RESERPIN DALAM TABLET OBAT
NIKEN WULANDARI
DEPATEMEN KIMIA
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM
INSTITUT PERTANIAN BOGOR
untuk menjamin mutu dan keamanannya. Salah satu jenis pengawasan mutu tersebut adalah analisis kadar senyawa aktif dalam proses pengendalian mutu obat. Penentuan kadar senyawa aktif memerlukan suatu metode analisis dengan ketelitian dan ketepatan yang cukup baik. Selain itu juga memenuhi kriteria lain seperti spesifisitas, linearitas, limit deteksi, limit kuantitasi, dan ketangguhan (robustness).
Metode spektrofotometri derivatif ultra-violet (SDUV) telah dikembangkan oleh Rachmanti (2006) untuk penentuan kadar reserpin dalam tablet obat hipertensi yang mengandung reserpin. Teknik ini dapat menjadi alternatif dalam pengawasan mutu produk obat hipertensi dengan senyawa aktif reserpin karena berbagai keuntungan yang dimilikinya seperti cepat, mudah, murah, dan tanpa adanya tahap pemisahan. Metode analisis penentuan reserpin dapat digunakan untuk analisis rutin jika telah tervalidasi.
Validasi metode analisis adalah suatu proses penilaian terhadap metode analisis tertentu berdasarkan percobaan laboratorium untuk membuktikan bahwa metode tersebut memenuhi persyaratan untuk digunakan (Harmita 2004). Proses ini bukan suatu proses tunggal, namun merupakan salah satu bagian dari prosedur analisis yang tidak dapat dipisahkan (Ermer & Miller 2005). Validasi metode dilakukan untuk metode yang baru dikembangkan, atau jika metode tersebut akan digunakan untuk kegiatan yang bersifat rutin, jika terjadi perubahan antara kondisi analisis dan kondisi pada saat validasi metode, dan jika terjadi perubahan metode dari metode standar.
Penentuan kadar reserpin dengan SDUV merupakan suatu metode baru yang dikembangkan untuk contoh tablet obat dengan matriks yang kompleks. Validasi metode SDUV dapat digunakan untuk mengetahui keabsahan data yang dihasilkan. Menurut ICH (1995), metode analisis dapat memberikan data yang dipercaya jika memenuhi beberapa parameter yang telah disyaratkan, yaitu spesifisitas, ketelitian (presisi), ketepatan (akurasi), linearitas, kisaran, limit deteksi, limit kuantitasi, dan ketangguhan. Untuk penentuan kadar senyawa aktif dalam produk obat, parameter ketang-guhan tidak perlu ditentukan, sedangkan parameter yang akan ditentukan dalam penelitian ini hanya meliputi linearitas, limit
SDUV untuk penentuan kadar reserpin dalam tablet obat Serpasil®.
TINJAUAN PUSTAKA
Reserpin
Reserpin (C33H40N2O9) (Gambar 1) meru-pakan salah satu jenis alkaloid indol yang digunakan untuk pengobatan penyakit hiper-tensi, menurunkan denyut jantung, terapi bagi penderita skizofrenia, dan obat penenang (Forney 2007). Reserpin dapat diperoleh dari ekstrak tumbuhan genus Rauwolfia, terutama spesies R. serpentina dan R. vomitoria. Sumber lain dari obat ini adalah berbagai
spesies tanaman dari genus Alstonia,
Excatavia, Ochrosia, Tanduzia, Vallesia, dan
Vinca (Apocynaceae), namun kadarnya sangat rendah (Hesse 2002).
N
Beberapa sifat fisik dan kimia dari reserpin adalah berwujud padatan (bubuk kristal) yang berwarna putih, tidak berbau dan menjadi berwarna kehitaman jika dibiarkan di udara terbuka. Reserpin tidak larut dalam air dan eter, sukar larut dalam etanol dingin, serta larut dalam kloroform dan asam asetat. (British Pharmacopoeia 1993).
(nm)
Spektrofotometri Derivatif Ultraviolet (SDUV)
Teknik SDUV telah diperkenalkan pada
tahun 1953 (Popovic et al. 2000) dan
mengalami perkembangan cukup pesat selama 25 tahun terakhir serta digunakan secara luas sebagai alat untuk analisis kuantitatif, pencirian, dan kendali mutu di bidang pertanian, farmasi, dan biomedis (Kazemipour
et al. 2002). Metode SDUV merupakan metode paling umum untuk penentuan secara serentak campuran biner suatu senyawa dengan spektrum yang bertumpang tindih (El Sayed & El Salem 2004).
Metode SDUV didasarkan pada spektrum turunan (derivatif) ke-n yang diperoleh dari spektrum serapan normal UV-Vis (ultraviolet dan sinar tampak) atau spektrum turunan ke-0 (Karpinska 2004). Spektrum derivatif diper-oleh dengan menggunakan persamaan mate-matika. Keuntungan dari cara ini adalah spektrum derivatif dapat dihitung dengan parameter yang berbeda dan teknik peng-halusan dapat digunakan untuk meningkatkan nisbah sinyal terhadap derau (noise) (Ojeda & Rojas 2004).
Menurut O’Haver (1979), spektro-fotometri derivatif merupakan pengukuran kemiringan garis spektrum, yaitu rerata perubahan absorbans terhadap panjang gelombang. Spektrofotometri derivatif tetap mempertahankan semua hukum (persamaan) spektrofotometri konvensional (Karpinska 2004). Hukum Beer pada spektrum normal, A = εbc, pada bentuk derivatif akan menjadi
Dn = n
panjang gelombang λ. Spektrum normal
diperoleh dari plot hubungan antara nilai absorbans (A) dan nilai panjang gelombang (λ), sedangkan spektrum turunan pertama merupakan plot hubungan antara dA/dλ dan nilai λ, nilai plot spektrum turunan pertama digunakan untuk menentukan d2A/dλ2 yang apabila diplot terhadap λ maka akan meng-hasilkan plot spektrum turunan kedua. Untuk
memperoleh spektrum turunan ke-n maka
dibuat plot hubungan antara dnA/dλn dan nilai
λ (Gambar 2) (Skujins 1986). Penerapan
hukum Lambert-Beer ini berdasarkan pada pemilihan kondisi optimum, termasuk pemilihan pita spektrum yang paling sesuai, orde derivatif yang cocok, serta metode pengukuran dan optimalisasi semua parameter instrumen yang penting (Popovic et al. 2000).
Gambar 2 Spektrum serapan normal dan turunannya (O’Haver 1979).
Analisis kuantitatif pada metode SDUV dilakukan dengan cara mengukur amplitudo spektrum derivatif. Jika konsentrasi contoh pada spektrum normal sebanding dengan absorbans, maka konsentrasi contoh pada spektrum derivatif sebanding dengan amplitudo. Metode pengukuran amplitudo ada beberapa cara, yaitu dari puncak ke puncak (p1, p2), puncak ke garis dasar (z), dan puncak tangen (t) (Gambar 3) (Popovic et al. 2000).
Gambar 3 Pengukuran amplitudo pada spek-trum derivatif (Popovic
et al. 2000).
Teknik SDUV mempunyai beberapa keuntungan
yang tidak dapat diperoleh dari teknik
lain. Beberapa di antaranya adalah mempercepat waktu analisis, mengurangi biaya yang dibutuhkan dan dapat digunakan untuk pemisahan senyawa campuran dengan spektrum yang bertumpang tindih tanpa membutuhkan tahap pemisahan analit dari matriksnya (Karpinska 2004). Selain itu, pergeseran garis dasar dapat dihilangkan dan efek kekeruhan dapat dikurangi (O’Haver 1979), meningkatkan selektivitas dan sensiti-vitas analisis komponen minor, meningkatkan resolusi spektrum, serta dapat menentukan
Spektrum normal
Spektrum turunan ke-1
Spektrum turunan ke-2 Absorbans
Amplitudo
Amplitudo
Panjang gelombang (nm)
A
m
pl
Validasi Metode Analisis
Validasi metode analisis adalah suatu proses penilaian terhadap metode analisis tertentu berdasarkan percobaan laboratorium untuk membuktikan bahwa metode tersebut memenuhi persyaratan untuk digunakan (Harmita 2004). Suatu metode yang baru dikembangkan untuk kegiatan rutin harus divalidasi sebelum digunakan. Selain itu, validasi metode dilakukan jika terjadi perubahan kondisi antara kondisi analisis dan kondisi pada saat validasi metode, atau terjadi perubahan metode dari metode standar. Beberapa manfaat validasi metode analisis adalah untuk mengevaluasi unjuk kerja suatu metode analisis, menjamin prosedur analisis, menjamin keakuratan dan kedapatulangan hasil prosedur analisis, dan mengurangi risiko penyimpangan yang mungkin timbul.
Terdapat beberapa rujukan validasi metode seperti United State Pharmacopoeia (USP), British Pharmacopoeia (BP), Association of Official Analytical Chemists (AOAC), International Conference on Harmo-nization (ICH), dan International Union of Pure and Applied Chemistry (IUPAC). Penelitian ini mengacu pada petunjuk validasi metode dari ICH (1995) yang dikhususkan untuk analisis obat-obatan. Menurut ICH (1995), prosedur analisis yang harus divalidasi meliputi beberapa jenis pengujian, yaitu identifikasi suatu senyawa tertentu, kuantitasi adanya pengotor, uji limit untuk mengen-dalikan keberadaan pengotor, serta uji kuantitatif komponen aktif atau komponen lain dalam produk obat-obatan. Selain itu, terdapat 8 parameter validasi metode analisis, yaitu spesifisitas, ketelitian, ketepatan, linearitas, kisaran, limit deteksi, limit kuan-titasi, dan ketangguhan, sedangkan parameter yang harus dipenuhi untuk validasi metode analisis produk obat-obatan meliputi spesifi-sitas, linearitas, kisaran, limit deteksi, limit kuantitasi, ketelitian, dan ketepatan.
Linearitas
Linearitas menunjukkan kemampuan suatu metode analisis untuk memperoleh hasil pengujian yang sesuai dengan konsentrasi analit dalam contoh pada kisaran konsentrasi tertentu (ICH 1995). Hal ini dapat dilakukan dengan cara membuat kurva kalibrasi dari beberapa set larutan standar yang telah diketahui konsentrasinya. Persamaan garis yang digunakan pada kurva kalibrasi diperoleh dari metode kuadrat terkecil, yaitu y
= a + bx. Persamaan ini akan menghasilkan
koefisien korelasi (r). Koefisien korelasi inilah yang digunakan untuk mengetahui linearitas suatu metode analisis. Penetapan linearitas minimum menggunakan lima konsentrasi yang berbeda. Nilai koefisien korelasi yang memenuhi persyaratan adalah lebih besar dari 0.9970 (ICH 1995 diacu dalam Chan 2004).
Linearitas juga dapat diketahui dari kemiringan garis, intersep, dan residual (Ermer & Miller 2005). Residual menyatakan besarnya penyimpangan yang terjadi antara nilai yang terukur (y) dan nilai teoretis yang dihitung dari persamaan regresi (ŷ). Plot antara residual dan konsentrasi dibuat untuk mengetahui distribusi residual secara statistik. Jika residual terdistribusi secara normal (rerata mendekati nol dan berbentuk linear), maka persamaan regresi dapat dikatakan mempunyai bentuk yang benar.
Limit Deteksi dan Limit Kuantitasi
Limit deteksi (LD) merupakan jumlah atau konsentrasi terkecil analit dalam contoh yang dapat dideteksi, namun tidak perlu diukur sesuai dengan nilai sebenarnya. Limit kuantitasi (LK) adalah jumlah analit terkecil dalam contoh yang dapat ditentukan secara kuantitatif pada tingkat ketelitian dan ketepatan yang baik. Limit kuantitasi merupakan parameter pengujian kuantitatif untuk konsentrasi analit yang rendah dalam matriks yang kompleks dan digunakan untuk menentukan adanya pengotor atau degradasi produk (ICH 1995). Limit deteksi dan limit kuantitasi dihitung dari rerata kemiringan garis dan simpangan baku intersep kurva standar yang diperoleh.
Ketelitian
Pengujian ketelitian dinyatakan dengan simpangan baku relatif (SBR). Menurut ICH (1995), kriteria nilai SBR yang dapat diterima adalah kurang dari 2%.
Ketepatan
Ketepatan suatu metode analisis didefinisikan sebagai kedekatan hasil yang diterima (baik sebagai nilai teoretis maupun sebagai nilai rujukan yang diterima) dengan nilai yang diperoleh dari hasil pengukuran (ICH 1995 diacu dalam Chan 2004). Ketepatan dinyatakan sebagai perolehan kembali yang ditentukan dengan cara menambahkan sejumlah tertentu standar dari analit yang akan diukur ke dalam contoh. Perolehan kembali (%) yang dapat diterima menurut ICH adalah 98–102%. ICH juga mensyaratkan minimum 9 kali pengukuran pada 3 tingkat konsentrasi yang berbeda.
BAHAN DAN METODE
Bahan dan Alat
Bahan-bahan yang digunakan adalah tablet obat Serpasil®, standar reserpin, etanol absolut, larutan H2SO4 0.25 M, larutan natrium nitrit 0.3% (b/v), dan larutan asam sulfamat 5% (b/v).
Alat-alat yang digunakan adalah spektro-fotometer UV-Vis 2800 Hitachi beserta kuvet 1 cm, neraca analitik, seperangkat komputer, peranti lunak UV Solutions versi 2.0 dan Microsoft Excel tahun 2003, pemanas, dan alat-alat kaca.
Pengukuran Konsentrasi Reserpin dengan Metode SDUV
Parameter validasi metode SDUV yang ditentukan meliputi linearitas, ketepatan, ketelitian, limit deteksi, dan limit kuantitasi. Penentuan kadar reserpin dilakukan pada kondisi optimum yang telah ditentukan pada penelitian sebelumnya (Rahmanti 2006). Kadar reserpin yang diperoleh dengan metode SDUV dibandingkan dengan metode rujukan British Pharmacopoeia (1993) (Lampiran 1).
Pembuatan Larutan Standar dan Contoh
Sebanyak 10 mg serbuk reserpin di-masukkan ke dalam gelas piala dan ditambahkan 20 ml etanol absolut hangat kemudian diaduk dengan pengaduk magnetik selama 20 menit. Larutan ini dimasukkan ke dalam labu takar 100 ml dan ditepatkan
dengan etanol. Larutan stok standar dengan konsentrasi 100 g/l diencerkan menjadi 10, 20, 30, 40, dan 50 g/ml.
Tablet obat sebanyak 0.5 g dihaluskan dan dimasukkan ke dalam gelas piala, kemudian ditambahkan 20 ml etanol absolut hangat dan dipanaskan pada suhu 50 ºC sambil diaduk dengan pengaduk magnetik selama 20 menit. Larutan ini dimasukkan ke dalam labu takar 25 ml dan ditepatkan dengan etanol.
Pengukuran Larutan Standar dan Contoh
Pengukuran larutan standar dan contoh dilakukan pada kondisi optimum yang telah ditentukan oleh Rachmanti (2006). Larutan standar dan contoh diukur serapannya dengan spektrofotometri UV-Vis pada panjang gelombang 200–350 nm dengan kecepatan pemayaran 200 nm/menit. Spektrum yang diperoleh kemudian diolah dengan perangkat lunak UV Solutions pada kondisi optimum sebagai berikut: orde turunan 1, orde peng-halusan 2, jumlah jendela 17, dan amplitudo diukur dari puncak spektrum ke garis dasar (z) pada panjang gelombang 277.4 nm.
Validasi Metode SDUV
Linearitas
Sebanyak 5 larutan standar disiapkan dengan konsentrasi 10, 20, 30, 40, 50 g/ml. Masing-masing konsentrasi dibuat sebanyak 6 ulangan kemudian setiap larutan diukur dengan menggunakan spektrofotometer UV-Vis pada kondisi optimum dan dibuat persamaan garisnya dengan metode regresi linear (y = a + bx). Peubah a menyatakan intersep dan b adalah kemiringan garis dari kelima larutan standar yang diukur. Linearitas kurva kalibrasi dilihat dari nilai koefisien korelasi (r).
Limit Deteksi (LD) dan Limit Kuantitasi (LK)
Persamaan linear yang diperoleh pada uji linearitas selanjutnya digunakan untuk meng-hitung limit deteksi dan limit kuantitasi. LD dan LK dihitung dari rerata kemiringan garis dan simpangan baku intersep kurva standar yang diperoleh dengan rumus sebagai berikut:
Sa = simpangan baku intersep kurva standar (n = 6)
b = rerata kemiringan garis kurva standar
Ketelitian
Larutan contoh yang telah disiapkan sesuai dengan prosedur di atas kemudian diukur dengan spektrofotometer UV-Vis sebanyak 6 ulangan pada hari yang sama (intraday) dan pada hari yang berlainan (interday) selama 5 hari berturut-turut. Ketelitian diukur dengan menghitung persentase simpangan baku relatif (%SBR) data dengan menggunakan rumus
s =
(
)
SBR = simpangan baku relatif
xi = kadar reserpin tiap ulangan
x = rerata kadar reserpin
n = jumlah ulangan
Ketepatan
Larutan standar reserpin disiapkan dengan konsentrasi 100 g/ml dan larutan stok contoh dibuat dengan konsentrasi 50 g/ml. Sebanyak 10 ml contoh dipindahkan ke dalam labu takar 50 ml dan ditambahkan larutan standar reserpin masing-masing sebanyak 10, 15, dan 17.5 ml kemudian volumenya ditepatkan dengan etanol. Masing-masing larutan dibuat 3 ulangan. Perolehan kembali dihitung dengan rumus
Perolehan kembali(%) =
c b a−
× 100%
dengan:
a = konsentrasi contoh + konsentrasi standar yang terukur
b = konsentrasi contoh
c = konsentrasi standar teoretis yang ditambahkan
Pengukuran Reserpin dengan Metode British Pharmacopoeia (1993)
Serbuk obat sebanyak 0.5 g dimasukkan ke dalam gelas piala kemudian ditambahkan 1 ml etanol, 1 ml H2SO4 0.25 M, dan ditambah-kan 5 ml etanol. Larutan ini dihangatditambah-kan agar
contoh obat mudah larut. Larutan didinginkan dan dimasukkan ke dalam labu takar 25 ml dan ditera dengan etanol. Larutan ini disebut larutan A.
Larutan A sebanyak 10 ml dimasukkan ke dalam gelas piala kemudian ditambahkan 2 ml H2SO4 0.25 M dan 2 ml larutan natrium nitrit 0.3% (b/v) segar. Campuran dipanaskan pada suhu 55 ºC selama 35 menit pada penangas air kemudian didinginkan. Larutan asam sulfamat 5% (b/v) sebanyak 1 ml ditambahkan ke dalam larutan contoh kemudian larutan contoh dimasukkan ke dalam labu takar 25 ml dan ditera dengan etanol. Larutan dibuat sebanyak 6 ulangan dan absorbans diukur pada λ = 389 nm menggunakan sel rujukan yang berisi 10 ml larutan A tanpa penambahan natrium nitrit.
HASIL DAN PEMBAHASAN
Kondisi Optimum Metode SDUV
Penentuan reserpin dalam tablet obat Serpasil® dengan metode SDUV dilakukan pada kondisi optimum yang telah ditentukan oleh Rahmanti (2006). Reserpin dalam obat dapat ditentukan dengan metode SDUV karena bentuk spektrum contoh dengan standar reserpin saling berimpit (bentuknya sama). Spektrum serapan normal (turunan ke-0) dibuat pada panjang gelombang 200–350 nm, yaitu daerah panjang gelombang sinar UV. Spektrum dibuat dengan kecepatan pemayaran 200 nm/menit sehingga spektrum yang diperoleh tetap akurat setelah diturunkan.
Amplitudo
Panjang gelombang (nm)
Gambar 4 Spektrum serapan reserpin pada kondisi optimum.
Keterangan:
Cokelat = standar reserpin 10 g/ml Biru tua = standar reserpin 20 g/ml
Linearitas metode SDUV ditentukan dengan cara membuat kurva hubungan antara amplitudo spektrum derivatif pada sumbu y
dan konsentrasi standar pada sumbu x.
Konsentrasi yang digunakan berkisar 10–50 g/ml. Pengujian parameter ini dilakukan sebanyak 6 ulangan. Tabel 1 menyajikan persamaan regresi linear dari masing-masing kurva standar (Lampiran 2) dan kurva standar rerata. Linearitas dinyatakan dengan koefisien korelasi (r). Berdasarkan hasil pengujian, diperoleh koefisien korelasi dari 6 ulangan berkisar 0.9997–0.9999, dan untuk kurva standar rerata sebesar 0.9999. Menurut ICH (1995 diacu dalam Chan 2004), nilai ini memenuhi syarat yang ditetapkan, yaitu 0.9970. Nilai koefisien korelasi yang tinggi menunjukkan hubungan yang linear antara sinyal detektor yang terukur dan jumlah reserpin dalam contoh.
Tabel 1 Koefisien korelasi dan persamaan regresi linear kurva standar (n = 6)
No. Persamaan Regresi Linear r
1 y = (1.38x + 2.58) × 10-4 0.9998
Persamaan regresi linear untuk kurva standar rerata adalah y = (1.42x + 2.94) × 10-4 (Gambar 5). Nilai intersep (a) dan batas galatnya (tSa) pada selang kepercayaan 95% sebesar 2.94 × 10-4 ± 7.27 × 10-5 (Tabel 2).
Nilai intersep menyatakan adanya pengaruh matriks pada larutan. Nilai intersep yang semakin jauh dari nol dipengaruhi oleh matriks dalam larutan yang semakin besar. Hal ini dapat mengganggu penentuan analit dalam contoh yang akan ditentukan. Persamaan regresi kurva standar rerata mempunyai intersep yang tidak jauh dari nol, yaitu 2.94 × 10-4 ± 7.27 × 10-5 sehingga matriks contoh tidak terlalu mengganggu penentuan kadar reserpin.
y = 0.000142x + 0.000294 r = 0.9999
Gambar 5 Kurva standar rerata reserpin pada konsentrasi 10–50 g/ml.
Nilai kemiringan garis menyatakan sensitivitas suatu metode. Nilai kemiringan garis yang kecil menunjukkan bahwa perubahan konsentrasi yang kecil tidak terlalu berpengaruh terhadap sinyal detektor yang dihasilkan, sehingga metode mempunyai sensitivitas yang kurang baik. Nilai kemiringan garis (b) dan batas galatnya (tSb) adalah 1.42 × 10-4 ± 2.19 × 10-6 (Tabel 2). Nilai ini cukup besar sehingga perubahan konsentrasi yang kecil akan berpengaruh terhadap perubahan sinyal detektor. Contoh perhitungan parameter statistika disajikan pada Lampiran 3.
Tabel 2 Parameter statistika kurva standar rerata (n = 6)
Parameter statistika Metode SDUV Persamaan regresi linear y = (1.42x + 2.94) × 10-4
Persamaan regresi yang diperoleh dari kurva kalibrasi metode mempunyai residual yang terdistribusi secara normal, sehingga dapat dikatakan bahwa penyimpangan yang terjadi tidak terlalu besar. Hal ini menunjukkan bahwa persamaan regresi dapat digunakan untuk menentukan kadar reserpin dalam contoh. Plot antara residual dan konsentrasi standar reserpin dapat dilihat pada Gambar 6.
Gambar 6 Hubungan antara konsentrasi reserpin dan residual kurva standar rerata.
Limit Deteksi dan Limit Kuantitasi
Limit deteksi (LD) dan limit kuantitasi (LK) ditentukan dari persamaan regresi linear kurva standar rerata hasil penentuan linearitas. Parameter ini ditentukan untuk mengetahui konsentrasi terendah pada saat sinyal antara blangko dan analit dapat dibedakan. Kedua parameter ini mempunyai nilai yang berbeda bergantung pada metode yang digunakan, walaupun menggunakan instrumen yang sama. Limit deteksi metode SDUV untuk penentuan reserpin dalam tablet obat sebesar 0.7158 g/ml. Nilai ini menunjukkan bahwa sinyal antara reserpin dan blangko dapat dibedakan pada konsentrasi terendah 0.7158
g/ml. Instrumen tidak dapat membedakan sinyal antara blangko dan reserpin pada konsentrasi di bawah nilai ini.
Limit kuantitasi ditentukan untuk mengetahui konsentrasi terendah yang dapat ditentukan oleh suatu metode pada tingkat ketelitian dan ketepatan yang baik. Nilai limit kuantitasi berdasarkan hasil penelitian adalah 2.1690 g/ml. Konsentrasi analit yang terukur di bawah nilai ini memberikan ketelitian dan ketepatan yang tidak baik. Perhitungan limit deteksi dan limit kuantitasi tertera pada
Lampiran 4.
Ketelitian
Ketelitian metode SDUV ditentukan sebanyak 6 ulangan, baik intraday maupun
interday. Ketelitian yang ditentukan adalah keterulangan karena dilakukan oleh operator, instrumen, peralatan, dan laboratorium yang sama. Keterulangan dilakukan untuk mengetahui adanya galat acak yang berasal dari penyiapan larutan maupun instrumen. Keterulangan intraday dapat digunakan untuk mengetahui adanya galat acak yang berasal dari penyiapan larutan, seperti penimbangan, pembuatan larutan, dan penyaringan. Hasil penelitian menunjukkan bahwa SBR intraday
untuk masing-masing hari bernilai kurang dari 2% (Tabel 3) dengan kadar reserpin rerata dalam 1 tablet obat untuk hari ke-1 hingga hari ke-5 secara berturut-turut adalah 0.2628; 0.2565; 0.2580; 0.2631; dan 0.2534 mg. Nilai SBR di bawah 2% menunjukkan bahwa galat acak yang berasal dari penyiapan larutan tidak memengaruhi hasil analisis secara nyata. Data dan perhitungan ketelitian untuk hari ke-1 hingga hari ke-5 ditunjukkan pada Lampiran 5.
Tabel 3 Simpangan baku relatif (%SBR)
untuk intraday maupun interday
Hari ke- Reserpin (mg) SBR (%)
Keterulangan interday dilakukan selama 5 hari berturut-turut. Hal ini dilakukan untuk mengetahui adanya galat acak yang berasal dari instrumen dan kondisi laboratorium pada hari yang berlainan. Nilai SBR untuk keterulangan interday adalah 1.61% dengan kadar reserpin rerata 0.2588 ± 0.0052 mg per tablet obat (Lampiran 6), sehingga dapat dikatakan bahwa galat acak yang berasal dari instrumen dan kondisi laboratorium yang berbeda tidak memengaruhi hasil analisis. Oleh sebab itu, metode SDUV mempunyai keterulangan intraday dan interday yang baik.
Ketepatan
dapat menunjukkan adanya galat sistematik yang dapat memengaruhi metode analisis. Galat sistematik dapat menyebabkan hasil analisis menjadi lebih besar atau lebih kecil. Beberapa contoh penyebab galat sistematik di antaranya adalah galat pada saat pengambilan contoh, kurva kalibrasi yang tidak linear, serta galat yang disebabkan oleh instrumen dan peralatan kaca yang digunakan (Harvey 2000).
Penentuan ketepatan dilakukan dengan menambahkan standar reserpin sebanyak 1.00, 1.5, dan 1.75 mg ke dalam contoh obat yang telah berisi 0.5 mg reserpin. Perolehan kembali yang diperoleh berkisar antara 107.72 dan 108.75% (Tabel 4). Menurut ICH (1995 diacu dalam Chan 2004), nilai ini berada di luar batas nilai yang diterima, yaitu 98–102%. Namun, menurut AOAC (1993), nilai ini berada pada kisaran yang dapat diterima, yaitu 80–110%, sehingga dapat dikatakan metode ini mempunyai ketepatan yang cukup baik. Perhitungan perolehan kembali ditunjukkan pada Lampiran 7.
Tabel 4 Perolehan kembali rerata metode SDUV
Standar Reserpin (mg) Perolehan
kembali (%) Ditambahkan Ditemukan
1.00 1.5875 108.75
1.50 2.1173 107.82
1.75 2.3851 107.72
Pengukuran Kadar Reserpin dengan Metode British Pharmacopoeia (1993)
Metode British Pharmacopoeia (1993) digunakan sebagai metode rujukan untuk menentukan kadar reserpin dalam tablet obat. Metode ini menggunakan teknik spektrofoto-metri UV-Vis konvensional, yaitu pengukuran absorbans contoh pada panjang gelombang tertentu. Pengukuran dimulai dengan mem-buat kurva standar pada konsentrasi 10–50
g/ml. Persamaan regresi kurva standar yang diperoleh adalah y = 0.01604x + 0.0374 dengan nilai r = 0.9999 (Gambar 7).
Kadar reserpin rerata dalam obat dengan menggunakan metode ini adalah 0.2375 mg dengan batas galat (selang kepercayaan 95%) 0.0025 mg. Perhitungan penentuan kadar reserpin dengan metode ini dapat dilihat pada Lampiran 8.
y = 0.01604x + 0.0374
Gambar 7 Persamaan regresi linear standar reserpin menggunakan metode British Pharmacopoeia (1993).
Perbandingan Metode SDUV dan British Pharmacopoeia (1993)
Kadar reserpin hasil penentuan dengan metode SDUV selanjutnya dibandingkan dengan metode rujukan. Hal ini dilakukan dengan cara melakukan uji statistika yang meliputi uji F dan uji t. Uji F dilakukan untuk membandingkan keragaman kedua metode. Kedua metode mempunyai keragaman yang tidak berbeda nyata karena Fhitung < Ftabel pada selang kepercayaan 95% (Tabel 5). Uji t
digunakan untuk membandingkan efektivitas dan efisiensi metode baik dari segi teoretis maupun dari segi teknis (biaya). Nilai thitung kedua metode lebih besar jika dibandingkan dengan nilai ttabel pada selang kepercayaan 95% (Tabel 5), sehingga kedua metode memberikan hasil yang berbeda nyata. Metode SDUV menghasilkan nilai yang lebih tinggi jika dibandingkan dengan metode British Pharmacopoeia (1993).
Tabel 5 Uji statistika metode SDUV dan British Pharmacopoeia (1993) Parameter
statistika Metode SDUV Metode BP 1993 Kurva standar y=(1.42x + 2.94)×10-4
y=0.01604x+0.0374
r 0.9999 0.9999
Kadar reserpin 0.2588 mg 0.2375 mg Batas galat 0.0052 0.0025
F hitung 1.3611
F tabel 5.0500
t hitung 9.0704
SIMPULAN DAN SARAN
Simpulan
Pengujian parameter validasi menunjuk-kan bahwa metode SDUV mempunyai keabsahan data yang kurang baik. Metode ini mempunyai linearitas yang baik dengan r = 0.9999, limit deteksi dan limit kuantifikasi berturut-turut adalah 0.7158 dan 2.1690
g/ml, ketelitian yang baik dengan nilai SBR di bawah 2%, namun mempunyai ketepatan yang kurang baik dengan perolehan kembali berkisar 107.72–108.75%. Uji statistika antara metode SDUV dan metode British
Pharma-copoeia (1993) memperlihatkan keragaman
yang sama (uji F) namun memberikan hasil yang berbeda nyata (uji t).
Saran
Perlu dilakukan penentuan ketidakpastian pengukuran dan sumber galat yang dapat mempengaruhi metode SDUV. Selain itu perlu digunakan metode pembanding yang lain seperti kromatografi cair kinerja tinggi dan metode lain untuk penentuan parameter ketepatan.
DAFTAR PUSTAKA
[AOAC] Association of Official Analytical Chemists. 1993. AOAC Peer – Verified Methods Program. Arlington: AOAC Inter national.
[AOAC] Association of Official Analytical
Chemistry. 2005. Official Methods of
Analysis of AOAC International. Ed ke-18. Maryland: AOAC International.
British Pharmacopoeia. 1993. British Phar-macopoeia Department of Health & Social Services for Northern Ireland. London: British Pharmacopoeia.
Chan CC. 2004. Potency Method Validation. Di dalam: Chan CC, Lee YC, Lam H,
Zhang XM, editor. Analytical Method
Validation and Instrument Performance Verification. New Jersey: John Wiley & Sons.
El-Sayed AAY, El-Salem NA. 2005. Recent development of derivative spectrophoto-metry and their analytical applications [ulasan]. Anal Sci 21:595-614.
Ermer J, Miller JH, editor. 2005. Method Validation in Pharmaceutical Analysis. Weinheim: Wiley-VCH.
Forney B. 2007. Reserpine For Veterinary Use. [terhubung berkala] www.wedge woodpharmacy.com [29 Jan 2007].
Harmita. 2004. Petunjuk pelaksanaan validasi
metode dan perhitungannya. Maj Ilmu
Kefarmasian, 1(3):117–135.
Harvey D. 2000. Modern Analytical
Chemistry: McGraw-Hill.
Hesse M. 2002. Alkaloids, Nature’s Curse or Blessing. Weinheim: Wiley-VCH.
[ICH] International Conference on Harmoni-zation. 1995. Validation of Analytical Prosedures: Methodology Q2B [terhubung berkala]. www.ich.org. [19 Des 2006]
[ICH] International Conference on Harmoni-zation. 1995. Validation of Analytical Prosedures: Text and Methodology Q2 (R1) [terhubung berkala]. www.ich.org. [19 Des 2006]
Karpinska J. 2004. Derivative spectrophoto-metry recent applications and directions of development [ulasan]. Talanta 64:801-822.
Kazemipour M, Noroozian E, Tehrani MS, Mahmoudian M. 2002. A new second-derivative spectrophotometry methods for the determination of permenthrin in shampoo. J Pharm Biomed Anal 30:1379-1384.
Lewis RJ. 2002. Hazardous Chemicals. Ed ke-5. New York: A John Willey & Sons.
O’Haver TC. 1979. Potential clinical applications of derivative and wavelength
modulation spectrometry. Clin Chem
25:1548-1553.
Ojeda CB, Rojas FS. 2004. Recent develop-ment in derivative ultraviolet/visible absorption spectrophotometry [ulasan].
Anal Chim Acta 518:1-24
Popovic GV, Pfendt LB, Stefanovic VM. 1999. Analytical application of derivative spectro-photometry. J Serb Chem Soc
Rachmanti WD. 2006. Metode cepat untuk kuantitasi reserpin dalam obat dan ekstrak
Rauwolfia serpentina secara spektro-fotometri derivatif ultraviolet [skripsi]. Bogor: Fakultas Matematika dan Ilmu Pengetahuan Alam, Institut Pertanian Bogor.
Singh DK, Srivastava B, Sahu A. 2004. Spectrophotometry determination of
Rauwolfia alkaloids: Estimation of reserpine in pharmaceuticals. Anal Sci
20:571-573.
Skujins S. 1986. Application of UV-Visible Derivative Spectrophotometry. Steinhau-sertasse: Varian AG.
[USP] United States Pharmacopoeia. 2006.
Lampiran 1 Diagram alir penelitian
Standar Reserpin dan Contoh obat
Metode Spektrofotometri Derivatif Ultraviolet
Metode Rujukan British Pharmacopoeia (1993)
Penyiapan standar dan contoh
Pengukuran larutan standar dan contoh pada kondisi
optimum
Valisasi Metode SDUV (Penentuan ketelitian, ketepatan, linearitas, limit deteksi, dan limit
kuantitasi)
Lampiran 2 Persamaan regresi linear kurva standar reserpin ulangan ke 1 (a), 2 (b), 3 (c), 4 (d), 5 (e), 6 (f)
(a)
y = 0.000138x + 0.000258 r = 0.9999
0 0.002 0.004 0.006 0.008
0 10 20 30 40 50
[Reserpin] (ppm) Amplitudo
(c)
y = 0.000136x + 0.000270 r = 0.9999
0 0.002 0.004 0.006 0.008
0 10 20 30 40 50
[Reserpin] (ppm) Amplitudo
(e)
y = 0.000144x + 0.000295 r = 0.9999
0 0.002 0.004 0.006 0.008
0 10 20 30 40 50
[Reserpin] (ppm) Amplitudo
(b)
y = 0.000141x + 0.000296 r = 0.9999
0 0.002 0.004 0.006 0.008
0 10 20 30 40 50
[Reserpin] (ppm) Amplitudo
(d)
y = 0.000146x + 0.000330 r = 0.9999
0 0.002 0.004 0.006 0.008
0 10 20 30 40 50
[Reserpin] (ppm) Amplitudo
(f)
y = 0.000145x + 0.000271 r = 0.9999
0 0.002 0.004 0.006 0.008
0 10 20 30 40 50
Lampiran 3 Parameter statistika kurva standar rerata metode SDUV
xi (xi- x)2 xi 2
yi ŷi (yi - ŷi) 2
10 400 100 0.00169 0.00171 4.00x10-10
20 100 400 0.00313 0.00313 0.00
30 0 900 0.00458 0.00455 9.00x10-10
40 100 1600 0.00595 0.00597 4.00x10-10
50 400 2500 0.00736 0.00739 9.00x10-10
Keterangan: xi= konsentrasi standar; yi = amplitudo yang terukur; ŷi = amplitudo teoritis
Persamaan regresi linear rerata: y = (1.42x + 2.94) × 10-4 Σxi2 = 5500
Σ (yi - ŷi) 2
= 2.60 × 10-9 Σ (xi- x)
2 = 1000
Simpangan baku regresi (Sr) =
2 1
2 ) (
− ∑
= −
n n
i yi yi )
= 2.94 × 10-5
Simpangan baku intersep (Sa) =
∑ −
∑
i(xi x) n
i xi r
S 2
2
= 3.08 × 10-5
Simpangan baku kemiringan (Sb) =
∑ −
i(xi x) r S
2
= 9.30 × 10-7
Selang kepercayaan intersep untuk α = 0.05 dan derajat bebas = 7 (ttabel = 2.36) yaitu
a ± tSa = 1.42 × 10-4 ± (2.36)(3.08 × 10-5)
= 1.42 × 10-4 ± 7.27 × 10-5
Selang kepercayaan kemiringan garis untuk α = 0.05 dan derajat bebas = 7 (ttabel = 2.36) yaitu
b ± tSb = 2.94 × 10-4 ± (2.36)(9.30 × 10-7)
Lampiran 4 Penentuan limit deteksi dan limit kuantitasi metode SDUV
LD = 3,3
b Sa
LK = 10
b Sa
=
0.000142 5 10 x 3.08 x
3.3 −
=
0.000142 5 10 x 3.08 x
10 −
= 0.7158 g/ml = 2.1690 g/ml
Keterangan:
LD = limit deteksi ( g/ml) LK = limit kuantitasi ( g/ml)
Sa = simpangan baku intersep kurva standar (n = 6)
Lampiran 5 Penentuan ketelitian intraday metode SDUV hari ke-1 hingga hari ke-5
Hari ke-1
Ulangan Bobot Obat (g) Amplitudo Kadar dalam
25 ml ( g/ml)
Kadar dalam 1 tablet (mg)
Contoh perhitungan ulangan 1:
y = (1.42x + 2.94) × 10-4
0.00393 = (1.42x + 2.94) × 10-4
x = 26.2394 g/ml
Kadar reserpin dalam 25 tablet obat
lanjutan Lampiran 5
Hari ke-2
Ulangan Bobot Obat (g) Amplitudo Kadar dalam
25 ml ( g/ml)
Kadar dalam 1 tablet (mg)
1 0.5006 0.00390 25.3944 0.2540
2 0.5006 0.00390 25.3944 0.2540
3 0.5002 0.00397 25.8873 0.2592
4 0.5001 0.00396 25.8169 0.2585
5 0.5005 0.00395 25.7465 0.2576
6 0.5008 0.00392 25.5352 0.2554
x = 0.2565
s = 0.0023
SBR = 0.89%
Batas galat (95%) = 0.0024
Hari ke-3
Ulangan Bobot Obat
(g) Amplitudo
Kadar dalam 25 ml ( g/ml)
Kadar dalam 1 tablet (mg)
1 0.5004 0.00398 25.9577 0.2586
2 0.5004 0.00397 25.8873 0.2579
3 0.5002 0.00401 26.1690 0.2608
4 0.5003 0.00393 25.6056 0.2552
5 0.5006 0.00392 25.5352 0.2543
6 0.5009 0.00402 26.2394 0.2612
x = 0.2580
s = 0.0028
SBR = 1.10%
Batas galat (95%) = 0.0030
Hari ke-4
Ulangan Bobot Obat (g) Amplitudo Kadar dalam
25 ml ( g/ml)
Kadar dalam 1 tablet (mg)
1 0.5007 0.00398 25.9577 0.2593
2 0.5003 0.00404 26.3803 0.2637
3 0.5004 0.00401 26.1690 0.2615
4 0.5007 0.00406 26.5211 0.2649
5 0.5005 0.00409 26.7324 0.2671
6 0.5005 0.00402 26.2394 0.2622
x = 0.2631
s = 0.0027
SBR = 1.04%
lanjutan Lampiran 5
Hari ke-5
Ulangan Bobot Obat (g) Amplitudo Kadar dalam
25 ml ( g/ml)
Kadar dalam 1 tablet (mg)
1 0.5003 0.00392 25.5352 0.2552
2 0.5003 0.00392 25.5352 0.2552
3 0.5007 0.00389 25.3239 0.2529
4 0.5002 0.00392 25.5352 0.2552
5 0.5002 0.00386 25.1127 0.2510
6 0.5009 0.00386 25.1127 0.2507
x = 0.2534
s = 0.0022
SBR = 0.85%
Batas galat (95%) = 0.0023
Lampiran 6 Penentuan ketelitian interday metode SDUV
Hari Kadar reserpin
tiap tablet (mg) SBR (%) 1 0.2628 1.32% 2 0.2565 0.89% 3 0.2580 1.10% 4 0.2631 1.04% 5 0.2534 0.85%
x = 0.2588
s = 0.0042
SBR = 1.61%
Lampiran 7 Penentuan perolehan kembali metode SDUV
Standar Reserpin (mg) Perolehan
kembali (%) Rerata (%) Batas Galat SBR (%)
Ditambahkan Terukur
1.0000 1.5804 108.04
108.75 0.66
1.0000 1.5946 109.46 1.77
1.0000 1.5875 108.75
1.5000 2.1161 107.74
107.82 0.13
1.5000 2.1161 107.74 0.34
1.5000 2.1196 107.98
1.7500 2.3839 107.65
107.72 0.11
1.7500 2.3875 107.86 0.29
1.7500 2.3839 107.65
Contoh perhitungan untuk jumlah standar yang ditambahkan = 1.00 mg (ulangan 1):
Perolehan kembali (%) =
c b a−
× 100%
=
mg 1.0000
mg 0.5000) (1.5804−
× 100%
= 108.04%
Keterangan:
a = konsentrasi contoh + konsentrasi standar yang terukur
b = konsentrasi contoh
c = konsentrasi standar teoritis yang ditambahkan
Simpangan baku (SB) =
(
)
1 2 1
− − = ∑
n x i x n
i = 0.0071 mg
Simpangan baku relatif (%) =
x SB
× 100%
=
mg 108.75
mg 0.71
× 100% = 0.66%
Batas galat =
n SB t ×
=
3 mg 0.71 4.3030
×
Lampiran 8 Penentuam kadar reserpin dengan metode British Pharmacopoeia(1993)
Serapan standar reserpin pada λ = 389 nm [Standar Reserpin]
( g/ml) Absorbans
10 0.199
20 0.365
30 0.527
40 0.684
50 0.843
y = 0.01604x + 0.0374 r = 0.9999
0 0.2 0.4 0.6 0.8 1
0 20 40 60
[Reserpin] (ppm) Absorbans
Kurva sandar reserpin metode British Pharmacopoeia(1993)
Serapan contoh obat pada λ = 389 nm
Ulangan Bobot obat (g) Absorbans Kadar dalam 25 ml larutan
Kadar dalam 1 obat (mg)
1 0.5008 0.414 23.4788 0.2347
2 0.5007 0.416 23.6035 0.2360
3 0.5004 0.426 24.2269 0.2424
4 0.5007 0.425 24.1646 0.2417
5 0.5007 0.413 23.4165 0.2342
6 0.5004 0.416 23.6035 0.2362
x = 0.2375
s = 0.0036
Batas galat (95%) = 0.0025
SBR = 1.51%
Contoh perhitungan ulangan 1: y = 0.01604x + 0.0374
0.460 = 0.01604x + 0.0374
x = 25.9938 g/ml
Kadar reserpin dalam 25 tablet obat
=
obat g 0.5 bobot
tablet 25 bobot larutan volume obat
g 0.5 dalam
kadar
×
×
=
g 0.5008
g 5.0071 ml
0.025 ppm
23.4788
×
×
lanjutan Lampiran 8
Kadar reserpin dalam 1 tablet =
25
tablet 25 dalam reserpin kadar
= 25
mg 5.8686
= 0.2347 mg
Simpangan baku =
(
)
1 2 1
− − = ∑
n x i x n
i = 0.0036 mg
Simpangan baku relatif (%) =
x s
× 100%
=
mg 0.2375
mg 0.0036
× 100% = 1.51%
Batas galat =
n s t ×
=
6 mg 0.0036 2.781×
Lampiran 9 Uji statistika metode SDUV dan metode British Pharmacopoeia (1993)
RESERPIN DALAM TABLET OBAT
NIKEN WULANDARI
DEPATEMEN KIMIA
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM
INSTITUT PERTANIAN BOGOR
untuk menjamin mutu dan keamanannya. Salah satu jenis pengawasan mutu tersebut adalah analisis kadar senyawa aktif dalam proses pengendalian mutu obat. Penentuan kadar senyawa aktif memerlukan suatu metode analisis dengan ketelitian dan ketepatan yang cukup baik. Selain itu juga memenuhi kriteria lain seperti spesifisitas, linearitas, limit deteksi, limit kuantitasi, dan ketangguhan (robustness).
Metode spektrofotometri derivatif ultra-violet (SDUV) telah dikembangkan oleh Rachmanti (2006) untuk penentuan kadar reserpin dalam tablet obat hipertensi yang mengandung reserpin. Teknik ini dapat menjadi alternatif dalam pengawasan mutu produk obat hipertensi dengan senyawa aktif reserpin karena berbagai keuntungan yang dimilikinya seperti cepat, mudah, murah, dan tanpa adanya tahap pemisahan. Metode analisis penentuan reserpin dapat digunakan untuk analisis rutin jika telah tervalidasi.
Validasi metode analisis adalah suatu proses penilaian terhadap metode analisis tertentu berdasarkan percobaan laboratorium untuk membuktikan bahwa metode tersebut memenuhi persyaratan untuk digunakan (Harmita 2004). Proses ini bukan suatu proses tunggal, namun merupakan salah satu bagian dari prosedur analisis yang tidak dapat dipisahkan (Ermer & Miller 2005). Validasi metode dilakukan untuk metode yang baru dikembangkan, atau jika metode tersebut akan digunakan untuk kegiatan yang bersifat rutin, jika terjadi perubahan antara kondisi analisis dan kondisi pada saat validasi metode, dan jika terjadi perubahan metode dari metode standar.
Penentuan kadar reserpin dengan SDUV merupakan suatu metode baru yang dikembangkan untuk contoh tablet obat dengan matriks yang kompleks. Validasi metode SDUV dapat digunakan untuk mengetahui keabsahan data yang dihasilkan. Menurut ICH (1995), metode analisis dapat memberikan data yang dipercaya jika memenuhi beberapa parameter yang telah disyaratkan, yaitu spesifisitas, ketelitian (presisi), ketepatan (akurasi), linearitas, kisaran, limit deteksi, limit kuantitasi, dan ketangguhan. Untuk penentuan kadar senyawa aktif dalam produk obat, parameter ketang-guhan tidak perlu ditentukan, sedangkan parameter yang akan ditentukan dalam penelitian ini hanya meliputi linearitas, limit
SDUV untuk penentuan kadar reserpin dalam tablet obat Serpasil®.
TINJAUAN PUSTAKA
Reserpin
Reserpin (C33H40N2O9) (Gambar 1) meru-pakan salah satu jenis alkaloid indol yang digunakan untuk pengobatan penyakit hiper-tensi, menurunkan denyut jantung, terapi bagi penderita skizofrenia, dan obat penenang (Forney 2007). Reserpin dapat diperoleh dari ekstrak tumbuhan genus Rauwolfia, terutama spesies R. serpentina dan R. vomitoria. Sumber lain dari obat ini adalah berbagai
spesies tanaman dari genus Alstonia,
Excatavia, Ochrosia, Tanduzia, Vallesia, dan
Vinca (Apocynaceae), namun kadarnya sangat rendah (Hesse 2002).
N
Beberapa sifat fisik dan kimia dari reserpin adalah berwujud padatan (bubuk kristal) yang berwarna putih, tidak berbau dan menjadi berwarna kehitaman jika dibiarkan di udara terbuka. Reserpin tidak larut dalam air dan eter, sukar larut dalam etanol dingin, serta larut dalam kloroform dan asam asetat. (British Pharmacopoeia 1993).
(nm)
Spektrofotometri Derivatif Ultraviolet (SDUV)
Teknik SDUV telah diperkenalkan pada
tahun 1953 (Popovic et al. 2000) dan
mengalami perkembangan cukup pesat selama 25 tahun terakhir serta digunakan secara luas sebagai alat untuk analisis kuantitatif, pencirian, dan kendali mutu di bidang pertanian, farmasi, dan biomedis (Kazemipour
et al. 2002). Metode SDUV merupakan metode paling umum untuk penentuan secara serentak campuran biner suatu senyawa dengan spektrum yang bertumpang tindih (El Sayed & El Salem 2004).
Metode SDUV didasarkan pada spektrum turunan (derivatif) ke-n yang diperoleh dari spektrum serapan normal UV-Vis (ultraviolet dan sinar tampak) atau spektrum turunan ke-0 (Karpinska 2004). Spektrum derivatif diper-oleh dengan menggunakan persamaan mate-matika. Keuntungan dari cara ini adalah spektrum derivatif dapat dihitung dengan parameter yang berbeda dan teknik peng-halusan dapat digunakan untuk meningkatkan nisbah sinyal terhadap derau (noise) (Ojeda & Rojas 2004).
Menurut O’Haver (1979), spektro-fotometri derivatif merupakan pengukuran kemiringan garis spektrum, yaitu rerata perubahan absorbans terhadap panjang gelombang. Spektrofotometri derivatif tetap mempertahankan semua hukum (persamaan) spektrofotometri konvensional (Karpinska 2004). Hukum Beer pada spektrum normal, A = εbc, pada bentuk derivatif akan menjadi
Dn = n
panjang gelombang λ. Spektrum normal
diperoleh dari plot hubungan antara nilai absorbans (A) dan nilai panjang gelombang (λ), sedangkan spektrum turunan pertama merupakan plot hubungan antara dA/dλ dan nilai λ, nilai plot spektrum turunan pertama digunakan untuk menentukan d2A/dλ2 yang apabila diplot terhadap λ maka akan meng-hasilkan plot spektrum turunan kedua. Untuk
memperoleh spektrum turunan ke-n maka
dibuat plot hubungan antara dnA/dλn dan nilai
λ (Gambar 2) (Skujins 1986). Penerapan
hukum Lambert-Beer ini berdasarkan pada pemilihan kondisi optimum, termasuk pemilihan pita spektrum yang paling sesuai, orde derivatif yang cocok, serta metode pengukuran dan optimalisasi semua parameter instrumen yang penting (Popovic et al. 2000).
Gambar 2 Spektrum serapan normal dan turunannya (O’Haver 1979).
Analisis kuantitatif pada metode SDUV dilakukan dengan cara mengukur amplitudo spektrum derivatif. Jika konsentrasi contoh pada spektrum normal sebanding dengan absorbans, maka konsentrasi contoh pada spektrum derivatif sebanding dengan amplitudo. Metode pengukuran amplitudo ada beberapa cara, yaitu dari puncak ke puncak (p1, p2), puncak ke garis dasar (z), dan puncak tangen (t) (Gambar 3) (Popovic et al. 2000).
Gambar 3 Pengukuran amplitudo pada spek-trum derivatif (Popovic
et al. 2000).
Teknik SDUV mempunyai beberapa keuntungan
yang tidak dapat diperoleh dari teknik
lain. Beberapa di antaranya adalah mempercepat waktu analisis, mengurangi biaya yang dibutuhkan dan dapat digunakan untuk pemisahan senyawa campuran dengan spektrum yang bertumpang tindih tanpa membutuhkan tahap pemisahan analit dari matriksnya (Karpinska 2004). Selain itu, pergeseran garis dasar dapat dihilangkan dan efek kekeruhan dapat dikurangi (O’Haver 1979), meningkatkan selektivitas dan sensiti-vitas analisis komponen minor, meningkatkan resolusi spektrum, serta dapat menentukan
Spektrum normal
Spektrum turunan ke-1
Spektrum turunan ke-2 Absorbans
Amplitudo
Amplitudo
Panjang gelombang (nm)
A
m
pl
Validasi Metode Analisis
Validasi metode analisis adalah suatu proses penilaian terhadap metode analisis tertentu berdasarkan percobaan laboratorium untuk membuktikan bahwa metode tersebut memenuhi persyaratan untuk digunakan (Harmita 2004). Suatu metode yang baru dikembangkan untuk kegiatan rutin harus divalidasi sebelum digunakan. Selain itu, validasi metode dilakukan jika terjadi perubahan kondisi antara kondisi analisis dan kondisi pada saat validasi metode, atau terjadi perubahan metode dari metode standar. Beberapa manfaat validasi metode analisis adalah untuk mengevaluasi unjuk kerja suatu metode analisis, menjamin prosedur analisis, menjamin keakuratan dan kedapatulangan hasil prosedur analisis, dan mengurangi risiko penyimpangan yang mungkin timbul.
Terdapat beberapa rujukan validasi metode seperti United State Pharmacopoeia (USP), British Pharmacopoeia (BP), Association of Official Analytical Chemists (AOAC), International Conference on Harmo-nization (ICH), dan International Union of Pure and Applied Chemistry (IUPAC). Penelitian ini mengacu pada petunjuk validasi metode dari ICH (1995) yang dikhususkan untuk analisis obat-obatan. Menurut ICH (1995), prosedur analisis yang harus divalidasi meliputi beberapa jenis pengujian, yaitu identifikasi suatu senyawa tertentu, kuantitasi adanya pengotor, uji limit untuk mengen-dalikan keberadaan pengotor, serta uji kuantitatif komponen aktif atau komponen lain dalam produk obat-obatan. Selain itu, terdapat 8 parameter validasi metode analisis, yaitu spesifisitas, ketelitian, ketepatan, linearitas, kisaran, limit deteksi, limit kuan-titasi, dan ketangguhan, sedangkan parameter yang harus dipenuhi untuk validasi metode analisis produk obat-obatan meliputi spesifi-sitas, linearitas, kisaran, limit deteksi, limit kuantitasi, ketelitian, dan ketepatan.
Linearitas
Linearitas menunjukkan kemampuan suatu metode analisis untuk memperoleh hasil pengujian yang sesuai dengan konsentrasi analit dalam contoh pada kisaran konsentrasi tertentu (ICH 1995). Hal ini dapat dilakukan dengan cara membuat kurva kalibrasi dari beberapa set larutan standar yang telah diketahui konsentrasinya. Persamaan garis yang digunakan pada kurva kalibrasi diperoleh dari metode kuadrat terkecil, yaitu y
= a + bx. Persamaan ini akan menghasilkan
koefisien korelasi (r). Koefisien korelasi inilah yang digunakan untuk mengetahui linearitas suatu metode analisis. Penetapan linearitas minimum menggunakan lima konsentrasi yang berbeda. Nilai koefisien korelasi yang memenuhi persyaratan adalah lebih besar dari 0.9970 (ICH 1995 diacu dalam Chan 2004).
Linearitas juga dapat diketahui dari kemiringan garis, intersep, dan residual (Ermer & Miller 2005). Residual menyatakan besarnya penyimpangan yang terjadi antara nilai yang terukur (y) dan nilai teoretis yang dihitung dari persamaan regresi (ŷ). Plot antara residual dan konsentrasi dibuat untuk mengetahui distribusi residual secara statistik. Jika residual terdistribusi secara normal (rerata mendekati nol dan berbentuk linear), maka persamaan regresi dapat dikatakan mempunyai bentuk yang benar.
Limit Deteksi dan Limit Kuantitasi
Limit deteksi (LD) merupakan jumlah atau konsentrasi terkecil analit dalam contoh yang dapat dideteksi, namun tidak perlu diukur sesuai dengan nilai sebenarnya. Limit kuantitasi (LK) adalah jumlah analit terkecil dalam contoh yang dapat ditentukan secara kuantitatif pada tingkat ketelitian dan ketepatan yang baik. Limit kuantitasi merupakan parameter pengujian kuantitatif untuk konsentrasi analit yang rendah dalam matriks yang kompleks dan digunakan untuk menentukan adanya pengotor atau degradasi produk (ICH 1995). Limit deteksi dan limit kuantitasi dihitung dari rerata kemiringan garis dan simpangan baku intersep kurva standar yang diperoleh.
Ketelitian
Pengujian ketelitian dinyatakan dengan simpangan baku relatif (SBR). Menurut ICH (1995), kriteria nilai SBR yang dapat diterima adalah kurang dari 2%.
Ketepatan
Ketepatan suatu metode analisis didefinisikan sebagai kedekatan hasil yang diterima (baik sebagai nilai teoretis maupun sebagai nilai rujukan yang diterima) dengan nilai yang diperoleh dari hasil pengukuran (ICH 1995 diacu dalam Chan 2004). Ketepatan dinyatakan sebagai perolehan kembali yang ditentukan dengan cara menambahkan sejumlah tertentu standar dari analit yang akan diukur ke dalam contoh. Perolehan kembali (%) yang dapat diterima menurut ICH adalah 98–102%. ICH juga mensyaratkan minimum 9 kali pengukuran pada 3 tingkat konsentrasi yang berbeda.
BAHAN DAN METODE
Bahan dan Alat
Bahan-bahan yang digunakan adalah tablet obat Serpasil®, standar reserpin, etanol absolut, larutan H2SO4 0.25 M, larutan natrium nitrit 0.3% (b/v), dan larutan asam sulfamat 5% (b/v).
Alat-alat yang digunakan adalah spektro-fotometer UV-Vis 2800 Hitachi beserta kuvet 1 cm, neraca analitik, seperangkat komputer, peranti lunak UV Solutions versi 2.0 dan Microsoft Excel tahun 2003, pemanas, dan alat-alat kaca.
Pengukuran Konsentrasi Reserpin dengan Metode SDUV
Parameter validasi metode SDUV yang ditentukan meliputi linearitas, ketepatan, ketelitian, limit deteksi, dan limit kuantitasi. Penentuan kadar reserpin dilakukan pada kondisi optimum yang telah ditentukan pada penelitian sebelumnya (Rahmanti 2006). Kadar reserpin yang diperoleh dengan metode SDUV dibandingkan dengan metode rujukan British Pharmacopoeia (1993) (Lampiran 1).
Pembuatan Larutan Standar dan Contoh
Sebanyak 10 mg serbuk reserpin di-masukkan ke dalam gelas piala dan ditambahkan 20 ml etanol absolut hangat kemudian diaduk dengan pengaduk magnetik selama 20 menit. Larutan ini dimasukkan ke dalam labu takar 100 ml dan ditepatkan
dengan etanol. Larutan stok standar dengan konsentrasi 100 g/l diencerkan menjadi 10, 20, 30, 40, dan 50 g/ml.
Tablet obat sebanyak 0.5 g dihaluskan dan dimasukkan ke dalam gelas piala, kemudian ditambahkan 20 ml etanol absolut hangat dan dipanaskan pada suhu 50 ºC sambil diaduk dengan pengaduk magnetik selama 20 menit. Larutan ini dimasukkan ke dalam labu takar 25 ml dan ditepatkan dengan etanol.
Pengukuran Larutan Standar dan Contoh
Pengukuran larutan standar dan contoh dilakukan pada kondisi optimum yang telah ditentukan oleh Rachmanti (2006). Larutan standar dan contoh diukur serapannya dengan spektrofotometri UV-Vis pada panjang gelombang 200–350 nm dengan kecepatan pemayaran 200 nm/menit. Spektrum yang diperoleh kemudian diolah dengan perangkat lunak UV Solutions pada kondisi optimum sebagai berikut: orde turunan 1, orde peng-halusan 2, jumlah jendela 17, dan amplitudo diukur dari puncak spektrum ke garis dasar (z) pada panjang gelombang 277.4 nm.
Validasi Metode SDUV
Linearitas
Sebanyak 5 larutan standar disiapkan dengan konsentrasi 10, 20, 30, 40, 50 g/ml. Masing-masing konsentrasi dibuat sebanyak 6 ulangan kemudian setiap larutan diukur dengan menggunakan spektrofotometer UV-Vis pada kondisi optimum dan dibuat persamaan garisnya dengan metode regresi linear (y = a + bx). Peubah a menyatakan intersep dan b adalah kemiringan garis dari kelima larutan standar yang diukur. Linearitas kurva kalibrasi dilihat dari nilai koefisien korelasi (r).
Limit Deteksi (LD) dan Limit Kuantitasi (LK)
Persamaan linear yang diperoleh pada uji linearitas selanjutnya digunakan untuk meng-hitung limit deteksi dan limit kuantitasi. LD dan LK dihitung dari rerata kemiringan garis dan simpangan baku intersep kurva standar yang diperoleh dengan rumus sebagai berikut:
Sa = simpangan baku intersep kurva standar (n = 6)
b = rerata kemiringan garis kurva standar
Ketelitian
Larutan contoh yang telah disiapkan sesuai dengan prosedur di atas kemudian diukur dengan spektrofotometer UV-Vis sebanyak 6 ulangan pada hari yang sama (intraday) dan pada hari yang berlainan (interday) selama 5 hari berturut-turut. Ketelitian diukur dengan menghitung persentase simpangan baku relatif (%SBR) data dengan menggunakan rumus
s =
(
)
SBR = simpangan baku relatif
xi = kadar reserpin tiap ulangan
x = rerata kadar reserpin
n = jumlah ulangan
Ketepatan
Larutan standar reserpin disiapkan dengan konsentrasi 100 g/ml dan larutan stok contoh dibuat dengan konsentrasi 50 g/ml. Sebanyak 10 ml contoh dipindahkan ke dalam labu takar 50 ml dan ditambahkan larutan standar reserpin masing-masing sebanyak 10, 15, dan 17.5 ml kemudian volumenya ditepatkan dengan etanol. Masing-masing larutan dibuat 3 ulangan. Perolehan kembali dihitung dengan rumus
Perolehan kembali(%) =
c b a−
× 100%
dengan:
a = konsentrasi contoh + konsentrasi standar yang terukur
b = konsentrasi contoh
c = konsentrasi standar teoretis yang ditambahkan
Pengukuran Reserpin dengan Metode British Pharmacopoeia (1993)
Serbuk obat sebanyak 0.5 g dimasukkan ke dalam gelas piala kemudian ditambahkan 1 ml etanol, 1 ml H2SO4 0.25 M, dan ditambah-kan 5 ml etanol. Larutan ini dihangatditambah-kan agar
contoh obat mudah larut. Larutan didinginkan dan dimasukkan ke dalam labu takar 25 ml dan ditera dengan etanol. Larutan ini disebut larutan A.
Larutan A sebanyak 10 ml dimasukkan ke dalam gelas piala kemudian ditambahkan 2 ml H2SO4 0.25 M dan 2 ml larutan natrium nitrit 0.3% (b/v) segar. Campuran dipanaskan pada suhu 55 ºC selama 35 menit pada penangas air kemudian didinginkan. Larutan asam sulfamat 5% (b/v) sebanyak 1 ml ditambahkan ke dalam larutan contoh kemudian larutan contoh dimasukkan ke dalam labu takar 25 ml dan ditera dengan etanol. Larutan dibuat sebanyak 6 ulangan dan absorbans diukur pada λ = 389 nm menggunakan sel rujukan yang berisi 10 ml larutan A tanpa penambahan natrium nitrit.
HASIL DAN PEMBAHASAN
Kondisi Optimum Metode SDUV
Penentuan reserpin dalam tablet obat Serpasil® dengan metode SDUV dilakukan pada kondisi optimum yang telah ditentukan oleh Rahmanti (2006). Reserpin dalam obat dapat ditentukan dengan metode SDUV karena bentuk spektrum contoh dengan standar reserpin saling berimpit (bentuknya sama). Spektrum serapan normal (turunan ke-0) dibuat pada panjang gelombang 200–350 nm, yaitu daerah panjang gelombang sinar UV. Spektrum dibuat dengan kecepatan pemayaran 200 nm/menit sehingga spektrum yang diperoleh tetap akurat setelah diturunkan.
Amplitudo
Panjang gelombang (nm)
Gambar 4 Spektrum serapan reserpin pada kondisi optimum.
Keterangan:
Cokelat = standar reserpin 10 g/ml Biru tua = standar reserpin 20 g/ml
Linearitas metode SDUV ditentukan dengan cara membuat kurva hubungan antara amplitudo spektrum derivatif pada sumbu y
dan konsentrasi standar pada sumbu x.
Konsentrasi yang digunakan berkisar 10–50 g/ml. Pengujian parameter ini dilakukan sebanyak 6 ulangan. Tabel 1 menyajikan persamaan regresi linear dari masing-masing kurva standar (Lampiran 2) dan kurva standar rerata. Linearitas dinyatakan dengan koefisien korelasi (r). Berdasarkan hasil pengujian, diperoleh koefisien korelasi dari 6 ulangan berkisar 0.9997–0.9999, dan untuk kurva standar rerata sebesar 0.9999. Menurut ICH (1995 diacu dalam Chan 2004), nilai ini memenuhi syarat yang ditetapkan, yaitu 0.9970. Nilai koefisien korelasi yang tinggi menunjukkan hubungan yang linear antara sinyal detektor yang terukur dan jumlah reserpin dalam contoh.
Tabel 1 Koefisien korelasi dan persamaan regresi linear kurva standar (n = 6)
No. Persamaan Regresi Linear r
1 y = (1.38x + 2.58) × 10-4 0.9998
Persamaan regresi linear untuk kurva standar rerata adalah y = (1.42x + 2.94) × 10-4 (Gambar 5). Nilai intersep (a) dan batas galatnya (tSa) pada selang kepercayaan 95% sebesar 2.94 × 10-4 ± 7.27 × 10-5 (Tabel 2).
Nilai intersep menyatakan adanya pengaruh matriks pada larutan. Nilai intersep yang semakin jauh dari nol dipengaruhi oleh matriks dalam larutan yang semakin besar. Hal ini dapat mengganggu penentuan analit dalam contoh yang akan ditentukan. Persamaan regresi kurva standar rerata mempunyai intersep yang tidak jauh dari nol, yaitu 2.94 × 10-4 ± 7.27 × 10-5 sehingga matriks contoh tidak terlalu mengganggu penentuan kadar reserpin.
y = 0.000142x + 0.000294 r = 0.9999
Gambar 5 Kurva standar rerata reserpin pada konsentrasi 10–50 g/ml.
Nilai kemiringan garis menyatakan sensitivitas suatu metode. Nilai kemiringan garis yang kecil menunjukkan bahwa perubahan konsentrasi yang kecil tidak terlalu berpengaruh terhadap sinyal detektor yang dihasilkan, sehingga metode mempunyai sensitivitas yang kurang baik. Nilai kemiringan garis (b) dan batas galatnya (tSb) adalah 1.42 × 10-4 ± 2.19 × 10-6 (Tabel 2). Nilai ini cukup besar sehingga perubahan konsentrasi yang kecil akan berpengaruh terhadap perubahan sinyal detektor. Contoh perhitungan parameter statistika disajikan pada Lampiran 3.
Tabel 2 Parameter statistika kurva standar rerata (n = 6)
Parameter statistika Metode SDUV Persamaan regresi linear y = (1.42x + 2.94) × 10-4
Persamaan regresi yang diperoleh dari kurva kalibrasi metode mempunyai residual yang terdistribusi secara normal, sehingga dapat dikatakan bahwa penyimpangan yang terjadi tidak terlalu besar. Hal ini menunjukkan bahwa persamaan regresi dapat digunakan untuk menentukan kadar reserpin dalam contoh. Plot antara residual dan konsentrasi standar reserpin dapat dilihat pada Gambar 6.
Gambar 6 Hubungan antara konsentrasi reserpin dan residual kurva standar rerata.
Limit Deteksi dan Limit Kuantitasi
Limit deteksi (LD) dan limit kuantitasi (LK) ditentukan dari persamaan regresi linear kurva standar rerata hasil penentuan linearitas. Parameter ini ditentukan untuk mengetahui konsentrasi terendah pada saat sinyal antara blangko dan analit dapat dibedakan. Kedua parameter ini mempunyai nilai yang berbeda bergantung pada metode yang digunakan, walaupun menggunakan instrumen yang sama. Limit deteksi metode SDUV untuk penentuan reserpin dalam tablet obat sebesar 0.7158 g/ml. Nilai ini menunjukkan bahwa sinyal antara reserpin dan blangko dapat dibedakan pada konsentrasi terendah 0.7158
g/ml. Instrumen tidak dapat membedakan sinyal antara blangko dan reserpin pada konsentrasi di bawah nilai ini.
Limit kuantitasi ditentukan untuk mengetahui konsentrasi terendah yang dapat ditentukan oleh suatu metode pada tingkat ketelitian dan ketepatan yang baik. Nilai limit kuantitasi berdasarkan hasil penelitian adalah 2.1690 g/ml. Konsentrasi analit yang terukur di bawah nilai ini memberikan ketelitian dan ketepatan yang tidak baik. Perhitungan limit deteksi dan limit kuantitasi tertera pada
Lampiran 4.
Ketelitian
Ketelitian metode SDUV ditentukan sebanyak 6 ulangan, baik intraday maupun
interday. Ketelitian yang ditentukan adalah keterulangan karena dilakukan oleh operator, instrumen, peralatan, dan laboratorium yang sama. Keterulangan dilakukan untuk mengetahui adanya galat acak yang berasal dari penyiapan larutan maupun instrumen. Keterulangan intraday dapat digunakan untuk mengetahui adanya galat acak yang berasal dari penyiapan larutan, seperti penimbangan, pembuatan larutan, dan penyaringan. Hasil penelitian menunjukkan bahwa SBR intraday
untuk masing-masing hari bernilai kurang dari 2% (Tabel 3) dengan kadar reserpin rerata dalam 1 tablet obat untuk hari ke-1 hingga hari ke-5 secara berturut-turut adalah 0.2628; 0.2565; 0.2580; 0.2631; dan 0.2534 mg. Nilai SBR di bawah 2% menunjukkan bahwa galat acak yang berasal dari penyiapan larutan tidak memengaruhi hasil analisis secara nyata. Data dan perhitungan ketelitian untuk hari ke-1 hingga hari ke-5 ditunjukkan pada Lampiran 5.
Tabel 3 Simpangan baku relatif (%SBR)
untuk intraday maupun interday
Hari ke- Reserpin (mg) SBR (%)
Keterulangan interday dilakukan selama 5 hari berturut-turut. Hal ini dilakukan untuk mengetahui adanya galat acak yang berasal dari instrumen dan kondisi laboratorium pada hari yang berlainan. Nilai SBR untuk keterulangan interday adalah 1.61% dengan kadar reserpin rerata 0.2588 ± 0.0052 mg per tablet obat (Lampiran 6), sehingga dapat dikatakan bahwa galat acak yang berasal dari instrumen dan kondisi laboratorium yang berbeda tidak memengaruhi hasil analisis. Oleh sebab itu, metode SDUV mempunyai keterulangan intraday dan interday yang baik.
Ketepatan
dapat menunjukkan adanya galat sistematik yang dapat memengaruhi metode analisis. Galat sistematik dapat menyebabkan hasil analisis menjadi lebih besar atau lebih kecil. Beberapa contoh penyebab galat sistematik di antaranya adalah galat pada saat pengambilan contoh, kurva kalibrasi yang tidak linear, serta galat yang disebabkan oleh instrumen dan peralatan kaca yang digunakan (Harvey 2000).
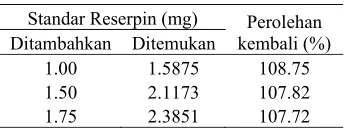
Penentuan ketepatan dilakukan dengan menambahkan standar reserpin sebanyak 1.00, 1.5, dan 1.75 mg ke dalam contoh obat yang telah berisi 0.5 mg reserpin. Perolehan kembali yang diperoleh berkisar antara 107.72 dan 108.75% (Tabel 4). Menurut ICH (1995 diacu dalam Chan 2004), nilai ini berada di luar batas nilai yang diterima, yaitu 98–102%. Namun, menurut AOAC (1993), nilai ini berada pada kisaran yang dapat diterima, yaitu 80–110%, sehingga dapat dikatakan metode ini mempunyai ketepatan yang cukup baik. Perhitungan perolehan kembali ditunjukkan pada Lampiran 7.
Tabel 4 Perolehan kembali rerata metode SDUV
Standar Reserpin (mg) Perolehan
kembali (%) Ditambahkan Ditemukan
1.00 1.5875 108.75
1.50 2.1173 107.82
1.75 2.3851 107.72
Pengukuran Kadar Reserpin dengan Metode British Pharmacopoeia (1993)
Metode British Pharmacopoeia (1993) digunakan sebagai metode rujukan untuk menentukan kadar reserpin dalam tablet obat. Metode ini menggunakan teknik spektrofoto-metri UV-Vis konvensional, yaitu pengukuran absorbans contoh pada panjang gelombang tertentu. Pengukuran dimulai dengan mem-buat kurva standar pada konsentrasi 10–50
g/ml. Persamaan regresi kurva standar yang diperoleh adalah y = 0.01604x + 0.0374 dengan nilai r = 0.9999 (Gambar 7).
Kadar reserpin rerata dalam obat dengan menggunakan metode ini adalah 0.2375 mg dengan batas galat (selang kepercayaan 95%) 0.0025 mg. Perhitungan penentuan kadar reserpin dengan metode ini dapat dilihat pada Lampiran 8.
y = 0.01604x + 0.0374
Gambar 7 Persamaan regresi linear standar reserpin menggunakan metode British Pharmacopoeia (1993).
Perbandingan Metode SDUV dan British Pharmacopoeia (1993)
Kadar reserpin hasil penentuan dengan metode SDUV selanjutnya dibandingkan dengan metode rujukan. Hal ini dilakukan dengan cara melakukan uji statistika yang meliputi uji F dan uji t. Uji F dilakukan untuk membandingkan keragaman kedua metode. Kedua metode mempunyai keragaman yang tidak berbeda nyata karena Fhitung < Ftabel pada selang kepercayaan 95% (Tabel 5). Uji t
digunakan untuk membandingkan efektivitas dan efisiensi metode baik dari segi teoretis maupun dari segi teknis (biaya). Nilai thitung kedua metode lebih besar jika dibandingkan dengan nilai ttabel pada selang kepercayaan 95% (Tabel 5), sehingga kedua metode memberikan hasil yang berbeda nyata. Metode SDUV menghasilkan nilai yang lebih tinggi jika dibandingkan dengan metode British Pharmacopoeia (1993).
Tabel 5 Uji statistika metode SDUV dan British Pharmacopoeia (1993) Parameter
statistika Metode SDUV Metode BP 1993 Kurva standar y=(1.42x + 2.94)×10-4
y=0.01604x+0.0374
r 0.9999 0.9999
Kadar reserpin 0.2588 mg 0.2375 mg Batas galat 0.0052 0.0025
F hitung 1.3611
F tabel 5.0500
t hitung 9.0704