BAB 2
TINJAUAN PUSTAKA 2.1 Defenisi Thalassemia
Thalassemia diartikan sebagai sekumpulan gangguan genetik yang
mengakibatkan berkurang atau tidak ada sama sekali sintesis satu atau lebih rantai globin (Ganie, 2004). Menurut Potts dan Mandleco, Thalassemia adalah gangguan genetik autosom resesif yang diturunkan, dengan karakteristik adanya gangguan
sintesis rantai hemoglobin. Thalassemia merupakan kelainan darah yang diturunkan dari orang tua kepada anak-anak melalui gen yang menyebabkan tubuh membuat sel darah merah sehat dan hemoglobin dalam jumlah yang lebih sedikit
daripada jumlah normal (NHLBI, 2012).
Penyakit kelainan darah ini menyebabkan sel darah merah di dalam pembuluh darah cepat hancur sehingga usia sel-sel darah merah menjadi lebih
pendek dan tubuh kekurangan darah. Jika pada orang sehat sel darah merah mampu bertahan hingga 120 hari, pada penderita thalassemia sel darah merah
hanya mampu bertahan kurang dari 120 hari (sekitar 20-30 hari) (Wijayaningsih, 2013).
Nama Thalassemia berasal dari gabungan dua kata Yunani yaitu thalassa
yang berarti lautan dan anaemia (‘weak blood”). Kata thalassa digunakan karena gangguan darah ini pertama kali ditemui pada pasien yang berasal dari
oleh seorang dokter di Detroit USA yang bernama Thomas B. Cooley pada tahun 1925 (Ganie, 2004).
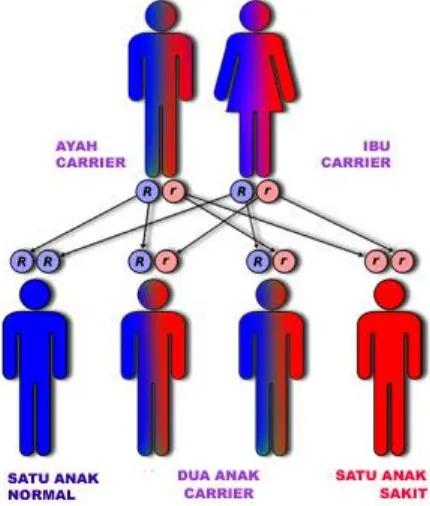
Mayoritas Thalassemia melibatkan rantai α ataupun β globin. Thalassemia diturunkan oleh orang tua yang carrier kepada anaknya. Sebagai contoh, jika ayah
dan ibu memiliki gen pembawa sifat Thalassemia (Thalassemia trait), maka kemungkinan anaknya untuk menjadi carrier Thalassemia adalah sebesar 50%, kemungkinan menjadi penderita Thalassemia mayor 25% dan kemungkinan
menjadi anak normal yang bebas Thalassemia hanya 25 % (Mambo, 2009). Keadaan tersebut dapat dilihat melalui gambar di bawah ini:
2.2 Klasifikasi Thalassemia
Thalassemia diklasifikasikan dalam dua kelompok utama sesuai rantai
globin yang terlibat, yaitu Thalassemia alfa dan Thalassemia beta (Muttaqin, 2009).
2.2.1 Thalassemia Alfa
Terdapat dua gen α globin pada tiap pasang kromosom 16. Genotip normal
α globulin digambarkan αα/αα (Permono, dkk, 2010). Kelainan ini terjadi akibat
adanya penurunan sintesis rantai alfa. Pada kebanyakan penderita di Asia dengan sindrom Thalassemia-α, defek biokimia primernya adalah berupa penghapusan
dari satu, dua, tiga, atau keempat gen globin α (Jones, 1995). Dikenal empat macam Thalassemia alfa berdasarkan banyaknya gen yang terganggu :
a. Silent Carrier (Pembawa Tersembunyi)
Merupakan delesi 1 rantai globin α. Kelainan hemoglobin sangat
minimal dan tidak memberikan gejala. Keadaan ini hanya dapat
dilihat dari pemeriksaan laboratorium secara molekuler (CAF, 2013).
b. Thalassemia Alfa Trait
Merupakan delesi 2 rantai globin α. Pada kelainan ini terjadi
anemia ringan dan eritrosit hipokromik, dapat menjadi carrier
c. Hemoglobin H Disease
Merupakan delesi 3 rantai globin α. Seseorang yang mengalami
kondisi ini akan menderita anemia sedang sampai berat, disertai dengan pembesaran limpa (CAF, 2013).
d. Thalassemia Alfa Mayor atau Hydrops Fetalis
Merupakan delesi 4 rantai globin α. Terjadi anemia yang parah dan kematian janin dalam kandungan. Selain itu, beberapa komplikasi
maternal termasuk preeklamsia, ante partum perdarahan, dll sering terjadi pada wanita hamil dengan kondisi ini (TIF, 2014). Biasanya
bayi akan meninggal dalam kandungan atau setelah dilahirkan karena kadar hemoglobin normal tidak mungkin terbentuk (CAF, 2013).
2.2.2 Thalassemia Beta
Merupakan Thalassemia yang sering terjadi, biasanya mempunyai tanda
dan gejala bervariasi. Thalassemia Beta dibagi atas : a. Thalassemia Beta Minor atau Trait
Pada jenis ini, penderita memiliki satu gen normal dan satu gen yang
bermutasi. Penderita mungkin mengalami anemia ringan yang ditandai dengan sel darah merah yang mengecil (mikrositer) (Kiswari, 2014).
b. Thalassemia Intermedia
Pada kondisi ini, kedua gen mengalami mutasi tetapi masih bisa
memproduksi sedikit rantai beta globin. Penderita biasanya mengalami anemia yang derajatnya tergantung dari mutasi gen yang terjadi
(Kiswari, 2014).
c. Thalassemia Mayor (Cooley‟s Anemia)
Pada kondisi ini, kedua gen mengalami mutasi sehingga tidak dapat
memproduksi rantai beta globin. Biasanya gejalanya muncul pada bayi ketika berumur 3 bulan berupa anemia yang berat (Kiswari, 2014).
Penderita Thalassemia mayor memerlukan transfusi darah yang rutin yang perawatan medis demi kelangsungan hidupnya. Jika dilakukan transfusi darah yang terus menerus akan terjadi penumpukan zat besi
yang berisiko terhadap kegagalan fungsi jantung, ginjal, hati, gonad atau disebut hemokromatosis. Pada Thalassemia mayor mempunyai
ciri anemia yang khas (CAF, 2013) diantaranya :
a.1 Pucat, anemia, kurus, hepatosplenomegali, dan icterus ringan, mulai nampak pada bayi berumur 3-6 bulan.
a.2 Pertumbuhan lambat (kerdil)
a.3 Hidung pesek tanpa pangkal hidung, jarak antara kedua mata lebar
dan tulang dahi lebar.
a.4 Kulit pucat kekuning-kuningan, jika sering dilakukan transfusi warna kulit menjadi kelabu karena penimbunan besi pada jaringan
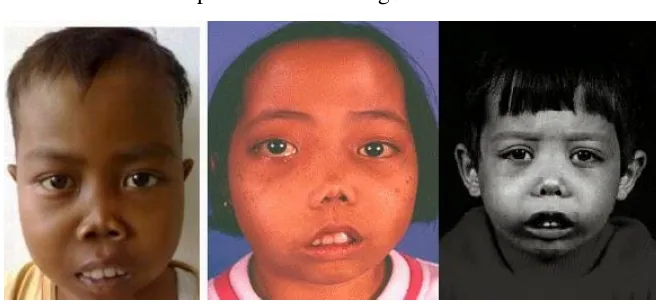
Keadaan tersebut dapat dilihat melalui gambar di bawah ini:
Gambar 2.2 KondisiAnak yang Menderita Thalassemia (dentosca.wordpress.com)
2.3 Patogenesis Thalassemia
Hemoglobin dewasa atau HbA mengandung dua rantai α dan dua rantai β, ditandai oleh dua gen globin β yang bertempat pada masing-masing dari dua
kromosom nomor 11. Sebaliknya, dua pasang gen α globin yang fungsional
berada pada setiap kromosom nomor 16 (Robbins, 1995).
Pada pasien dengan Thalassemia terjadi penurunan sintesis rantai globin
(alfa dan beta) sehingga menyebabkan anemia karena hemoglobinisasi eritrosit yang tidak efektif. Eritrosit yang normalnya dapat hidup sampai dengan 120 hari,
menjadi mudah rusak dan umur sel darah merah menjadi kurang dari 100 hari. Pasien dengan Thalassemia alfa disebabkan karena penurunan sintesis globin α. Setiap orang normal dewasa mempunyai 4 kopi rantai hemoglobin,
alfa, maka disebut sebagai pembawa yang tersembunyi (Silent Carrier), jika hanya terdapat 2 gen globin alfa disebut Trait Thalassemia Alfa (Thalassemia
Minor), jika hanya terdapat 1 gen globin alfa dinyatakan mempunyai penyakit hemoglobin H, dan jika tidak memiliki sama sekali gen globin alfa maka dapat
berakibat fatal pada bayi, yang dapat menyebabkan kematian.
Thalassemia beta terjadi akibat penurunan atau tidak adanya rantai globin β, hal ini disebabkan karena adanya mutasi. Penurunan rantai beta menyebabkan
rantai alfa tidak stabil sehingga berakibat pada kerusakan membrane eritrosit. Eritrosit mudah rusak sebelum waktunya sehingga dapat menyebabkan anemia
berat. Di sisi lain pemecahan hemoglobin akan menghasilkan zat besi yang kemudian akan terjadi penimbunan pada hati, kulit, dan limpa dan pada jangka waktu yang lama menimbulkan komplikasi yaitu kegagalan fungsi organ seperti
hati, endokrin, dan jantung (Tartowo, dkk, 2008).
2.4 Gambaran Klinis Thalassemia
Kurangnya oksigen dalam aliran darah menyebabkan tanda-tanda dan gejala Thalassemia. Kekurangan oksigen terjadi karena tubuh tidak membuat seldarah merah dan hemoglobin yang sehat dalam jumlah yang cukup. Tingkat
keparahan gejala tergantung pada tingkat keparahan kelainan yang terjadi.
Rantai α terdapat pada Hb F (fetal haemoglobin) dan Hb A (adult
globin hanya dalam jumlah yang sangat kecil, sehingga hemoglobin dalam darah masih dapat bekerja secara normal.
Penderita Thalassemia alfa trait dan Thalassemia beta trait dapat mengalami anemia ringan. Namun, sebagian besar dari penderita Thalassemia
jenis ini tidak memiliki tanda-tanda atau gejala. Anemia ringan dapat membuat penderitanya mudah merasa lelah.
Hampir semua anak dengan Thalassemia β homozigot dan heterozigot
memperlihatkan gejala klinis sejak lahir, seperti gagal tumbuh, kesulitan makan, infeksi berulang, dan badan yang lemah. Bayi nampak lebih pucat dan didapatkan
splenomegaly (Permono, dkk, 2010).
Seseorang yang menderita Thalassemia beta intermedia mengalami anemia ringan sampai sedang. Penderita Thalassemia jenis ini juga mungkin memiliki
masalah kesehatan lainnya, seperti:
a. Pertumbuhan dan perkembangan yang terhambat. Anemia dapat
memperlambat pertumbuhan dan perkembangan anak.
b. Permasalahan pada tulang. Thalassemia dapat menyebabkan permasalahan pada perkembangan sumsum tulang. Sumsum tulang adalah zat spons
dalam tulang yang berfungsi untuk membuat sel-sel darah. Ketika sumsum tulang mengembang, ukuran tulang menjadi lebih luas dari biasanya. Hal
tersebut memungkinkan tulang menjadi rapuh dan mudah patah.
c. Pembesaran limpa. Limpa adalah organ yang membantu tubuh melawan infeksi dan menghilangkan materi yang tidak diinginkan. Ketika seseorang
ukuran limpa menjadi lebih besar dari biasanya. Hal ini menyebabkan anemia berat. Jika ukuran limpa menjadi terlalu besar, maka harus
dilakukan tindakan operasi pengangkatan limpa tersebut.
Penderita hemoglobin H disease dan Thalassemia beta mayor
(Cooley‟s Anemia) dapat mengalami anemia dengan tingkat yang berat. Tanda dan gejala biasanya terjadi dalam 2 tahun pertama kehidupan. Penderita akan mengalami anemia berat dan masalah kesehatan lainnya,
seperti: a. Wajah pucat
b. Lemas
c. nafsu makan menurun
d. Urin berwarna lebih pekat (tanda bahwa sel-sel darah merah yang rusak)
e. Pertumbuhan dan perkembangan yang terhambat f. Warna kekuningan pada kulit atau putih mata
g. Pembesaran limpa, hati, atau jantung
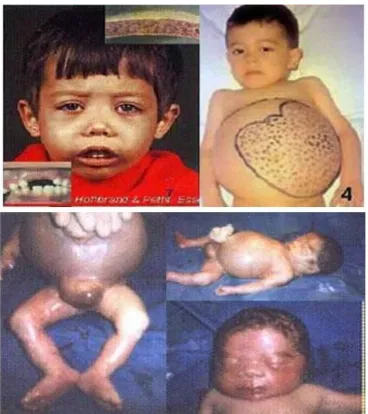
h. Masalah tulang (terutama dengan tulang di wajah) (NHLBI, 2012). Keadaan tersebut dapat dilihat melalui gambar di bawah ini:
2.5 Komplikasi Thalassemia
Pengobatan yang semakin maju sekarang ini memungkinkan para
penderita Thalassemia untuk hidup lebih lama lagi. Namun, pengobatan dan perawatan tersebut juga mengakibatkan efek samping yang membuat para
penderita Thalassemia mengalami komplikasi. Komplikasi yang dialami penderita Thalassemia tersebut yakni :
a. Jantung dan Liver
Transfusi darah secara teratur merupakan perawatan standar untuk penderita Thalassemia. Namun, transfusi darah tersebut dapat
menyebabkan peningkatan jumlah zat besi dalam darah.. Hal ini dapat merusak organ dan jaringan, terutama jantung dan hati. Penyakit jantung yang disebabkan oleh kelebihan zat besi adalah
penyebab utama kematian pada seseorang yang menderita Thalassemia. Penyakit jantung tersebut diantaranya gagal jantung, aritmia (detak
jantung tidak teratur), dan serangan jantung (NHLBI, 2012). b. Infeksi
Di antara para penderitaThalassemia, infeksi merupakan penyebab
utama penyakit dan penyebab paling umum kedua kematian dari para penderita Thalassemia. Para penderita Thalassemia yang telah
Infeksi dapat terjadi karena berbagai alasan. Pada usia bayi, tanpa transfusi adekuat, anak dengan anemia rentan terhadap infeksi bakteri.
Infeksi pneumokokus, Haemophilus, dan meningokokus mungkin terjadi jika sudah dilakukan splenektomi dan penisilin profilaktik tidak
diberikan.Transfusi virus melalui transfusi darah dapat terjadi. Penyakit hati pada Thalassemia tersering disebabkan oleh hepatitis C, tetapi hepatitis B juga sering ditemukan. Virus imunodefisiensi
manusia (HIV) telah ditularkan kepada beberapa pasien melalui transfusi darah (Hoffbrand & Moss, 2013).
c. Osteoporosis
Banyak dari para penderita Thalassemia yang memiliki masalah pada tulang, termasuk osteoporosis. Osteoporosis merupakan suatu
kondisi dimana tulang lemah, rapuh dan mudah patah (NHLBI, 2012).
2.6 Epidemiologi Thalassemia
2.6.1 Distribusi dan Frekuensi Thalassemia Berdasarkan Orang
Berdasarkan penelitian Dwi Sarwani S.S., dkk, di Yayasan Talasemia Indonesia Cabang Banyumas tahun 2012, terdapat 51,6% penderita Thalassemia
berjenis kelamin laki-laki dan 48,4% berjeniskelamin perempuan. Menurut penelitian Anggraini di Rumah Sakit Hasan Sadikin Bandung melaporkan bahwa
penderita Thalassemia berjenis kelamin laki-laki sebanyak 32 orang dan berjenis kelamin perempuan sebanyak 25 orang (Rejeki, dkk, 2012).
Pada penelitian yang dilakukan Sandra Bulan yang melibatkan 55
dr. Kariadi Semarang, menunjukkan bahwa sebagian besar penderita Thalassemiabeta mayor yang menjadi subyek penelitian berjenis kelamin wanita
30 (54,5%). Rerata umur subyek penelitian 9,8 ± 3,40 tahun. Sebaran umur merata di semua kelompok umur. Rerata umur subyek penelitian saat didiagnosis
menderita Thalassemia beta mayor adalah 2,7±2,47 tahun. Umur pertama didiagnosa sebagian besar berumur 0-1 tahun sebesar 29 (52,7%) (Bulan, 2009). Proporsi penderita Thalassemia Beta tertinggi adalah pada kelompok umur ≤ 15
tahun sebesar 84,5% (Lazuana, 2014).
Menurut penelitian Humris-Pleyte tahun 2001 di Rumah Sakit Cipto
Mangunkusuomo Jakarta, ditemukan bahwa dari 192 kasus Thalassemia yang diteliti terdapat sebanyak 59,4% kasus diagnosanya sudah dapat ditegakkan sebelum anak berusia 1 tahun, 33,3% kasus pada saat anak berusia 1-2 tahun, dan
7,3% kasus diagnosisnya ditegakkan sebelum anak berusia 2 tahun (Suprianto, 2007). Menurut data di Unit Thalassemia RSCM, berdasarkan jumlah total pasien
Thalassemia yang menjalani terapi di RSCM tahun 2011 terdapat 719 penderita (47,93%) berada pada rentang usia 6-15 tahun dan mendapatkan transfusi satu kali setiap bulan (Rahayu, 2012).
2.6.2 Distribusi dan Frekuensi Thalassemia Berdasarkan Tempat
Penyakit Thalassemia tersebar luas di daerah Mediterania seperti Italia,
Yunani, Afrika bagian utara, kawasan Timur Tengah, India Selatan, Sri Lanka sampai kawasan Asia Tenggara termasuk Indonesia, daerah ini dikenal sebagai kawasan Thalassemia. Frekuensi Thalassemia di Asia Tenggara antara 3-9%
menjadikan Thalassemia menyebar luas di seluruh belahan dunia, termasuk Eropa Utara, dimana Thalassemia yang sebelumnya tidak ditemukan hingga menjadi
masalah kesehatan serius bagi penduduknya (Eleftheriou, 2007).
Thalassemia Alfa dijumpai dalam jumlah yang besar di Asia Tenggara
(Thailand, Semenanjung Melayu, dan Indonesia), daerah Mediterania, Timur Tengah dan Afrika Barat. Thalassemia beta mempunyai distribusi yang luas di dunia ini. Sering dijumpai di daerah sekitar Mediterania dan beberapa bagian dari
Timur Tengah, India, Pakistan, dan Asia Tenggara. Di daerah-daerah ini frekuensi pembawa gen Thalassemia beta bervariasi antara 2 dan 30% (Jones, 1995).
Secara umum, prevalensi penyakit keturunan di Kalimantan Timur adalah 8,3%, untuk penyakit Thalassemia sendiri sebesar 0,2% (Balitbangkes Depkes RI, 2009). Menurut data dari RS Hasan Sadikin Bandung, jumlah penderita
Thalassemia di seluruh Jawa Barat mencapai sekitar 2000 orang (RSHS Bandung, 2014).Menurut penelitian Ratna Akbari Ganie tahun 2004, diketahui bahwa
populasi di kota Medan mempunyai prevalensi carrier Thalassemia 7,69%, dimana prevalensi carrier Thalassemia α sebesar 3,35%, selebihnya adalah
carrier Thalassemia β dan Hb-E (Ganie, 2004).
2.6.3 Distribusi dan Frekuensi Thalassemia Berdasarkan Waktu
Lie-Injo Luan Eng dan Yo Kian Tjai tahun 1955 melaporkan adanya
300 penderita Thalassemia.Manurung (1978) dari bagian Ilmu Kesehatan Anak FK Universitas Sumatera Utara Medan telah melaporkan 13 kasus (Ganie, 2005).
Data yang diperoleh dari Perhimpunan Yayasan Talasemia Indonesia menunjukkan bahwa hingga Juni 2008, di RSCM telah merawat 1.433 pasien.
Sejak 2006 sampai 2008 rata-rata pasien baru Thalassemia meningkat sekitar 8%.Data sampai bulan Juli 2011 tercatat 1.500 pasien di Unit Thalassemia RSCM. Data dari klinik Thalassemia menyatakan, di RS Hasan Sadikin Bandung, pada
2013 tercatat 600-700 penderita thalassemia yang menjalani transfusi darah, dan sekira 450 dari pasien tersebut adalah anak ( RSHS Bandung, 2014).
Jumlah penderita Thalassemia di Yayasan Talasemia Indonesia cabang Banyumas terus meningkat, pada tahun 2008 terdapat 44 penderita, pada tahun 2009 meningkat 32,3% menjadi 65 penderita. Pada tahun 2010, penderita
Thalassemia meningkat lagi 53,85% menjadi 100 penderita dan tahun 2011 meningkat menjadi 63% (Rejeki, dkk, 2012).
2.6.4 Determinan Thalassemia Genetik
Thalassemia merupakan penyakit yang diturunkan dari orang tua
kepada anak-anak melalui gen. Thalassaemia adalah gangguan gen tunggal yang menurun dari orang tua kepada anaknya secara autosomal resesif.
Penyakit 'autosomal' dapat menyerang laki-laki maupun perempuan. 'Resesif' berarti bahwa anak dapat memperoleh kelainan gen dari ayah maupun ibunya, yang apabila diturunkan dari keduanya dapat berakibat
Penyakit ini diturunkan melalui gen yang disebut sebagai gen alfa globin dan gen beta globin yang terletak pada kromosom 16 dan
kromosom 11. Pada manusia, kromosom selalu ditemukan berpasangan. Kelainan sebelah gen globin disebut carrier Thalassemia. Seorang carrier
Thalassemia tampak sehat, sebab masih ada sebelah gen globin yang normal dan dapat berfungsi dengan baik. Seorang carrier Thalassemia biasanya tidak memerlukan pengobatan. Kelainan gen globin yang terjadi
pada kedua kromosom disebut Thalassemia mayor (homozigot). Kedua belah gen yang mengalami kelainan berasal dari kedua orang tua yang
masing-masing carrier Thalassemia.
Pada proses pembuahan, anak hanya mendapat sebelah gen globin dari ibunya dan sebelah lagi dari ayahnya. Bila kedua orang tuanya
masing-masing carrier Thalasemia, maka pada setiap pembuahan akan terdapat beberapa kemungkinan. Kemungkinan pertama, anak
mendapatkan gen globin yang berubah (gen Thalassemia) dari ayah dan ibunya, sehingga anak akan menderita Thalassemia. Sedangkan bila anak hanya mendapat sebelah gen Thalassemia dari ibu atau ayahnya, maka
anak akan menjadi carrier Thalassemia. Kemungkinan lainnya adalah anak mendapatkan gen globin normal dari kedua orang tuanya, sehingga
2.7 Pencegahan Thalassemia 2.7.1 Pencegahan Primer
a. Edukasi
Edukasi masyarakat tentang Thalassemia memegang peranan yang
sangat penting dalam program pencegahan. Masyarakat harus diberi pengetahuan tentang penyakit yang bersifat genetik dan diturunkan, terutama tentang Thalassemia yang frekuensi carriernya cukup tinggi di
masyarakat.
Pendidikan genetika harus diajarkan di sekolah, demikian pula
pengetahuan tentang gejala awal Thalassemia. Media massa harus dapat berperan lebih aktif dalam menyebarluaskan informasi tentang Thalassemia, meliputi gejala awal, cara penyakit diturunkan, dan cara
pencegahannya (HTA Indonesia, 2010). b. Skrining carrier
Skrining Thalassemia ditujukan untuk menjaring individu carrier Thalassemia pada suatu populasi, idealnya dilakukan sebelum memiliki anak. Skrining ini bertujuan untuk mengidentifikasi individu dan pasangan
carrier, dan menginformasikan kemungkinan mendapat anak dengan
Thalassemia dan pilihan yang dapat dilakukan untuk menghindarinya.
Skrining dapat dilakukan di sekolah, klinik dokter keluarga, klinik keluarga berencana, klinik antenatal, saat pranikah, atau pada saat bayi baru lahir. Pendekatan genetik klasik dalam mendeteksi carrier
dibanding dengan skrining populasi. Bila ada individu yang teridentifikasi sebagai carrier, maka skrining pada anggota keluarga yang lain dapat
dilakukan (HTA Indonesia, 2010). c. Konseling Genetik Pra-Nikah
Konseling genetik pra-nikah ditujukan untuk pasangan pra-nikah terutama pada populasi yang berisiko tinggi agar memeriksakan diri apakah mereka mengemban sifat genetik tersebut atau tidak. Konseling ini
juga ditujukan kepada mereka yang memiliki kerabat penderita Thalassemia.
Tujuan utama dari konseling pra-nikah adalah untuk mencegah terjadinya perkawinan antar carrier. Hal ini mengingat bahwa mereka berpeluang 25% untuk mendapatkan anak dengan Thalassemia mayor. Jika
pasangan antar carrier tetap memutuskan untuk menikah, mereka dianjurkan untuk tidak mempunyai anak atau melakukan pre-natal
diagnosis pada awal kehamilan (Ganie, 2004). d. Pre-Natal Diagnosis
Tujuan dari pre-natal diagnosis adalah untuk mengetahui sedini
mungkin apakah janin yang dikandung menderita Thalassemia atau tidak. Diagnosis pre-natal pada Thalassemia dapat dilakukan pada usia 8-10
2.7.2 Pencegahan Sekunder a. Transfusi Darah (NHLBI, 2012)
Transfusi darah adalah perawatan utama bagi orang-orang yang menderita Thalassemia. Perawatan ini bertujuan untuk memberikan sel-sel darah merah
yang sehat bagi penderita. Penderita beta Thalassemia mayor (anemia Cooley) membutuhkan transfusi darah secara teratur (setiap 2 minggu sekali ataupun 1 bulan sekali). Transfusi ini membantu para penderita Thalassemia untuk
mempertahankan hemoglobin normal dan kadar sel darah merah. Transfusi darah membuat para penderita Thalassemia merasa lebih sehat,
sehingga dapat menikmati kegiatan normal, dan hidup sampai dewasa. Transfusi darah yang teratur perlu dilakukan untuk mempertahankan hemoglobin penderita di atas 10 g/dL.
b. Medikamentosa (Permono, dkk, 2006)
b.1 Transfusi darah secara teratur dapat menyebabkan penumpukan zat besi dalam
darah. Kondisi ini disebut kelebihan zat besi. Kondisi tersebut dapat merusak hati, jantung, dan bagian lain dari tubuh. Untuk mencegah kerusakan ini, maka dilakukan suatu bentuk terapi khelasi zat besi untuk membuang kelebihan zat
besi dari tubuh. Pemberian terapi khelasi zat besi (deferoxamine) diberikan setelah kadar ferritin serum sudah mencapai 1000 mg/l atau sekitar 10-20 kali
transfusi darah.
b.3 SuplemenAsam Folat
Asam folat adalah vitamin B yang membantu membangun sel-sel darah
merah yang sehat. Pemberian asam folat 2-5 mg/hari dapat memenuhi kebutuhan tubuh.
b.4 Vitamin E 200-400 IU setiap hari sebagai antioksidan dapat memperpanjang umur sel darah merah.
c. Splenektomi
Ketika limpa menjadi terlalu aktif dan mulai menghancurkan sel-sel darah merah, transfusi menjadi semakin dan terus semakin kurang efektif. Kemudian
menjadi perlu suatu pembedahan untuk mengangkat limpa tersebut. Operasi ini disebut splenektomi (Vullo, dkk, 1995).
Splenektomi dapat menurunkan kebutuhan sel darah merah sampai 30%
pada pasien yang indeks transfusinya (dihitung dari penambahan PRC yang diberikan selama setahun dibagi berat badan dalam kg pada pertengahan tahun)
melebihi 200 ml/kg/tahun. Karena adanya risiko infeksi, splenektomi sebaiknya ditunda hingga penderita mencapai usia 5 tahun. Sedikitnya 2-3 minggu sebelum dilakukan splenektomi, penderita Thalassemia sebaiknya di
vaksinasi dengan vaksin pneumococcal dan Haemophlus influenzae type B dan sehari setelah operasi diberi penisilin profilaksis. Bila terjadi reaksi alergi,
Splenektomi dilakukan dengan indikasi :
1. Limpa yang terlalu besar, sehingga membatasi gerak penderita,
menimbulkan peningkatan tekanan intra abdominal dan bahaya terjadinya ruptur.
2. Hipersplenisme ditandai dengan peningkatan kebutuhan transfusi darah atau kebutuhan suspensi eritrosit (PRC) melebihi 250 ml/kg berat badan dalam satu tahun (Permono, B, dkk, 2006).
2.7.3 Pencegahan Tersier
Pencegahan tersier adalah suatu usaha untuk mengurangi ketidakmampuan
dan mengadakan rehabilitasi bagi penderita Thalassemia. Pencegahan tersier bagi penderita Thalassemia adalah dengan mendirikan suatu organisasi atau perhimpunan orang tua penderita Thalassemia tersebut. Para penderita
Thalassemia membutuhkan dana yang cukup besar untuk tetap dapat bertahan hidup melalui transfusi darah yang rutin yang harus dibarengi dengan pemberian
agen pengkhelat besi yang memadai. Oleh karena itu keberadaan suatu organisasi maupun LSM yang berkaitan dengan penyakit Thalassemia ini sangat dibutuhkan kehadirannya untuk memudahkan penghimpunan dana dan sebagai sarana untuk
saling bertukar informasi tentang Thalassemia.
Perhimpunan Thalassemia Indonesia saat ini telah ada di Jakarta dengan
nama Yayasan Thalassemia Indonesia Pusat, dan sudah memiliki cabang di beberapa kota, yakni Bandung, Yogyakarta, Semarang, Purwokerto, dan Cianjur. Organisasi serupa juga perlu dikembangkan di daerah lainnya. Partisipasi orang
penderita yang kurang mampu. Selain itu, memlalui wadah ini para orang tua penderita diharapkan dapat sering bertemu untuk bertukar, informasi, pikiran,
serta pengalaman dalam mengatasi masalah kesehatan dan psikologis putra-putri mereka.
Perkumpulan seperti ini jika dikelola dengan baik dapat memberikan dukungan moral kepada orang tua, agar mereka tidak merasa frustasi dan sendiri dalam menghadapi masalah berat berkaitan dengan penyakit Thalassemia yang
diderita anaknya (Ganie, 2005). 2.8 Kerangka Konsep
Berdasarkan latar belakang dan tinjauan pustaka di atas, maka kerangka konsep dari penelitian ini digambarkan sebagai berikut :
KARAKTERISTIK PENDERITA THALASSEMIA
1. Sosiodemografi Umur
Jenis Kelamin Suku
Agama Pendidikan Daerah Asal 2. Keluhan Utama 3. Jenis Thalassemia 4. Penatalaksanaan Medis