UJI VALIDASI METODE
ZERO CROSSING
SPEKTROFOTOMETRI DERIVATIF PADA PENETAPAN
KADAR KAFEIN DAN PARASETAMOL
DALAM SEDIAAN TABLET
SKRIPSI
Diajukan Sebagai Salah Satu Syarat Untuk Memperoleh Gelar Sarjana Farmasi Pada Fakultas Farmasi
Universitas Sumatera
OLEH:
NURUL ROSITA
NIM 121524019
PROGRAM EKSTENSI SARJANA FARMASI
FAKULTAS FARMASI
UNIVERSITAS SUMATERA UTARA
MEDAN
UJI VALIDASI METODE
ZERO CROSSING
SPEKTROFOTOMETRI DERIVATIF PADA PENETAPAN
KADAR KAFEIN DAN PARASETAMOL
DALAM SEDIAAN TABLET
SKRIPSI
Diajukan Sebagai Salah Satu Syarat Untuk Memperoleh Gelar Sarjana Farmasi Pada Fakultas Farmasi
Universitas Sumatera Utara
OLEH:
NURUL ROSITA
NIM 121524019
PROGRAM EKSTENSI SARJANA FARMASI
FAKULTAS FARMASI
UNIVERSITAS SUMATERA UTARA
MEDAN
Dra. Siti Nurbaya, M.Si., Apt. NIP 195008261974122001 PENGESAHAN SKRIPSI
UJI VALIDASI METODE
ZERO CROSSING
SPEKTROFOTOMETRI DERIVATIF PADA PENETAPAN
KADAR KAFEIN DAN PARASETAMOL
DALAM SEDIAAN TABLET
OLEH:NURUL ROSITA NIM 121524019
Dipertahankan di Hadapan Panitia Penguji Skripsi Fakultas Farmasi Universitas Sumatera Utara
Pada Tanggal: 20 Juni 2015
Pembimbing I, Panitia Penguji,
Prof. Dr. Muchlisyam, M.Si., Apt. NIP 195006221980021001
Pembimbing II, Prof
NI195
Medan, Juli 2015 Fakultas Farmasi
Universitas Sumatera Utara Wakil Dekan I,
Prof. Dr. Julia Reveny, M.Si., Apt. NIP 195807101986012001
Panitia Penguji,
Prof. Dr. Ginda Haro, M.Sc., Apt. NIP 195108161980031002
Pembimbing II,
Dra. Tuty Roida Pardede, M.Si., Apt. NIP 195401101980032001
Dra. Sudarmi, M.Si., Apt. NIP 1954091019832001
KATA PENGANTAR
Puji dan syukur kehadirat Tuhan Yang Maha Esa, karena limpahan rahmat
dan kasih_Nya, sehingga penulis dapat menyelesaikan skripsi ini yang berjudul “Uji Validasi Metode Zero Crossing Spektrofotometri Derivatif Pada Penetapan
Kadar Kafein dan Parasetamol Dalam Sediaan Tablet”. Skripsi ini diajukan sebagai salah satu syarat untuk memperoleh gelar sarjana farmasi pada Fakultas Farmasi Universitas Sumatera Utara.
Pada kesempatan ini penulis juga mengucapkan terima kasih yang tulus dan ilkhas kepada Ibu Prof. Dr. Julia Reveny, M.Si., Apt., selaku Wakil Dekan Fakultas Farmasi USU Medan yang telah memberikan fasilitas sehingga penulis
dapat menyelesaikan pendidikan demi memperoleh gelar sarjana farmasi. Bapak Dr. Muchlisyam, M.Si., Apt., dan Ibu Dra. Tuty Roida Pardede, M.Si., Apt., selaku
Pembimbing yang telah memberikan waktu, bimbingan, dan nasehat selama penelitian hingga selesainya penyusunan skripsi ini. Bapak Drs. Chairul Azhar Dalimunthe, M.Si., Apt., selaku Penasehat Akademik yang telah memberikan
bimbingan kepada penulis selama masa perkuliahan. Bapak dan Ibu staf pengajar Fakultas Farmasi USU Medan yang telah mendidik selama perkuliahan. Bapak dan Ibu Staf Laboratorium Penelitian yang telah memberikan fasilitas, petunjuk
dan membantu selama penelitian.
Terima kasih kepada Bapak Prof. Dr. Ginda Haro, M.Sc., Apt., Ibu Dra. Sudarmi, M.Si., Apt., dan Ibu Dra. Siti Nurbaya, M.Si., Apt., selaku dosen penguji yang telah memberikan kritik, saran, dan arahan kepada penulis dalam
menyelesaikan skripsi ini.
Ibunda tercinta, Baharuddin S.Pd dan Anita yang tiada hentinya memberi semangat dan doa bagi kesuksesan penulis, juga kepada Suami tercinta Cheryl
Bayu Zachary dan anakku tersayang M. Al Azzam Mudrik Mutawakil serta Kakak dan Adikku, Briptu Badrul Rahman, Anharuddin, Suryani, Ana Maulida dan Ahmad Thaybi yang selalu memberi doa, motivasi dan semangatnya. Buat
teman-teman seperjuangan di Laboratorium Penelitian: Aisyah, kak Tina, Dadang, Yudi, Tya, dan Husna serta teman-teman: Fauzul, Dara, Cut Nuri, Rini, Maiza, Kak
Mus, Marjan dan teman-teman stambuk 2012 lainnya yang tidak dapat penulis sebutkan satu-persatu namanya, terima kasih telah memberi bantuan, dukungan dan motivasi dalam menyelesaikan penulisan skripsi ini.
Penulis menyadari bahwa dalam penulisan skripsi ini masih jauh dari kesempurnaan, oleh karena itu dengan segala kerendahan hati, penulis menerima
kritik dan saran demi kesempurnaan skripsi ini. Akhir kata, penulis berharap semoga skripsi ini dapat memberi manfaat bagi kita semua.
Medan, Juni 2015
Penulis,
UJI VALIDASI METODE
ZERO CROSSING
SPEKTROFOTOMETRI DERIVATIF PADA PENETAPAN
KADAR KAFEIN DAN PARASETAMOL DALAM SEDIAAN
TABLET
ABSTRAK
Latar Belakang: Banyak obat yang menggunakan berbagai macam zat aktif, seperti obat analgesik. Sehingga muncul kesulitan untuk menganalisis kadar masing-masing komponen. Oleh karena itu diperlukan suatu metode untuk menganalisis masing-masing komponen tersebut, misalnya untuk menganalisis kadar campuran parasetamol dan kafein dalam sediaan tablet. Spektrofotometri derivatif merupakan metode transformasi dari spektrofotometri konvensional yang dikembangkan untuk analisis kuantitatif multikomponen senyawa aktif pada suatu sediaan.
Tujuan Penelitian: Untuk menguji validasi metode zero crossing spektrofotometri
derivatif kandungan parasetamol dan kafein dalam sediaan tablet.
Metode: Pengambilan sampel secara purposif terhadap sediaan tablet Paramex® dan Saridon® dan kemudian menentukan jumlah kandungan parasetamol dan kafein dengan spektrofotometri derivatif metode zero crossing pada serapan derivat kedua dalam pelarut HCl 0,1N.
Hasil: Hasil penelitian menunjukkan bahwa kadar parasetamol dalam sediaan tablet Paramex® yang dianalisis sebesar 100,13% ± 0,8455% dan sediaan tablet Saridon® sebesar 98,21% ± 5,1002% dan kadar kafein dalam sediaan tablet Paramex® sebesar 101,39% ± 5,7119% dan sediaan tablet Saridon® sebesar 98,18% ± 3,8038%. Dapat disimpulkan bahwa hasil penetapan kadar campuran parasetamol dan kafein dalam tablet memenuhi persyaratan sesuai dengan persyaratan yang tertera pada United States Pharmacopoeia, (USP 30 NF 25, 2007). % perolehan kembali parasetamol sebesar 99,93% dan kafein sebesar 101,77%. RSD parasetamol sebesar 3,5% dan kafein sebesar 3,5%.
Kesimpulan: Berdasarkan hasil penelitian yang dilakukan maka metode spektrofotometri derivatif yang digunakan memenuhi persyaratan akurasi dan presisi dan dapat digunakan untuk menetapkan kandungan parasetamol dan kafein dalam sediaan tablet.
VALIDATION TEST WITH ZERO CROSSING METHODE OF DERIVATIVE SPECTROFOTOMETRY FOR DETERMINATION OF
CAFFEINE AND PARACETAMOL IN TABLET
ABSTRACT
Background: Many drugs that use various active substance, such as analgesics. Hence the difficulty to analyze the levels of each component. Therefore we need a method to analyze each of these components, for example, to analyze the levels of a mixture of paracetamol and caffein in tablets. The purpose of this research is to developmentof derivative spectrophotometry method with the zero crossing for determination of paracetamol and caffein mixture in tablet. Derivative spectrophotometry is a transformation method from conventional spectrophotometric that developed for quantitative analysis of multicomponent active compounds in a preparation.
Objective: The aim of this study was to test the validation of derivative spectrophotometry method in estimating the content of paracetamol and caffeine in tablets.
Method: The method of research was done by purposive sampling to Paramex® and Saridon® in tablets and then determine the amount of paracetamol and caffeine content using derivative spectrophotometric with zero crossing method to second derivative in the solven HCl 0.1N.
Result: The results of research was exhibited that paracetamol in Paramex® that were analyzed are 100.13% ± 0.8455% and Saridon® are 98.21% ± 5.1002% and caffeine in Paramex® are 101.39% ± 5.7119% and Saridon® are 98.18% ± 3.8038%. Therefore, it suggested was result of determination of paracetamol and caffeine mixture in tablet the requirement of the United States Pharmacopoeia, (USP 30 NF 25, 2007). % Recovery of paracetamol are 99.93% and caffeine are 101.77%. RSD of paracetamol are 3.5% and caffeine are 3.5%.
Conclusion: Based on the results of research that derivative spectrophotometry method fulfilled the requirements of accuracy and precision and can be used to determinate the content of paracetamol and caffeine in tablets.
DAFTAR ISI
Halaman
JUDUL ... i
HALAMAN PENGESAHAN ... iii
KATA PENGANTAR ... . iv
ABSTRAK ... vi
ABSTRACT ... vii
DAFTAR ISI ... viii
DAFTAR TABEL ... xii
DAFTAR GAMBAR ... xiii
DAFTAR LAMPIRAN ... xix
BAB I PENDAHULUAN ... 1
1.1 Latar Belakang ... 1
1.2 Perumusan Masalah ... 3
1.3 Hipotesis ... 3
1.4 Tujuan Penelitian ... 4
1.5 Manfaat Penelitian ... 4
BAB II TINJAUAN PUSTAKA ... ... 5
2.1. Uraian Bahan ... 5
2.1.1 Parasetamol ... 5
2.1.2. Kafein ... 6
2.2. Spektrofotometri Ultraviolet - Visibel ... 7
2.2.2. Pembagian Metode Analisis Spektrofotometri
Ultraviolet – Visibel ... 7
2.2.3. Proses Penyerapan Radiasi pada Spektrofotometri Ultraviolet – Visibel ... 7
2.2.4. Kegunaan Spektrofotometri Ultraviolet – Visibel ... 10
2.2.5. Komponen Spektrofotometri Ultraviolet – Visibel ... 11
2.3. Spektrofotometri Derivatif ... 12
2.3.1. Pengertian Spektrofotometri Derivatif ... 12
2.3.2. Metode Spektrofotometri Derivatif ... 14
2.3.3. Kegunaan Spektrofotometri Derivatif ... 15
2.4. Validasi Metode Analisis ... 16
2.4.1. Akurasi ( Kecermatan ) ... 17
2.4.2. Presisi ( Keseksamaan ) ... 18
2.4.3. Batas Deteksi dan Batas Kuantitasi ... 18
2.4.4. Linieritas ... 19
2.4.5. Rentang ... 19
BAB III METODE PENELITIAN ... 20
3.1 Waktu dan Tempat Penelitian ... 20
3.2 Alat ... 20
3.3 Bahan ... 20
3.4 Pengambilan Sampel ... 20
3.5 Prosedur Penelitian ... 21
3.5.2 Pembuatan Larutan Induk Baku ... 21
3.5.2.1 Pembuatan Larutan Induk Baku Parasetamol ... 21
3.5.2.2 Pembuatan Larutan Induk Baku Kafein ... 21
3.5.3 Pembuatan Spektrum Serapan Maksimum ... 21
3.5.3.1 Pembuatan Spektrum Serapan Maksimum Parasetamol ... 21
3.5.3.2 Pembuatan Spektrum Serapan Maksimum Kafein ... 22
3.5.4 Pembuatan Spektrum Serapan Derivatif ... 22
3.5.4.1 Pembuatan Spektrum Serapan Derivatif Parasetamol ... 22
3.5.4.2 Pembuatan Spektrum Serapan Derivatif Kafein ... 22
3.5.5 Penentuan Zero Crossing ... 23
3.5.6 Penentuan Panjang Gelombang ( ) Analisis ... 23
3.5.7 Pembuatan dan Penentuan Linieritas Kurva Kalibrasi... 23
3.5.7.1 Pembuatan Kurva Kalibrasi dan Penentuan Kurva Kalibrasi Parasetamol ... .... 23
3.5.7.2 Pembuatan Kurva Kalibrasi dan Penentuan Kurva Kalibrasi Kafein ... 25
3.5.8 Penentuan Kadar Parasetamol dan Kafein dalam Sediaan Tablet .... ... 25
3.5.9 Uji Validasi ... 26
3.5.9.1 Uji Akurasi ... 26
3.5.9.2 Uji Presisi ... 26
BAB IV HASIL DAN PEMBAHASAN ... 29
4.1 Hasil Penentuan Kurva Serapan Maksimum ... 29
4.2 Hasil Penentuan Kurva Serapan Derivatif Parasetamol ... 30
4.3 Hasil Penentuan Kurva Serapan Derivatif Kafein ... 32
4.4 Hasil Penentuan Zero Crossing ... 34
4.4.1 Zero Crossing Derivat Kedua Pada Parasetamol
dan Kafein ...
344.5 Hasil Penentuan Panjang Gelombang Analisis ... 36
4.6 Hasil Penentuan Linieritas Kurva Kalibrasi Parasetamol dan Kafein ... 44
4.6.1 Kurva Kalibrasi Parasetamol dan Kafein... 44
4.7 Hasil Penentuan Kadar Parasetamol dan Kafein Pada Sediaan Tablet ... 45
4.8 Hasil Uji Validasi ... 46
4.8.1 Hasil Uji Akurasi ... 46
4.8.2 Hasil Uji Presisi ... 48
4.8.3 Batas Deteksi dan Batas Kuantitasi ... 48
BAB V KESIMPULAN DAN SARAN ... 49
5.1 Kesimpulan ... 49
5.2 Saran ... 50
DAFTAR PUSTAKA ... 51
DAFTAR TABEL
Halaman
Tabel 2.1. Perbedaan antara transisi n→π* dan transisi π→π* ... 10 Tabel 4.1. Panjang gelombang analisis dan absorbansi pada derivat
kedua ... 43 Tabel 4.2. Kadar parasetamol dan kafein pada sediaan tablet ... 46 Tabel 4.3. Perolehan kembali parasetamol dan kafein dengan
metode penambahan baku standar pada sediaan tablet
DAFTAR GAMBAR
Halaman
Gambar 2.1. Rumus Struktur Parasetamol ... 5
Gambar 2.2. Rumus Struktur Kafein ... 6
Gambar 2.3. Kurva Serapan Derivat Pertama sampai Derivat Keempat . 13
Gambar 2.4. Kurva Aplikasi Metode Evaluasi Spektrum Derivatif ... 14
Gambar 2.5. Kurva Sederhana Aplikasi Zero Crossing ... 15
Gambar 4.6. Kurva Serapan Maksimum Parasetamol 6,5 µ g/mL ... 29
Gambar 4.7. Kurva Serapan Maksimum Kafein 8,6 µg/mL... 29
Gambar 4.8. Kurva Tumpang Tindih Serapan Parasetamol dalam berbagai konsentrasi ... 30
Gambar 4.9. Kurva Tumpang Tindih Serapan Derivat Pertama Parasetamol dalam berbagai konsentrasi ... 31
Gambar 4.10. Kurva Tumpang Tindih Serapan Derivat Kedua Parasetamol dalam berbagai konsentrasi ... 31
Gambar 4.11. Kurva Tumpang Tindih Serapan Kafein dalam berbagaikonsentrasi ... 32
Gambar 4.12. Kurva Tumpang Tindih Serapan Derivat Pertama Kafein dalam berbagai konsentrasi ... 33
Gambar 4.13 Kurva Tumpang Tindih Serapan Derivat Kedua Kafein dalam berbagai konsentrasi ... 33
Gambar 4.14. Zero Crossing Parasetamol Derivat Pertama dengan = 217,37 nm; 242,83 nm; dan 312,32 nm; ... 34
Gambar 4.15. Zero Crossing Parasetamol Derivat Kedua dengan = 225,59 nm; 257,30 nm; dan 277,91 nm; ... 35
Gambar 4.16. Zero Crossing Kafein Derivat Pertama dengan = 245,18 nm; 272,73 nm; dan 311,52 nm ... 35
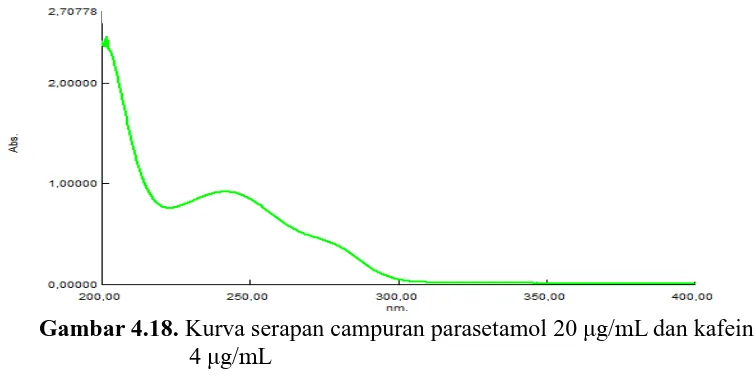
Gambar 4.18. Kurva Serapan Campuran Parasetamol 20 µ g/mL
dan Kafein 4 µ g/mL ... 37 Gambar 4.19. Kurva Tumpang Tindih Serapan Parasetamol 20 µg/mL
dan Kafein 4 µg/mL ... 37 Gambar 4.20. Kurva Tumpang Tindih Serapan Parasetamol 20 µg/mL
dan Kafein 4 µ g/mL dan Campuran Parasetamol 20 µg/mL dan Kafein 4 µ g/mL ... 37
Gambar 4.21. Kurva Serapan Derivatif Pertama Campuran
Parasetamol 20 µ g/mL dan Kafein 4 µ g/mL ... 38 Gambar 4.22. Kurva Serapan Derivatif Kedua Campuran
Parasetamol 20µg/mL dan Kafein 4 µ g/mL ... 38
Gambar 4.23. Kurva Tumpang Tindih Serapan Derivatif Pertama Parasetamol 20 µ g/mL dan Kafein 4 µ g/mL ... 38
Gambar 4.24. Kurva Tumpang Tindih Serapan Derivatif Kedua Parasetamol 20 µ g/mL dan Kafein 4 µ g/mL ... 39 Gambar 4.25. Kurva Tumpang Tindih Serapan Derivatif Pertama
Parasetamol 20 µ g/mL, Kafein 4 µg/mL dan Campuran Parasetamol 20 µ g/mL dan Kafein 4 µ g/mL ... 39 Gambar 4.26. Kurva Tumpang Tindih Serapan Derivatif Kedua
Parasetamol 20 µg/mL, Kafein 4 µ g/mL dan
Campuran Parasetamol 20 µ g/mL dan Kafein 4 µg/mL ... 39
Gambar 4.27. Zero Crossing derivatif Pertama Parasetamol 20 µ g/mL .... 40 Gambar 4.28. Zero Crossing derivatif Kedua Parasetamol 20 µ g/mL
pada = 215,6 nm ... 40 Gambar 4.29. Zero Crossing derivatif Pertama Kafein 4 µ g/mL
pada
= 225,6 nm ... 40
Gambar 4.30. Zero Crossing derivatif Kedua Kafein 4 µ g/mL pada = 225,6 nm ... 41
Gambar 4.31. Panjang Gelombang Analisis Derivatif
Pertama Parasetamol dan Kafein ... 41 Gambar 4.32. Panjang Gelombang Analisis Derivatif Kedua
Gambar 4.33. Panjang Gelombang Analisis Derivatif Kedua Kafein ... 43 Gambar 4.34. Kurva Kalibrasi Parasetamol Pada Panjang
Gelombang 215,6 nm ... 44 Gambar 4.35.Kurva Kalibrasi Kafein Pada Panjang Gelombang 225,6
DAFTAR LAMPIRAN
Halaman
Lampiran 1. Gambar Sampel Sediaan Tablet ... 53
Lampiran 2. Komposisis Sampel Sediaan Tablet ... 54
Lampiran 3. Gambar Alat Spektrofotometer Ultraviolet (UV) ... 55
Lampiran 4. Perhitungan Pembuatan HCl 0,1N ... 56
Lampiran 5. Bagan Alir Prosedur Penelitian ... 57
Lampiran 6. Kurva Serapan Parasetamol Baku dan Kafein Baku ... 62
Lampiran 7. Kurva Serapan Derivat Kedua Parasetamol Baku dan Kafein Baku ... 67
Lampiran 8. Kurva Serapan Panjang Gelombang Analisis Parasetamol dan Kafein ... 72
Lampiran 9. Data Kalibrasi Parasetamol BPFI, Persamaan Regresi dan Koefisien Korelasi ... 74
Lampiran 10. Data Kalibrasi Kafein BPFI, Persamaan Regresi dan Koefisien Korelasi ... 76
Lampiran 11. Perhitungan Batas Deteksi (Limit of Detection, LOD) dan Batas Kuantitasi (Limit of Qua ntita tion, LOQ) Parasetamol ... 78
Lampiran 12. Perhitungan Batas Deteksi (Limit of Detection, LOD) dan Batas Kuantitasi (Limit of Quantitation, LOQ) Kafein ... 79
Lampiran 13. Kurva Serapan Derivat Kedua Sampel ... 80
Lampiran 14. Hasil Analisis Kadar Parasetamol dan Kafein dalam Sampel ... 86
Lampiran 15. Contoh Perhitungan Kadar Parasetamol dan Kafein dalam Sampel Paramex® ... 87
Lampiran 16. Contoh Perhitungan Kadar Parasetamol dan Kafein dalam Sampel Saridon® ... 91
Lampiran 18. Perhitungan Statistik Kadar Parasetamol dan Kafein
dalam Sampel Saridon ® ... 98
Lampiran 19. Kurva Serapan Uji Perolehan Kembali Parasetamol dan Kafein dalam Sampel Paramex ® ... 102
Lampiran 20. Hasil Uji Perolehan Kembali Parasetamol dan Kafein dalam Sampel Paramex® ... 107
Lampiran 21. Contoh Perhitungan Uji Perolehan Kembali dengan menggunakan Sampel Paramex® ... 108
Lampiran 22. Perhitungan Simpangan Baku Relatif (Relative Standard Devia tion, RSD) Persen Perolehan Kembali Parasetamol . 113
Lampiran 23. Perhitungan Simpangan Baku Relatif (Relative Standard Devia tion, RSD) Persen Perolehan Kembali Kafein ... 114
Lampiran 24. Daftar Nilai Distribusi t ... 115
Lampiran 25. Sertifikat Pengujian Parasetamol ... 116
UJI VALIDASI METODE
ZERO CROSSING
SPEKTROFOTOMETRI DERIVATIF PADA PENETAPAN
KADAR KAFEIN DAN PARASETAMOL DALAM SEDIAAN
TABLET
ABSTRAK
Latar Belakang: Banyak obat yang menggunakan berbagai macam zat aktif, seperti obat analgesik. Sehingga muncul kesulitan untuk menganalisis kadar masing-masing komponen. Oleh karena itu diperlukan suatu metode untuk menganalisis masing-masing komponen tersebut, misalnya untuk menganalisis kadar campuran parasetamol dan kafein dalam sediaan tablet. Spektrofotometri derivatif merupakan metode transformasi dari spektrofotometri konvensional yang dikembangkan untuk analisis kuantitatif multikomponen senyawa aktif pada suatu sediaan.
Tujuan Penelitian: Untuk menguji validasi metode zero crossing spektrofotometri
derivatif kandungan parasetamol dan kafein dalam sediaan tablet.
Metode: Pengambilan sampel secara purposif terhadap sediaan tablet Paramex® dan Saridon® dan kemudian menentukan jumlah kandungan parasetamol dan kafein dengan spektrofotometri derivatif metode zero crossing pada serapan derivat kedua dalam pelarut HCl 0,1N.
Hasil: Hasil penelitian menunjukkan bahwa kadar parasetamol dalam sediaan tablet Paramex® yang dianalisis sebesar 100,13% ± 0,8455% dan sediaan tablet Saridon® sebesar 98,21% ± 5,1002% dan kadar kafein dalam sediaan tablet Paramex® sebesar 101,39% ± 5,7119% dan sediaan tablet Saridon® sebesar 98,18% ± 3,8038%. Dapat disimpulkan bahwa hasil penetapan kadar campuran parasetamol dan kafein dalam tablet memenuhi persyaratan sesuai dengan persyaratan yang tertera pada United States Pharmacopoeia, (USP 30 NF 25, 2007). % perolehan kembali parasetamol sebesar 99,93% dan kafein sebesar 101,77%. RSD parasetamol sebesar 3,5% dan kafein sebesar 3,5%.
Kesimpulan: Berdasarkan hasil penelitian yang dilakukan maka metode spektrofotometri derivatif yang digunakan memenuhi persyaratan akurasi dan presisi dan dapat digunakan untuk menetapkan kandungan parasetamol dan kafein dalam sediaan tablet.
VALIDATION TEST WITH ZERO CROSSING METHODE OF DERIVATIVE SPECTROFOTOMETRY FOR DETERMINATION OF
CAFFEINE AND PARACETAMOL IN TABLET
ABSTRACT
Background: Many drugs that use various active substance, such as analgesics. Hence the difficulty to analyze the levels of each component. Therefore we need a method to analyze each of these components, for example, to analyze the levels of a mixture of paracetamol and caffein in tablets. The purpose of this research is to developmentof derivative spectrophotometry method with the zero crossing for determination of paracetamol and caffein mixture in tablet. Derivative spectrophotometry is a transformation method from conventional spectrophotometric that developed for quantitative analysis of multicomponent active compounds in a preparation.
Objective: The aim of this study was to test the validation of derivative spectrophotometry method in estimating the content of paracetamol and caffeine in tablets.
Method: The method of research was done by purposive sampling to Paramex® and Saridon® in tablets and then determine the amount of paracetamol and caffeine content using derivative spectrophotometric with zero crossing method to second derivative in the solven HCl 0.1N.
Result: The results of research was exhibited that paracetamol in Paramex® that were analyzed are 100.13% ± 0.8455% and Saridon® are 98.21% ± 5.1002% and caffeine in Paramex® are 101.39% ± 5.7119% and Saridon® are 98.18% ± 3.8038%. Therefore, it suggested was result of determination of paracetamol and caffeine mixture in tablet the requirement of the United States Pharmacopoeia, (USP 30 NF 25, 2007). % Recovery of paracetamol are 99.93% and caffeine are 101.77%. RSD of paracetamol are 3.5% and caffeine are 3.5%.
Conclusion: Based on the results of research that derivative spectrophotometry method fulfilled the requirements of accuracy and precision and can be used to determinate the content of paracetamol and caffeine in tablets.
BAB I
PENDAHULUAN
1.1. Latar Belakang
Berbagai sediaan obat yang terdapat di pasaran mengkombinasikan dua atau lebih zat aktif dalam satu sediaan, salah satunya adalah obat analgesik.
Analgesik merupakan obat yang meredakan rasa nyeri tanpa mengakibatkan kehilangan kesadaran. Kombinasi analgesik yang banyak ditemukan adalah
parasetamol dengan kafein. Penambahan kafein bertujuan untuk meningkatkan efikasi dari analgesik (Tan dan Rahardja, 2007).
Bentuk sediaan farmasi seperti tablet harus memenuhi beberapa
persyaratan sesuai dengan standar yang ada pada acuan misalnya pada farmakope. Salah satu persyaratan tersebut adalah persyaratan kadar. Pesyaratan kadar untuk
sediaan tablet campuran parasetamol dan kafein menurut (USP 30 NF 25, 2007) yaitu mengandung parasetamol dan kafein tidak kurang dari 90,0% dan tidak lebih dari 110,0 % dari jumlah yang tertera pada etiket.
Sekarang ini spektrofotometri ultraviolet-Visible (UV-Vis) berkembang sejalan dengan perkembangan ilmu pengetahuan, sehingga dapat digunakan untuk menetapkan kadar campuran, yaitu melalui aplikasi metode spektrofotometri
derivatif. Spektrofotometri derivatif merupakan metode manipulatif terhadap
spektrum pada spektrofotometri ultraviolet (Connors, 1982).
Spektofotometri derivatif adalah spektrofotometri ultraviolet yang mentransformasikan spektrum serapan menjadi spektrum derivatif pertama, kedua
transformasi dari spektrofotometri konvensional yang di dikembangkan untuk analisis kuantitatif multikomponen senyawa aktif pada suatu sediaan.
Spektrofotometri derivatif dapat digunakan untuk senyawa yang memiliki matriks kompleks, sehingga penentuan baik secara kuantitatif maupun kualitatif dapat dilakukan tanpa harus melakukan pemisahan antara analit dengan matriksnya.
Pemisahan kafein dari matriks dapat menjadi sumber kesalahan analisis dan memperpanjang waktu analisa. Oleh karena itu, diperlukan metode lain yang lebih
cepat, murah dengan tingkat ketelitian dan ketepatan yang tinggi, serta dapat mengatasi efek matriks tanpa harus memisahkannya terlebih dahulu (Nurhidayati, 2007).
Spektrofotometri derivatif memiliki beberapa keuntungan dibandingkan dengan spektrofotometri konvensional antara lain gambaran struktur yang lembut
dari spektrum serapan dan gambaran ini dapat lebih jelas bila spektrum derivatif ditingkatkan. Spektrofotometri derivatif juga dapat menganalisis secara kuantitatif satu komponen dalam campuran bahan penyerap yang rumit (Munson, 1991).
Selain itu metode spektrofotometri derivatif relatif lebih sederhana, alat dan biaya operasionalnya relatif lebih murah dan waktu analisisnya lebih cepat.
Metode yang digunakan untuk penetapan kadar parasetamol dan kafein
dari spektrum derivatif adalah metode zero crossing. Metode ini merupakan
penentuan panjang gelombang analisis dimana senyawa tersebut mempunyai serapan nol dan menjadi panjang gelombang analisis untuk zat lain dalam campurannya (Hayun, dkk., 2006). Selain dalam bidang farmasi, spektrofotometri
derivatif telah diaplikasikan secara luas di dalam analisis klinik dan biokimia
Berbagai peneliti telah melakukan penelitian dengan menggunakan spektrofotometri derivatif pada satu zat atau lebih, sebagai contoh penetapan kadar
parasetamol dan kafein dalam tablet kombinasi parasetamol dan kafein secara spektrofotometri ultraviolet sinar tampak (Naid dan Pakaya, 2011). Penetapan kadar triprolidina hidroklorida dan pseudoefedrina hidroklorida dalam tablet
(Hayun, dkk., 2006; Dongoran, 2011). Penetapan kadar reserpin dalam obat (Wulandari, 2006).
Menurut Harmita (2004), untuk menguji validasi metode dilakukan uji akurasi (ketepatan) dengan parameter persen perolehan kembali dan metode penambahan baku (standard addition method) dan uji presisi (ketelitian) dengan
parameter Relative Standard Deviation (RSD).
Berdasarkan uraian diatas maka penulis tertarik untuk melakukan uji
validasi metode zero crossing spektrofotometri derivatif pada penetapan kadar parasetamol dan kafein dalam sediaan tablet.
1.2 Perumusan Masalah
a. Apakah campuran dari parasetamol dan kafein dalam sediaan tablet dapat ditetapkan kadarnya dengan menggunakan metode spektrofotometri derivatif ?
b. Apakah kadar campuran parasetamol dan kafein dalam sediaan tablet
memenuhi persyaratan (USP 30 NF 25, 2007).
c. Apakah hasil uji validasi terhadap metode spektrofotometri derivatif untuk menganalisa campuran parasetamol dan kafein dalam sediaan tablet dapat
1.3 Hipotesis
Berdasarkan perumusan masalah maka dibuat hipotesis sebagai berikut:
a. Campuran dari parasetamol dan kafein dalam sediaan tablet dapat ditetapkan kadarnya dengan menggunakan metode spektrofotometri
derivatif.
b. Kadar campuran parasetamol dan kafein dalam sediaan tablet memenuhi persyaratan (USP 30 NF 25, 2007).
c. Hasil uji validasi terhadap metode spektrofotometri derivatif untuk
menganalisa kadar campuran parasetamol dan kafein dalam sediaan tablet dapat memenuhi syarat pengujian.
1.4 Tujuan Penelitian
a. Untuk mengetahui apakah campuran dari parasetamol dan kafein dalam sediaan tablet dapat ditetapkan kadarnya dengan menggunakan metode
spektrofotometri derivatif.
b. Untuk mengetahui kadar campuran parasetamol dan kafein dalam sediaan tablet memenuhi persyaratan (USP 30 NF 25, 2007).
c. Untuk mengetahui hasil uji validasi terhadap metode spektrofotometri derivatif dalam menganalisa kadar campuran parasetamol dan kafein
dalam sediaan tablet dapat memenuhi syarat pengujian.
1.5 Manfaat Penelitian
Manfaat penelitian adalah untuk mengetahui kadar parasetamol dan kafein
BAB II
TINJAUAN PUSTAKA
2.1 Uraian Bahan
2.1.1. Parasetamol
Parasetamol merupakan metabolit dari fenasetin yang dahulunya paling
banyak digunakan sebagai analgetik. Khasiatnya analgetik dan antipiretik, tetapi tidak antiradang. Dewasa ini pada umumnya dianggap sebagai zat antinyeri yang
paling aman, juga untuk pengobatan swamedikasi (pengobatan mandiri). Efek analgetisnya diperkuat oleh kodein dan kafein dengan kira-kira 50% (Tan dan Rahardja, 2007).
Gambar 2.1. Rumus struktur parasetamol (Ditjen POM, 2014)
Rumus Molekul : C8H9NO2
Berat Molekul : 151,16
Nama Kimia : 4-Hidroksiasetanilida
Kandungan : Tidak kurang dari 98,5% dan tidak lebih dari
101,0% C8H9NO2, dihitung terhadap zat anhidrat
Pemerian : Serbuk hablur putih, tidak berbau, dan rasa sedikit pahit.
2.1.2. Kafein
Kafein adalah salah satu jenis alkaloid yang banyak terdapat di
daun teh (Camellia sinensis), biji kopi (Coffea arabica ), dan biji coklat (Theobroma cacao). Kafein memiliki efek farmakologi yang bermanfaat secara klinis, seperti menstimulasi susunan saraf pusat, relaksasi otot polos terutama otot
polos bronkus, dan stimulasi otot jantung . Efek samping dari penggunaan kafein secara berlebihan (overdosis) dapat menyebabkan gugup, gelisah, tremor,
insomnia, mual, dan kejang (Tan dan Rahardja, 2007).
Gambar 2.2. Rumus Struktur Kafein (Ditjen POM, 2014)
Rumus Molekul : C8H10N4O2
Berat Molekul : 194,19
Nama Kimia : 1,3,7- trimetilxantine
Kandungan : Tidak kurang dari 98,5% dan tidak lebih dari
101,0% C8H10N4O2, dihitung terhadap zat anhidrat.
Pemerian : Serbuk putih; bentuk jarum mengkilat;
biasanya menggumpal; tidak berbau; rasa pahit.
Kelarutan : Agak sukar larut dalam air, dalam etanol, mudah larut dalam kloroform; sukar larut dalam eter.
2.2 Spektrofotometri Ultraviolet-Visibel
2.2.1 Pengertian Spektrofotometri Ultraviolet-Visibel
Spekrofotometri ultraviolet-visibel merupakan salah satu teknik analisis
spektrofotometri yang menggunakan sumber radiasi elektromagnetik sinar ultraviolet dan sinar tampak dengan memakai instrumen spektrofotometer
(Gandjar dan Rohman, 2007). Sinar ultraviolet memiliki panjang gelombang antara 200-400 nm sedangkan sinar tampak memiliki panjang gelombang antara 400-800 nm (Moffat, dkk., 2005).
2.2.2 Pembagian Metode Analisis Spektrofotometri Ultraviolet-Visibel
Spektrofotometri ultraviolet-visibel dibagi atas empat metode analisis
yaitu analisis zat tunggal, analisis multikomponen, spektrofotometri perbedaan
(Difference Spectrophotometry), dan spektrofotometri derivatif (Moffat, dkk., 2005).
2.2.3 Proses Penyerapan Radiasi pada Spektrofotometer Ultraviolet-Visibel
Radiasi di daerah ultraviolet atau visibel diserap melalui eksitasi elektron yang terlibat dalan ikatan antara atom-atom pembentuk molekul (Gandjar dan
Rohman, 2007; Watson, 2005).
Jika suatu berkas radiasi dikenakan pada larutan sampel maka intensitas sinar radiasi yang diteruskan dapat diukur besarnya. Radiasi yang diserap oleh cuplikan ditentukan dengan membandingkan intensitas sinar yang diteruskan
sama dengan energi yang dibutuhkan untuk menyebabkan perubahan energi. Kekuatan radiasi juga mengalami penurunan dengan adanya penghamburan dan
pemantulan cahaya, akan tetapi penurunan hal ini sangat kecil dibandingkan dengan proses penyerapan (Gandjar dan Rohman, 2007).
Sinar ultraviolet dan sinar tampak (visibel) memberikan energi yang cukup
untuk terjadinya transisi elektron (Gandjar dan Rohman, 2007). Sebagai contoh, molekul organik sederhana yang mempunyai dua jenis ikatan karbon-karbon
seperti pada etilen. Ikatan π lebih lemah dari ikatan dan akibatnya elektron π lebih tinggi energinya dari elektron (Munson, 1984). Elektron yang energinya tertinggi dalam molekul, berada dalam tingkat energi elektron dasar, terdapat
dalam orbital , π, atau n, masing-masing mempunyai keadaan tereksitasi sesuai dengan energi elektron terendah. Transisi elektron yang terkait dengan absorbsi radiasi ultraviolet dan sinar tampak adalah → *, n→ *, n→π*, dan π→π*
(Satiadarma, dkk., 2004).
Penyerapan radiasi ultraviolet dan sinar tampak dibatasi oleh sejumlah
gugus fungsional (yang disebut dengan kromofor) yang mengandung elektron valensi dengan tingkat energi eksitasi yang relatif rendah. Elektron yang terlibat pada penyerapan radiasi ultraviolet dan visibel ini ada tiga, yaitu elektron sigma,
elektron phi, dan elektron bukan ikatan (non bonding electron) (Gandjar dan
Rohman, 2007).
1. Transisi → *
Energi yang diperlukan untuk transisi ini besarnya sesuai dengan energi
sinar yang frekuensinya terletak di antara ultraviolet vakum (kurang dari 180 nm). Jenis transisi ini terjadi pada daerah ultraviolet vakum sehingga kurang begitu bermanfaat untuk analisis dengan cara spektrofotometri ultraviolet-visibel.
2. Transisi n→ *
Jenis transisi ini terjadi pada senyawa organik jenuh yang mengandung
atom-atom yang memiliki elektron bukan ikatan (elektron n). Energi yang diperlukan untuk transisi jenis ini lebih kecil dibandingkan transisi → *
sehingga sinar yang diserap pun mempunyai panjang gelombang lebih panjang,
yakni sekitar 150-250 nm. Kebanyakan transisi ini terjadi pada panjang gelombang kurang dari 200 nm.
3. Transisi n→π* dan transisi π→π*
Untuk memungkinkan terjadinya transisi ini, maka molekul organik harus mempunyai gugus fungsional yang tidak jenuh sehingga ikatan rangkap dalam
gugus tersebut memberikan orbital phi yang diperlukan. Jenis transisi ini merupakan transisi yang paling cocok untuk analisis sebab dengan panjang
Tabel 2.1. Perbedaan antara transisi n→π* dan transisi π→π*
Transisi n→π* Transisi π→π*
Absorptivitas molar ( ) antara
10-100 Lcm-1mol-1
Absorptivitas molar ( ) antara
1000-10000 Lcm-1mol-1
Biasanya pelarut yang polar
menyebabkan pergeseran biru atau
hypsocromic shift (pergeseran pita serapan ke arah panjang gelombang yang lebih pendek)
Biasanya pelarut yang polar
menyebabkan pergeseran merah atau
ba thocromic shift (pergeseran pita serapan ke arah panjang gelombang yang lebih panjang)
(Gandjar dan Rohman, 2007)
2.2.4 Kegunaan Spektrofotometri Ultraviolet-Visibel
Data spektrum ultraviolet-visibel secara tersendiri tidak dapat digunakan untuk identifikasi kualitatif obat karena rentang daerah radiasi yang relatif sempit
hanya dapat menghasilkan sedikit sekali puncak absorbsi maksimum dan
minimum. Akan tetapi jika digabung dengan cara lain seperti spektrofotometri inframerah, resonansi magnet inti, dan spektrometri massa, maka dapat digunakan untuk maksud identifikasi kualitatif suatu senyawa tersebut. Penggunaannya
terbatas pada konfirmasi identitas dengan menggunakan parameter panjang gelombang maksimum, nilai absorptivitas, nilai absorptivitas molar, nilai
Hukum Lambert-Beer menjadi dasar aspek kuantitatif spektrofotometri ultraviolet-visibel. Menurut Hukum Lambert-Beer, serapan berbanding lurus
terhadap konsentrasi dan ketebalan sel, yang dapat ditulis dengan persamaan :
A = a.b.c (g/liter) atau A = . b. c (mol/liter) atau A = A1
1.b.c (g/100 ml)
Dimana: A = serapan
a = absorptivitas
b = ketebalan sel
c = konsentrasi
= absorptivitas molar
A11 = absorptivitas spesifik
2.2.5 Komponen Spektrofotometer Ultraviolet-Visibel
Biasanya spektrofotometer telah mempunyai software untuk mengolah data yang dapat dioperasikan melalui komputer yang telah terhubung dengan spektrofotometer (Moffat, dkk., 2005).
Komponen spektrofotometer UV-Vis adalah sebagai berikut:
1. Sumber-sumber lampu: lampu deuterium digunakan untuk daerah UV pada panjang gelombang dari 190-350 nm, sementara lampu halogen kuarsa atau
lampu tungsten digunakan untuk daerah visibel pada panjang gelombang antara 350- 900 nm.
3. Optik-optik: dapat didesain untuk memecah sumber sinar melewati 2 kompartemen.
4. Detektor: digunakan sebagai alat yang menerima sinyal dalam bentuk radiasi elektromagnetik, mengubah, dan meneruskannya dalam bentuk sinyal listrik ke rangkaian sistem penguat elektronika. Respon tiap jenis detektor terhadap
bagian dari spektrum radiasi tidak sama, sehingga setiap spektrofotometer menggunakan detektor yang paling cocok untuk daerah pengukurannya
(Satiadarma, dkk., 2004; Gandjar dan Rohman, 2007).
2.3 Spektrofotometri Derivatif
2.3.1 Pengertian Spektrofotometri Derivatif
Spektrofotometri derivatif merupakan transformasi spektrum serapan
menjadi spektrum derivatif pertama, kedua, atau spektrum derivatif orde lebih tinggi (Ditjen POM, 1995). Spektrofotometri derivatif merupakan metode manipulatif terhadap spektrum pada spektrofotometri ultraviolet-visibel
(Moffat, dkk., 2005).
Pada spektrofotometri konvensional, spektrum serapan merupakan plot serapan (A) terhadap panjang gelombang ( ). Pada spektrofotometri derivatif, plot A lawan , ditransformasikan menjadi plot dA/d lawan untuk derivatif pertama,
dan d2A/ d 2lawan untuk derivatif kedua, dan seterusnya.
A = f(λ), order nol
dA/dλ = f (λ), order pertama
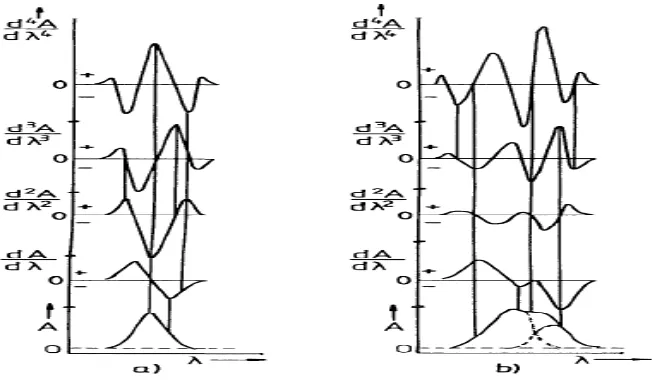
Gambar 2.3. Spektrum serapan normal sampai derivat keempat (Talsky, 1994)
Gambar (a) menunjukkan spektrum serapan normal yang diderivatisasi sampai spektrum derivat keempatnya, sedangkan Gambar (b) menunjukkan spektrum yang saling tumpang tindih yang diderivatisasi mulai dari spektrum
serapan normal hingga spektrum derivat keempat (Talsky, 1994).
Spektrum derivatif merupakan sebuah plot perubahan serapan dengan
panjang gelombang. Spektrum derivatif pertama dilambangkan dengan dA/dλ, spektrum derivatif kedua dilambangkan dengan dA2/d 2, dan seterusnya (Ditjen POM, 1995). Hal ini dapat dilihat dari persamaan menurut hukum
Lambert-Beer berikut ini :
dA/dλ =
bc x d cm dA ) 1 %, 1 (
dA2/dλ2 =
bc x d cm A d 2 2 ) 1 %, 1 (
dn =
2.3.2 Metode Spektrofotometri Derivatif
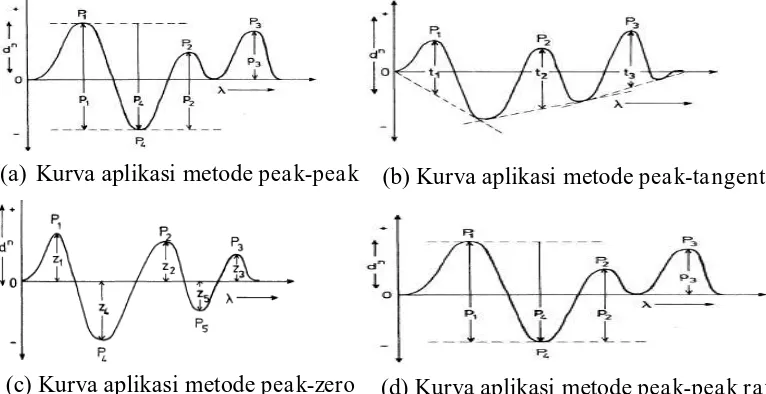
Ada empat metode umum yang digunakan untuk evaluasi spektra pada spektrofotometri derivatif yaitu metode peak-peak, metode peak-tangent, metode
pea k-zero (zero crossing), metode peak-peak ratio (rasio spektra) (Talsky, 1994;
Nurhidayati, 2007).
Pada metode peak-peak, absorbsinya diukur dari puncak maksimum sampai minimum yang ditunjukkan P1, P2, dan P3 pada gambar (a) sedangkan pada
metode peak-tangent, absorbsinya diukur dari puncak maksimum sampai
pertengahan puncak minimum yang dapat ditunjukkan pada t1, t2, dan t3 pada
gambar (b). Pada metode peak-zero, absorbsinya diukur dari puncak maksimum sampai titik nol kurva yang ditunjukkan pada z1, z2, z3, z4, dan z5 pada gambar (c)
sedangkan pada metode peak-peak ratio, absorbsinya diukur sebagai
perbandingan antara P1 dengan P2 yang ditunjukkan pada gambar (d) (Talsky,
1994). Kurva aplikasi metode evaluasi spektra derivatif dapat dilihat pada Gambar
Gambar 2.4. Kurva aplikasi metode evaluasi spektra derivative (Talsky, 1994) (a) Kurva aplikasi metode peak-peak (b) Kurva aplikasi metode peak-tangent
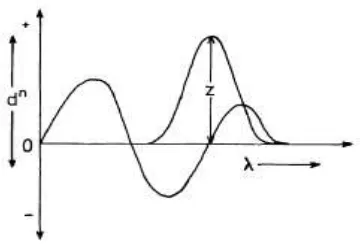
[image:33.596.121.504.512.709.2]Metode zero crossing merupakan metode yang paling umum digunakan dalam pemilihan panjang gelombang analisis untuk campuran biner. Panjang
gelombang zero crossing adalah panjang gelombang dimana senyawa tersebut mempunyai serapan nol dan menjadi panjang gelombang analisis untuk zat lain dalam campurannya. Pengukuran pada metode zero crossing tiap komponen
[image:34.596.221.401.320.443.2]dalam campuran merupakan fungsi tunggal konsentrasi dari yang lainnya (Nurhidayati, 2007). Kurva sederhana aplikasi zero crossing dapat dilihat pada
Gambar 2.5.
Gambar 2.5. Kurva sederhana aplikasi zero crossing (Talsky, 1994)
2.3.3 Kegunaan Spektrofotometri Derivatif
Metode spektrofotometri derivatif dapat digunakan untuk analisis
kuantitatif zat dalam campuran yang spektrumnya mungkin tersembunyi dalam suatu bentuk spektrum besar yang saling tumpang tindih dengan mengabaikan
proses pemisahan zat yang bertingkat-tingkat. Dalam bidang farmasi, karena terkait terapi, penetapan kadar obat adalah kontrol kualitas pada industri farmasi. Metode spektrofotometri derivatif adalah teknik analisis dengan kemampuan
Sebagai contoh yaitu penetapan kadar campuran pseuoefedrin HCl, triprolidin HCl dan dekstrometorfan HBr (Watson, 2005), penetapan kadar
parasetamol dalam tablet kombinasi parasetamol dengan kofein secara sinar tampak (Naid, 2011), penetapan kadar efedrin dan zat warna dalam sediaan sirup (Cairns, 2008).
Beberapa keuntungan dari spektrofotometri derivatif antara lain yaitu spektrum derivatif memberikan gambaran struktur yang terinci dari spektrum
serapan dan gambaran ini makin jelas dari spektum derivatif pertama ke derivatif keempat. Selain itu dapat dilakukan analisis kuantitatif suatu komponen dalam campuran dengan panjang gelombangnya saling berdekatan. Bila dibandingkan
dengan kromatografi cair kinerja tinggi (KCKT), metode spektrofotometri derivatif relatif lebih sederhana, alat dan biaya operasionalnya lebih murah dan
waktu analisisnya lebih cepat (Nurhidayati, 2007).
Kekurangan utama dari teknik ini adalah ketergantungannya pada parameter instrumentasi, seperti kecepatan pemindaian dan slit width. Kondisi
instrumen saat pengukuran spektrum serapan normal memiliki banyak pengaruh pada bentuk dan intensitas dari spektrum derivatifnya (Ojeda dan Rojas, 2013).
2.4.Validasi Metode Analisis
Validasi metode adalah suatu proses yang menunjukkan bahwa prosedur
analitik telah sesuai dengan penggunaan yang dikehendaki. Proses validasi metode untuk prosedur analitik dimulai dengan pengumpulan data validasi oleh pelaksana guna mendukung prosedur analitiknya. Validasi metode yang sempurna hanya
Hasil validasi metode dapat digunakan untuk memutuskan kualitas, reabilitas, dan konsistensi dari hasil analisis. Adapun karakteristik dalam validasi
metode menurut (USP 30 NF 25, 2007) yaitu akurasi, presisi, spesifisitas, batas deteksi, batas kuantitasi, linieritas, rentang, dan kekuatan/ketahanan.
2.4.1 Akurasi
Akurasi adalah kedekatan nilai hasil uji yang diperoleh melalui metode analisis dengan nilai yang sebenarnya. Akurasi dinyatakan dengan persen
perolehan kembali (% recovery). Akurasi dapat ditentukan dengan dua metode, yaitu spiked-placebo recovery atau metode simulasi dan standard addition method (metode penambahan baku). Pada metode spiked-placebo recovery, analit murni
ditambahkan (spiked) ke dalam campuran bahan pembawa sediaan farmasi, lalu campuran tersebut dianalisis dan jumlah analit yang dianalisis dibandingkan
dengan jumlah analit yang telah diketahui konsentrasinya dapat ditambahkan langsung ke dalam sediaan farmasi. Metode ini dinamakan metode penambahan baku atau standard addition method (USP 30 NF 25, 2007, Ermer dan McB.
Miller, 2005, Harmita, 2004).
Menurut Harmita (2004), dalam metode penambahan baku, sejumlah sampel yang dianalisis ditambah analit dengan konsentrasi biasanya 80% sampai 120% dari kadar analit yang diperkirakan, dicampur, dan dianalisis kembali. Selisih
kedua hasil dibandingkan dengan kadar yang sebenarnya. Dalam kedua metode tersebut, persen perolehan kembali dinyatakan sebagai rasio antara hasil yang diperoleh dengan hasil yang sebenarnya.
2.4.2 Presisi
Presisi adalah ukuran keterulangan metode analisis, termasuk di antaranya
kemampuan instrumen dalam melakukan hasil analisis yang reprodusibel. Presisi dinyatakan sebagai standar deviasi relatif. Berdasarkan rekomendasi ICH (the
Interna tiona l Conference on the Ha rmonisa tion), karakteristik presisi ada tiga tingkatan, yaitu keterulangan (repeatability), presisi antara (intermediate precision), dan reprodusibilitas (reproducibility). Keterulangan dilakukan dengan
cara menganalisis sampel yang sama oleh analis yang sama menggunakan instrumen yang sama dalam periode waktu yang singkat. Presisi antara dikerjakan oleh analis yang berbeda sedangkan reprodusibilitas dikerjakan oleh analis yang
berbeda dan di laboratorium yang berbeda (USP 30 NF 25, 2007; Satiadarma, dkk., 2004).
2.4.3 Spesifisitas
Spesifitas adalah suatu ukuran seberapa mampu metode tersebut mengukur analit saja dengan adanya senyawa-senyawa lain yang terkandung di dalam
sampel (Watson, 2005). Secara umum, spesifitas dapat ditunjukkan oleh minimalnya gangguan oleh senyawa lain terhadap hasil analisis misalnya mendapatkan hasil yang sama dengan atau tanpa senyawa pengganggu.
Pendekatan tidak langsung adalah lewat pengamatan karakteristik akurasi dari
metode tersebut. Bila akurasi metode telah dapat diterima maka metode tersebut otomatis telah masuk kriteria sebagai metode yang spesifik (Ermer dan McB. Miller, 2005).
2.4.4 Batas Deteksi dan Batas Kuantifikasi
sampel yang masih dapat dideteksi, meskipun tidak dapat dikuantifikasi. Batas deteksi merupakan batas uji yang spesifik menyatakan apakah analit di atas atau
dibawah nilai tertentu (Gandjar dan Rohman, 2007).
Batas Kuantifikasi didefinisikan sebagai konsentrasi analit terendah dalam
sampel yang dapat ditentukan dengan presisi dan akurasi yang dapat diterima pada kondisi operasional metode yang digunakan (Gandjar dan Rohman, 2007).
2.4.5 Linieritas
Linieritas adalah kemampuan suatu metode untuk memperoleh nilai hasil uji
langsung atau setelah diolah secara metematika proporsional dengan konsentrasi
analit dalam sampel dalam batas rentang konsentrasi tertentu (Satiadarma, dkk., 2004). Linieritas dapat ditentukan secara langsung dengan
pengukuran analit pada konsentrasi sekurang-kurangnya lima titik konsentrasi
yang mencakup seluruh rentang konsentrasi kerja (Ermer dan McB. Miller, 2005).
2.4.6 Rentang
Rentang adalah interval antara batas konsentrasi tertinggi dan terendah analit yang terbukti dapat ditentukan menggunakan prosedur analisis, dengan presisi, akurasi, dan linieritas yang baik. Rentang biasanya dinyatakan dalam
BAB III
METODOLOGI PENELITIAN
3.1 Waktu dan Tempat Penelitian
Penelitian ini termasuk jenis deskriptif dan penelitian ini dilaksanakan di
Laboratorium Penelitian Fakultas Farmasi Universitas Sumatera Utara pada bulan
Januari 2015.
3.2 Alat
Alat–alat yang digunakan dalam penelitian adalah spektrofotometer
inframerah (Shimadzu), spektrofotometer ultraviolet (UV-1800 Shimadzu) yang
dilengkapi dengan komputer, sonikator (Branson 1510), neraca analitik (Mettler
Toledo), kuvet, lumpang dan alu, alat-alat gelas dan alat-alat lainnya yang
diperlukan dalam penyiapan sampel dan larutan.
3.3 Bahan
Bahan–bahan yang digunakan dalam penelitian adalah HCl 0,1N, Baku
Parasetamol (BPFI), Baku Kafein (BPFI), tablet merek dagang Paramex®
(Konimex), dan Saridon® (Bayer).
3.4 Pengambilan Sampel
Pengambilan sampel dilakukan secara purposif yaitu tanpa
membandingkan antara satu tempat dengan tempat yang lain, karena sampel
dianggap homogen dan berasal dari nomor batch yang sama. Sampel yang digunakan adalah tablet dengan merek dagang Paramex® (Konimex), dan
3.5 Prosedur Penelitian
3.5.1 Pembuatan Pereaksi
Diencerkan 8,5 mL HCl 37% dengan 1 liter akuades (Ditjen POM, 1995).
Perhitungan pembuatan pereaksi dapat dilihat pada Lampiran 4 halaman 56.
3.5.2 Pembuatan Larutan Induk Baku
3.5.2.1 Pembuatan Larutan Induk Baku Parasetamol
Ditimbang 25 mg baku parasetamol kemudian dimasukkan ke dalam labu
tentukur 25 mL, dilarutkan dengan HCl 0,1 N hingga larut, dicukupkan volume
dengan HCl 0,1 N sampai garis tanda (LIB I) untuk mendapatkan larutan dengan
konsentrasi 1000 g/mδ (δIB I). Dari larutan LIB I dipipet 2,5 mL dimasukkan ke
dalam labu tentukur 25 mL, dicukupkan dengan HCl 0,1 N sampai garis tanda
(LIB II) untuk mendapatkan larutan dengan konsentrasi 100 g/mδ (LIB II).
3.5.1.2 Pembuatan Larutan Induk Baku Kafein
Ditimbang 25 mg baku kafein kemudian dimasukkan ke dalam labu
tentukur 25 mL, dilarutkan dengan HCl 0,1 N hingga larut, dicukupkan volume
dengan HCl 0,1 N sampai garis tanda (LIB I) untuk mendapatkan larutan dengan
konsentrasi 1000 g/mδ (δIB I). Dari larutan LIB I dipipet 2,5 mL dimasukkan ke
dalam labu tentukur 25 mL, dicukupkan dengan HCl 0,1 N sampai garis tanda
(LIB II) untuk mendapatkan larutan dengan konsentrasi 100 g/mδ (LIB II).
3.5.3 Pembuatann Spektrum Serapan Maksimum
3.5.3.1 Pembuatan Spektrum Serapan Maksimum Parasetamol
Dipipet 0,65 mL Larutan Induk Baku II (LIB II) parasetamol
(konsentrasi = 100 g/mδ), dimasukkan ke dalam labu tentukur 10 mL,
sehingga diperoleh larutan dengan konsentrasi 6,5 g/mδ, kemudian diukur
serapan pada panjang gelombang 200 – 400 nm.
3.5.3.2 Pembuatan Spektrum Serapan Maksimum Kafein
Dipipet 0,86 mL Larutan Induk Baku II (LIB II) kafein (konsentrasi
100 g/mδ), dimasukkan ke dalam labu tentukur 10 mL, diencerkan dengan HCl
0,1N hingga garis tanda, lalu dikocok sampai homogen sehingga diperoleh larutan
dengan konsentrasi 8,6 g/mδ, kemudian diukur serapan pada panjang
gelombang 200 – 400 nm.
3.5.4 Pembuatan Spektrum Serapan Derivatif
3.5.4.1 Pembuatan Spektrum Serapan Derivatif Parasetamol
Dipipet Larutan Induk Baku II parasetamol (100 g/mδ) sebanyak 0,5 mL;
1,0 mL; 1,5 mL; 2,0 mL; dan 2,5 mL. Masing-masing dimasukkan ke dalam labu
tentukur 10 mL, diencerkan dengan HCl hingga garis tanda. Lalu dikocok sampai
homogen sehingga diperoleh larutan dengan konsentrasi 5; 10; 15; 20; dan 25
g/mδ. Kemudian dibuat spektrum serapan biasa (tanpa diderivatkan), spektrum
serapan derivat pertama dan derivat kedua pada panjang gelombang 200 - 400 nm
dengan ∆ = 2 nm.
3.5.4.2 Pembuatan Spektrum Serapan Derivatif Kafein
Dipipet Larutan Induk Baku II kafein (konsentrasi = 100 g/mδ) sebanyak
0,4 mL; 0,6 mL; 0,8 ml; 1,0 mL; dan 1,2 mL. Masing-masing dimasukkan ke
dalam labu tentukur 50 mL, diencerkan dengan HCl0,1N hingga garis tanda. Lalu
dikocok sampai homogen sehingga diperoleh larutan dengan konsentrasi 4; 6; 8;
10; dan 12 g/mδ. Kemudian dibuat spektrum serapan biasa (tanpa diderivatkan),
200 - 400 nm dengan ∆ = 2 nm.
3.5.5 Penentuan Zero Crossing
Penentuan zero crossing diperoleh dengan menumpang tindihkan atau
mengoverlappingkan spektrum serapan pada masing-masing derivat dari berbagai konsentrasi larutan. Zero Crossing tiap spektrum derivat dari masing-masing zat
ditunjukkan oleh panjang gelombang yang memiliki serapan nol pada berbagai
konsentrasi.
3.5.6 Penentuan Panjang Gelombang (λ) Analisis
Dibuat larutan parasetamol dengan konsentrasi 20 g/mδ, kafein dengan
konsentrasi 4 g/mδ, dan larutan campuran kedua zat itu sehingga di dalamnya
terdapat parasetamol dengan konsentrasi 20 g/mδdan kafein dengan konsentrasi
4 g/mδ. Kemudian dibuat spektrum serapan derivat pertama dari masing-masing
larutan zat tunggal dan dari campuran zat. Spektrum serapan derivat pertama dari
larutan zat tunggal dan campuran keduanya ditumpang tindihkan. Demikian juga
untuk spektrum serapan derivat kedua. Yang dipilih untuk menjadi panjang
gelombang analisis adalah pada saat serapan senyawa pasangannya nol dan
serapan maksimum zat itu dan campurannya hampir sama atau persis sama,
karena pada panjang gelombang tersebut dapat secara selektif mengukur serapan
zat tersebut.
3.5.7 Pembuatan dan Penentuan Linieritas Kurva Kalibrasi
3.5.7.1 Pembuatan Kurva Kalibrasi dan Penentuan Linearitas Kurva
Kalibrasi Parasetamol
Dipipet Larutan Induk Baku II parasetamol (konsentrasi = 100 g/mδ)
dimasukkan ke dalam labu tentukur 10 mL, diencerkan dengan HCl 0,1N hingga
garis tanda. Lalu dikocok sampai homogen sehingga diperoleh larutan dengan
konsentrasi 5; 10; 15; 20; dan 25 g/mδ. Kemudian diukur serapan pada derivat
kedua (∆ = 2 nm) pada panjang gelombang 215,6 nm. Kemudian dilakukan
analisis hubungan antara konsentrasi dengan serapan, sehingga diperoleh
persamaan regresi linear y = ax + b, dan berdasarkan nilai serapan pada panjang
gelombang 215,6 nm, dilakukan pula perhitungan limit deteksi/ limit of detection
(LOD) dan limit kuantitasi/limit of quantitation (LOQ).
Untuk menentukan batas deteksi (LOD) dan batas kuntitasi (LOQ) dapat digunakan rumus :
Keterangan :
SB = simpangan baku
LOD = batas deteksi
LOQ = batas kuantitasi
2 ) ( 2
n Yi Y SB Slope SB x LOD33.5.7.2 Pembuatan Kurva Kalibrasi dan Penentuan Linearitas Kurva
Kalibrasi Kafein
Dipipet Larutan Induk Baku II kafein (100 g/mδ) sebanyak 0,4 mL;
0,6 mL; 0,8 mL; 1,0 mL; dan 1,2 mL. Masing-masing dimasukkan ke dalam labu
tentukur 10 mL, diencerkan dengan HCl 0,1N hingga garis tanda. Lalu dikocok
sampai homogen sehingga diperoleh larutan dengan konsentrasi 4; 6; 8; 10; dan
12 g/mδ. Kemudian diukur serapan pada derivat kedua (∆ = 2 nm) pada
panjang gelombang 225,61 nm. Kemudian dilakukan analisis hubungan antara
konsentrasi dengan serapan, sehingga diperoleh persamaan regresi linear
y = ax + b, dan berdasarkan nilai serapan pada panjang gelombang 225,6 nm,
dilakukan pula perhitungan limit deteksi/ limit of detection (LOD) dan limit kuantitasi/limit of quantitation (LOQ). Perhitungan menentukan batas deteksi
(LOD) dan batas kuantitasi (LOQ) seperti rumus diatas.
3.5.8 Penentuan Kadar Parasetamol dan Kafein dalam Sediaan Tablet
Dua puluh tablet merek dagang yang mengandung parasetamol 250 mg dan
kafein 50 mg ditimbang, lalu digerus dalam lumpang sampai halus dan homogen.
Kemudian ditimbang seksama sejumlah serbuk setara dengan 50 mg parasetamol.
Kemudian dari berat analit yang ditimbang setara 50 mg parasetamol ini dihitung
kesetaraan kafein yang terkandung di dalamnya (penimbangan serbuk sebanyak
6 kali pengulangan), dimasukkan ke dalam labu tentukur 50 mL, ditambahkan
HCl 0,1N sampai garis tanda sambil dikocok. Larutan kemudian dihomogenkan
dengan pengaduk ultrasonik selama 15 menit. Larutan tersebut kemudian disaring,
lebih kurang 10 mL filtrat pertama dibuang. Filtrat selanjutnya ditampung.
tentukur 10 mL dan diencerkan dengan HCl 0,1N hingga garis tanda
(konsentrasi = 20 g/mδ parasetamol dan konsentrasi = 4 g/mδ untuk kafein)
dan diukur serapannya pada serapan/derivat kedua pada panjang gelombang 215,6
nm dan 225,6 nm.
3.5.9 Uji Validasi
3.5.9.1 Uji Akurasi
Uji akurasi dilakukan dengan metode penambahan bahan baku yaitu
dengan membuat 3 konsentrasi analit sampel dengan rentang spesifik 80%, 100%,
120%. Dimana pada masing-masing rentang spesifik digunakan 70% analit dan
30% baku yang akan ditambahkan (Harmita, 2004).
Kemudian campuran sampel dan baku diukur serapannya pada panjang
gelombang 200-400 nm, selanjutnya spektrum serapan ditransformasikan menjadi
spektrum serapan derivat kedua dengan Δ = 2 nm pada panjang gelombang
analisis parasetamol dan kafein masing – masing 215,6 nm dan 225,6 nm.
Persen perolehan kembali dapat dihitung dengan rumus:
% perolehan kembali = CF − CA
A
∗
×
100 %Keterangan:
CF = Konsentrasi sampel setelah penambahan bahan baku
CA = Konsentrasi sampel sebelum penambahan bahan baku
C*A = Jumlah baku yang ditambahkan
3.5.9.2 Uji Presisi
Uji presisi (keseksamaan) ditentukan dengan parameter RSD (Relative
RSD =
Untuk menghitung Standar Deviasi (SD) digunakan rumus :
Keterangan :
RSD = standar deviasi relatif
SD = standar deviasi
X = kadar rata-rata parasetamol atau kafein dalam sampel
3.5.10 Analisis Data Statistik
Analisis data secara statistik menggunakan uji t. Untuk mengetahui apakah
data diterima atau ditolak digunakan rumus seperti di bawah ini :
t hitung =
n SD X X /
Dasar penolakan data jika thitung ≥ ttabel dan thitung≤ -ttabel.
Untuk mencari kadar sebenarnya dengan taraf kepercayaan 99% dengan derajat
kebebasan dk= n-1, digunakan rumus :
µ = X ± t(1-1/2α)dk x
n SD
Keterangan :
µ = interval kepercayaan
X = kadar rata-rata sampel
X = kadar sampel
t = harga t tabel sesuai dengan dk = n-1
α = tingkat kepercayaaan
dk = derajat kebebasan
SD = standar deviasi
BAB IV
HASIL DAN PEMBAHASAN
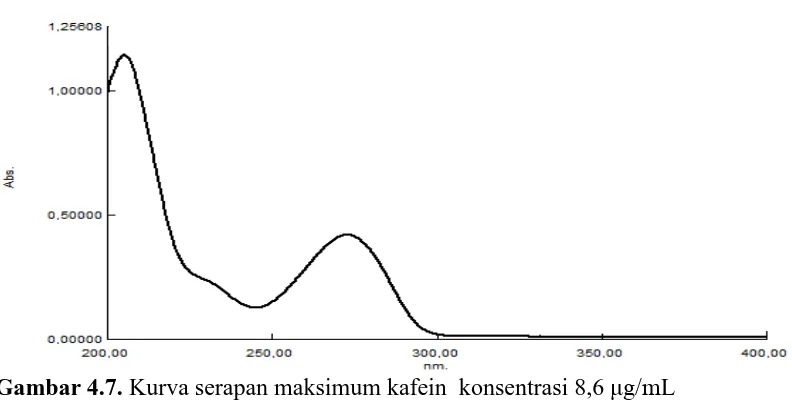
4.1 Hasil Penentuan Kurva Serapan Maksimum
Penentuan kurva serapan maksimum dilakukan pada panjang gelombang
200 − 400 nm. Pengukuran untuk parasetamol dilakukan pada konsentrasi
6,5 g/mδ dan untuk kafein pada konsentrasi 8,6 g/mδ. Berdasarkan hasil
penelitian diperoleh panjang gelombang parasetamol dengan konsentrasi
6,5 g/mδ adalah pada = 244,0 nm (Gambar 4.6) dan kafein dengan konsentrasi
8,6 g/mδ adalah pada = 272,6 nm (Gambar 4.7).
Gambar 4.7. Kurva serapan maksimum kafein konsentrasi 8,6 g/mL
4.2 Hasil Penentuan Kurva Serapan Derivatif Parasetamol
Kurva serapan larutan baku parasetamol dibuat dengan dengan konsentrasi
5 g/mL; 10 g/mL; 15 g/mL; 20 g/mL; dan 25 g/mL. Kemudian diukur
serapan pada panjang gelombang 200 – 400 nm. Kurva serapan baku parasetamol
dapat dilihat pada Lampiran 6 halaman 62 - 64. Kurva serapan baku parasetamol
selanjutnya ditransformasikan menjadi kurva serapan derivat pertama dan kedua
dengan Δ = 2 nm. Kurva serapan derivat kedua parasetamol dapat dilihat pada
Lampiran 7 halaman 67 - 69. Kemudian kurva serapan derivat kedua dari masing -
masing konsentrasi ditumpang tindihkan. Kurva tumpang tindih serapan baku
parasetamol dan kurva tumpang tindih serapan derivat pertama dan kedua
parasetamol dari konsentrasi masing – masing dapat dilihat pada Gambar 4.8, 4.9
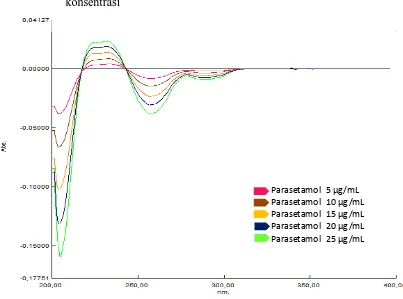
Gambar 4.8. Kurva tumpang tindih serapan parasetamol dalam berbagai konsentrasi
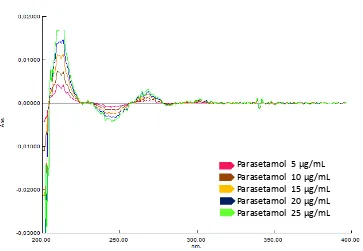
Gambar 4.9. Kurva tumpang tindihserapan derivat pertama parasetamol dalam berbagai konsentrasi.
Parasetamol 5 μg/mL Parasetamol 10 μg/mL Parasetamol 15 μg/mL Parasetamol 20 μg/mL Parasetamol 25 μg/mL
[image:50.596.117.520.385.684.2]Gambar 4.10. Kurva tumpang tindihserapan derivat kedua parasetamol dalam berbagai konsentrasi.
4.3 Hasil Penentuan Kurva Serapan Derivatif Kafein
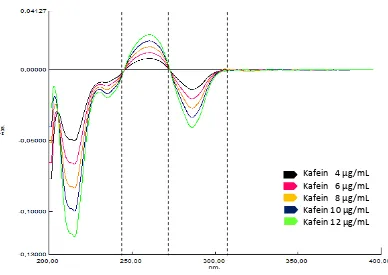
Kurva serapan baku kafein dibuat dengan konsentrasi 4 g/mL; 6 g/mL;
8 g/mL; 10 g/mL; dan 12 g/mL. Kemudian diukur serapan pada panjang
gelombang 200 – 400 nm. Kurva serapan baku kafein dapat dilihat pada
Lampiran 6 halaman 64 - 66. Kurva serapan baku kafein selanjutnya
ditransformasikan menjadi kurva serapan derivat pertama dan kedua dengan Δ = 2
nm. Kurva serapan derivat kedua kafein dapat dilihat pada Lampiran 7 halaman
69 - 71 Kemudian kurva serapan derivat kedua dari masing – masing konsentrasi
ditumpang tindihkan. Kurva tumpang tindih serapan baku kafein dan kurva
tumpang tindih serapan derivat pertama dan kedua kafein dari masing–masing
konsentrasi masing-masing dapat dilihat pada Gambar 4.11, 4.12 dan 4.13.
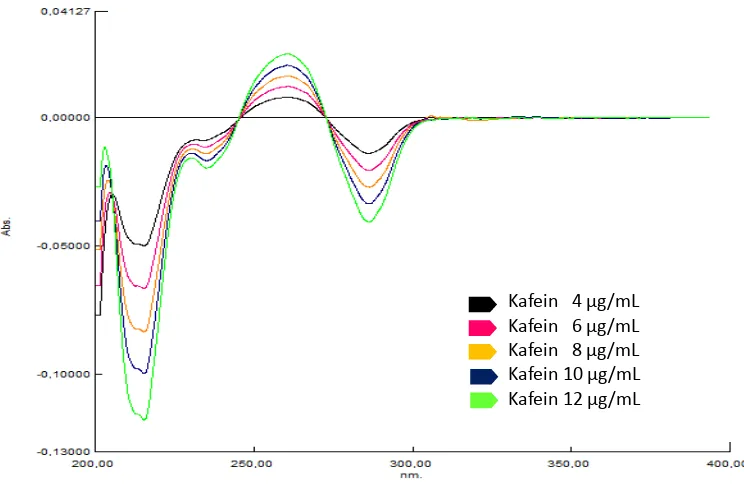
Gambar 4.11. Kurva tumpang tindihserapan kafein dalam berbagai konsentrasi.
Gambar 4.12. Kurva tumpang tindihserapan derivat pertama kafein dalam berbagai konsentrasi.
Kafein 4 μg/mL Kafein 6 μg/mL Kafein 8 μg/mL Kafein 10 μg/mL Kafein 12 μg/mL
[image:52.596.117.489.405.650.2]Gambar 4.13. Kurva tumpang tindihserapan derivat kedua kafein dalam berbagai konsentrasi.
4.4 Hasil Penentuan Zero Crossing
4.4.1 Zero Crossing Derivat Kedua pada Parasetamol dan Kafein
Penentuan zero crossing derivat pertama dan kedua diperoleh dengan menumpang tindihkan dari masing-masing konsentrasi pada parasetamol dan
kafein. Zero crossing pada serapan derivat pertama dan kedua parasetamol dan kafein ditunjukkan oleh panjang gelombang yang memiliki serapan nol pada
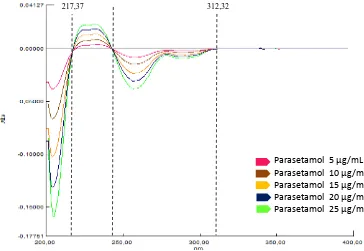
berbagai konsentrasi. Zero crossing serapan derivat petama parasetamol 217,37 nm; 242,83 nm; dan 312,32 nm dan serapan derivat kedua pada parasetamol
diperoleh dengan panjang gelombang 225,59 nm; 257,30 nm; 277,91 nm
sedangkan serapan derivat pertama kafein 245,18 nm; 272,73 nm; 311,52 nm dan
serapan derivat kedua kafein dengan panjang gelombang 215,60 nm; 229,68 nm;
dan kedua parasetamol dan kafein dapat dilihat pada Gambar 4.14 – 4.17.
Gambar 4.14. Zero crossing parasetamol pada derivat pertama
Gambar 4.15. Zero crossing parasetamol pada derivat kedua
Parasetamol 5 μg/mL Parasetamol 10 μg/mL Parasetamol 15 μg/mL Parasetamol 20 μg/mL Parasetamol 25 μg/mL 225,59
nm
232,61
257,30 277,91
217,37 312,32
245,18 272,73 311,52
[image:54.596.118.517.414.687.2]Gambar 4.16. Zero crossing Kafein pada derivat pertama
Gambar 4.17. Zero crossing Kafein pada derivat kedua
4.5 Hasil Penentuan Panjang Gelombang Analisis
Penentuan panjang gelombang analisis dilakukan dengan membuat larutan
parasetamol dengan konsentrasi 20 g/mL, kafein dengan konsentrasi 4 g/mL,
Kafein 4 μg/mL Kafein 6 μg/mL Kafein 8 μg/mL Kafein 10 μg/mL Kafein 12 μg/mL 215,60 229,6
235,99
307,6 286,09 260,48
dan larutan campuran parasetamol dan kafein sehingga di dalamnya terdapat
parasetamol dengan konsentrasi 20 g/mL dan kafein dengan konsentrasi
4 g/mL. Kemudian dibuat spektrum serapan derivat pertama dan kedua dari
masing-masing larutan parasetamol, kafein dan campuran parasetamol dengan
kafein, selanjutnya ditumpang tindihkan.Yang dipilih untuk menjadi panjang
gelombang analisis adalah pada saat serapan senyawa pasangannya nol dan
serapan senyawa yang lain dan campurannya memiliki nilai serapan positif,
hampir sama atau persis sama. Dilihat Gambar 4.27 dan 4.29 spektrum derivat
pertama tidak ditemukan zero crossing maka dilanjutkan ke derivat kedua Gambar 4.28 dan 4.30. Kurva serapan campuran parasetamol dan kafein, kurva serapan
tumpang tindih parasetamol dan kafein, kurva serapan tumpang tindih
parasetamol, kafein dan campuran parasetamol dan kafein pada derivat pertama
[image:56.596.114.492.447.636.2]dan kedua masing-masing dapat dilihat pada Gambar 4.18 – 4.33
Gambar 4.19. Kurva serapan tumpang tindih parasetamol 20 g/mL dan kafein 4 g/mL
Gambar 4.20. Kurva serapan tumpang tindih parasetamol 20 g/mL dan kafein 4 g/mL dan campuran keduanya
Gambar 4.21. Kurva serapan derivat pertama campuran parasetamol 20 g/mδ
dan kafein 4 g/mδ
Parasetamol 20 g/mδ Kafein 4 g/mδ
Parasetamol 20 g/mδ Kafein 4 g/mδ
Campuran Parasetamol 20
[image:57.596.116.475.87.240.2]Gambar 4.22. Kurva serapan derivat kedua campuran parasetamol 20 g/mδ dan
kafein 4 g/mδ
Gambar 4.23. Kurva tumpang tindih serapan derivat pertama parasetamol 20
g/mδ dan kafein 4 g/mδ
Gambar 4.24. Kurva tumpang tindih serapan derivat kedua parasetamol
20 g/mδ, kafein 4 g/mδ
Parasetamol 20 g/mδ Kafein 4 g/mδ
[image:58.596.117.507.122.459.2] [image:58.596.121.486.500.667.2]a
Gambar 4.25. Kurva tumpang tindih serapan derivat pertama parasetamol
[image:59.596.126.476.312.481.2]20 g/mδ, kafein 4 g/mδdan campuran keduanya
Gambar 4.26. Kurva tumpang tindih serapan derivat kedua parasetamol
20 g/mδ, kafein 4 g/mδdan campuran keduanya
Gambar 4.27. Zero crossing derivat pertama parasetamol Parasetamol 20 g/mδ Kafein 4 g/mδ
Campuran Parasetamol 20
g/mδ dan Kafein 4 g/mδ Parasetamol 20 g/mδ Kafein 4 g/mδ
Campuran Parasetamol 20
g/mδ dan Kafein 4 g/mδ
Parasetamol 20 g/mδ Kafein 4 g/mδ
[image:59.596.115.474.488.684.2]Gambar 4.28. Zero crossing derivat kedua parasetamol
Gambar 4.29. Zero crossing derivat pertama kafein
Parasetamol 20 g/mδ Kafein 4 g/mδ
Kafein 4 g/mδ Parasetamol 20 g/mδ
225,60