i SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm)
Program Studi Ilmu Farmasi
Oleh :
Yunita Dwi Wulansari NIM : 078114113
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
iv
As long as we do our best
There's no need to regret anything
As long as we keep struggling
In the end...
we are the winner no matter
(Anonim)
Karya ini kupersembahkan untuk yang tersayang,
Mama, Papa, Kakak, dan Adikku,
vii
yang berjudul Validasi Metode Kromatografi Lapis Tipis (KLT)-Densitometri pada Penetapan Kadar Kurkumin Dalam Sediaan Cair Obat Herbal Terstandar (OHT) Kiranti®. Skripsi ini disusun sebagai salah satu syarat untuk memperoleh gelar Sarjana Farmasi (S.Farm).
Selama perkuliahan, penelitian dan penyusunan skripsi, penulis banyak mendapat bantuan dari berbagai pihak baik berupa bimbingan, dukungan, semangat, kritik maupun saran. Oleh karena itu, pada kesempatan ini penulis ingin menyampaikan ucapan terima kasih yang sebesar-besarnya kepada
1. Ipang Djunarko, M.Si, Apt selaku Dekan Fakultas Farmasi Sanata Dharma. 2. Christine Patramurti, M.Si., Apt selaku dosen pembimbing sekaligus dosen
pembimbing akademik, atas bimbingan, masukan, perhatian, semangat, dan motivasi yang diberikan baik selama perkuliahan, penelitian maupun penyusunan skripsi ini.
3. Jeffry Julianus, M.Si., selaku dosen penguji atas segala arahan, masukan, kritik, dan saran yang telah diberikan kepada penulis
4. Yohanes Dwiatmaka, M.Si., selaku dosen penguji atas segala arahan, masukan, kritik, dan saran yang telah diberikan kepada penulis.
viii peneliti bekerja di laboratorium.
8. Segenap dosen dan karyawan atas segala ilmu dan pengalaman yang diberikan, sehingga berguna dalam penyusunan skripsi ini.
9. Eliz dan Veny, sebagai rekan kerja penulis atas segala dukungan, kebersamaan, bantuan, dan semangat yang diberikan baik selama penelitian maupun saat penyusunan naskah skripsi.
10. Tere, Seno, Lilis, Upil, Lala, Pakdhe, K3n, Benny, dan Pace, sebagai teman seperjuangan di Laboratorium Kimia Analisis Instrumental atas semangat dan kebersamaannya
11. Lia, atas dukungan, semangat, bantuan, nasehat, kebersamaan, dan pengalaman berharga, baik selama perkuliahan, penelitian, maupun penyusunan skripsi.
12. Dinar, Anin, Fifi, dan Dika, atas dukungan dan bantuan yang diberikan kepada penulis.
13. Yudi, Yemi, Veny, Lilis, dan Devina sebagai teman yang sering satu kelompok atas pengetahuan dan pengalaman yang diberikan.
14. Mas Bayu, atas ilmu dan pengalaman yang telah dibagikan sehingga sangat membantu penulis dalam penyusunan skripsi.
ix 1 bulan di Ngaran, Bantul.
18. Teman-teman FST 07 atas pengalaman, suka duka, kekompakan dan kebersamaan yang pasti tak akan terlupakan.
19. Teman-teman angkatan 2007, atas pengalaman dan kebersamaannya selama ini.
20. Semua pihak yang tidak dapat disebutkan satu persatu oleh penulis, terimakasih atas bantuan yang diberikan selama ini sehingga penulis bisa menyelesaikan skripsi ini.
Penulis menyadari skripsi ini masih memiliki banyak kekurangan. Oleh karena itu, penulis mengharapkan kritik dan saran untuk membantu penulis dalam perkembangan selanjutnya.
x
HALAMAN PENGESAHAN ... HALAMAN PERSEMBAHAN ... PERNYATAAN KEASLIAN KARYA ... LEMBAR PERSETUJUAN PUBLIKASI KARYA………. PRAKATA... BAB I PENGANTAR ... A. Latar Belakang ...
1. Permasalahan... 2. Keaslian penelitian ... 3. Manfaat penelitian ... B. Tujuan Penelitian ... BAB II PENELAAHAN PUSTAKA ...
xi
E. Kromatografi Lapis Tipis... 1. Tinjauan umum... BAB III METODE PENELITIAN ...
A. Jenis Rancangan Penelitian... B. Variabel Penelitian... C. Definisi Operasional... D. Bahan Penelitian... E. Alat Penelitian... F. Tata Cara Penelitian... 1. Pembuatan pelarut (metanol pH 4)... 2. Pembuatan fase gerak ... 3. Pembuatan larutan baku kurkumin ... 4. Penetapan panjang gelombang maksimum ...
xii 6. Akurasi pengukuran baku dalam matriks sampel ... BAB IV HASIL DAN PEMBAHASAN...
A. Pembuatan metanol pH 4... B. Pembuatan Fase gerak ... C. Pembuatan Larutan Baku ... D. Penetapan Panjang Gelombang Maksimum... E. Pengamatan NilaiRetardation Factor(Rf) dan
xiii
A. Kesimpulan... B. Saran... DAFTAR PUSTAKA... LAMPIRAN... BIOGRAFI PENULIS...
xiv Tabel III.
Tabel IV. Tabel V.
Tabel VI. Tabel VII. Tabel VIII. Tabel IX.
Kriteria CV yang dapat diterima……….... Data replikasi kurva baku kurkumin……….. Data replikasi kurva baku kurkumin dengan penyesuaian nilai AUC……… Perbandingan nilai Rfbaku dan sampel, serta nilai resolusi…..
Data %recovery……….
DataCoefficient of Variation(CV)……… Recoverydan CV baku kurkumin dalam matriks sampel……..
22 39
xv Logo Obat Herbal Terstandar………... Kiranti Sehat Datang Bulan……….. Gugus metilen aktif kurkumin……….. Spektra baku kurkumin konsentrasi 50 ppm, 100 ppm, dan 175 ppm………... Gugus kromofor dan auksokrom kurkumin... Baku Kukumin 100 ppm... Interaksi kurkumin dengan fase gerak……….. Interaksi hidrogen kurkumin dengan fase diam………….…... Hubungan antara konsentrasi dengan AUC/100 (replikasi III)………. Rfbaku kurkumin konsentrasi 100 ppm……… Rfsampel replikasi I………. Rangekonsentrasi kurkumin……….. Kromatogram sampel tanpa penambahan baku kurkumin……. Kromatogram sampel dengan penambahan baku kurkumin…..
xvi Kromatogram seri baku kurkumin replikasi III…………...… Kromatogram validasi metode………. Contoh perhitungan kadar kurkumin………... Persamaan kurva baku dan gambar kurva baku kurkumin….. Nilai AUC dan contoh perhitunganrecoverykurkumin…….. Contoh perhitungan CV kurkumin……….. Kromatogram sampel tanpa penambahan baku kurkumin…... Kromatogram sampel dengan penambahan baku kurkumin… Contoh perhitungan resolusi……… Nilai AUC sampel dan sampel yang diadisi baku kurkumin... Contoh perhitunganrecoverybaku kurkumin dalam sampel.. Perhitungan CV kadar baku kurkumin dalam sampel……….
xvii
tradisional yang mengandung kurkumin adalah sediaan cair obat herbal terstandar merk ‘Kiranti”. Penetapan kadar kurkumin dapat dilakukan dengan metode KLT-densitometri. Oleh karena itu perlu dilakukan validasi metode terlebih dahulu untuk mengetahui metode yang digunakan dapat memberikan hasil yang dapat dipercaya.
Penelitian ini merupakan penelitian noneksperimental-deskriptif. Dalam penelitian ini kurkumin dan senyawa-senyawa lain dalam sampel dipisahkan dengan metode KLT dengan fase diam silika gel G 60 dan fase gerak kloroform : asam asetat glasial (95:5), serta dengan jarak pengembangan sejauh 10 cm. Setelah pemisahan senyawa dengan metode KLT, kemudian dilakukan analisis kuantitatif dengan densitometer.
Parameter validasi yang diteliti adalah selektivitas, linearitas, akurasi, presisi, danrange. Hasil penelitian menunjukkan metode ini memiliki selektivitas dan linearitas yang baik pada konsentrasi 50-175 ppm (r=0,9999), nilai recovery dan CV berturut-turut untuk konsentrasi kurkumin 50 ppm; 100 ppm; dan 175 ppm 98,95-101,10% dan 1,7%; 98,61-101,79% dan 0,7%; 100,18-103,83% dan 0,9%. Berdasarkan hasil tersebut, maka metode KLT-densitometri ini memiliki validitas yang baik untuk menetapkan kadar kurkumin dalam sampel.
xviii
Medicine Kiranti . The determination of curcumin can be done with TLC-densitometry method. For that reason, it is important to get validation method first to know the appropriate method can be used to get trustable result.
This research is noneksperimental-descriptive research. In this research, curcumin and other compounds in the sample are separated using TLC method with the stationary phase silica gel G 60 and a mobile phase of chloroform: glacial acetic acid (95:5), along with range development for 10 cm. Then quantitative analysis with densitometer can be done.
Validation parameter which observed were specificity, linearity, accuracy, precision, and range. The result showed that this method has good specificity and linearity in the concentration 50-175 ppm (r=0,9999), recovery and CV value consecutively for the curcumin concentration 50 ppm, 100 ppm, and 175 ppm were 98,95-101,10% and 1,7%; 98,61-101,79% and 0,7%; 100,18-103,83% and 0,9%. With this result, it can be concluded that this TLC-densitometry method has good validity for quantitative analysis of curcumin in the sample.
BAB I PENGANTAR
A. Latar Belakang
Penggunaan bahan alam, baik sebagai obat maupun tujuan lain cenderung meningkat, terlebih dengan adanya isuback to natureserta krisis berkepanjangan yang mengakibatkan turunnya daya beli masyarakat. Penggunaan obat-obatan tradisional dipercaya oleh masyarakat memiliki efek samping yang relatif kecil bila dibandingkan obat sintesis (Sari, 2006). Walaupun demikian bukan berarti tanaman obat atau obat tradisional tidak memiliki efek samping yang merugikan, bila tidak disertai dengan ketepatan dosis (Katno dan Pramono,2010).
Kebijakan Badan Pengawasan Obat dan Makanan mengenai obat herbal adalah meningkatkan penjaminan keamanan obat herbal dengan mendorong perkembangan obat herbal sampai ke tingkat fitofarmaka atau setidaknya obat herbal terstandar (OHT). Perkembangan ini bertujuan untuk menjamin keamanan obat herbal sehingga obat herbal dapat dimasukkan dalam pengobatan formal.
OHT adalah obat herbal yang menggunakan bahan baku yang telah terstandar dan khasiatnya telah dibuktikan dengan uji praklinis. Standarisasi bahan baku diperlukan agar dapat diperoleh bahan baku yang seragam yang akhirnya dapat menjamin efek farmakologi tanaman tersebut (Hanani, 2010).
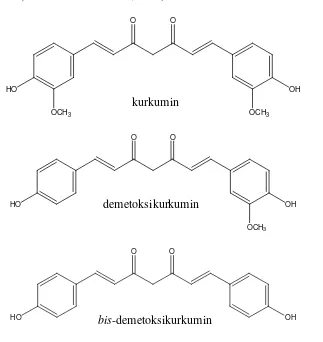
Salah satu senyawa yang berasal dari tanaman dan banyak digunakan sebagai bahan obat atau campuran obat tradisional adalah kurkumin. Kurkumin banyak terdapat pada tanaman Curcuma xanthorriza (temulawak) dan Curcuma
domesticae Val. (kunyit). Dalam tanaman Curcuma domesticae, kurkumin terdapat bersama dengan demetoksikurkumin dan bis-demetoksikurkumin, yang dikenal dengan nama kurkuminoid. Kurkumin memiliki stabilitas yang rendah, dimana kurkumin bersifat fotosensitif dan mudah terdegradasi dalam larutan. Oleh karena itu, perlu penjaminan mutu terhadap produk yang memiliki kandungan kurkumin karena dikhawatirkan kurkumin dalam produk terdegradasi selama proses distribusi dan penyimpanannya.
Salah satu obat tradisional yang mengandung kurkumin sebagai komposisi terbesarnya adalah sediaan cair OHT Kiranti®. Produk ini cukup banyak digunakan oleh masyarakat. Bahan baku yang digunakan dalam produk ini adalah simplisia. Standarisasi simplisia yang dilakukan meliputi: penetapan kadar minyak atsiri, penetapan kadar air, kadar abu larut air, kadar abu yang tidak larut air, kadar abu larut asam, kadar sari larut air, kadar sari larut etanol, susut pengeringan, dan bahan organik asing (Direktorat Jenderal Pengawas Obat dan Makanan, 1995a). Standarisasi yang dilakukan belum mencakup analisis kadar zat aktif pada produk jadi. Oleh karena itu, penetapan kadar zat aktif dalam OHT Kiranti® perlu dilakukan untuk mengetahui jumlah zat aktif yang nantinya mempengaruhi khasiat dari OHT tersebut.
selanjutnya dapat ditetapkan kadarnya dengan metode densitometri. KLT cocok untuk analisis obat di laboratorium farmasi, karena metodenya sederhana, cepat dalam pemisahan, sensitif, kecepatan pemisahan tinggi, dan memerlukan jumlah cuplikan yang sangat sedikit (Khopkar, 1990).
Penelitian ini merupakan rangkaian dalam penelitian penetapan kadar kurkumin dengan metode KLT-Densitometri yang meliputi optimasi, validasi metode, dan aplikasinya pada sediaan OHT yang beredar di pasaran. Dalam hal ini peneliti mengambil bagian pada tahap validasi metode KLT-Densitometri untuk penetapan kadar kurkumin dalam sediaan cair OHT.
Metode penetapan kadar kurkumin dalam sediaan OHT ini menggunakan sistem yang diperoleh dari hasil optimasi yang dilakukan pada rangkaian penelitian ini. Suatu metode analisis harus divalidasi ketika suatu metode menggunakan sistem baru yang belum divalidasi sebelumnya. Validasi ini bertujuan untuk memberikan jaminan bahwa metode analisis dengan sistem yang digunakan tersebut memenuhi parameter-parameter validasi yang meliputi selektivitas, linearitas, akurasi, presisi, dan range sehingga dapat memberikan hasil analisis yang valid atau dapat dipercaya. Oleh karena itu, validasi metode merupakan tahapan yang penting untuk dilakukan sebelum metode ini diaplikasikan untuk analisis kadar kurkumin dalam OHT Kiranti®.
1. Permasalahan
KLT-Densitometri memenuhi parameter-parameter validasi yaitu selektivitas, linearitas, akurasi, presisi, danrange?
2. Keaslian penelitian
Sejauh sepengetahuan penulis, penelitian validasi metode penetapan kadar kurkumin dalam sediaan cair OHT Kiranti® dengan metode KLT-densitometri belum pernah dilakukan sebelumnya. Penelitian mengenai penetapan kadar kurkumin dengan metode KLT yang pernah dilakukan yaitu, penetapan kadar baku kurkumin (E-Merck) dengan metode KLT-densitometri dengan fase diam silika gel GF 254 dan fase gerak kloroform:etanol:air suling (25:0,96:0,04) (Martono, 1996), penetapan kadar kurkuminoid secara simultan dalam sampel Curcuma menggunakan metode high performance thin layer chromatography (HPTLC) (Gupta, Gupta, and Kumar, 1999), standarisasi mutu dengan HPTLC terhadap adanya kurkuminoid dalam Curcuma longa L. (Paramasivam, Aktar, Poi, Banerjee, and Bandyopahyay, 2008).
Penelitian yang dilakukan penulis, menggunakan sistem KLT dengan fase gerak kloroform : asam asetat glasial (95:5). Sejauh sepengetahuan penulis sistem KLT yang digunakan tersebut belum pernah digunakan dalam penelitian sebelumnya.
3. Manfaat penelitian
b. Manfaat praktis. Hasil penelitian diharapkan dapat memberikan informasi mengenai selektivitas, linearitas, akurasi, presisi, dan range metode penetapan kadar kurkumin dalam sediaan cair OHT Kiranti® secara KLT-Densitometri.
B. Tujuan
BAB II
PENELAAHAN PUSTAKA
A. Kurkumin
Kurkumin merupakan senyawa yang banyak terdapat dalam tanaman kunyit
(Curcuma longaL.)dan temulawak (Curcuma xanthorrhizaRoxb.) (Tonnesen, 1989;
Van der Goot, 1997). Di alam kurkumin umumnya ditemukan bersama demetoksikurkumin dan bis-demetoksikurkumin, yang dikenal dengan kurkuminoid (Tonnesen dan Karlsen, 1985).
O
Gambar 1. Struktur molekul kurkuminoid (Aggarwal, Bhatt, Ichikawa, Ahn, Sethi, Sandur, Natarajan, Seeram, dan Shishodia, 2006)
kurkumin
demetoksikurkumin
bis-demetoksikurkumin
Kurkumin (1,7 – bis(4’hidroksi-3 metoksifenil)-1,6 heptadien, 3, 5-dion (Jaruga, 1998; Pan, 1999) memiliki berat molekul 368,126 g/mol (Tonnesen and Karlsen, 1983). Kurkumin tergolong senyawa diarilheptanoid dengan rumus molekul C21O6H2O (Tonnesen and Karlsen, 1985). Strukturnya yang rigid dan planar (adanya sistem konjugasi) membuat afinitas kurkumin terhadap lipid bilayer menjadi besar, dan juga bertanggung jawab terhadap warna kuning yang ada (Nakayama, 1997). Panjang gelombang 425 nm diketahui sebagai panjang gelombang serapan maksimum kurkumin dimana menghasilkan sensitivitas pengukuran paling baik (Paramasivamet al., 2008).
Kurkuminoid mempunyai aktivitas antiinflamasi (Kohli, Ali, Ansari, and Raheman, 2005). Kurkuminoid menghambat senyawa eicosanoid seperti prostaglandin, tromboksan dan prostasiklin dengan cara menghambat aktivitas enzim cyclooxygenase (COX). Kurkuminoid juga menghambat pembentukan senyawa leukotrien dengan menghambat aktivitas enzim lipooxygenase (LOX) (Kohli et al., 2005). Dari tiga senyawa kurkuminoid, kurkumin mempunyai aktivitas antiinflamasi yang paling kuat dibandingkan senyawa turunannya (Agnam, Samhoedi, Timmerman, Venie, Sugiyanto, and Goot, 1995).
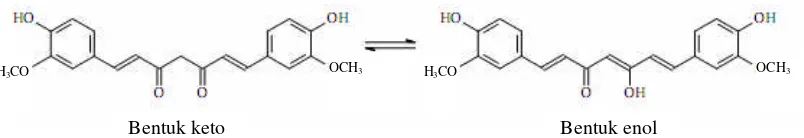
Kurkumin memiliki 2 bentuk tautomer, yaitu bentuk enol dan keto (gambar 2). Dalam larutan, kurkumin terutama berada dalam bentuk enol.
Gambar 2.Tautomerisasi keto-enol kurkumin (Stankovic, 2004).
Bentuk keto Bentuk enol
OCH3 OCH3 H3CO
Sifat kimia kurkuminoid yang menarik adalah sifat perubahan warna akibat perubahan pH lingkungan. Dalam susana asam, kurkuminoid berwarna kuning atau kuning jingga, sedangkan dalam suasana basa berwarna merah (Tonnesen and Karlsen, 1985).
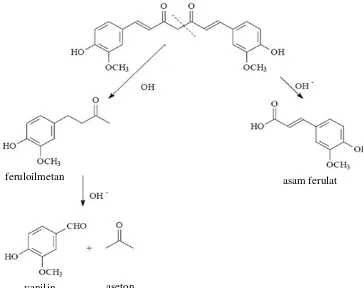
Stabilitas kurkumin sangat dipengaruhi oleh pH lingkungan. Dalam larutan berair dengan pH basa, kurkumin mengalami reaksi degradasi pada gugus metilen aktif pada senyawa tersebut. Degradasi ini terjadi bila kurkumin berada dalam lingkungan pH 8,5 – 10,0 dalam waktu yang relatif lama, walaupun hal ini tidak berarti bahwa dalam waktu yang relatif singkat tidak terjadi degradasi kurkumin (Tonnesen and Karlsen, 1985). Kurkumin dapat mengalami degradasi membentuk asam ferulat dan feruloilmetan. Feruloilmetan dapat terhidrolisis menghasilkan vanillin dan aseton (Stankovic, 2004).
Gambar 3.Reaksi degradasi kurkumin (Stankovic, 2004).
feruloilmetan asam ferulat
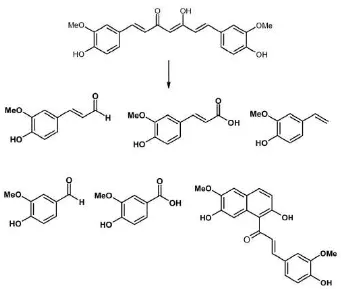
Instabilitas kurkumin juga dipengaruhi oleh adanya cahaya yang menyebabkan terjadinya degradasi fotokimia senyawa tersebut (Van der Goot, 1997; Supardjan, dan Meiyanto, 2002) dan oleh sinar ultraviolet (Bermawie, Rahardjo, Wahyuno, and Ma’mun, 2005).
Gambar 4.Poduk fotodegradasi kurkumin (Tonnesen and Greenhill, 1992).
B. Sediaan Cair Oral
Sediaan cair oral obat tradisional memiliki beberapa persyaratan yang harus dipenuhi antara lain: keseragaman volume, angka lempeng total tidak lebih dari 10, angka kapang khamir tidak lebih dari 10, mikroba patogen negatif, aflatoksin lebih lebih dari 30 bagian per juta (bpj), bahan tambahan, wadah dan penyimpanan, penandaan (Kementerian Kesehatan RI, 1994).
C. Obat Herbal Terstandar (OHT)
Merupakan sediaan obat bahan alam yang telah dibuktikan keamanan dan
khasiatnya secara ilmiah dengan uji pra klinik dan bahan bakunya telah distandarisasi.
OHT harus memenuhi kriteria: aman, klaim khasiat dibuktikan praklinik,dan telah
dilakukan standarisasi bahan baku yang digunakan dalam produk jadi (Badan
Pengawas Obat dan Makanan Republik Indonesia, 2004).
Gambar 5. Logo obat herbal terstandar
(Badan Pengawas Obat dan Makanan Republik Indonesia, 2004)
Bahan baku yang digunakan dalam produk jadi dapat berupa simplisia.
Simplisia adalah bahan alamiah yang digunakan sebagai obat tradisional dan belum
mengalami pengolahan apapun juga, kecuali dinyatakan lain merupakan bahan yang
dikeringkan (Badan Pengawas Obat dan Makanan Republik Indonesia, 2005).
Standarisasi simplisia meliputi: penetapan kadar minyak atsiri, penetapan kadar abu,
penetapan kadar abu yang tidak larut dalam asam, penetapan kadar abu yang larut air,
dalam air, penetapan kadar sari yang larut dalam etanol, penetapan bahan organik
asing, dan penetapan kadar tanin (Direktorat Jenderal Pengawas Obat dan Makanan, 1995a).
D. Kiranti®
Kiranti Sehat Datang Bulan® merupakan OHT yang memiliki indikasi untuk mengatasi rasa nyeri, perasaan letih, bau badan tak sedap saat haid, keputihan dan membuat tubuh tetap fit sekaligus segar (Anonim, 2010).
Gambar 6. Kiranti Sehat Datang Bulan®(Anonim, 2010). Komposisi Kiranti Sehat Datang Bulan® terdiri dari: Curcumae domesticae Rhizoma(30 g), Tamarindi Pulpa (6 g), Kaempferiae Rhizoma (3 g), Arengae pinnata Fructose (3 g), Zingiberis Rhizoma (0,8 g), Paullinia cupana (0,23 g),Cinnamomi Cortex(0,1 g) dan air (hingga 150 mL) (Anonim, 2010).
E. Kromatografi Lapis Tipis
1. Tinjauan Umum
Kromatografi didefinisikan sebagai prosedur pemisahan zat terlarut oleh
lebih, salah satu diantaranya bergerak secara berkesinambungan dalam arah tertentu
dan didalamnya zat-zat itu menunjukkan perbedaan mobilitas disebabkan adanya
perbedaan dalam adsorpsi, partisi, kelarutan, tekanan uap, ukuran molekul atau
kerapatan muatan ion. Dengan demikian masing-masing zat dapat diidentifikasi atau
ditetapkan dengan metode analitik (Direktorat Jenderal Pengawas Obat dan Makanan,
1995b).
Teknik kromatografi umum membutuhkan zat terlarut terdistribusi di antara
dua fase, satu diantaranya diam (fase diam), yang lainnya bergerak (fase gerak). Fase
gerak membawa zat terlarut melalui media, hingga terpisah dari zat terlarut lainnya,
yang terelusi lebih awal atau lebih akhir. Umumya zat terlarut dibawa melalui media
pemisah oleh aliran pelarut berbentuk cairan atau gas yang disebut eluen. Fase diam
dapat bertindak sebagai adsorben, seperti halnya adsorben alumina yang diaktifkan,
silika gel, dan resin penukar ion, atau dapat bertindak melarutkan zat terlarut sehingga
partisi antara fase diam dan fase gerak. Dalam proses terakhir ini suatu lapisan cairan
dalam suatu penyangga yang inert berfungsi sebagai fase diam (Direktorat Jenderal
Pengawas Obat dan Makanan, 1995b).
Kromatografi lapis tipis (KLT) bersama-sama dengan kromatografi kertas
dengan berbagai macam variasinya pada umumnya merupakan kromatografi planar.
Kromatografi lapis tipis dalam pelaksanannya lebih mudah dan lebih murah dibandingkan kromatografi kolom. Demikian juga peralatan yang digunakan. Dalam kromatografi lapis tipis, peralatan yang digunakan lebih sederhana dan dapat dikatakan bahwa hampir semua laboratorium dapat melaksanakan setiap saat secara cepat (Rohman, 2009).
Pemilihan pelarut yang digunakan untuk senyawa yang akan dianalisis dengan metode KLT, harus dapat melarutkan analit dengan sempurna, mudah menguap, viskositas rendah, serta dapat membasahi lapisan penyerap (Sherma and Fried, 1996).
Deteksi bercak pemisahan pada KLT dapat dilakukan dengan cara-cara berikut:
a. menyemprot lempeng KLT dengan reagen kromogenik yang akan bereaksi
secara kimia dengan seluruh solut yang mengandung gugus fungsional tertentu
sehingga bercak menjadi berwarna. Kadang-kadang bercak dipanaskan terlebih
dahulu untuk mempercepat reaksi pembentukan warna dan intensitas warna
bercak
b. mengamati lempeng di bawah lampu ultraviolet dengan panjang gelombang 254
atau 366 nm untuk menampakkan solut sebagai bercak yang gelap atau bercak
yang berfluoresensi terang pada dasar yang berfluoresensi
c. menyemprot lempeng dengan asam sulfat pekat atau asam nitrat pekat lalu
dipanaskan untuk mengoksidasi solut-solut organik yang akan nampak sebagai
bercak hitam kecoklatan
e. melakukan scanning pada permukaan lempeng dengan densitometer (Gandjar
dan Rohman, 2007).
Pada kromatografi planar, senyawa yang berbeda dalam campuran sampel menempuh jarak yang berbeda sesuai dengan seberapa kuat mereka berinteraksi dengan fase diam dibandingkan dengan fase gerak. Semakin polar solut maka semakin tertahan kuat ke dalam adsorben polar (silika gel). Solut-solut non polar tidak mempunyai afinitas atau mempunyai sedikit afinitas terhadap adsorben polar, sementara solut-solut yang terpolarisasi memiliki afinitas yang kecil terhadap adsorben polar disebabkan adanya interaksi dipol atau interaksi-interaksi yang diinduksi oleh dipol. Solut-solut polar, terutama yang mampu membentuk ikatan hidrogen, akan terikat kuat pada adsorben karenanya butuh fase gerak yang cukup polar untuk mengelusinya. Berikut adalah urutan polaritas solut-solut organik: alkana < alkena < aromatis < eter < ester < keton dan aldehid < tiol < amin dan amida < alkohol < fenol < asam-asam organik (Gandjar dan Rohman, 2007).
Retardation factor (Rf) merupakan parameter karakteristik KLT. Harga Rf didefinisikan sebagai perbandingan antara jarak senyawa dari titik awal dan jarak tepi muka pelarut dari titik awal (Roth, 1994). Angka Rf berjangka antara 0,00 sampai 1,00 dan hanya dapat ditentukan dua desimal (Stahl, 1985).
Rf=
Rstd yang didefinisikan sebagai perbandingan antara jarak yang digerakkan oleh senyawa yang tidak diketahui dengan jarak yang digerakkan oleh senyawa standar yang diketahui (Hardjono, 1983). Nilai Rxdapat dihitung:
Rx,a
(Dean, 1995). Pengekoran noda kromatogram terjadi apabila proses pemisahan yang terjadi tidak sempurna. Terlalu tingginya konsentrasi komponen yang ditentukan juga merupakan salah satu penyebab terjadinya kromatogram yang berekor. Penyebab pengekoran yang lain adalah ketidakjenuhan chamber. Ketidaktepatan pemilihan fase gerak terhadap jenis fase diam dan macam sampel yang dianalisis juga merupakan penyebab pengekoran kromatogram yang lainnya (Mulja dan Suharman, 1995).
2. Sistem KLT
Adanya air dari atmosfer yang diserap oleh permukaan silika gel mampu mendeaktifkan permukaan silika gel karena air akan menutup sisi aktif silika gel. Hal seperti ini dapat diatasi dengan memanaskan plat pada suhu 105ºC (Gandjar dan Rohman, 2007).
b. Fase gerak. Fase gerak adalah medium angkut yang terdiri atas satu atau beberapa pelarut. Fase gerak bergerak di dalam fase diam yaitu lapisan berpori karena ada gaya kapiler. Yang digunakan adalah pelarut bertingkat mutu analitik dan bila diperlukan sistem pelarut multikomponen, maka harus berupa suatu campuran sederhana mungkin terdiri atas maksimum tiga komponen (Stahl, 1985).
Pemilihan sistem pelarut untuk mencapai sistem pemisahan yang diperlukan mungkin melibatkan beberapa percobaan, tetapi pilihan pelarut cukup terbatas dengan pertimbangan interferensi respon detektor atau kerusakan yang mungkin terjadi dari fase diam (Dean, 1995).
Berikut adalah beberapa petunjuk dalam memilih dan mengoptimasi fase gerak:
1) fase gerak harus mempunyai kemurnian yang sangat tinggi karena KLT merupakan teknik yang sensitif
F. Densitometri
Densitometri merupakan metode analisis instrumental yang mendasarkan pada interaksi radiasi elektromagnetik dengan analit yang merupakan bercak pada plat KLT. Densitometri lebih dititikberatkan untuk analisis kuantitatif analit-analit dengan kadar kecil, yang mana diperlukan pemisahan terlebih dahulu dengan KLT (Rohman, 2009).
KLT-densitometri merupakan salah satu dari metode analisa kuantitatif. Penetapan kadar suatu senyawa dengan metode ini dilakukan dengan mengukur kerapatan bercak senyawa yang dipisahkan dengan cara KLT. Pada umumnya pengukuran kerapatan bercak tersebut dibandingkan dengan kerapatan bercak senyawa standar yang dielusi bersama-sama (Hardjono, 1985).
Metode densitometri mempunyai cara kerja yang sederhana dan cepat. Pada metode densitometri diperlukan adsorbens dan fase gerak yang murni. Untuk memperoleh hasil yang baik umumnya digunakan adsorbens siap pakai yang telah mengalami pra pencucian (Gritter, 1991).
Teknik pengukuran dapat didasarkan atas pengukuran intensitas sinar yang diserap (absorbansi), intensitas sinar yang dipantulkan (reflaktansi) atau intensitas sinar yang difluoresensikan (fluoresensi) (Mintarsih, 1990).
Pada beberapa alat TLC scanner sudah dilengkapi alat pemroses data atau mikro komputer, sehingga integrasi luas puncak atau tinggi puncak tersebut dapat langsung direkam atau dicatat sebagai data sekaligus dengan kromatogramnya dan dapat pula direkam dan dicatat langsung sebagai kadarnya, melalui teknik pemrogaman tertentu. Penelusuran bercak dapat dilakukan secara horizontal maupun vertikal (scanning horizontal atau scanning vertical). Penelusuran bercak secara horisontal dapat dilakukan satu persatu, atau apabila satu pelat bercak yang diperoleh segaris semua maka dapat dilakukan penelusuran untuk semua bercak sekaligus. Sedangkan cara penelusuran vertikal, hanya dapat dilakukan satu per satu. Pada penelusuran bercak horisontal dengan penelusuran beberapa bercak sekaligus hanya dapat dilakukan apabila bercak-bercak tesrsebut benar-benar berada dalam satu baris. Cara ini akan mengalami kesulitan jika bercak yang sangat dekat dengan bercak yang akan ditetapkan, karena ada kemungkinan bercak yang tidak diinginkan ikut tertetapkan (Mintarsih, 1990).
Pelat yang digunakan untuk KLT pada densitometri sebaiknya digunakan pelat buatan pabrik, karena pada pelat buatan sendiri fase diamnya kurang rata, sehingga akan mempengaruhi hasil penelusuran dengan densitometri, yaitu berupa puncak yang lebar dan kasar. Puncak yang lebar disebabkan kurang kompaknya fase diam, puncak yang kasar disebabkan permukaan pelat kurang rata (Mintarsih, 1990).
Ada dua cara penetapan dengan alat densitometer. Pertama, setiap kali penetapan ditotolkan sediaan baku dari senyawa yang bersangkutan dan dielusi bersama dalam satu lempeng, kemudian Area Under Curve (AUC) sampel dibandingkan dengan AUC zat baku. Yang kedua, dengan membuat kurva hubungan antara jumlah zat baku dengan AUC. Kurva baku diperoleh dengan membuat totolan zat baku pada pelat KLT dengan bermacam-macam konsentrasi (minimal tiga macam konsentrasi). Bercak yang diperoleh dicari AUC dengan densitometer. Dari kurva baku diperoleh persamaan : y = bx + a, dimana x adalah banyaknya zat yang ditotolkan dan y adalah AUC (Supardjan, 1987).
G. Validasi Metode 1. Tinjauan Umum
Suatu metode analisis harus divalidasi untuk melakukan verifikasi bahwa parameter-parameter kinerjanya cukup mampu untuk mengatasi problem analisis, karenanya suatu metode harus divalidasi, ketika:
a. metode baru dikembangkan untuk mengatasi problem analisis tertentu
b. metode yang sudah baku direvisi untuk menyesuaikan perkembangan, atau karena munculnya suatu problem yang mengarahkan bahwa metode tersebut harus direvisi
c. penjaminan mutu yang mengindikasikan bahwa metode baku telah berubah seiring dengan berjalannya waktu
d. metode baku digunakan di laboratorium yang berbeda, dikerjakan oleh analis yang berbeda, atau dikerjakan dengan alat yang berbeda
e. untuk mendemonstrasikan kesetaraan antar 2 metode, seperti antara metode baru dan metode baku (Swartz dan Krull, 1997).
Tujuan utama validasi metode adalah untuk menghasilkan hasil analisis yang paling baik. Untuk memperoleh hasil tersebut, semua variabel yang terkait dengan metode analisis harus dipertimbangkan seperti prosedur pengambilan sampel, tahap penyiapan sampel, jenis penyerap yang digunakan pada kromatografi, fase gerak, dan sistem deteksinya. Banyaknya parameter yang harus divalidasi tergantung pada tujuan analisis (Adamovics, 1997).
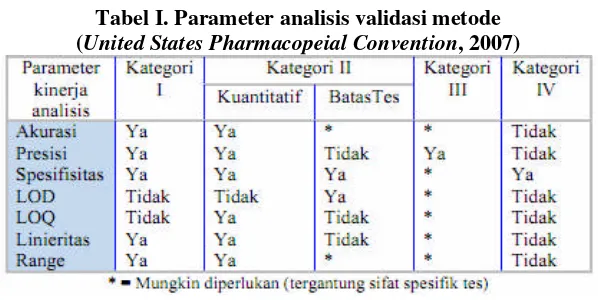
Menurut The United States Pharmacopeia 30th tahun 2007, metode analisis dapat dibedakan menjadi 4 kategori, yaitu:
b. Kategori II. Mencakup prosedur analisis kualitatif dan kuantitatif yang digunakan untuk menganalisis impurities dalam sediaan farmasi.
c. Kategori III. Mencakup prosedur analisis yang digunakan untuk menentukan karakteristik penampilan suatu sediaan farmasi, misalnya disolusi dan pelepasan obat.
d. Kategori IV (tes identifikasi)
Tabel I. Parameter analisis validasi metode (United States Pharmacopeial Convention, 2007)
2. Parameter Validasi
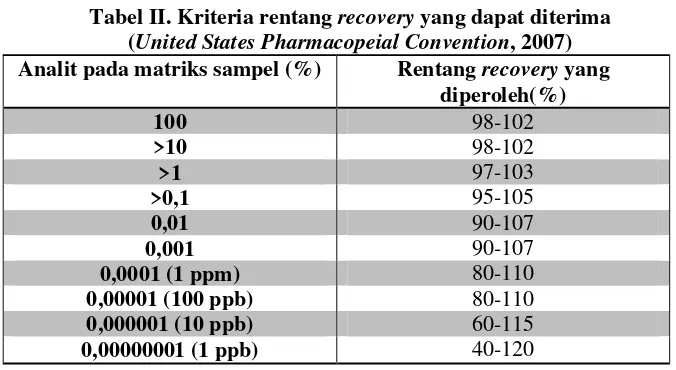
Tabel II. Kriteria rentangrecoveryyang dapat diterima (United States Pharmacopeial Convention, 2007) Analit pada matriks sampel (%) Rentangrecoveryyang
diperoleh(%)
0,0001 (1 ppm) 80-110
0,00001 (100 ppb) 80-110
0,000001 (10 ppb) 60-115
0,00000001 (1 ppb) 40-120
b. Presisi. Presisi suatu metode analisis merupakan sejumlah pencaran hasil yang diperoleh dari analisis berulang kali pada suatu sampel homogen. Presisi umumnya dinyatakan dalam coefficient of variation (CV) atau koefisien variasi (KV) (Mulja dan Hanwar, 2003).
Tabel III. Kriteria CV yang dapat diterima (United States Pharmacopeial Convention, 2007)
Kadar analit CV(%)
≥1 % 2,5
> 0,1 5
1 ppm 16
1 ppb 32
d. Batas Deteksi (Limit of Detection atau LOD). Batas deteksi didefinisikan sebagai konsentrasi analit terendah dalam sampel yang masih dapat dideteksi, meskipun tidak selalu dapat dikuantifikasi. LOD merupakan batas uji yang secara spesifik menyatakan apakah analit di atas atau di bawah nilai tertentu. Sebagai contoh, batas deteksi merupakan banyaknya sampel yang menunjukkan respon (S) 3 kali terhadap derau (N) atau LOD = 3 S/N (Swartz and Krull, 1997).
e. Batas Kuantifikasi (Limit of Quantification atau LOQ). Batas kuantifikasi didefinisikan sebagai konsentrasi analit terendah dalam sampel yang dapat ditentukan dengan presisi dan akurasi yang dapat diterima pada kondisi operasional metode yang digunakan (Rohman, 2009). Batas kuantifikasi didefinisikan sebagai konsentrasi analit terendah dalam sampel yang dapat ditentukan dengan presisi dan akurasi yang dapat diterima pada kondisi operasional metode yang digunakan. Kadang-kadang rasio signal to noise 10:1 digunakan untuk menentukan LOQ (Rohman, 2009).
Selektivitas pada metode kromatografi ditunjukkan melalui nilai resolusi (Harmita, 2004). Dalam teknik pemisahan, daya pisah (resolusi) antara analit yang dituju dengan pengganggu lainnya harus > 1,5 (Swartz and Krull, 1997).
g. Range(kisaran). Menurut ICH, kisaran suatu prosedur analisis adalah interval antara konsentrasi (jumlah) analit pada level atas dan pada level bawah dalam suatu sampel, yang mana dapat ditunjukkan bahwa prosedur analisis mempunyai level akurasi, presisi, dan linearitas yang sesuai.
F. Landasan Teori
Kurkumin banyak terdapat dalam tanamanCurcuma longa danCurcuma xanthorrhiźa. Kurkumin di alam biasanya terdapat bersama bis-demetoksikurkumin dan demetoksikurkumin, yang dikenal dengan nama kurkuminoid. Kurkumin merupakan salah satu senyawa yang terkandung dalam banyak obat tradisional. Salah satu kategori obat tradisional adalah obat herbal terstandar, dimana obat tradisional ini telah melalui standarisasi bahan baku dan uji praklinis.
Metode KLT dapat memisahkan beberapa campuran senyawa karena adanya perbedaan interaksi antara senyawa-senyawa tersebut dengan fase diam dan fase gerak yang digunakan. Bercak analit hasil pemisahan KLT dapat dianalisis kuantitatif dengan metode densitometri.
linearitas, dan range. Metode dikatakan valid apabila memenuhi persyaratan parameter validasi.
G. Hipotesis
BAB III
METODE PENELITIAN
A. Jenis dan Rancangan Penelitian
Penelitian yang dilakukan bersifat noneksperimental deskriptif karena tidak terdapat manipulasi dan perlakuan terhadap subjek uji.
B. Variabel Penelitian
1. Variabel bebas adalah sistem KLT yang telah dioptimasi, yaitu jenis dan komposisi fase gerak.
2. Variabel tergantung adalah parameter validasi yaitu selektivitas, linearitas akurasi, presisi, danrange.
3. Variabel pengacau terkendali adalah:
a. pelarut, untuk mengatasinya digunakan pelarutpro analisis yang memiliki kemurnian tinggi.
b. pH pelarut, untuk mengatasinya dilakukan pengaturan pH pelarut, yaitu pada pH 4.
c. cahaya, untuk mengatasinya pengerjaan dilakukan di ruangan dengan intensitas cahaya yang terbatas serta dengan penggunaan alumunium foil.
C. Definisi Operasional
1. Sistem KLT yang digunakan dalam penelitian adalah fase diam berupa silika gel G 60 dan fase gerak berupa campuran kloroform dan asam asetat glasial (95:5).
2. Kadar kurkumin dinyatakan dalam satuanpart per million(ppm).
3. Parameter validasi yang digunakan adalah selektivitas, linearitas, akurasi, presisi, danrange.
D. Bahan Penelitian
Bahan-bahan yang digunakan dalam penelitian ini adalah baku kurkumin (hasil sintesis Prof. Dr. Sudibyo Martono, M.S., Apt. yang telah dikonformasi strukturnya dengan metode spektroskopi H-NMR dan Mass Spectra, serta memiliki titik lebur 181,2-182,40C), metanol p.a. EMSURE® ACS, ISO, Reag. Ph. Eur (E. Merck), kloroform p.a. EMSURE® ACS, ISO, Reag. Ph. Eur (E. Merck), asam asetat glasial EMPARTA®ACS (E. Merck), plat KLT silika gel G 60(E. Merck), dan sediaan cair OHT Kiranti®.
E. Alat Penelitian
DOA-P104-BN), dan alat-alat gelas yang umum digunakan dalam analisis (Pyrex).
F. Tata Cara Penelitian
1. Pembuatan pelarut (metanol pH 4)
Dibuat campuran metanol dan asam asetat glasial dengan perbandingan 9:1.
2. Pembuatan fase gerak
Fase gerak yang digunakan dalam penelitian menggunakan campuran kloroform : asam asetat glasial (95:5). Fase gerak dibuat dalam labu ukur 100 mL kemudian digojog.
3. Pembuatan larutan baku kurkumin
a. Pembuatan larutan stok kurkumin 1000 ppm. Sejumlah lebih kurang 10,0 mg baku kurkumin ditimbang seksama kemudian dilarutkan dalam metanol pH 4 hingga volume tepat 10,0 mL.
b. Pembuatan seri larutan baku. Sebanyak 0,250 mL; 0,375 mL; 0,500 mL; 0,625 mL; 0,750 mL; dan 0,875 mL larutan stok kurkumin diambil dan dimasukkan ke dalam labu ukur 5 ml kemudian diencerkan dengan metanol pH 4 hingga tanda, sehingga didapatkan konsentrasi 50 ppm, 75 ppm, 100 ppm, 125 ppm, 150 ppm, dan 175 ppm.
4. Penetapan panjang gelombang maksimum
diam silika gel G 60 dan setelah kering dikembangkan dalam bejana kromatografi yang telah dijenuhi dengan fase gerak. Setelah mencapai jarak rambat 10 cm, plat dikeluarkan dari bejana dan dikeringkan. Plat hasil pengembangan kemudian secepatnya discanning panjang gelombang serapan maksimumnya dengan densitometer.
5. Pembuatan kurva baku dan pengamatan nilai Retardation Factor (Rf) kurkumin
Seri larutan baku konsentrasi 50 ppm, 75 ppm, 100 ppm, 125 ppm, 150 ppm, dan 175 ppm masing-masing ditotolkan dengan volume penotolan 3 µL pada plat KLT dengan fase diam silika gel G 60 dan setelah kering dikembangkan dalam bejana kromatografi yang telah dijenuhi dengan fase gerak. Setelah mencapai jarak rambat 10 cm, plat dikeluarkan dari bejana dan dikeringkan. Plat hasil pengembangan kemudian secepatnya diukur AUC dan tinggi peaknya dengan densitometer. Replikasi dilakukan sebanyak 3 kali dan pilih persamaan kurva baku yang paling baik. Selain itu dilihat pula nilai Rf dari masing-masing seri baku kurkumin.
6. PenentuanrecoverydanCoefficient of Variations(CV) baku
7. Penentuan recovery dan Coefficient of Variations (CV) baku dalam matriks sampel
a. Pembuatan larutan sampel (LS). Sejumlah lebih kurang 15,0 mL sampel ditambah metanol pH 4 hingga volume 25,0 mL. Larutan sampel kemudian didegasing selama 15 menit dan disaring dengan kertas saring. Larutan hasil penyaringan ditambah metanol pH 4 hingga volume 25,0 mL.
b. Pembuatan larutan sampel dengan penambahan baku kurkumin (LSK). Sejumlah 0,125 mL larutan sampel kurkumin dimasukkan dalam labu takar 5 mL, kemudian ditambahkan 4,5 mL LSdan ditambahkan metanol pH 4 hingga tanda. Replikasi dilakukan sebanyak 5 kali.
c. Pengembangan dan pengukuran. LSdan LSKdiberi perlakuan seperti pada poin F.5. Setelah itu dihitung kadar baku kurkumin dalam sampel menggunakan persamaan kurva baku yang telah dibuat pada poin F.5. Kadar baku kurkumin dalam sampel adalah selisih kadar LSK dengan kadar LS. Selanjutnya dihitungrecoverydan CVnya.
G. Analisis Hasil
1. Selektivitas
Selektivitas ditentukan dengan membandingkan nilai Rf baku dan Rf sampel. Selain itu selektivitas juga ditunjukkan dengan nilai resolusi ≥ 1,5. Resolusi dapat dihitung dengan cara berikut:
2. Linearitas
Linearitas dilihat dari harga r (koefisien korelasi) hasil pengukuran seri baku kurkumin. Suatu metode dapat dikatakan memiliki linearitas yang baik jika r > 0,999.
3. Akurasi
Akurasi metode analisis dinyatakan denganrecoveryyang dapat dihitung dengan cara berikut:
4. Presisi
Presisi metode analisis dinyatakan dengan CV (koefisien variasi) yang dapat dihitung dengan cara berikut:
5. Range
Rangemerupakan interval konsentrasi analit yang memenuhi persyaratan linearitas, akurasi, dan presisi.
6. Akurasi pengukuran baku dalam matriks sampel
Nilai recovery baku kurkumin dalam matriks sampel dapat dihitung dengan rumus berikut:
BAB IV
HASIL DAN PEMBAHASAN
A. Pembuatan Metanol pH 4
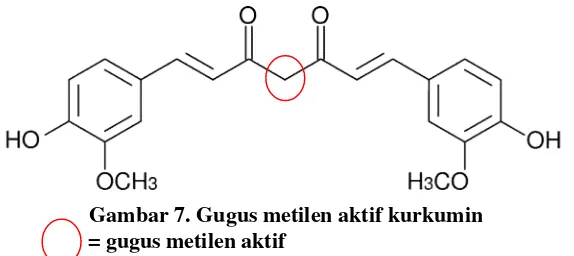
Kurkumin merupakan senyawa yang tidak stabil dalam pH basa, karena dapat terdegradasi pada gugus metilen aktifnya, sehingga terbentuk asam ferulat dan feruloilmetan. Apabila kurkumin terdegradasi, maka kadar yang diperoleh akan berkurang sehingga tidak dapat merepresentasikan kadar kurkumin yang sebenarnya.
Gambar 7. Gugus metilen aktif kurkumin = gugus metilen aktif
Oleh karena itu, untuk menjaga stabilitas kurkumin selama pengerjaan, maka digunakan metanol pH 4 sebagai pelarut. pH pelarut yang digunakan ini, diperoleh berdasarkan hasil optimasi yang dilakukan pada rangkaian penelitian ini. Metanol sendiri memiliki pH 5, sehingga untuk menurunkan pHnya hingga 4, dilakukan penambahan asam asetat glasial sebanyak 1 bagian pada setiap 9 bagian metanol.
B. Pembuatan Fase Gerak
Pembuatan fase gerak pada penelitian ini menggunakan fase gerak yang diperoleh dari hasil optimasi yang dilakukan pada rangkaian penelitian ini, yaitu kloroform : asam asetat glasial (95:5). Pembuatan fase gerak dengan jenis dan komposisi tersebut bertujuan agar didapatkan polaritas fase gerak yang sesuai sehingga dapat memisahkan kurkumin dengan senyawa lain dalam sampel secara optimal. Selain itu, pemilihan asam asetat glasial sebagai salah satu komposisi fase gerak, juga bertujuan untuk memberikan suasana asam untuk menjaga stabilitas kurkumin. Sistem kromatografi pada penelitian ini merupakan kromatografi fase normal, karena fase gerak pada penelitian ini bersifat non polar, sedangkan fase geraknya, yaitu silika gel bersifat polar.
C. Pembuatan Larutan Baku
kurva baku yang diperoleh dapat digunakan untuk penetapan kadar analit dalam sampel.
D. Penetapan Panjang Gelombang Maksimum Kurkumin
Penetapan panjang gelombang maksimum kurkumin dilakukan agar didapatkan panjang gelombang dimana kurkumin memberikan respon yang maksimum, sehingga sensitivitas pengukurannya tinggi, serta memberikan hasil yang reprodusibel pada pengulangan pengukuran. Oleh karena itu, dengan pengukuran pada panjang gelombang maksimum, diharapkan dapat meminimalkan kesalahan pada pengukuran.
Gambar 8. Spektra baku kurkumin konsentrasi 50ppm, 100 ppm, dan 175 ppm (λ maks=425 nm)
Kurkumin memiliki gugus kromofor yang panjang serta auksokrom, sehingga dapat memberikan serapan pada panjang gelombang visibel.
Gambar 9. Gugus kromofor dan auksokrom pada kurkumin = kromofor
= auksokrom
175 ppm
100 ppm
E. Pengamatan nilaiRetardation Factor(Rf) dan Pembuatan Kurva Baku Kurkumin
Pengamatan nilai Rf merupakan parameter analisis kualitatif yang nantinya digunakan untuk mengetahui ada tidaknya analit dalam sampel. Pengamatan nilai Rfmenggunakan konsentrasi tengah seri larutan baku kurkumin, yaitu 100 ppm. Dari hasil pengamatan, diperoleh nilai Rfbaku kurkumin 0,52.
Gambar 10. Baku kurkumin 100 ppm (nilai Rf=0,52)
OCH3
Gambar 11. Interaksi kurkumin dengan fase gerak
O Si O
Gambar 12. Interaksi hidrogen kurkumin dengan fase diam
Pada gambar 11 dan 12 dapat dilihat bahwa interaksi kurkumin dengan fase geraknya lebih dominan. Adanya interaksi hidrogen dan interaksi dipol-dipol antara asam asetat glasial dengan kurkumin serta interaksi dipol-dipol kloroform dengan kurkumin akan menyebabkan lepasnya interaksi hidrogen antara kurkumin dengan permukaan silika gel. Hal ini akan menyebabkan kurkumin dapat terelusi oleh fase gerak dan mencapai jarak rambat tertentu. Interaksi yang sesuai antara kurkumin dengan fase diam dan fase gerak akan menghasilkan nilai Rfyang baik, yaitu antara 0,2-0,8.
Selain dari analisis interaksi senyawa terhadap fase diam dan fase geraknya. Nilai Rf suatu senyawa juga dapat dijelaskan dari polaritas senyawa tersebut dan kesesuaiaannya terhadap polaritas dari fase gerak yang digunakan. Fase gerak yang digunakan yaitu campuran antara kloroform dan asam asetat glasial (95:5) dengan indeks polaritas campuran 4,205. Dari hasil percobaan indeks polaritas ini memiliki kesesuaian polaritas dengan kurkumin, sehingga kurkumin dapat terelusi oleh fase gerak dan mencapai jarak rambat tertentu.
Pembuatan kurva baku kurkumin dilakukan 3 replikasi. Hal ini bertujuan untuk mendapatkan nilai koefisien korelasi yang paling baik. Koefisien korelasi menunjukkan korelasi hubungan antara konsentrasi dengan respon pengukuran, baik itu Area Under Curve (AUC) atau tinggi peak. Respon yang menunjukkan nilai korelasi yang paling baik terhadap konsentrasi akan digunakan dalam pembuatan persamaan kurva baku.
Tabel IV. Data replikasi kurva baku kurkumin Baku kurkumin
Replikasi I Replikasi II Replikasi III
Seri
51,5 7009,9 183 49,5 6528,6 145,5 50 6600 149,6
77,25 9403,3 252,1 74,25 8785,8 211,9 75 8864,1 216,9
103 11159,2 272,2 99 11108,2 253 100 11298,6 257,7
128,75 13535,4 341,5 123,75 13577,8 330,9 125 13455,5 336,9 154,5 15461,5 355,8 148,5 15698 358,6 150 15774,6 364,6 180,25 18444 449,5 173,25 17818,8 449,5 175 18201,1 397,7
A 2509,47
B 86,2372 1,900 B 91,9564 2,0190 B 92,4502 2,0146
r 0,9976 0,9821 r 0,9997 0,9890 r 0,9999 0,9886
Pada tabel IV dapat dilihat bahwa hubungan antara kadar dengan AUC menunjukkan korelasi yang lebih baik dibandingkan hubungan antara kadar dengan tinggi peak. Korelasi yang baik ini dilihat dari nilai r yang paling mendekati 1. Oleh karena itu, respon pengukuran yang akan digunakan adalah AUC.
Tabel V. Data replikasi kurva baku kurkumin dengan penyesuaian nilai AUC Baku kurkumin
Replikasi I Replikasi II Replikasi III
Seri baku
51,5 70,099 49,5 65,286 50 66
77,25 94,033 74,25 87,858 75 88,641
103 111,592 99 111,082 100 112,986
128,75 135,354 123,75 135,778 125 134,555
154,5 154,615 148,5 156,98 150 157,746
180,25 184,44 173,25 178,188 175 182,011
A 25,0948 A 20,1123 A 19,6500
B 0,8624 B 0,9196 B 0,9245
r 0,9976 r 0,9997 r 0,9999
Gambar 13. Hubungan antara konsentrasi kurkumin dengan AUC/100 (replikasi III)
F. Validasi Metode Analisis
Validasi metode analisis dilakukan untuk membuktikan bahwa metode analisis yang digunakan memenuhi persyaratan validitas sehingga memberikan hasil analisis yang dapat dipercaya. Validasi dilakukan dengan 3 seri konsentrasi sebanyak 5 replikasi. Konsentrasi yang digunakan merupakan konsentrasi rendah, sedang, dan tinggi dari konsentrasi seri baku, yaitu 50 ppm, 100 ppm, dan 175 ppm. Pemilihan ketiga seri konsentrasi ini adalah untuk mewakili setiap konsentrasi dari seri baku, yaitu 50 ppm sampai 175 ppm. Parameter yang digunakan dalam penentuan validitas metode ini adalah selektivitas, linearitas, akurasi, presisi, danrange.
1. Selektivitas
analit dari matriks sampel, dilakukan dengan mengekstraksi sampel dengan metanol pH 4. Ekstraksi dilakukan dengan menggunakan ultrasonikator. Gelombang ultrasonik yang dihasilkan akan memberikan energi atau getaran yang akan mendorong kurkumin keluar dari serbuk simplisia yang tersuspensi dalam sampel, kemudian adanya metanol akan dapat menarik dan melarutkan kurkumin. Hal ini disebabkan karena kurkumin memiliki kelarutan yang baik dalam metanol. Namun banyak senyawa-senyawa dalam sampel yang juga memiliki kelarutan yang baik dalam metanol, sehingga dapat ikut terekstraksi bersama kurkumin, seperti demetoksikurkumin danbis-demetoksikurkumin, serta minyak atsiri. Oleh karena itu, selektivitas yang baik dari metode diperlukan untuk mengukur analit secara akurat tanpa terganggu oleh senyawa-senyawa lain yang terdapat dalam sampel.
Tabel VI. Perbandingan nilai Rfbaku dan sampel, serta nilai resolusi
Konsentrasi seri larutan baku kurkumin (ppm)
Rfbaku Replikasi
sampel Rfsampel Resolusi
50 0,53 1 0,50 2,4
75 0,53 2 0,50 2,25
100 0,52 3 0,50 2,4
125 0,53 4 0,51 2,53
150 0,53 5 0,54 2,57
175 0,54
Rata-rata 0,53 0,51 2,43
Pada tabel VI dapat dilihat bahwa Rf baku dan analit dalam sampel menunjukkan nilai yang identik, dimana nilai Rf rata-rata dari baku adalah 0,53 dan Rf rata-rata dari analit dalam sampel adalah 0,51. Dari gambar 14 dan 15 dapat dilihat nilai Rfyang identik antara baku dan sampel. Sedangkan untuk nilai resolusi, pada gambar 15 dapat dilihat bahwa pada kromatogram sampel terdapat 3peakdan nilai rata-rata resolusi antarapeaknomor 3 denganpeakterdekat (peak nomor 2) adalah 2,43. Berdasarkan nilai Rf dan resolusi yang diperoleh tersebut, maka metode KLT-Densitometri ini dikatakan memenuhi parameter selektivitas dalam menetapkan kadar kurkumin.
Gambar 15. Rfsampel replikasi 1 = 0,50
2. Linearitas
Linearitas suatu metode ditunjukkan oleh nilai koefisien korelasi (r) dari kurva baku, dimana nilai r ini menunjukkan korelasi hubungan antara konsentrasi dengan respon pengukuran, dalam hal ini AUC. Suatu metode dikatakan memiliki linearitas yang baik apabila nilai r > 0,999 (Yuwono and indrayanto, 2005).
Berdasarkan data yang diperoleh dari hasil pembuatan kurva baku diperoleh nilai r untuk replikasi II = 0,9997 dan replikasi III = 0,9999. Nilai r yang kurva baku repliasi II dan III tersebut memenuhi persyaratan, sehingga dapat dikatakan metode KLT-densitometri ini memiliki linearitas yang baik dalam menetapkan kadar kurkumin.
3. Akurasi
Tabel VII. Data %recovery
Recovery(%) Kadar
kurkumin Replikasi I Replikasi II Replikasi III Replikasi IV Replikasi V
50 ppm 101,10 100,11 99,00 100,75 98,95
100 ppm 101,64 100,77 101,79 100,10 98,61
175 ppm 101,99 100,18 102,72 103,83 100,96
Berdasarkan hasil yang diperoleh, nilai recovery yang masuk pada rentang 98-102% adalah konsentrasi level rendah hingga sedang. Pada level konsentrasi tinggi (175 ppm), metode ini memiliki nilai recoveryantara 100,18-103,83%, nilairecoveryyang diperoleh tidak memenuhi persyaratan akurasi yang baik yaitu 98-102% (Harmita, 2004). Oleh karena itu, metode ini dikatakan memiliki akurasi yang baik pada kadar 50 ppm dan 100 ppm, sehingga dapat digunakan untuk menetapkan kadar kurkumin pada level tersebut.
4. Presisi
Presisi merupakan parameter yang menggambarkan kedekatan hasil pengukuran satu dengan lainnya dalam kondisi analisis yang sama. Presisi dinyatakan dengan nilai Coefficient of Variation (CV) atau Relative Standar Deviation(RSD). Kriteria presisi yang baik yaitu nilai CV≤ 2% (Harmita, 2004).
Tabel VIII. DataCoefficientof Variation (CV)
Kadar kurkumin (ppm) SD CV (%)
50 ppm 50,2883 0,8552 1,7
100 ppm 100,5458 0,7125 0,7
175 ppm 179,4245 1,5902 0,9
kadar kurkumin pada level konsentrasi tersebut menggunakan metode ini akan memberikan kedekatan hasil pengukuran.
5. Range
Rangemerupakan interval antara konsentrasi analit pada level bawah dan level atas dalam suatu sampel, yang masih memenuhi parameter linearitas, akurasi, dan presisi.
Gambar 16.Rangekonsentrasi kurkumin
Pada gambar 16 dapat dilihat bahwarangekonsentrasi metode ini adalah 50-100 ppm. Range ini menunjukkan area analisis yang memenuhi parameter linearitas, akurasi, dan presisi.
G. Penentuan Akurasi dan Presisi Baku Kurkumin dalam Sampel Penentuan akurasi dan presisi baku kurkumin dalam sampel dilakukan dengan menambahkan baku kurkumin ke dalam matriks sampel. Namun sebelum melihat akurasi dan presisi baku kurkumin dalam sampel, adisi baku kurkumin pada sampel juga dilakukan untuk memastikan bahwapeak dengan nilai Rf yang identik terhadap baku kurkumin memang merupakan peak senyawa kurkumin.
Linearitas dan presisi
Hal ini dapat diketahui dengan melihat apakah terjadi penambahan luas area pada peak yang dimaksud ketika dilakukan penambahan baku ke dalam sampel. Apabila luas area pada peak tersebut bertambah ketika baku kurkumin ditambahkan maka dapat dipastikan bahwa peak tersebut merupakan peak kurkumin. Berikut adalah gambar peak dari sampel tanpa penambahan baku dan sampel yang ditambah baku:
Gambar17. Kromatogram sampel tanpa penambahan baku kurkumin
Setelah dapat dipastikan bahwa peak dengan nilai Rf yang identik tersebut merupakan kurkumin, maka dilakukan penentuan akurasi dan presisi baku kurkumin dalam sampel. Hal ini dilakukan untuk mengetahui apakah metode ini masih dapat mengukur respon baku kurkumin dalam matriks sampel secara akurat dan seksama. Akurasi dan presisi baku yang ditambahkan dapat dilihat pada tabel IX.
Tabel IX.Recoverydan CV baku kurkumin dalam matriks sampel Replikasi Recovery(%) CV (%)
I 101,28
II 99,47
III 101,83
IV 96,99
V 103,55
2,5
49
A. Kesimpulan
Metode KLT-densitometri dengan instrumen Camag TLC Scanner 3 CAT. No. 027.6485 SER. No.160602, fase diam silika gel G 60, fase gerak kloroform:asam asetat glasial (95:5), volume penotolan 3,0 μL, dan jarak pengembangan 10 cm memiliki selektivitas dan linearitas yang baik akurasi yang baik pada konsentrasi 50-100 ppm, presisi yang baik pada konsentrasi 50-175 ppm, serta range antara 50-100 ppm. Berdasarkan hasil tersebut, maka metode KLT-densitometri ini memiliki validitas yang baik untuk menetapkan kadar kurkumin dalam sampel OHT Kiranti®.
B. Saran
DAFTAR PUSTAKA
Adamovics, 1997, Chromatographic Analysis of Pharmaceuticals,2nd Edition, Marcel Dekker, New York.
Aggarwal, B., Bhatt, D., Ichikawa, H., Ahn, K.S., Sethi, G., Sandur, S.K., Natarajan, C., Seeram, N., and, Shishodia, S., 2006, Curcumin-Biological and medicinal Properties, 299-300, http://www.indsaff.com/ 10%20Curcumin%20biological.pdf , diakses tanggal 25 Oktober 2010. Agnam, N., Samhoedi, H., Timmerman, H., Venie, U. A., Sugiyanto, Goot, H.,
1995,The Relationship Between Structure And Inhibition Of Lipoxygenase Activity of Curcumin Derivatives In International Symposium On Curcumin Pharmacochemistry ISCP, Yogyakarta.
Anonim, 2010, Kiranti Sehat Datang Bulan, http://diarykiranti.com/kiranti-funfact-sdb/, diakses tanggal 25 Oktober 2010.
Aulton, 1988, Pharmaceutics: The Science of Dossage Form Design, edisi II, Churchill Livingstone, New York, pp. 310,335.
Badan Pengawas Obat dan Makanan Republik Indonesia, 2004, Keputusan Kepala Badan Pengawas Obat dan Makanan Republik Indonesia No. HK 00.05.4.2411 tentang Ketentuan Pokok Pengelompokkan dan Penandaan Obat Bahan Alam indonesia, Badan Pengawas Obat dan Makanan Republik Indonesia, Jakarta.
Badan Pengawas Obat dan Makanan Republik Indonesia, 2005, Pedoman Cara Pembuatan Obat tradisional yang Baik, Badan Pengawas Obat dan Makanan Republik Indonesia, Jakarta.
Bermawie, N., Rahardjo, M., Wahyuno, D., Ma’mun, 2005,Status Teknologi Dan Panen Tanaman Kunyit Dan Temulawak Sebagai Penghasil Kurkumin, 85, 96, Balai Penelitian Tanaman Obat dan Aromatik, Bogor.
Dean, J., 1995, Analytical Chemistry Handbook,Mc Graw-Hill Inc., USA, pp.4.98, 4.113.
Direktorat Jenderal Pengawasan Obat dan Makanan RI, 1995a, Materia Medika Indonesia, edisi VI, Departemen Kesehatan RI, Jakarta, pp.148-152.
Drakeiron, 2008, Info Pentagamavunon-0, http://drakeiron.wordpress.com, diakses tanggal 25 Oktober 2010.
Gritter, J.R., Bobbit, J.M., dan Scharting, A.E., 1991, Pengantar Kromatografi, diterjemahkan oleh Kosasih Pamawinata, Edisi II, Penerbit ITB, Bandung. Gandjar, G.I., dan Rohman, A, 2007, Kimia Farmasi Analisis, Pustaka Pelajar,
Yogyakarta, pp.362-363.
Gupta, A. P., Gupta, M. M., and Kumar, S., 1999,Simultaneous determination of curcuminoids in curcuma samples using high performance thin layer chromatography, http://203.190.147.121/bitstream/123456789/80/1/J Journal_Liquid_Chromatography_Related_Technologies_22_1561.pdf, diakses tanggal 15 Februari 2010.
Hanani, E., 2010, Standarisasi Simplisia dan Ekstrak Daun Handeuleum (Graptophyllum Pictum), Laporan Penelitian, Universitas Indonesia, Jakarta.
Harborne, 1973, Phtochemical methods, diterjemahkan oleh Kosasih Padmawinata dan Iwang Soediro, Terbitan II, hal.10-11, Institut Teknologi Bandung, Bandung.
Hardjono, 1983,Kromatografi, Laboratorium Analisa Kimia Fisika Pusat, UGM, Yogyakarta, pp.32-34.
Harmita, 2004, Petunjuk Pelaksanaan Validasi Metode dan Cara Perhitungannya, Departemen Farmasi FMIPA UI, Depok, pp.5-25.
Katno, dan Pramono, 2010, Tingkat Manfaat dan Keamanan Tanaman Obat dan Obat Tradisional, Laporan Penelitian, Fakultas Farmasi Universitas Gadjah Mada, Yogyakarta.
Kementerian Kesehatan RI, 1994, Keputusan Menteri Kesehatan RI Nomor:661/MENKES/VII/1994, Departemen Kesehatan RI, Jakarta.
Khopkar, 1990, Concepts of Analytical Chemistry, diterjemahkan oleh Sapto Raharjo, Universitas Indonesia Press, Jakarta.
Kohli, K., Ali, J., Ansari, M. J., and Raheman, Z., 2005, Curcumin : A Natural Anti- inflammatory Agent, 141 -142, In Indian Journal of Pharmacology, Jamie Hamdard University, New Delhi.
Mintarsih, 1990, Penetapan Kadar Alkaloid Kininda dalam Akar, Batang, dan Daun Chinchona Succirubra Pavon et Klotzsch dari Daerah Kaliurang Secara Spektrodensitometri (TLC-Scanner),Skripsi, Universitas Gadjah Mada, Yogyakarta.
Mulja, H.M. dan Suharman, 1995, Analisis Instrumental, Airlangga University Press, Surabaya, pp.102.
Mulja, H.M dan Hanwar, 2003, Pinsip Cara Berlaboratorium yang Baik (Good Laboratory Practice),Majalah Farmasi Airlangga, Vol.III, No.2, 71-76. Nakayama, T., 1997, Affinities of Dietary Phenolic Antioxidants for Lipid
Bilayers, in Shahidi, F., Ho, Chi-Tang. (Eds.), Phytochemicals and Phytopharmaceutical, 355-356, AOCS Press, USA.
Paramasivam, M., Aktar, W., Poi, R., Banerjee, H., Bandyopahyay, A., 2008, Occurrence of curcuminoids in Curcuma longa: A quality standardization by HPTLC, http://www.banglajol.info/index.php/BJP/article/, diakses tanggal 15 Februari 2010
Rohman, 2009, Kromatografi Untuk Analisis Obat, Graha Ilmu, Yogyakarta, pp.45, 47, 53, 217.
Roth, H.J., 1994, Pharmaceutical Analysis, diterjemahkan oleh Sarjono Kisman, Slamet Ibrahim, Cetakan 2, Gajah Mada University Press, Yogyakarta. Sari, L.O., 2006, Pemanfaatan Obat Tradisional dengan Pertimbangan Manfaat
dan Keamanannya, Majalah Ilmu Kefarmasian, Vol.III, No.1, 1-7.
Sherma, J., and, Fried B., 1996, Handbook of Thin Layer Chromatography, 2nd Edition, Marcel Dekker, Inc., pp.20.
Stahl, 1985,Drugs Analysis by Chromatography and Microscopy, diterjemahkan oleh Kosasih Padmawinata, Institut Teknologi Bandung, Bandung.
Stankovic, I., 2004, Curcumin: Chemical and Technical Assestment, ftp://ftp.fao.org/es/esn/jecfa/cta/CTA_61_Curcumin.pdf, diakses tanggal 15 Februari 2010.
Sudjadi, 1988, Metode Pemisahan, cetakan pertama, Penerbit Kanisius, Yogyakarta, pp.167-175.
Swartz and Krull, 1997, Chromatographic Analysis of Pharmaceuticals, 2nd Edition, Marcel Dekker, USA.
Tonnesen, H. H., 1989, Studies On Curcumin And Curcuminoids, Catalytic Effect Of Demethoxy And Bis-demethoxycurcumin On The Peroxydation Of Linoleic Acid By 15- Lipoxygenase,Internal Journal Pharmacy, Vol. XV, 51, 179-181
Tonnesen, H. H. and Greenhill, J. V., 1992, Studies on curcumin andcurcuminoids.. Curcumin as a reducing agent and as a radical scavenger,Int. J. Pharm., XXII87
Tonnesen, H. H., and Karlsen, 1983, Curcuminoid and It’s Compounds, Journal Chromatography, Vol. 4, 259 -376.
Tonnesen, H.H., and Karlsen, 1985, Studies On Curcumin and Curcuminoids Alkaline Degradation of Curcuming Z.Lebens, Unters, Forsch, 180 : 132-134.
United States Pharmacopeial Convention, 2007,The United States Pharmacopeia, 30thedition, United States Pharmacopeial Convenction Inc., New York. Van der Goot, H., 2002, The Chemistry and Qualitative Structure – Activity
Relationships Of Curcumin In Recent Development In Curcumin Pharmacochemistry, Procedings of The International Symposium on Curcumin Pharmacochemistry, 1995, Edited By Suwijyo Pramono, Aditya Media, Yogyakarta.
Lampiran 2. Data Penimbangan Bahan 1. Baku Kurkumin
Kurkumin (g) Replikasi I Replikasi II Replikasi III
Berat kertas 13,966 15,001 14,785
Berat kertas + zat 13,976 15,011 14,795 Berat kertas + zat 13,9764 15,0112 14,7952 Berat kertas + sisa 13,9661 15,0013 14,7852
Berat zat 0,0103 0,0099 0,0100
2. Validasi Metode Kurkumin
Replikasi Kurkumin (g)
I II III IV V
Berat kertas 14,675 14,253 13,728 14,852 15,227 Berat kertas +
zat 14,685 14,263 13,738 14,862 15,237
Berat kertas +
zat 14,6853 14,2633 13,7386 14,8622 15,2374 Berat kertas +
sisa 14,6753 14,2531 13,7287 14,8523 15,2371 Berat zat 0,0100 0,0102 0,0099 0,0099 0,0103
3. Validasi Metode Kurkumin dalam Matriks Sampel Kurkumin (g)
Berat kertas 15,752
Berat kertas + zat 15,762 Berat kertas + zat 15,7623 Berat kertas + sisa 15,7524
Berat zat 0,0099
2. Seri 2 (75 ppm)
3. Seri 3 (100 ppm)
5. Seri 5 (150 ppm)
6. Seri 6 (175 ppm)
Lampiran 4. Kromatogram validasi metode 1. Konsentrasi rendah (50 ppm)
Replikasi II
Replikasi III
Replikasi V
2. Konsentrasi sedang (100 ppm)
Replikasi I
Replikasi III
Replikasi IV
3. Konsentrasi tinggi (175 ppm)
Replikasi I
Replikasi II
Replikasi IV
Replikasi V
Lampiran 5. Contoh perhitungan kadar kurkumin 1. Skema pembuatan
Timbang lebih kurang seksama 10,0 mg kurkumin ↓
Larutkan dengan metanol pH 4 ad hingga 10,0 ml ↓
Pipet 0,25 ml; 0,375 ml; 0,5 ml; 0,625 ml; 0,75 ml; dan 0,875 ml ↓
Encerkan dengan metanol pH 4 ad hingga 5,0 ml 2. Perhitungan seri kadar kurkumin
Bobot kurkumin hasil penimbangan = 0,0103 g = 10,3 mg
Kadar seri larutan baku kurkumin : C1.V1 = C2.V2
1030 ppm x 0,25 ml = C2 x 5 ml C2 = 51,5 ppm
Baku kurkumin
Replikasi I Replikasi II Replikasi III
Seri
51,5 7009,9 183 49,5 6528,6 145,5 50 6600 149,6
77,25 9403,3 252,1 74,25 8785,8 211,9 75 8864,1 216,9
103 11159,2 272,2 99 11108,2 253 100 11298,6 257,7
128,75 13535,4 341,5 123,75 13577,8 330,9 125 13455,5 336,9 154,5 15461,5 355,8 148,5 15698 358,6 150 15774,6 364,6 180,25 18444 449,5 173,25 17818,8 449,5 175 18201,1 397,7
A 2509,47
B 86,2372 1,900 B 91,9564 2,0190 B 92,4502 2,0146
r 0,9976 0,9821 r 0,9997 0,9890 r 0,9999 0,9886
Dilakukan penyesuaian nilai AUC agar nilai tanαmendekati 1 Baku kurkumin
Replikasi I Replikasi II Replikasi III
Seri baku
51,5 70,099 49,5 65,286 50 66
77,25 94,033 74,25 87,858 75 88,641
103 111,592 99 111,082 100 112,986
128,75 135,354 123,75 135,778 125 134,555
154,5 154,615 148,5 156,98 150 157,746
180,25 184,44 173,25 178,188 175 182,011
A 25,0948 A 20,1123 A 19,6500
B 0,8624 B 0,9196 B 0,9245
r 0,9976 r 0,9997 r 0,9999
Lampiran 6. Persamaan kurva baku dan gambar kurva baku kurkumin 1. Persamaan kurva baku yang digunakan adalah replikasi 3, yaitu :
2. Gambar kurva baku kurkumin
Lampiran 7. Nilai AUC dan contoh perhitunganrecoverykurkumin 1. AUC kurkumin
AUC/100 Kadar
kurkumin Replikasi I Replikasi II Replikasi III Replikasi IV Replikasi V
50 ppm 66,386 66,85 64,956 65,752 66,761
100 ppm 112,693 118,79 112,817 112,087 113,547
175 ppm 184,658 184,968 184,713 185,956 187,885
2. Contoh perhitunganrecovery Replikasi I
Perhitungan kadar teoritis
- Bobot kurkumin hasil penimbangan = 0,0100 g = 10 mg
- Kadar stok kurkumin = 10 mg/10 ml = 1000 mg/1000ml = 1000 ppm C1.V1=C2.V2
Perhitungan kadar terukur y = 0,9245x+19,6500
Kadar rendah→ 66,386 = 0,9245x + 19,6500 x = 50,5527 ppm
Kadar sedang→ 112,693 = 0,9245x + 19,6500 x = 100,6414 ppm
Kadar tinggi→ 184,658 = 0,9245x + 19,6500 x = 178,4835 ppm
kurkumin Replikasi I Replikasi II Replikasi III Replikasi IV Replikasi V
50 ppm 101,10 100,11 99,00 100,75 98,95
100 ppm 101,64 100,77 101,79 100,10 98,61
175 ppm 101,99 100,18 102,72 103,83 100,96
Lampiran 8. Contoh perhitungan CV kurkumin Kadar kurkumin (ppm) SD CV (%)
50 ppm 50,2883 0,8552 1,7
100 ppm 100,5458 0,7125 0,7 175 ppm 179,4245 1,5902 0,9
2. Replikasi II
3. Replikasi III
5. Replikasi V
Lampiran 10. Kromatogram sampel dengan penambahan baku 1. Replikasi I
3. Replikasi III
4. Replikasi IV
Lampiran 11. Contoh perhitungan resolusi
Diketahui: Rfkurkumin = 0,50 Rfpeakterdekat = 0,32 Perhitungan:
Resolusi (Rs) =
= 2,4 (replikasi I) Replikasi II→ 2,25
Replikasi III→ 2,4 Replikasi IV→ 2,53 Replikasi V→ 2,57
Lampiran 12. Nilai AUC sampel dan sampel yang diadisi baku kurkumin AUC/100
Lampiran 13. Contoh perhitunganrecoverybaku kurkumin dalam sampel
Perhitungan kadar teoritis
Bobot kurkumin = 0,0099 g = 9,9 mg
Kadar stok kurkumin = 9,9 mg/10 ml = 990 mg/1000 ml = 990 ppm C1.V1= C2.V2
C2= 24,75 ppm Replikasi I
Perhitungan kadar baku + sampel terukur y = 0,9245x + 19,65
93,055 = 0,9245x +19,65 x = 79,3997 ppm
Perhitungan kadar sampel terukur y = 0,9245x + 19,65
69,88 = 0,9245x +19,65 x = 54,3321 ppm
Perhitungan kadar baku terukur
Kadar terukur baku = kadar (baku + sampel) terukur - kadar sampel terukur = 79,3997 – 54,3321
= 25,0676 ppm
Perhitunganrecoverykadar baku terukur Recovery=
=
= 102,34 %
Lampiran 14. Tabel perhitungan CV kadar baku kurkumin dalam sampel
Replikasi (ppm) (ppm) SD CV (%)
BIOGRAFI PENULIS