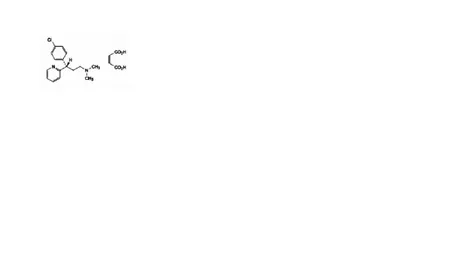Disusun Oleh: Disusun Oleh: Wanda
Wanda Indriani Indriani Wibowo Wibowo 098114003098114003 Kenny
Kenny Ryan Ryan Limanto Limanto 098114006098114006 Bernadetta
Bernadetta Arum Arum Wijayanti Wijayanti 098114007098114007 Rachelia
Rachelia Octavia Octavia 098114008098114008 Johanes
Johanes Putra Putra Wicaksono Wicaksono 098114010098114010 Dina
Dina Christin Christin 098114015098114015 Jenny
Jenny Marina Marina 098114016098114016
KELOMPOK A KELOMPOK A11 LABORATORIUM BIOANALISIS LABORATORIUM BIOANALISIS FAKULTAS FARMASI FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA UNIVERSITAS SANATA DHARMA
YOGYAKARTA YOGYAKARTA
2011 2011
REVIEW JURNAL REVIEW JURNAL Kromatografi
Kromatografi
Kromatografi merupakan teknik analisis yang paling sering digunakan pada Kromatografi merupakan teknik analisis yang paling sering digunakan pada analisis senyawa dalam sediaan farmasi maupun cairan biologis. Berdasarkan alat analisis senyawa dalam sediaan farmasi maupun cairan biologis. Berdasarkan alat yang digunakan, kromatografi dapat dibagi menjadi empat, antara lain: (a) yang digunakan, kromatografi dapat dibagi menjadi empat, antara lain: (a) kromatografi kertas; (b) kromatografi lapis tipis; (c) kromatografi cair kinerja kromatografi kertas; (b) kromatografi lapis tipis; (c) kromatografi cair kinerja tinggi; dan (d) kromatografi gas. Dalam penelitian ini akan dibahas lebih lanjut tinggi; dan (d) kromatografi gas. Dalam penelitian ini akan dibahas lebih lanjut tentang kromatografi cair kinerja tinggi (KCKT) yang juga dikenal sebagai HPLC tentang kromatografi cair kinerja tinggi (KCKT) yang juga dikenal sebagai HPLC (( High Performance Liquid Chromatog High Performance Liquid Chromatographyraphy) ) (Gandjar, (Gandjar, 2010).2010).
Kromatografi merupakan teknik yang mana solut atau zat-zat terlarut Kromatografi merupakan teknik yang mana solut atau zat-zat terlarut terpisah oleh perbedaan kecepatan elusi, dikarenakan solut-solut ini melewati terpisah oleh perbedaan kecepatan elusi, dikarenakan solut-solut ini melewati suatu kolom kromatografi. Pemisahan solut-solut ini diatur oleh distribusi solut suatu kolom kromatografi. Pemisahan solut-solut ini diatur oleh distribusi solut dalam fase gerak dan fase diam. Instrumentasi KCKT pada dasarnya terdiri atas dalam fase gerak dan fase diam. Instrumentasi KCKT pada dasarnya terdiri atas delapan komponen pokok yaitu: (1) wadah fase gerak, (2) sistem penghantaran delapan komponen pokok yaitu: (1) wadah fase gerak, (2) sistem penghantaran fase gerak; (3) alat untuk memasukkan sampel; (4) kolom; (5) detektor; (6) wadah fase gerak; (3) alat untuk memasukkan sampel; (4) kolom; (5) detektor; (6) wadah penampung buangan fase
penampung buangan fase gerak; (7) gerak; (7) tabung penghubung, dan tabung penghubung, dan (8) suatu (8) suatu komputerkomputer atau
atauintegrator integrator atau perekam (Gandjar, 2010). atau perekam (Gandjar, 2010). Liquid chromatography-mass spectrometry
Liquid chromatography-mass spectrometry (LC-MS atau alternatif KC-SM) (LC-MS atau alternatif KC-SM) adalah teknik
adalah teknik kimia analisiskimia analisis yang menggabungkan kemampuan pemisahan fisikyang menggabungkan kemampuan pemisahan fisik dari kromatografi cair (KCKT) dengan kemampuan analisis (detektor) dari kromatografi cair (KCKT) dengan kemampuan analisis (detektor) spektrometer massa. LC-MS adalah teknik yang banyak digunakan untuk berbagai spektrometer massa. LC-MS adalah teknik yang banyak digunakan untuk berbagai aplikasi yang memiliki sensitivitas dan spesifisitas sangat tinggi. Pada umumnya aplikasi yang memiliki sensitivitas dan spesifisitas sangat tinggi. Pada umumnya aplikasi LC-MS berorientasi pada deteksi dan identifikasi potensi spesifik suatu aplikasi LC-MS berorientasi pada deteksi dan identifikasi potensi spesifik suatu senyawa terhadap keberadaan senyawa lainnya (dalam campuran yang kompleks). senyawa terhadap keberadaan senyawa lainnya (dalam campuran yang kompleks).
Pendahuluan Pendahuluan
Pada penelitian ini, dilakukan pengembangan dan validasi metode analisis Pada penelitian ini, dilakukan pengembangan dan validasi metode analisis KC-MS yang sederhana, spesifik, cepat, sensitif untuk menetapkan kadar KC-MS yang sederhana, spesifik, cepat, sensitif untuk menetapkan kadar chlorpheniramine maleate
chlorpheniramine maleate (RS-CPM) dalam plasma manusia. Tujuan dari (RS-CPM) dalam plasma manusia. Tujuan dari penetapan
penetapan kadar kadar obat obat dalam dalam plasma plasma adalah adalah mengetahui mengetahui bioavailabilitas bioavailabilitas dandan bioekivalensi dari suatu obat.
Gambar 1. StrukturDexchlor pheni rami ne maleate
Dexchlorpheniramine maleate (RS-CPM) atau yang dikenal juga sebagai 3-(4-chlorophenyl)-N,N-dimethyl-3-(2-pyridyl) propylamine monomaleate merupakan obat antihistamin yang poten. Dalam penggunaannya, CPM banyak digunakan untuk meringankan gejala penyakit tertentu, seperti demam dan alergi. Struktur dari CPM yang mempunyai atom C kiral yang mengakibatkan CPM terdapat dalam bentuk sinister (berotasi berlawanan arah jarum jam) dan rectus (berotasi searah jarum jam). Berdasarkan penelitian yang ada, CPM yang mempunyai aktivitas farmakologi adalah S-CPM (dextrorotatory S-enantiomer ). Di dalam Farmakope Eropa III ( European Pharmacopoeia III ), metode KCKT
maksimum yang diijinkan dalam sampel yang diuji sebesar 2% b/b. Dari studi farmakokinetika yang telah dilakukan, diketahui bahwa kadar CPM dalam plasma sangatlah rendah, yaitu berada pada level maksimal konsentrasi sebesar 6,2 ng/mL dan 7,0-8,2 ng/ mL untuk pemberian oral dengan dosis tunggal 2 mg dan 4 mg. Dari penelitian terdahulu, banyak metode yang dapat digunakan untuk menetapkan kadar RS-CPM, antara lain GC (Gas Chromatography), GC-MS (Gas Chromatography-Mass Spectrometry), dan KCKT ( High Performance Liquid Chromatography).
Pada penelitian ini, dilakukan pengembangan dan validasi metode analisis KCKT-MS yang sederhana, spesifik, cepat, sensitif untuk menetapkan kadar S-CPM dalam plasma manusia. Pengembangan dari metode ini digunakan untuk menetapkan kadar S-CPM dalam plasma setelah pemberian dosis tunggal tablet yang mengandung S-CPM sebanyak 6 mg pada subjek uji pria yang sehat. Dari dapat digunakan untuk menentukan kemurnian dari S-CPM, dimana kadar R-CPM
metode ini, dipeoleh nilai LOQ sebesar 1,0 ng/ mL dengan waktu pengerjaan selama 5 menit.
Gambar 2. StrukturSimvastatin (STA)
Alat dan Bahan
Dalam penelitian ini, standar S-CPM yang digunakan mempunyai kemurnian 99,67%. Standar internal simvastatin (STA) yang digunakan mempunyai kemurnian 98,44%. Perlu ditambahkan standar internal sebagai faktor koreksi sebab zat analit yang diuji memiliki konsentrasi yang sangat kecil dalam sampel (plasma). Pemilihan simvastatin sebagai standar internal dikarenakan alasan sebagai berikut:
1. terpisah sempurna dari peak senyawa yang dianalisis dan peak lain, 2. memiliki waktu retensi yang mirip dengan sampel,
3. tidak terdapat dalam sampel awal,
4. dapat me-mimic analit disetiap tahap preparasi sampel,
5. tidak harus memiliki kemiripan secara kimiawi dengan analit dan 6. stabil dan tidak bereaksi dengan sampel atau fase gerak.
Ada berbagai macam cairan biologis yang dapat digunakan, meliputi darah, serum, plasma, urin, keringat, saliva, empedu, air susu dan air mata. Cairan biologis yang digunakan dalam penelitian ini merupakan plasma. Perbedaan plasma dan serum adalah pada plasma diberikan antikoagulan (berupa heparin), sedangkan pada serum tidak. Pemilihan plasma sebagai sampel biologis
disebabkan karena protein (yang mengikat obat) dalam plasma tidak mengendap sehingga obat masih berada di dalam plasma dan dapat diukur kadarnya.
Untuk pembuatan larutan stok S-CPM dan standar internal (STA), analit dilarutkan dengan asetonitril sehingga dihasilkan larutan dengan konsentrasi 1000 µg/ mL. Asetonitril digunakan sebagai pelarut S-CPM dan standar internal supaya kedua analit yang akan ditetapkan kadarnya berada dalam bentuk bebas sehingga bersifat lebih non-polar, dapat larut dalam pelarut organik dan dapat dipisahkan dari protein (senyawa endogen). Senyawa endogen harus dipisahkan dari larutan uji supaya tidak mengganggu kinerja alat yang digunakan. Senyawa endogen (seperti protein) memiliki ukuran yang besar sehingga dapat menyumbat kolom dan mengganggu pembacaan serapan yang dihasilkan.
Larutan stok sekunder dan kerja dibuat dengan cara mengencerkan larutan stok dengan campuran pelarut air: asetonitril (50:50 v/v). Larutan stok kerja yang telah dibuat digunakan untuk membuat kurva baku dan uji kontrol kualitas dari sampel yang akan ditetapkan kadarnya. Pada percobaan ini, kurva kalibrasi digunakan untuk melihat adanya korelasi antara respon yang dihasilkan (AUC) dan konsentrasi, dimana semakin tinggi konsentrasi maka AUC yang dihasilkan akan semakin besar juga. Kurva kalibrasi dibuat dengan delapan seri konsentrasi larutan baku S-CPM dalam rentang 1,0 – 150,0 ng/mL.
Kontrol kualitas sampel berfungsi sebagai kontrol dalam validasi metode analisis untuk sampel. Kontrol ini dilakukan dengan cara menambahkan S-CPM yang diketahui konsentrasinya ke dalam blanko plasma. Sampel kemudian dihomogenkan dengan vortex dan disimpan pada suhu -70 ± 2oC. Sampel disimpan pada suhu rendah (-70 ± 2oC) supaya terjadi inaktivasi enzim yang mungkin terdapat di dalam plasma. Enzim tersebut dapat mendegdradasi obat yang akan diuji.
Sampel disiapkan dengan cara ekstraksi cair-cair. Sebanyak 0,5 mL alikuot plasma darah manusia dicampur dengan 0,1 mL larutan standar kerja internal (STA dengan konsentrasi 2500,0 ng/ mL) dan ditambahkan sebanyak 1,0 mL buffer borat dengan pH 9,00, lalu dicampurkan. Tujuan penambahan buffer borat dengan pH 9,00 adalah agar CPM berada dalam suasana basa sehingga CPM lebih
mudah larut dalam pelarut organik yaitu etil asetat sedangkan protein masuk ke dalam fase air. Larutan tersebut dihomogenkan dengan vortex dan diekstraksi dengan menggunakan etil asetat sebanyak 3 x 2 mL dengan tujuan untuk memperoleh pemisahan yang sempurna. Tidak dilakukan dalam satu kali ekstraksi karena dikhawatirkan masih ada CPM yang belum terlarut dalam etil asetat. Diambil fase etil asetat karena senyawa yang akan ditetapkan kadarnya memiliki kelarutan yang besar pada etil asetat (lapisan atas). Larutan yang terdapat pada lapisan atas diuapkan sehingga diperoleh residu. Residu tersebut kemudian dilarutkan dalam fase gerak dengan volume yang kecil untuk memperoleh konsentrasi yang lebih pekat. Hal ini dikarenakan sampel yang digunakan dalam penelitian ini merupakan trace analysis sample, dimana kmsentrasi sampel yang
akan ditetapkan kadarnya sangat kecil sehingga perlu dilakukan pemekatan.
Instrumentasi Alat
Pada analisis Dexchlorpheniramine maleate dalam plasma darah digunakan instrumen berupa Kromatografi Cair Kinerja Tinggi (KCKT) dengan detektor spektrometer massa. Penggunaan KCKT didasarkan pada pemisahan yang relatif baik, cepat, sensitif, dan spesifik dibandingkan dengan Kromatografi Cair (KC)
karena KCKT merupakan teknik pemisahan yang diberikan tekanan tinggi.
Kombinasi Kromatografi Cair - Spektrometer Massa (KC-SM) memiliki beberapa kelebihan yaitu:
1. dapat digunakan untuk menganalisis molekul obat dan metabolitnya dengan BM yang rendah maupun tinggi,
2. SM memberikan identifikasi yang baik sebagai detektor pada KCKT karena bobot molekul merupakan salah satu determinasi yang spesifik untuk tiap molekul dan jika informasi ini digabungkan dengan strukturnya, dapat memberikan identifikasi yang baik
3. SM memiliki selektifitas yang tinggi terkait dengan kemampuannya dalam identifikasi dan
4. selektifitas SM digunakan untuk menandai standar internal dan analit, ditambah dengan sensitifitas, akurasi, dan presisi determinasi kuantitatif yang baik.
Fase gerak yang digunakan pada penetapan kadar S-CPM yaitu metanol:amonium asetat (90:10) dengan kecepatan aliran 0,5 mL/ menit. Dalam percobaan ini, waktu pengerjaan yang dibutuhkan hanya enam menit. Hal ini dikarenakan dalam waktu enam menit, standar dan sampel sudah terpisah dengan baik. Pemilihan fase gerak disesuaikan berdasarkan kepolaran analit yang akan
dianalisis. Kecepatan alir yang digunakan adalah 0,5 mL/ menit karena pada kecepatan alir tersebut diperoleh pemisahan yang baik. Hal ini ditandai dengan resolusi peakyang dihasilkan lebih besar dari 1,5.
Pengembangan Metode
Hal ini bertujuan untuk mengembangkan dan memvalidasi metode uji yang sederhana, cepat, dan sensitif untuk melakukan kuantifikasi S-CPM. Metode ini cocok untuk menentukan farmakokinetika dari suatu senyawa uji. Untuk mencapai tujuan tersebut, diperlukan optimasi ekstraksi sampel, deteksi parameter dan kromatografi. Larutan standar S-CPM dianalisis menggunakan KC-SM dengan sistem injeksi langsung yang kemudian diperiksa dengan bantuan ESI dan APCI. Berdasarkan data spektra massa yang diperoleh, berat molekul untuk S-CPM adalah 274,9.
Dalam KCKT, tujuan dari optimasi fase gerak adalah untuk mendapatkan resolusi peak yang baik dan simetris baik analit dan standar internal (SI). Prinsip pemisahan pada KCKT adalah molekul yang terlarut dalam fase gerak akan melewati kolom yang merupakan fase diam. Molekul yang memiliki ikatan yang kuat dengan kolom akan cenderung bergerak lebih lambat dibanding molekul yang berikatan lemah dengan kolom. Dengan demikian, berbagai macam tipe molekul dapat dipisahkan berdasarkan kepolaran dan pergerakan pada kolom. Fase gerak yang akan dioptimasi adalah amonium asetat, asam asetat, atau kombinasi dari keduanya pada konsentrasi yang berbeda. Dari hasil penelitian, diperoleh bahwa campuran dari asetonitril-air (mengandung 10 mL amonium asetat dan 0,5% asam asetat) (90:10 v/v) dapat digunakan sebagai fase gerak. Hal ini dikarenakan komposisi fase gerak tersebut memberikan pemisahan yang baik. Junlah buffer yang ditambahkan (amonium asetat dan asam asetat) perlu dioptimasi untuk mempertahankan bentuk peak karena jumlah buffer
mempengaruhi derajat ionisasi dan fragmentasi saat dideteksi dengan spektrometer massa.
Selain itu perlu dilakukan optimasi kolom dan setelah dilakukan pembandingan terhadap beberapa jenis kolom, maka digunakan kolom
Phenomenex (Luna)-ODS (100x4,6 mm, i.d. 5 µm) dengan flow rate 0,5 mL/ menit untuk dapat menghasilkan bentuk peak yang baik dan waktu proses yang diijinkan adalah selama 2 menit. Ekstraksi pada tahap preparasi sampel sangat mempengaruhi hasil pemisahan dan deteksi dari spektometer massa. Hal ini dikarenakan adanya pengotor dapat menyumbat kolom dan bahan yang mudah menguap dapat mengganggu saat deteksi. Oleh karena itu dilakukan optimasi dengan menggunakan enam macam pelarut organik, antara lain dietil eter, etil asetat, heksana, diklorometan, kloroform, dan butil metil eter, serta campuran dari pelarut-pelarut organik tersebut dengan kombinasi dan rasio yang berbeda. Dari
hasil optimasi, etil asetat menghasilkan kromatogram yang paling baik sehingga etil asetat digunakan sebagai pelarut untuk mengekstraksi sampel.
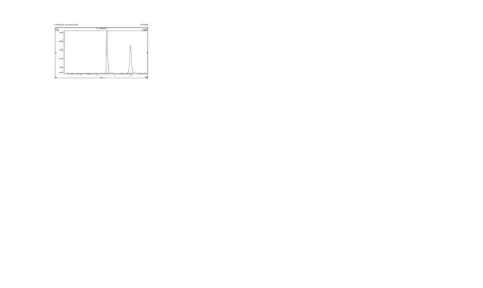
Gambar 4. Kromatogram hasil pemisahan pada blangko
Validasi
Penetapan kadar S-CPM dalam sampel plasma yang diambil dari sukarelawan dilakukan pada kondisi kromatografi yang optimal. Parameter validasi seperti akurasi, presisi (keterulangan dan reprodusibilitas), linearitas dan rentang, sensitivitas (LOD dan LOQ), robustness, stabilitas, selektifitas/ spesifitas dan uji kesesuaian sistem perlu dievaluasi terlebih dahulu. Hasil dari validasi ditunjukkan pada tabel berikut :
Tabel 1. Studi presisi S-CPM dari hasil pengukuran (ng/ mL)
Larutan baku yang telah ditambahkan dengan standar internal, blanko tanpa penambahan standar internal, blanko dengan penambahan standar internal, kurva
kalibrasi, uji kualitas kontrol sampel dianalisis dan direkam kromatogramnya. Fase gerak yang digunakan untuk penelitian memberikan pemisahan yang baik antara sampel, standar internal dan senyawa endogen. Dari kromatogram yang dihasilkan tidak tampak adanya gangguan pada waktu retensi sampel dan standar internal.
Gambar 5. Kromatogram hasil pemisahan matriks biologis (plasma)
Akurasi
Akurasi adalah ukuran yang menunjukkan derajat kedekatan hasil analisis dengan kadar analit yang sebenarnya. Akurasi ini dinyatakan dengan persen (%) perolehan kembali (recovery) analit yang ditambahkan. Akurasi harus diukur dengan menggunakan minimal lima determinasi per konsentrasi dan minimal tiga konsentrasi dalam kisaran konsentrasi yang diharapkan adalah dianjurkan. Nilai rata-rata harus berada dalam jarak 15% dari nilai yang sebenarnya kecuali pada LLOQ, di mana seharusnya tidak menyimpang lebih dari 20%. Deviasi rata-rata dari nilai yang sebenarnya berfungsi sebagai ukuran akurasi (Anonim, 2001).
Kisaran batas akurasi harus berada dalam rentang linier. Akurasi khas dari pemulihan zat obat dalam campuran harus sekitar 98-102%. Nilai-nilai keakuratan data di luar rentang pemulihan harus dipertimbangkan (Anonim, 2001). Sedangkan menurut Mulja dan Hanwar, akurasi untuk kadar obat yang besar adalah sebesar 95-105% dan untuk bioanalisis rentang 80-120% masih bisa diterima (Mulja dan Hanwar, 2003).
Pada penelitian ini, akurasi dari metode ini ditentukan oleh recovery relatif dan absolut (Tabel 1). Nilai persentase recovery absolut S-CPM berkisar antara 89,06% sampai 91,32 dan nilai recovery relatif berkisar antara 88.07 % sampai 91,33%. Apabila penelitian ini mengikuti parameter dari Chan, maka penelitian ini dapat dikatakan belum memiliki akurasi yang baik sebab Chan dalam bukunya
menyebutkan bahwa akurasi zat obat dalam campuran harus berada dalam range antara 98-102%. Namun berbeda apabila penelitian ini mengikuti parameter dari Mulja dan Hanwar karena dalam bukunya, Mulja dan Hanwar mengatakan bahwa akurasi untuk bioanalisis dalam rentang 80-120% masih dapat diterima dimana akurasi yang dalam penelitian ini dinyatakan dalam persen recovery masih masuk dalam range tersebut baik recovery absolut maupun untuk recovery relatifnya.
Presisi
Presisi adalah ukuran yang menunjukkan derajat kesesuaian antara hasil uji individual, diukur melalui penyebaran hasil individual dari rata-rata jika prosedur diterapkan secara berulang-ulang pada sampel-sampel yang diambil dari campuran yang homogen (Harmita, 2004). Presisi dinyatakan dalam koefisien variasi (KV). Suatu metode dapat dikatakan baik apabila memiliki KV <2% (Harmita, 2004).
Akurasi harus diukur dengan menggunakan minimal lima determinasi tiap konsentrasi dan minimal tiga konsentrasi dalam kisaran konsentrasi yang diharapkan. Nilai rata-rata harus berada dalam jarak 15% dari nilai yang sebenarnya kecuali pada LLOQ, dimana seharusnya tidak menyimpang lebih dari 20%. Deviasi dari rata-rata dari nilai yang sebenarnya berfungsi sebagai ukuran akurasi (Anonim, 2001). Pada penelitian ini, digunakan tiga macam konsentrasi dalam sampel yaitu 25, 75, 125 ng/ mL. Hasil ini dapat dikatakan sudah memiliki presisi yang cukup baik apabila mengikuti parameter yang disebutkan oleh Harmita karena % CV yang terdapat pada penelitian ini rata-rata kurang dari 2%, hanya satu sampel yang memiliki konsentrasi lebih dari 2% yaitu sampel pada kondisi long-term stability dengan % CV = 2,07. Dapat disimpulkan bahwa metode yang dikembangkan cukup akurat dan dapat dipercaya.
Tabel 2. Uji stabilitas S-CPM pada plasma
Spesifisitas
Spesifisitas atau selektivitas suatu metode adalah kemampuan yang hanya mengukur zat tertentu saja secara cermat dan seksama dengan adanya komponen lain yang mungkin ada dalam matriks sampel. Selektivitas seringkali dapat dinyatakan sebagai derajat penyimpangan metode yang dilakukan terhadap sampel yang mengandung bahan yang ditambahkan berupa cemaran, hasil urai, senyawa sejenis, senyawa asing lainnya, dan dibandingkan terhadap hasil analisis sampel yang tidak mengandung bahan lain yang ditambahkan ( Harmita, 2004).
Spesifisitas metode ini diuji sebanyak enam kali, dimana blanko plasma yang telah ditambah dengan standar dan sampel dibandingkan dengan kromatogram dari larutan baku. Dari hasil yang diperoleh, dapat disimpulkan bahwa metode ini bersifat selektif. Hal ini dikarenakan kromatogram yang
dihasilkan tidak mengalami gangguan waktu retensi akibat adanya senyawa endogen pada matriks biologis yang digunakan.
Linearitas
Linearitas merupakan kemampuan suatu metode (pada rentang tertentu) untuk mendapatkan hasil uji yang berupa variasi data (misal: absorban, luas area
kromatogram) yang secara langsung proporsional terhadap konsentrasi analit di dalam sampel (Anonim, 2007).
Menurut International Conference on Harmonisation (ICH), rentang linearitas yang baik adalah + 20% dari konsentrasi nominal. Untuk memperoleh rentang tersebut, paling tidak digunakan lima tingkat konsentrasi. Dalam penelitian ini, linearitas menunjukkan bahwa metode yang digunakan linear dalam rentang konsentrasi S-CPM yang spesifik. Kurva kalibrasi diplotkan antara respon faktor dan konsentrasi larutan standar. Linearitas ditemukan pada rentang 1 sampai 150 ng/mL. Kurva kalibrasi dibuat selama 11 hari yang berbeda dari 4 minggu untuk menentukan variabilitas slopedan intersep.
Tabel 3. Hasil analisis statistik studi bioekuivalensi dari sampel dan standar
dexchl orphenir amine maleat .
Dari tabel 3, terlihat bahwa tidak ada perbedaan yang signifikan dari slopes danintercepts yang melewati rentang konsentrasi optimum.
L imi t of Detection (LOD) dan L imi t of Quantititation (LOQ)
LOD adalah konsentrasi analit terendah dalam sampel yang masih dapat diukur pada kondisi percobaan tertentu tetapi tidak perlu secara kuantitatif. Penentuan LOD pada metode instrumental didasarkan pada signal to noise ratio yaitu dengan cara membandingkan hasil pengukuran analit yang telah diketahui konsentrasinya terhadap respon blanko. Konsentrasi analit yang mampu memberikan respon 2-3 kali respon blanko inilah yang kemudian ditetapkan sebagai LOD. Penentuan LOD dapat pula didasarkan pada standar deviasi yang diperoleh dari pengukuran sejumlah blanko yang kemudian dikalikan dengan faktor sebesar dua atau tiga (Anonim, 1995).
LOQ adalah konsentrasi terendah dari analit dalam sampel yang masih dapat dikuantifikasi dengan presisi dan akurasi yang baik pada kondisi percobaan tertentu dari suatu metode. LOQ merupakan parameter uji kuantitatif untuk senyawa berkadar rendah dalam sampel yang mengandung bahan-bahan lainnya
seperti bahan pengotor dalam serbuk obat dan hasil degradasi dari suatu produk obat jadi. Penentuan LOQ pada metode instrumental biasanya didasarkan pada standar deviasi yang diperoleh dari pengukuran sejumlah blanko yang kemudian dikalikan dengan suatu faktor sebesar sepuluh (Anonim, 1995).
Lower Limit of Quantification (LLOQ) adalah jumlah terkecil dari analit dalam sampel yang dapat dikuantifikasi dan memberikan akurasi dan presisi yang baik. LLOQ menunjukkan sensitivitas dari metode tersebut dimana pada
konsentrasi standar terendah akan memberikan koefisien variasi < 20%. Standar terendah pada kurva kalibrasi dapat diterima sebagai LLOQ jika kondisi berikut terpenuhi:
1. Respon analit (LLOQ) harus minimal lima kali respon blanko.
2. Peak analit (respon) harus dapat diidentifikasi dan memiliki keterulangan yang berbeda, dengan presisi kurang dari 20% koefisien variasi (CV) dan akurasi 80
sampai 120%.
Dalam penelitian ini ditemukan batas deteksi (LOD) sebesar 0,25 ng/ mL dan batas kuantifikasi (LOQ) sebesar 1,00 ng/ mL untuk S-CPM. Hasil ini menunjukkan bahwa metode yang dikembangkan sensitif. LOD dan LOQ dipengaruhi oleh kondisi pemisahan (kolom, reagen, dan instrumentasi dan data sistem), perubahan instrumen (misalnya sistem pompa dan detektor) dan penggunaan pelarut yang bukan grade KCKT dapat menghasilkan perubahan
dalam signal-to-noise ratio.
Kekasaran (Ruggedness ) dan Ketahanan (Robustness )
Kekasaran merupakan tingkat reprodusibilitas hasil yang diperoleh di bawah kondisi percobaan yang beragam dan diekspresikan sebagai persen relative standar deviation (% RSD). Kondisi-kondisi ini meliputi laboratorium, analis, alat, reagen dan waktu percobaan yang berbeda. Kekasaran dapat diketahui jika metode telah digunakan berulang kali. Strategi untuk menentukan kekasaran suatu metode akan bervariasi tergantung pada kompleksitas metode dan waktu tersedia untuk melakukan validasi. Penentuan kekerasan metode dapat dibatasi oleh kondisi-kondisi percobaan yang kritis, seperti pengecekan pengaruh kolom
kromatografinya yang berbeda (pabrik dan jenisnya sama) atau pengaruh operasional metode pada laboratorium yang berbeda (Gandjar, 2010).
Ketahanan merupakan kapasitas metode untuk tetap tidak terpengaruh oleh adanya variasi parameter metode yang kecil. Ketahanan dievaluasi dengan melakukan variasi parameter-parameter metode seperti: persentase pelarut organik, pH, kekuatan ionik, suhu dan sebagainya. Ketahanan suatu metode dievaluasi dengan cara membuat variasi parameter-parameter penting dalam metode tersebut secara sistematis lalu mengukur pengaruhnya pada pemisahan (Gandjar, 2010).
Dalam penelitian ini tidak ada perubahan yang signifikan dalam parameter kromatografi yang diamati (operator, instrumen, reagen dan kolom dari jenis yang sama) dan kondisi optimum (pH, rasio fase gerak dan aliran) yang berubah.
Uji Stabilitas
Uji ini digunakan untuk memastikan bahwa sampel, reagen dan baku yang digunakan stabil pada waktu tertentu supaya diperoleh hasil-hasil analisis yang reprodusibel dan reliabel. Stabilitas sampel plasma yang telah di- spiking dengan baku, diuji freeze-thaw cycles (sebanyak tiga kali siklus); stabilitas jangka pendek dan panjang pada suhu kamar dalam tiga hari dan dalam empat minggu pada suhu -70oC. Hasil dari percobaan dapat diperlihatkan pada tabel 2. Untuk larutan standar, stabilitas dari larutan diuji pada suhu kamar dan keadaan membeku dalam waktu enam minggu dan empat minggu. Hasil rerata konsentrasi yang diperoleh dari hasil pengukuran dibandingkan dengan teoritisnya. Dari hasil tersebut, dapat disimpulkan bahwa sampel dapat disimpan dalam keadaan beku untuk satu bulan tanpa mengalami degradasi. Sampel stabil pada jangka pendek dan larutan sampel dapat disimpan pada suhu kamar tanpa mengalami degradasi.
Uji Kesesuaian Sistem
Uji kesesuaian sistem bertujuan untuk menjamin bahwa suatu metode dapat menghasilkan akurasi dan presisi yang dapat diterima (Gandjar, 2010). Uji kesesuaian sistem dapat dilihat dari efisiensi kolom (jumlah plat teoritis), nilai
resolusi dan simetrisitas dari puncak yang dihasilkan. Dari hasil percobaan, diperoleh jumlah plat teoritis sebesar 18432 hingga 22987 dan nilai resolusinya sebesar 2,46. Nilai plat teoritis yang besar tersebut menunjukkan bahwa pemisahan memiliki efisiensi yang baik karena nilai resolusi yang diperoleh lebih dari 1,5. Hal ini menunjukkan pemisahan yang dihasilkan telah mencapai base line. Hasil ini digunakan untuk mengevaluasi kesesuaian dari sistem yang digunakan untuk menetapkan kadar S-CPM.
Dari hasil penelitian, dapat disimpulkan bahwa metode yang digunakan ini telah mempunyai akurasi, presisi, selektivitas dan linearitas yang baik untuk penetapan kadar S-CPM dalam plasma dan dapat digunakan untuk mengetahui bioavailabilitas dan bioekivalensi dari obat tersebut.
Aplikasi dalam pengembangan metode
Metode penetapan kadar dari CPM dalam plasma ini digunakan untuk studi bioekivalensi pada obat. Obat yang tak berlabel, untuk dua kali pemakaian, untuk pemakaian dua periode, dosis tunggal yang dibandingkan antara sampel yang mengandung 6 mg CPM dan standar. Pengujian bioekivalensi dari obat dilakukan pada laki-laki yang sehat, dewasa dan memenuhi standar GCP (Good Clinical Practice) dan aturan FDA. Studi ini dilakukan pada 24 orang pria yang sehat dan
umurnya di antara 18-45 tahun. Sampel darah dari sukarelawan diambil sebanyak 6 mL sebelum pemberian dosis, pada saat 0,5; 1,0; 1,5; 2; 3; 4; 6; 8; 12; 18; 24 jam setelah pemberian obat. Sampel yang diperoleh disimpan dalam wadah yang
mengandung natrium sitrat sebagai antikoagulan dan disentrifugasi selama 3000 rpm selama 15 menit dan pada suhu 15oC. Plasma yang ada kemudian disimpan pada tube yang telah dilabeli. Kemudian sampel disimpan dalam freezer pada
suhu -70±5oC hingga dianalisis dengan metode KC-SM yang telah divalidasi. Parameter farmakokinetika yang dianalisis antara lain konsentrasi maksimal obat dalam plasma (Cmax), waktu saat obat pada konsentrasi maksimal (Tmax), AUC dari konsentrasi obat dalam plasma sebelum diberikan obat hingga waktu pengukuran terakhir (AUC0-t), AUC konsentrasi obat dalam plasma sebelum diberikan obat hingga obat habis terabsorbsi (AUC0-∞), kecepatan eliminasi dan
waktu paruh eliminasi obat. Berdasarkan hasil uji statistik dengan taraf kepercayaan 90%, disimpulkan Cmax, AUC0-t dan AUC0-∞ yang kemudian dibandingkan dengan standar, bioekivalensi dari obat mempunyai rentang dari 80,0-125,0% untuk Cmax, AUC0-t dan AUC0-∞. Rerata (±SD) konsentrasi dari konsentrasi obat maksimal dalam plasma dari standar sebesar 22.1150 (4.4148) ng/ mL dan untuk obat yang diuji sebesar 25,3421 (2,0605) ng/ mL.
Tabel 4. Profil farmakokinetika dari hasil pengujian.
Daftar Pustaka
Anonim, 1995, Farmakope Indonesia, edisi IV, Departemen Kesehatan Republik Indonesia, Jakarta
Anonim, 2001, Analytical Method Validation and Instrument Performance Verification, John Wiley and Sons, Inc., New Jersey, pp. 118-120.
Anonim, 2007, The United States Pharmacopeia, Edisi 30, United States Pharmacopeia Convention, Inc., USA
Gandjar, I.G., 2010, Kimia Farmasi Analisis, Penerbit Pustaka Pelajar, Yogyakarta, pp. 466-472.
Harmita, 2004, Petunjuk Pelaksanaan Validasi dan Cara Perhitungannya, http://jurnal.farmasi.ui.ac.id/pdf/2004/v01n03/harmita010301.pdf, diakses tanggal 1 November 2011
Mulja, M., dan Hanwar, D., 2003, Prinsip-prinsip Cara Berlaboratorium yang Baik (Good Laboratory Practice), Majalah Farmasi Indonesia Airlangga,