian akurasi dengan kadar rata-rata
% recovery 100,34%. Hasil ini dapat diterima karena memenuhi syarat uji akurasi,
bahwa rentang rata-rata % recovery ialah 98-102%. Maka dapat disimpulkan
bahwa metode ini mempunyai akurasi yang baik (Epshtein, 2004). No
Luas Area Konsentrasi ( µg/ml ) Reco
very
1 80 9,504 61456 86300 22,384 31,983 100,99
2 80 9,504 60392 84580 21,973 31,461 99,83
3 80 9,504 60711 85495 22,096 31,672 100,75
4 100 11,880 75613 106208 27.854 39,675 99,50
5 100 11,880 75815 107083 27,932 40,013 101,69
6 100 11,880 75740 106563 27,903 39,812 100,24
7 120 14,252 89023 125907 33,035 47,286 99,.99
8 120 14,252 90444 127100 33,584 47,747 99,38
9 120 14,252 90164 127325 33,476 47,834 100,75
Kadar rata – rata (%) Recovery = 100,34
Hasil uji presisi dengan parameter RSD (Relative Standard Deviasi)
diperoleh 0,75%, persyaratan nilai RSD yang ditentukan adalah < 2%. Maka dapat
disimpulkan bahwa metode analisis mempunyai presisi yang baik (Harmita,
2004).
Batas deteksi dan batas kuantitasi dihitung dari persamaan regresi yang
diperoleh dalam kurva kalibrasi. Dari hasil perhitungan diperoleh nilai LOD
BAB V
KESIMPULAN DAN SARAN
5.1 Kesimpulan
Hasil optimasi fase gerak pada penetapan kadar flukonazol dalam sediaan
kapsul diperoleh perbandingan fase gerak asetonitril:air (45:55), laju alir 1 ml/
menit, menggunakan kolom Shimadzu VP-ODS (250 x 4,6 mm) dan dideteksi
pada panjang gelombang 260 nm. Metode ini memberikan hasil uji validasi yang
memenuhi syarat.
Kadar flukonazol dalam kapsul yang dianalisis dari sediaan kapsul dengan
nama dagang dan generik yang terdapat di pasaran dengan kondisi kromatografi
yang terpilih diperoleh hasil yang memenuhi persyaratan kadar pada USP
Revision Bulletin 33th Edition yaitu mengandung flukonazol tidak kurang dari 90,0
% dan tidak lebih dari 110,0 % dari jumlah yang tertera pada etiket.
5.2 Saran
Disarankan agar dilakukan penelitian lebih lanjut terhadap penetapan
kadar flukonazol dalam sediaan farmasi lainnya secara KCKT dengan fase gerak
DAFTAR PUSTAKA
Anonim. (2010). Fluconazole. Tanggal akses 23 Desember 2012.
Berridge, J.C. (1985). Techniques for the Automated Optimization of HPLC Separations. Britain: John Wiley & Sons Ltd. Halaman 1-4.
Corrêa, J.C.R., Soarres, C.D.V., dan Salgado, H.R.N. (2012). Development and Validation of Dissolution Test for Fluconazole Capsules by HPLC and Derivative UV Spectrophotometry. Chromatography Research International Article. Kanada: Hindawi Publishing Corporation. Halaman
1-8.
Ditjen POM. (1995). Farmakope Indonesia. Edisi IV. Jakarta: Departemen
Kesehatan RI. Halaman 1002.
Dong, M.W. (2005). Modern HPLC for Practicing Scientists. Chichester: John wiley & Sons Ltd. Halaman 39-42, 84-86.
Épshtein, N.A. (2004). Validation of HPLC Techniques for Pharmaceutical Analysis. Pharmaceutical Chemistry Journal. 38(4): 212 – 228
Gandjar, I.G., dan Rohman, A. (2007). Kimia Farmasi Analisis. Yogyakarta:
Pustaka Pelajar. Halaman 323, 378-382, 393-397, 465-470.
Gritter, R.J., Bobbit, J.M., dan Schwarting, A.E. (1985). Introduction of
Chromatography. Penerjemah Kosasih Padmawinata. Pengantar
Kromatografi. Edisi Ketiga. Bandung: Penerbit ITB. Halaman 186-239.
Harmita. (2004). Petunjuk Pelaksanaan Validasi Metode dan Cara Perhitungannya. Review Artikel. Majalah Ilmu Kefarmasian. Vol 1(3):
117-135.
Johnson, E.L., dan Stevenson, R. (1978). Basic Liquid Chromatography.
Penerjemah Kosasih Padmawinata. Dasar Kromatografi Cair. Bandung:
Penerbit ITB. Halaman 16, 278-279.
McMaster, M.C. (2007). HPLC A Practical User’s Guide. Edisi Kedua. New
Jersey: John Wiley and Sons Inc. Halaman 7.
Meyer, V.R. (2004). Practical High Peformance Liquid Chromatography. Edisi
Kedua. Chichester: John Wiley & Sons Ltd. Halaman 17-56.
Miller, J.M. (2005). Chromatography-Concepts and Contrasts. Chichester: John
Moffat, A.C., Osselton, M.D., dan Widdop, B. (2005). Clarke‘s Analysis Of Drug And Poisons. Edisi Ketiga. London: Pharmaceutical Press. Electronic
version.
Rohman, A. (2009). Kromatografi untuk Analisis Obat. Cetakan Pertama.
Yogyakarta: Graha Ilmu. Halaman 111-122 dan 222-240.
Sadasivudu, P., Shastri, N., dan Sadanandam, M. (2009). Development and Validation of RP-HPLC and UV Methods of Analysis for Fluconazole in Pharmaceutical Solid Dosage Forms. International Journal of ChemTech Research 4(1): 1131-1136.
Setiabudi, R., dan Bahry, B. (2007). Obat Jamur. Dalam: Farmakologi dan
Terapi. Edisi kelima. Bagian Farmakologi FKUI. Jakarta: Universitas Indonesia Press. Halaman 571-578.
Sudjana. (2005). Metode Statistika. Edisi VI. Bandung: Tarsito. Halaman 168.
Tan, T.H., dan Rahardja, K. (2007). Obat-Obat Penting Khasiat, Penggunaan,
dan Efek-Efek Sampingnya. Edisi Keenam. Jakarta: PT. Elex Media
Komputindo. Halaman 101-107.
USP Conventional Inc. (2006). The United States Pharmacopeia. Edisi Kedua
puluh sembilan. United States: Electronic Version. Halaman 911
USP Conventional Inc. (2010). The United States Pharmacopeia Revision Buletin.
Edisi Ketiga puluh tiga. United States: Electronic Version.
Wibisono, Y. (2005). Metode Statistik. Yogyakarta: Gajah Mada University Press.
Lampiran 1 Kromatogram Penyuntikan Larutan Flukonazol untuk
Mencari Komposisi Fase Gerak Asetonitril:Air yang Optimum pada Analisis
Perbandingan fase gerak asetonitril:air (50:50) dengan laju alir 1 ml/menit
Perbandingan fase gerak asetonitril:air (40:60) dengan laju alir 1 ml/menit.
Lampiran 2 Kromatogram Larutan Flukonazol Baku pada Pembuatan Kurva Kalibrasi
A
Perbandingan fase gerak asetonitril:air (45:55) dengan laju alir 1 ml/menit, konsentrasi 12,0 µg/ml.
B