SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S. Farm.)
Program Studi Ilmu Farmasi
Oleh :
Kristian Bayu Kuncoro NIM : 068114060
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
i
VALIDASI METODE DAN PENETAPAN KADAR PARASETAMOL DALAM JELLY SECARA HIGH PERFORMANCE LIQUID
CHROMATHOGRAPHY (HPLC) FASE TERBALIK MENGGUNAKAN TEKNIK PREPARASI PEMANASAN
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S. Farm.)
Program Studi Ilmu Farmasi
Oleh :
Kristian Bayu Kuncoro NIM : 068114060
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
ii
VALIDASI METODE DAN PENETAPAN KADAR PARASETAMOL DALAM JELLY SECARA HIGH PERFORMANCE LIQUID
CHROMATHOGRAPHY (HPLC) FASE TERBALIK MENGGUNAKAN TEKNIK PREPARASI PEMANASAN
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S. Farm.)
Program Studi Ilmu Farmasi
Oleh :
Kristian Bayu Kuncoro NIM : 068114060
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
v
SKRIPSI INI KUPERSEMBAHKAN UNTUK
MAMAH PAPAH TERCINTA,
yang sangat menyayangiku
KAKAKKU,
yang kuat dan bersemangat
YASHINTA WIDYANIGTYAS,
aku bersyukur bisa mengenalmu lebih
vii PRAKATA
Puji dan syukur penulis haturkan kepada Tuhan Yang Maha Kasih atas segala berkat dan karunia-Nya sehingga penulis dapat menyelesaikan skripsi yang berjudul “Validasi Metode dan Penetapan Kadar Parasetamol Dalam Jelly Secara
High Performance Liquid Chromathography (HPLC) Fase Terbalik
Menggunakan Teknik Preparasi Pemanasan” yang disusun sebagai salah satu syarat untuk mencapai gelar Sarjana Farmasi di Universitas Sanata Dharma Yogyakarta.
Dalam penulisan skripsi ini, penulis mendapatkan bantuan dari banyak pihak. Pada kesempatan ini penulis ingin menyampaikan penghargaan dan ucapan terima kasih kepada :
1. Rita Suhadi, M.Si., Apt. selaku Dekan Fakultas Farmasi Universitas Sanata Dharma atas ide awal penelitian yang berawal dari PKM, atas bimbingannya selama penulis melakukan proses pembelajaran di Fakultas Farmasi Universitas Sanata Dharma.
2. Christine Patramurti, M.Si., Apt. selaku dosen pembimbing atas ide luar biasa mengenai penelitian, atas perhatian, dukungan, arahan, serta semangat yang diberikan kepada penulis baik selama penelitian maupun penyusunan skripsi ini.
viii
4. Dra. M. M. Yetty Tjandrawati, M.Si. selaku dosen penguji atas segala arahan, masukan, kritik, diskusi dan semangat yang diberikan kepada penulis.
5. Mas Bimo, Pak Parlan, Mas Kunto, dan Pak Otok atas bantuannya selama peneliti bekerja di laboratorium Kimia Analisis Instrumental.
6. Segenap dosen pengajar, staf sekretariatan serta laboran Fakultas Farmasi Universitas Sanata Dharma atas dukungan dan bantuannya dalam menyelesaikan skripsi ini.
7. Kho Jimmy Iwan Tamara, selaku rekan kerja penulis sebelum penelitian, selama penelitian, dan penyusunan naskah skripsi atas kebersamaannya di saat susah dan gembira, kita jalani bersama.
8. Seluruh staf dan karyawan Perpustakaan Universitas Sanata Dharma Yogyakarta (Kampus III Paingan) khususnya pak Totok yang sangat membantu penulis selama berada di perpustakaan. Dan terima kasih atas pelayanan serta fasilitas terbaik yang di berikan.
9. Om Sigit, atas berbagi pengalaman berharganya selama bekerja di Industri Farmasi yang dapat memberikan semangat untuk menyelesaikan skripsi dan semangat untuk mencintai pekerjaan.
10. Sahabat-sahabatku, Bernardus Tatag, Felicia Satya C, Lulu Lunggati atas proses pendewasaan, semangat, kasih sayang dan dukungan serta kebersamaan yang telah dilalui dalam suka dan duka bersama penulis.
xi INTISARI
Telah dilakukan validasi metode dan penetapan kadar parasetamol dalam jelly secara HPLC fase terbalik menggunakan teknik preparasi pemanasan dengan tujuan untuk mengetahui akurasi, presisi, spesifisitas, dan linearitas dari metode HPLC sehingga dapat digunakan untuk menetapkan kadar parasetamol dalam jelly.
Penelitian ini merupakan penelitian non eksperimental dekriptif. Tahap pendahuluan dalam penelitian ini adalah pembuatan jelly parasetamol kemudian mengubah sistem jelly yang semi padat dan sangat viskos menjadi cair dengan menggunakan teknik pemanasan pada suhu 50ºC selama 30 menit. Selanjutnya, parasetamol di analisis secara kuantitatif dengan menggunkan metode HPLC fase terbalik dengan fase diam kolom packing Kromasil 100-5 C18 panjang kolom 25 cm, internal diameter 4,6 mm, perbandingan fase gerak metanol:aquabides (90:10), kecepatan alir 1 ml/menit, dan detektor UV pada λ pengamatan 247,4 nm yang telah tervalidasi.
Hasil penelitian menunjukkan nilai rata-rata % recovery 100,3991%, CV 0,6654%, dan koefisien korelasi (r) 0,99905. Rata-rata kadar parasetamol dalam 10 sampel adalah 111,1855 mg. Sehingga metode penetapan kadar parasetamol dalam jelly secara High Performance Liquid Chromatography (HPLC) fase terbalik menggunakan teknik preparasi pemanasan memiliki validitas yang baik dengan kadar parasetamol yang sesuai dengan persyaratan.
xii ABSTRACT
Has been perform validation method and determining the concentration of paracetamol in jelly by reversed phase HPLC uses heating preparation technique with aim to knowing the accuracy, precision, specificity, and linearity from HPLC method, finally can be used to determination the concentration of paracetamol in jelly
This research is descriptive non experimental research. The preliminary stage in this research was making paracetamol jelly, and then change jelly system that semi solid and very viskos to be liquid with uses heating technique at 50ºC temperature during 30 minutes. Next, paracetamol be analyzed by quantitatife with uses reversed phase HPLC with stationery phase column packing Kromasil 100-5 column’s length 25 cm, internal diameter 4,6 mm, the mobile phase comparison metanol:aquabidest (90:10), flow rate 1 ml/minute, and UV detector UV at λ observation 247,4 nm has been validated
Result of research indicate % recovery average value 100,3991%, CV 0,6654%, and coefficient of corelation (r) 0,99905. The average of paracetamol concentration in 10 sampels is 111,1855 mg. So, method of determining the concentration of paracetamol in jelly by High Performance Liquid Chromatography (HPLC) reversed phase fase uses heating preparation technique have good validity with parasetamol concentration that suitable with requirement.
xiii DAFTAR ISI
HALAMAN SAMPUL ... i
HALAMAN JUDUL... ii
HALAMAN PERSETUJUAN PEMBIMBING ... iii
HALAMAN PENGESAHAN... iv
HALAMAN PERSEMBAHAN ... v
HALAMAN PERSETUJUAN PUBLIKASI... vi
PRAKATA... vii
PERNYATAAN KEASLIAN KARYA ... x
INTISARI... xi
1. Perumusan Masalah ... 3
2. Keaslian Penelitian... 3
3. Manfaat Penelitian ... 3
B. Tujuan Penelitian... 4
xiv
A. Parasetamol... 5
1. Stabilitas Suhu Parasetamol ... 7
2. Stabilitas pH Parasetamol ... 7
B.Bentuk Sediaan Gel………….. ... 8
1. Definisi dan Klasifikasi Gel ... 8
2. Mekanisme Pembentukan Gel secara umum ... 9
3. Analisis Sediaan Gel dan Perusakan Sistem Gel ... 10
C.Jelly... 10
D. Senyawa Eksipien Penyusun Jelly... 11
E.Karagenin... 11
1. Definisi dan sifat dasar karagenin ... 11
2. Pembentukan gel dengan gelling agent karagenin... 12
3. Kelarutan ... 14
4. Stabilitas pH ... 15
F. Bubuk Konnyaku... 16
G.Asam sitrat ... 16
H.Frukto oligosakarida ... 17
I. Vitamin D ... 18
J. Kalsium... 18
K.Pewarna makanan karmoisin CI 14720 ... 19
L.Spektrofotometri Ultraviolet... 19
M. High Performance Liquid Chromatography... 22
xv
2. Komponen-komponen HPLC... 23
a. Pompa (Pump)... 23
b. Injektor (Injector)... 23
c. Kolom (Column) ... 24
d. Detektor (Detector) ... 25
3. Kromatografi Partisi Fase Balik... 25
a. Kolom... 26
b. Fase gerak ... 26
4. Injeksi sampel... 27
5. Waktu retensi ... 27
6. Profil puncak dan Pelebaran puncak ... 28
a. Penyebab pertama : Difusi Eddy... 28
b. Penyebab kedua : Distribusi aliran... 29
c. Penyebab ketiga : Difusi molekul sampel dalam fase gerak... 30
d. Penyebab keempat :Perpindahan massa antara fase gerak, fase gerak yang stagnan, dan fase diam ... 31
7. Persamaan Van Deemter ... 32
8. Faktor-faktor yang digunakan untuk evaluasi kinerja kolom ... 34
a. Efisiensi kolom ... 34
b. Faktor asimetri (faktor pengekoran)... 36
N. Validitas Metode Analisis Instrumental... 37
1. Akurasi ... 37
xvi
3. Linieritas dan rentang... 39
4. Spesifisitas ... 39
O. Landasan Teori... 41
P. Hipotesis... 42
BAB III. METODE PENELITIAN ... 43
A.Jenis dan Rancangan Penelitian... 43
B.Variabel dan Definisi Operasional ... 43
1. Klasifikasi Variabel... 43
2. Definisi Operasional ... 43
C.Bahan Penelitian ... 44
D.Alat Penelitian ... 44
E.Tata Cara Penelitian... 45
1. Pembuatan fase gerak... 45
2. Pembuatan larutan baku parasetamol... 45
3. Penetapan λ maksimum parasetamol ... 45
4. Pembuatan kurva baku dan penentuan waktu retensi parasetamol ... 46
5. Validasi metode analisis... 46
6. Penetapan kadar sampel ... 47
a. Pembuatan larutan jelly tanpa parasetamol... 47
b. Pembuatan dan penyiapan sampel jelly parasetamol ... 47
c. Destruksi jelly ... 48
F. Analisis hasil... 49
xvii
a. Akurasi ... 49
b. Presisi ... 49
c. Linearitas ... 49
d. Spesifisitas ... 49
2. Analisis kuantitatif ... 50
BAB IV. HASIL DAN PEMBAHASAN ... 51
A.Penyiapan Fase Gerak ... 51
B.Optimasi Metode HPLC ... 53
1. Penentuan panjang gelombang serapan maksimum menggunakan spektrofotometer ultraviolet ... 53
2. Pengamatan waktu retensi dan pembuatan kurva baku parasetamol... 57
C.Analisis Validasi Metode... 63
1. Akurasi ... 65
2. Presisi ... 66
3. Spesifisitas ... 67
4. Linieritas ... 71
5. Range ... 71
D.Penetapan Kadar Parasetamol dalam Campuran ... 72
1. Pembuatan jelly parasetamol... 72
2. Penyiapan Sampel ... 72
3. Destruksi sampel dan isolasi analit dari sampel... 73
BAB V. KESIMPULAN DAN SARAN... 78
xviii
B.Saran ... 78
DAFTAR PUSTAKA ... 79
LAMPIRAN... 83
xix
DAFTAR TABEL
Tabel I. Senyawa eksipien penyusun jelly ... 11
Tabel II. Daya kelarutan karaginan pada berbagai media pelarut... 15
Tabel III. Stabilitas karaginan dalam berbagai media pelarut... 16
Tabel IV. Kriteria rentang recovery yang dapat diterima (Harmita, 2004) ... 38
Tabel V. Kriteria KV yang dapat diterima ... 38
Tabel VI. Parameter analitik ... 40
Tabel VII. Data Kurva Baku Parasetamol... 62
Tabel VIII. Data Validasi Metode Analisis... 65
Tabel IX. Data % recovery... 66
Tabel X. Data CV... 67
Tabel XI. Perbandingan pengamatan waktu retensi seri larutan baku parasetamol dengan sampel... 68
xx
DAFTAR GAMBAR
Gambar 1. Struktur kimia parasetamol ... 5
Gambar 2. Reaksi hidrolisis parasetamol... 7
Gambar 3. Struktur kimia kappa karaginan ... 12
Gambar 4. Mekanisme pembentukan gel karaginan ... 13
Gambar 5. Struktur kimia asam sitrat ... 17
Gambar 6. Struktur kimia vitamin D... 18
Gambar 7. Struktur kimia karmoisin... 19
Gambar 8. Diagram blok HPLC... 23
Gambar 9. Difusi Eddy dalam kromatografi kolom... 29
Gambar 10. Difusi distribusi aliran dalam kromatografi ... 30
Gambar 11. Pelebaran pita oleh difusi longitudinal... 30
Gambar 12. Struktur pori molekul fase diam... 31
Gambar 13. Perpindahan massa antara fase diam dan fase gerak... 32
Gambar 14. Kurva Van Deemter ... 32
Gambar 15. Cara mengukur tR, σt, Wh/2, Wb suatu puncak kromatogram... 35
Gambar 16. Menghitung besarnya TF pada kromatogram ... 36
Gambar 17. Gugus kromofor parasetamol ... 54
Gambar 18. Gugus auksokrom parasetamol ... 54
xxi
Gambar 20. Spektrum serapan parasetamol (λmaks = 247,2 nm) pada konsentrasi 5,0 ppm replikasi 2... 55 Gambar 21. Kromatogram tR larutan baku parasetamol 5,0 ppm ... 58 Gambar 22. Gugus non polar pada parasetamol ... 59 Gambar 23. Interaksi antara gugus non polar dari parasetamol (benzen) dengan
fase diam Oktadekil (C18)... 59 Gambar 24. Interaksi antara gugus parasetamol dengan fase gerak campuran
metanol:air (90:10) ... 60 Gambar 25. Kurva Baku Parasetamol C vs AUC ... 63 Gambar 26. tR seri baku parasetamol konsentrasi 8,0 ppm = 2,525 menit ... 69 Gambar 27. tR sampel replikasi 6 dengan konsetrasi 7,4587 ppm = 2,542
xxii
DAFTAR LAMPIRAN
Lampiran 1. Sertifikat Analisis Parasetamol... 84 Lampiran 2. Kemasan jelly (Nutrijel) ... 85 Lampiran 3. Data Penimbangan Baku Parasetamol... 86 Lampiran 4. Skema Pembuatan larutan baku Parasetamol dan contoh perhitungan
kadar larutan baku yang digunakan... 87 Lampiran 5. Kromatogram Larutan Baku Parasetamol ... 89 Lampiran 6. Data Penentuan Kurva Baku Parasetamol ... 95 Lampiran 7. Data Validasi Metode ... 96 Lampiran 8. Kromatogram Validasi Metode ... 97 Lampiran 9. Data Penimbangan Sampel Parasetamol ... 106 Lampiran 10. Skema Pembuatan sampel dan contoh perhitungan kadar
BAB I PENDAHULUAN
A. Latar Belakang
Penelitian ini merupakan satu rangkaian pengembangan penelitian dari Widyaningtyas dkk (2008) mengenai “Formulasi dan Penetapan Kadar Sediaan Parasetamol Dalam Bentuk Jelly untuk meningkatkan Kepatuhan Anak Minum
Obat”. Dalam penelitian ini penulis lebih menekankan pada validasi metode dan
penetapkan kadar parasetamol tiap kemasan jellynya.
Farmasis sebagai bagian health care team harus selalu meningkatkan
kemampuannya tidak hanya dalam hal menjamin penyediaan dan pemberian
informasi obat yang berkualitas, tetapi juga berupaya untuk menginovasi bentuk sediaan obat yang praktis, nyaman, manjur dan aman sehingga sediaan obat dapat diterima oleh pasien, khususnya anak-anak dengan rasa dan bau yang lebih sedap, bentuk yang lebih menarik, maupun bentuk sediaan yang dapat dikombinasikan dengan makanan dapat digunakan untuk mengurangi kejadian ”gagal menerima obat oleh pasien atau yang disebut failure to receive drug”. Salah satu bentuk sediaan obat semi solid yaitu jelly dapat dikembangkan sebagai obat analgesik antipiretik dengan parasetamol sebagai zat aktif di dalamnya (Handajani, 2006).
Jelly merupakan sistem semipadat terdiri dari suspensi yang dibuat dari partikel anorganik yang kecil atau molekul organik yang besar terpenetrasi oleh suatu cairan (Anonim, 1995).
Pada proses pembuatan jelly dan pencampuran obat ke dalam jelly dengan proses pemanasan di khawatirkan dapat mengakibatkan perubahan sifat fisika, kimia, dan klinis dari zat aktifnya, yaitu parasetamol. Sifat fisika yang dapat berubah yaitu stabilitas sediaan, sedangkan perubahan sifat kimia dapat diketahui dengan melakukan pengujian kadar zat aktif yang terdapat dalam sediaan racikan tersebut. Dari penelitian Novianti P (2004) dan Arisandi W.S., (2008) mengenai pengaruh suhu dan pH terhadap kadar parasetamol dalam sediaan yang mengandung parasetamol, mengemukakan bahwa suhu dan pH mempengaruhi kadar parasetamol meskipun tidak berbeda secara signifikan. Karena adanya pengaruh suhu yang tinggi yaitu 60ºC, di khawatirkan dapat mendegradasi zat aktif sehingga kadar parasetamol yang ada di dalam sediaan dapat berkurang.
1. Perumusan Masalah :
Berdasarkan latar belakang di atas, maka dapat disusun permasalahan sebagai berikut:
a. Apakah metode HPLC fase terbalik menggunakan teknik preparasi pemanasan mempunyai validitas yang baik untuk menetapkan kadar parasetamol dalam jelly yang didasarkan pada parameter akurasi, presisi, sensitivitas, dan linearitas?
b. Berapakah kadar parasetamol dalam jelly, dan apakah kadar parasetamol tersebut sesuai dengan kadar penggunaan untuk anak yaitu 120mg/kemasan jelly?
2. Keaslian Penelitian
Sejauh penelusuran pustaka penulis, penelitian tentang validasi penetapan kadar parasetamol menggunakan metode HPLC fase terbalik telah banyak dilakukan. Tetapi validasi metode dan penetapan kadar parasetamol dalam jelly secara High Performance Liquid Chromatograph (HPLC) dengan menggunakan teknik preparasi pemanasan belum pernah dilakukan sebelumnya.
3. Manfaat Penelitian
a. Manfaat Teoritis
Penelitian ini diharapkan dapat memberikan informasi bahwa parasetamol dapat di campurkan dalam jelly sebagai alternatif bentuk sediaan yang sesuai dan nyaman bagi anak, dan untuk membuktikan bahwa kadar parasetamol tidak mengalami perubahan selama berada dalam jelly.
b. Manfaat Metodologis.
Penelitian ini diharapkan dapat memberikan informasi bahwa metode HPLC fase terbalik dengan teknik preparasi pemanasan dapat digunakan untuk menetapkan kadar parasetamol dalam jelly dengan validitas yang memenuhi persyaratan.
c. Manfaat Praktis.
Penelitian ini diharapkan dapat memberikan informasi mengenai kualitas jelly parasetamol dan perkembangan bentuk sediaan lain yang cocok dan nyaman bagi anak-anak.
B. Tujuan Penelitian
1. Untuk memberikan informasi bahwa metode HPLC fase terbalik dengan teknik preparasi pemanasan dapat digunakan untuk penetapan kadar parasetamol dalam jelly dengan validitas metode yang baik
BAB II
TINJAUAN PUSTAKA
A. Parasetamol
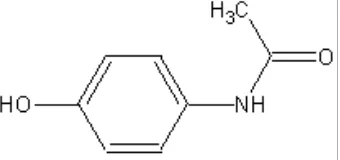
Parasetamol atau 4’-hidroksiasetanilida dengan bobot molekul 151,16 mengandung tidak kurang dari 98,0 % dan tidak lebih dari 101,0 % C8H9NO2, dihitung terhadap zat anhidrat. Pemerian serbuk hablur putih, tidak berbau, rasa sedikit pahit, kelarutan : larut dalam air mendidih dan dalam natrium hidroksida 1 N, mudah larut dalam etanol (Anonim, 1995). Struktur kimia dari parasetamol dapat dilihat pada gambar 1.
Gambar 1. Struktur kimia parasetamol
Parasetamol memiliki jarak lebur 169oC-172oC. Kelarutannya adalah 1 gram dapat larut kira-kira 70 ml air pada suhu 25oC, 1 g larut dalam 20 ml air mendidih, dalam 70 ml alkohol, dalam 13 ml aseton, dalam 50 ml kloroform, dalam 40 ml gliserin dan dalam 9 ml propilenglikol. Tidak larut dalam benzen dan eter dan larut dalam alkali hidroksida. Larutan jenuh mempunyai pH kira-kira 6 dan pKa 9,51 (Connors,et al.,1986).
Serapan maksimum parasetamol pada daerah ultraviolet di larutan asam adalah 254 nm (A 1%, 1cm = 668) dan dalam larutan basa adalah 257 nm (A 1%, 1cm = 715) (Clarke, 1986). A 1%, 1cm atau serapan jenis adalah serapan dari
larutan 1 % zat terlarut dalam sel dengan ketebalan 1 cm (Anonim, 1995). Serapan parasetamol pada panjang gelombang maksimum lebih kurang 244 nm, terhadap air sebagai blangko (Anonim, 1995).
Parasetamol merupakan metabolit fenasetin dengan efek antipiretik yang telah digunakan sejak tahun 1893. Efek antipiretik ditimbulkan oleh gugus aminobenzen. Parasetamol juga digunakan sebagai analgesik. Namun penggunaan parasetamol untuk meredakan demam (antipiretik) tidak seluas penggunaannya sebagai analgesik. Efek analgesik dari parasetamol yaitu meredakan rasa nyeri ringan hingga sedang (Wilmana, 1995).
Dosis untuk nyeri dan demam oral 2-3 dd 0,5-1 g, maksimal 4 g/hari, pada penggunaan kronis maksimal 2,5 g/hari. Anak-anak: 4-6 dd 10 mg/kg, yakni rata-rata usia 3-12 bulan 60 mg, 1-4 tahun 120-180 mg, 4-6 tahun 180 mg, 7-12 tahun 240-360 mg, 4-6 kali sehari. Dosis rektal 20mg/ kg setiap kali, dewasa 4 dd 0,5-1 g, anak-anak usia 3-12 bulan 2-3 dd 120 mg, 1-4 tahun 2-3 dd 240 mg, 4-6 tahun 4 dd 240 mg dan 7-12 tahun 2-3 dd 0,5 g (Rahardja, 2007).
Jalur utama degradasi yang menyebabkan asetaminofen tidak stabil adalah peristiwa hidrolisis yang memecah parasetamol menjadi p-aminofenol dan asam asetat (Connors,et al.,1986). Reaksi hidrolisis parasetamol dapat dilihat pada gambar 2.
Gambar 2. Reaksi hidrolisis parasetamol
1. Stabilitas Suhu Parasetamol
Stabilitas suatu obat perlu di uji untuk mengetahui apakah suatu obat masih layak untuk dikonsumsi atau tidak. Stabilitas obat tergantung dari beberapa faktor, antara lain temperatur. Semua obat pada dasarnya akan rusak apabila disimpan dalam temperatur yang tinggi. Semakin naik suhu penyimpanan maka waktu paruh (t1/2) dan waktu kadaluwarsa (t90) semakin kecil. Dengan demikian menyatakan bahwa dengan semakin naiknya suhu penyimpanan, parasetamol akan mengalamani degradasi sehingga kadarnya berkurang (Novianti, 2004).
2. Stabilitas pH Parasetamol
terdegradasi melalui peristiwa hidrolisis, sehingga perlu dirancang suatu sediaan parasetamol agar mendekati pH optimumnya (Arisandi, 2008).
B. Bentuk Sediaan Gel 1. Definisi dan klasifikasi Gel
Gel merupakan sistem semipadat terdiri dari suspensi yang dibuat dari partikel anorganik yang kecil atau molekul organik yang besar terpenetrasi oleh suatu cairan (Anonim, 1995). Barry (1983) mendefinisikan gel sebagai sistem dua komponen dari sediaan semipadat yang kaya akan cairan. Pada gel yang polar, polimer alam atau sintetik yang digunakan pada konsentrasi rendah (biasanya di bawah 10%) membentuk matriks tiga dimensi melalui cairan hidrofilik. Sistem yang terbentuk mungkin jernih ataupun keruh, karena gelling agent yang digunakan tidak terlarut sempurna atau terbentuknya agregat (Barry, 1983).
Secara umum, ada dua sistem klasifikasi gel. Klasifikasi pertama membagi gel berdasarkan gelling agent-nya, yaitu (i) inorganik yang merupakan sistem dua fase, (ii) organik yang merupakan sistem satu fase. Klasifikasi kedua membagi gel berdasarkan solvennya yaitu (i) hidrogel (inorganik, gum alam dan sintetik, serta organik), (ii) organogel (tipe hidrokarbon, lemak minyak atau hewan, organogel hidrofilik) ( Allen, 2002 ).
aqueous-alcoholic dan gelling agent. Polimer organik yang biasa digunakan adalah asam poliakrilat (carbopol), natrium karboksimetilselulosa, atau selulosa non ionik lainnya.
Hidrogel akan memberikan efek mendinginkan karena evaporasi pelarut. Hidrogel mudah diaplikasikan dan memberi kelembaban secara instan tetapi pada penggunaan jangka panjang akan membuat kulit kering. Dengan demikian, diperlukan humectant seperti gliserol (Buchmann, 2001). Salah satu alasan penggunaan hidrogel adalah pelarut yang digunakan dalam pembuatan obat mempunyai kompatibilitas yang baik terhadap jaringan biologis tubuh (Zatz dan Kushla, 1996).
Dispersi hidrolipid merupakan tipe khusus dari emulsi yang merupakan sistem dispersi dari fase dispers lipofilik medium dispers hidrofilik. Konsentrasi lipid berkisar antara 2-20%. Pada prinsipnya, dispersi hidrolipid merupakan sistem termodinamik yang tidak stabil sehingga dibutuhkan polimer yang dapat menstabilkan koloid liofilik dalam medium berair. Ukuran droplet minyak yang terdispersi berkisar antara 20-50 µm (Buchmann, 2001).
2. Mekanisme Pembentukan Gel secara umum
3. Analisis Sediaan Gel dan Perusakan Sistem Gel
Gel tersusun atas sejumlah kecil komponen padatan yang terdispersi dalam sejumlah besar cairan. Komponen padat dari gel membentuk jaringan tiga dimensi yang membentuk rigiditas gel. Oleh sebab itu, meskipun sebagian besar komponennya berupa cairan, gel memiliki kemampuan untuk mempertahankan bentuknya dengan pemberian sedikit tekanan. Padatan yang lazim digunakan dalam gel adalah polimer meskipun beberapa gel tersusun atas padatan inorganik. Contoh polimer yang biasa digunakan sebagai gelling agent antara lain carbomer, poloxamer, CMC-Na. Hidroxy Propyl Methyl Cellulose (HPMC), dan karaginan (Swarbick and Boylan, 1992).
Untuk memperoleh analit, yang merupakan komponen gel, system disperse dari gel perlu dipisahkan terlebih dahulu. Salah satu cara untuk merusak sistem gel adalah dengan cara pendidihan. Kenaikan suhu pada sistem menyebabkan jumlah tumbukan antara partikel-partikel solid dengan molekul-molekul air bertambah banyak. Menyebabkan lepasnya elekrolit yang teradsorpsi pada permukaan koloid (Sugianto, 2006).
C. Jelly
beraroma buah-buahan. Sifat dasar dari makanan ini rendah lemak dan tinggi serat (Kusumawardini, 2006).
D. Senyawa Eksipien Penyusun Jelly Tabel I. Senyawa Eksipien Penyusun Jelly Karagenin
Pewarna makanan karmoisin CI 14720
E. Karagenin 1. Definisi dan sifat dasar karagenin
Karagenin atau disebut juga karagenan merupakan suatu istilah untuk polisakarida yang diperoleh melalui ekstraksi alkali (dan modifikasi) dari alga merah (Rhodophyceae) kebanyakan berasal dari genus Chondrus, Euchema, Gigartina, dan Iridaea. Rumput laut yang berbeda menghasilkan karagenan yang berbeda pula (Chaplin, 2007).
Karagenan dijual dalam bentuk bubuk, warnanya bervariasi dari putih sampai kecoklatan bergantung dari bahan mentah dan proses yang digunakan. Ukuran karagenan umumnya sebesar 60 mesh. Karagenan tidak dapat larut dalam pelarut organik seperti alkohol, eter dan minyak. Kelarutan dalam air bergantung pada struktur karagenan, media, dan suhu. Umumya, gel karagenan harus dipanaskan sementara non-gel karagenan dapat larut dalam air dingin (Kelco, 2007).
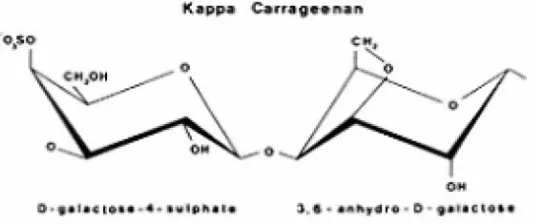
Sifat dasar karaginan terdiri dari tiga tipe karaginan yaitu kappa, iota dan lambda karaginan. Tipe karaginan yang paling banyak dalam aplikasi pangan adalah kappa karaginan. Sifat-sifat karaginan meliputi kelarutan, viskositas, pembentukan gel dan stabilitas pH (Towle, 1973). Berikut struktur kimia kappa karaginan dapat dilihat pada Gambar 3
Gambar 3. Struktur kimia kappa karaginan
2. Pembentukan gel dengan gelling agent karagenin
Sifat pembentukan gel ini beragam dari satu jenis hidrokoloid ke jenis lain, tergantung pada jenisnya. Gel mempunyai sifat seperti padatan, khususnya sifat elastis dan kekakuan.
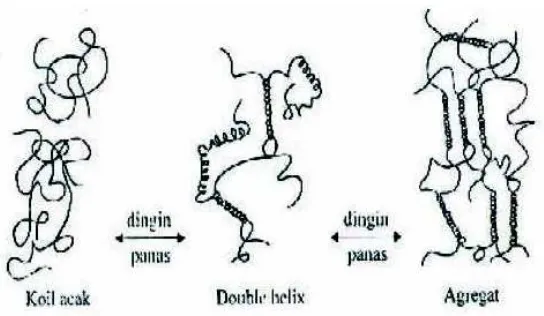
Kappa-karaginan dan iota-karaginan merupakan fraksi yang mampu membentuk gel dalam air dan bersifat reversible yaitu meleleh jika dipanaskan dan membentuk gel kembali jika di dinginkan. Proses pemanasan dengan suhu yang lebih tinggi dari suhu pembentukan gel akan mengakibatkan polimer karaginan dalam larutan menjadi random coil (acak). Bila suhu diturunkan, maka polimer akan membentuk struktur double helix (pilinan ganda) dan apabila penurunan suhu terus dilanjutkan polimer-polimer ini akan terikat silang secara kuat dan dengan makin bertambahnya bentuk heliks akan terbentuk agregat yang bertanggung jawab terhadap terbentuknya gel yang kuat (Glicksman, 1969). Jika diteruskan, ada kemungkinan proses pembentukan agregat terus terjadi dan gel akan mengerut sambil melepaskan air. Proses terakhir ini disebut sineresis (Fardiaz 1989). Mekanisme pembentukan gel karaginan dapat dilihat pada Gambar 4.
Kemampuan pembentukan gel pada kappa dan iota karaginan terjadi pada saat larutan panas yang dibiarkan menjadi dingin karena mengandung gugus 3,6-anhidrogalaktosa. Adanya perbedaan jumlah, tipe dan posisi gugus sulfat akan mempengaruhi proses pembentukan gel. Kappa karaginan dan iota karaginan akan membentuk gel hanya dengan adanya kation-kation tertentu seperti K+, Rb+ dan Cs+. Kappa karaginan sensitif terhadap ion kalium dan membentuk gel kuat dengan adanya garam kalium, sedangkan iota karaginan akan membentuk gel yang kuat dan stabil bila ada ion Ca2+, akan tetapi lambda karaginan tidak dapat membentuk gel (Glicksman, 1983). Potensi membentuk gel dan viskositas larutan karaginan akan menurun dengan menurunnya pH, karena ion H+ membantu proses hidrolisis ikatan glikosidik pada molekul karaginan (Angka dan Suhartono, 2000).
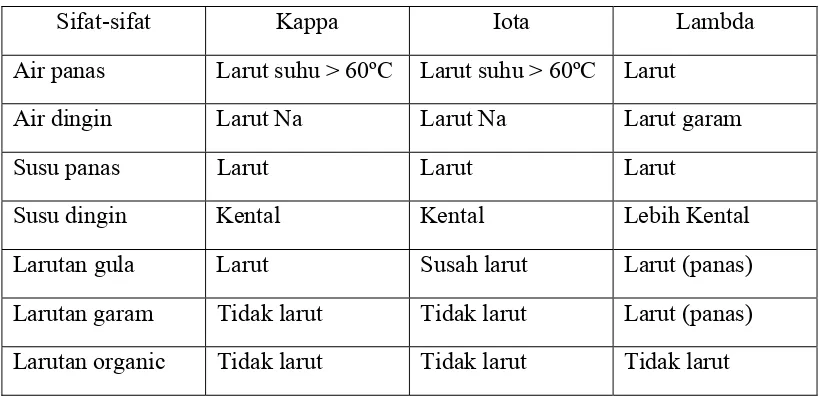
3. Kelarutan
Tabel II. Daya Kelarutan karaginan pada berbagai media pelarut
Sifat-sifat Kappa Iota Lambda
Air panas Larut suhu > 60ºC Larut suhu > 60ºC Larut
Air dingin Larut Na Larut Na Larut garam
Susu panas Larut Larut Larut
Susu dingin Kental Kental Lebih Kental
Larutan gula Larut Susah larut Larut (panas) Larutan garam Tidak larut Tidak larut Larut (panas) Larutan organic Tidak larut Tidak larut Tidak larut
4. Stabilitas pH
Karaginan dalam larutan memiliki stabilitas maksimum pada pH 9 dan akan terhidrolisis pada pH dibawah 3,5. Pada pH 6 atau lebih umumnya larutan karaginan dapat mempertahankan kondisi proses produksi karaginan (cPKelco ApS, 2004). Hidrolisis asam akan terjadi jika karaginan berada dalam bentuk larutan, hidrolisis akan meningkat sesuai dengan peningkatan suhu. Larutan karaginan akan menurun viskositasnya jika pHnya diturunkan dibawah 4,3 (Imeson, 2003).
Tabel III. Stabilitas karaginan dalam berbagai media pelarut
Stabilitas Kappa Iota Lamda
pH netral
dan alkali Stabil Stabil Stabil
Ph asam
Konnyaku berbahan dasar alami dari umbi tanaman konjac yaitu konyaku potato dan calcium hydroxide atau oxide calcium yang diekstrak dari kulit telur (Evimeinar, 2006). Konnyaku merupakan makanan alami yang terdiri dari 97% air dan 3% glukomanan, yaitu serat makanan. Konnyaku juga kaya akan mineral dan rendah kalori serta tidak mengandung lemak. Konnyaku tidak dapat tercerna oleh sistem pencernaan (Anonim, 2009).
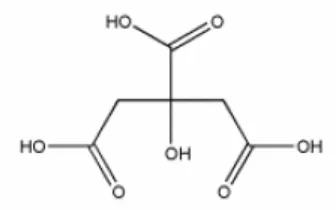
G. Asam sitrat
Merupakan senyawa intermedier dari asam organik yang berbentuk
kristal atau serbuk putih. Asam sitrat ini mudah larut dalam air, spriritus, dan
etanol, tidak berbau, rasanya sangat asam, serta jika dipanaskan akan meleleh,
kemudian terurai yang selanjutnya terbakar sampai menjadi arang. Asam sitrat
juga terdapat dalam sari buah-buahan seperti nanas, jeruk, lemon, markisa. Asam
ini dipakai untuk meningkatkan rasa asam (mengatur tingkat keasaman) pada
berbagai pengolahan minum, produk air susu, selai, jeli, dan lain-lain. Asam sitrat
proses kristalisasi dalam madu, gula-gula (termasuk fondant), dan juga untuk
mencegah pemucatan berbagai makanan, misalnya buah-buahan kaleng dan ikan.
Larutan asam sitrat yang encer dapat digunakan untuk mencegah pembentukan
bintik-bintik hitam pada udang. Penggunaan maksimum dalam minuman adalah
sebesar 3 gram/liter sari buah (Anonim, 2007).
Pemerian : hablur bening, tidak berwarna atau serbuk hablur granul
sampai halus, putih tidak berbau, rasa sangat asam. Bentuk hidrat mekar dalam
udara kering.
Kelarutan : sangat mudah larut dalam air, mudah larut dalam etanol, agak
sukar larut dalam eter (Anonim,1995). Struktur kimia dari asam sitrat dapat dilihat pada gambar 5.
Gambar 5. Struktur kimia asam sitrat
H. Frukto oligosakarida
Fruktooligosakarida (FOS) merupakan prebiotik yang banyak digunakan
saat ini. Prebiotik merupakan istilah yang dikaitkan dengan kesehatan saluran
cerna. Prebiotik sendiri didefinisikan sebagai komponen bahan makanan yang
tidak dicerna oleh sistem pencernaan, namun bila dikonsumsi oleh manusia
saluran cerna secara selektif, dan yang terpenting harus memperlihatkan efek
positif terhadap kesehatan (Anonim, 2007).
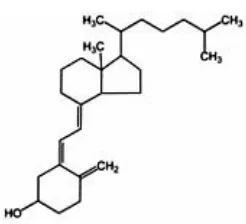
I.Vitamin D
Vitamin D yang juga sering disebut ergocalciferolum atau ergocalciferol
merupakan sumber vitamin D pada sediaan jelly. Vitamin D merupakan serbuk
hablur putih, tidak berbau, dapat terpengaruh oleh cahaya dan udara. Kelarutan:
tidak larut dalam air, larut dalam etanol, dalam kloroform, dan dalam minyak
lemak. Titik didih antara 115ºC dan 117ºC (Anonim,1995). Struktur kimia dari
vitamin D dapat dilihat pada gambar 6.
Gambar 6. Struktur kimia vitamin D
J. Kalsium
Kalsium adalah mineral yang amat penting bagi manusia, antara lain bagi
metabolisme tubuh, penghubung antar syaraf, kerja jantung, dan pergerakan otot.
keseimbangan cairan tubuh, mencegah osteoporosis (keropos tulang), mencegah penyakit jantung, menurunkan resiko kanker usus (Anonim, 2006).
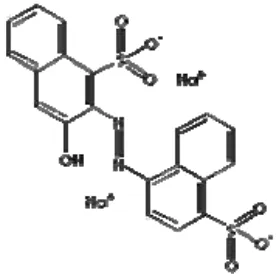
K. Pewarna makanan karmoisin CI 14720
Zat pewarna sintetis, secara umum dapat dibagi ke dalam dua golongan,
yaitu zat pewarna asam dan zat pewarna dasar. Contoh pewarna dari jenis asam adalah amaranth dan karmoisin. Karmoisine yang disebut juga azorubine, atau CI 14.720 adalah zat pewarna sintetis berwarna merah yang digunakan untuk pewarna makanan atau sediaan obat-obatan dan termasuk pewarna azo. Biasanya berbentuk garam dinatrium dari asam sulfat (Anonim, 2009). Struktur kimia dari karmoisin CI 14720 dapat dilihat pada gambar 7.
Gambar 7. Struktur kimia karmoisin
L. Spektrofotometri Ultraviolet
Serapan cahaya molekul dalam daerah spektrum ultraviolet dan visibel tergantung pada struktur elektronik dari molekul. Spektrofotometri ultraviolet dan visibel dari senyawa-senyawa organik berkaitan erat dengan transisi-transisi diantara tingkatan-tingkatan tenaga elektronik. Disebabkan karena hal ini, maka serapan radiasi ultraviolet dan visibel sering dikenal sebagai spektroskopi elektronik. Transisi-transisi tersebut biasanya antara orbital ikatan atau orbital pasangan elektron bebas dan orbital non ikatan tak jenuh atau orbital non ikatan. Panjang gelombang serapan merupakan ukuran dari pemisahan tingkatan-tingkatan tenaga dari orbital yang bersangkutan. Dalam praktek, spektrofotometri ultraviolet digunakan terbatas pada sistem-sistem terkonjugasi. Meskipun demikian terdapat keuntungan yang selektif dari serapan ultraviolet, yaitu gugus-gugus karakteristik dapat dikenal dalam molekul yang sangat kompleks (Sastrohamidjojo, 2002).
Molekul selalu mengabsorbsi cahaya elektromagnetik jika frekuensi cahaya ini sama dengan getaran molekul tersebut. Elektron terikat dan elektron yang tidak terikat akan tereksitasi pada suatu daerah frekuensi yang sesuai dengan cahaya ultraviolet dan cahaya tampak. Spektrum absorbsi daerah ini adalah sekitar 220-800 nm dan dinyatakan dengan spektrum elektron. Suatu spektrum ultraviolet meliputi daerah bagian ultraviolet, spektrum visibel bagian daerah sinar tampak (Roth dan Blaschke, 1994).
berbagai senyawa diukur sampai kadar dibawah 1 ppm (Silverstein, 1981). Sensitifitas yang bagus, hasil yang cukup akurat, serta pengerjaan yang relatif sederhana menjadikan spektrofotometri ultraviolet sebagai metode analisis yang digunakan secara luas (Schinner, 1982).
Absorbansi cahaya ultraviolet mengakibatkan transisi elektronik, yaitu promosi elektron dari orbital keadaan dasar yang berenergi rendah ke orbital keadaan tereksitasi berenergi lebih tinggi. Panjang gelombang ultraviolet bergantung pada mudahnya promosi elektron. Molekul-molekul yang memerlukan banyak energi untuk promosi elektron akan menyerap pada panjang gelombang yang lebih pendek. Molekul yang memerlukan energi lebih sedikit akan menyerap pada panjang gelombang yang lebih panjang (Fessenden dan Fessenden, 1997).
Untuk penentuan kadar spektrofotometri, yang ditentukan adalah absorbsi maksimum kurva absorbsi. Jika absorbsi ini untuk penentuan kadar adalah sangat rendah atau senyawa mula-mula mengabsorbsi dibawah 220 nm, maka seringkali senyawa diubah menjadi suatu zat warna melalui reaksi kimia dan absorbsi ditentukan dalam daerah sinar tampak (visibel) (Roth dan Blaschke.,1994).
M.High Performance Liquid Chromatography
HPLC merupakan kondisi kromatografi yang fase geraknya dialirkan menuju kolom secara cepat dengan bantuan tekanan dari pompa dan hasilnya dapat dideteksi dengan detektor (Hendayana, 2006). Tujuan dari HPLC adalah memperoleh hasil pemisahan yang baik dalam waktu relatif singkat (Mulja dan Suharman, 1995).
1. Kelebihan HPLC
High Performance Liquid Chromatography (HPLC) atau High Pressure
Liquid Chromatography (HPLC) merupakan salah satu metode kimia dan
2. Komponen-komponen HPLC
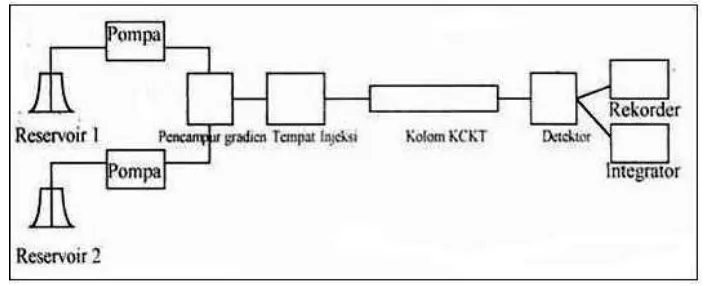
HPLC merupakan teknis analisis yang paling sering digunakan dalam analisis farmasi untuk pemisahan, identifikasi, dan determinasi dalam campuran yang kompleks. Komponen-komponen penting dari HPLC dapat dilihat pada Gambar 8 Diagram Blok HPLC berikut ini :
Gambar 8. Diagram blok HPLC
a. Pompa (Pump). Fase gerak dalam HPLC adalah suatu cairan yang bergerak melalui kolom. Ada dua tipe pompa yang digunakan, yaitu kinerja konstan (constant pressure) dan pemindahan konstan (constant displacement). Pemindahan konstan dapat dibagi menjadi dua, yaitu: pompa reciprocating dan pompa syringe. Pompa reciprocating menghasilkan suatu aliran yang berdenyut teratur (pulsating), oleh karena itu membutuhkan peredam pulsa atau peredam elektronik untuk menghasilkan garis dasar (base line) detektor yang stabil, bila detektor sensitif terhadapan aliran. Keuntungan utamanya ialah ukuran reservoir tidak terbatas. Pompa syringe memberikan aliran yang tidak berdenyut, tetapi reservoirnya terbatas (Putra, 2004).
model umum yaitu stopped flow dan solvent flowing. Ada tiga tipe dasar injektor yang dapat digunakan pertama stop-flow (aliran dihentikan) injeksi dilakukan pada kinerja atmosfir, sistem tertutup, dan aliran dilanjutkan lagi. Teknik ini bisa digunakan karena difusi di dalam cairan kecil dan resolusi tidak dipengaruhi. Kedua adalah septum dimana septum yang digunakan pada HPLC sama dengan yang digunakan pada Kromtografi Gas. Injektor ini dapat digunakan pada kinerja sampai 60-70 atmosfir. Tetapi septum ini tidak tahan dengan semua pelarut-pelarut kromatografi cair. Partikel kecil dari septum yang terkoyak (akibat jarum injektor) dapat menyebabkan penyumbatan. Dan yang ketiga adalah loop valve
dimana tipe injektor ini umumnya digunakan untuk menginjeksi volume lebih besar dari 10 μl dan dilakukan dengan cara otomatis (dengan menggunakan adaptor yang sesuai, volume yang lebih kecil dapat diinjeksikan secara manual). Pada posisi LOAD, sampel di isi kedalam loop pada kinerja atmosfir, bila VALVE difungsikan, maka sampel akan masuk ke dalam kolom (Putra, 2004).
Kolom umumnya dibuat dari stainless steel dan biasanya di operasikan pada temperatur kamar, tetapi bisa juga digunakan temperatur lebih tinggi, terutama untuk kromatografi penukar ion dan kromatografi eksklusi. Pengepakan kolom tergantung pada model HPLC yang digunakan (Liquid Solid
Chromatography, LSC; Liquid Liquid Chromatography, LLC; Ion Exchange
Chromatography, IEC, Exclution Chromatography, EC).
d. Detektor (Detector). Suatu detektor dibutuhkan untuk mendeteksi adanya komponen sampel di dalam kolom (analisis kualitatif) dan menghitung kadarnya (analisis kuantitatif). Detektor yang baik memiliki sensitifitas yang tinggi, gangguan (noise) yang rendah, kisar respons linier yang luas, dan memberi respons untuk semua tipe senyawa. Suatu kepekaan yang rendah terhadap aliran dan fluktuasi temperatur sangat diinginkan, tetapi tidak selalu dapat diperoleh.
Detektor HPLC yang umum digunakan adalah detektor UV 254 nm. Variabel panjang gelombang dapat digunakan untuk mendeteksi banyak senyawa dengan range yang lebih luas. Detektor indeks refraksi juga digunakan secara luas, terutama pada kromatografi eksklusi, tetapi umumnya kurang sensitif jika dibandingkan dengan detektor UV. Detektor-detektor lainnya antara lain detektor Fluorometer-Detektor Spektrofotometer Massa, detektor lonisasi nyala-Detektor Refraksi lndeks, detektor Elektrokimia-Detektor Reaksi Kimia.
3. Kromatografi Partisi Fase Balik
dalam kondisi yang terdiri atas dua pelarut yang tidak bercampur dan keseluruhan kondisi di biarkan seimbang, solut akan tersebar antara kedua fase itu menurut persamaan :
K adalah koefesien distribusi, Cs adalah konsentrasi solut dalam fase diam dan Cm adalah konsentrasi solut dalam fase gerak (Skoog et al.,1994). Hal-hal yang harus diperhatikan dalam pemilihan metode kromatografi partisi fase balik adalah :
a. Kolom. Kolom yang digunakan pada jenis kromatografi ini ialah kemasan fase terikat. Fase diam yang biasa digunakan pada kromatografi partisi fase balik adalah oktadesilsilan (ODS). Selain ODS, dikenal pula silika dengan substitusi oktil (C8) (Munson,1991).
b. Fase gerak. Fase gerak pada HPLC sangat berpengaruh pada tambatan sampel dan pemisahan komponen dalam campuran. Pada fase balik, kandungan utama fase geraknya adalah air. Pelarut yang dapat campur dengan air seperti metanol, etanol, asetonitril, dan tetrahidrofuran ditambahkan untuk mengatur kepolaran fase gerak pada fase balik HPLC.
Dalam kasus ini, ukuran kolom sama, tetapi silika di modifikasi menjadi
non polar melalui pelekatan rantai-rantai hidrokarbon panjang pada
permukaannya secara sederhana baik berupa atom karbon 8 atau 18. Sebagai
contoh, pelarut polar digunakan berupa campuran air dan alkohol seperti metanol.
polar dalam campuran yang melalui kolom. Atraksi yang terjadi tidak akan sekuat
atraksi antara rantai-rantai hidrokarbon yang berlekatan pada silika (fase diam)
dan molekul-molekul polar dalam larutan. Oleh karena itu, molekul-molekul polar
dalam campuran akan menghabiskan waktunya untuk bergerak bersama dengan
pelarut. Senyawa-senyawa non polar dalam campuran akan cenderung
membentuk atraksi dengan gugus hidrokarbon karena adanya dispersi gaya van
der Waals. Senyawa-senyawa ini juga akan kurang larut dalam pelarut karena
membutuhkan pemutusan ikatan hidrogen sebagaimana halnya senyawa-senyawa
tersebut berada dalam molekul-molekul air atau metanol misalnya. Oleh
karenanya, senyawa-senyawa ini akan menghabiskan waktu dalam larutan dan
akan bergerak lambat dalam kolom. Ini berarti bahwa molekul-molekul polar akan
bergerak lebih cepat melalui kolom. Fase balik HPLC adalah bentuk yang biasa
digunakan dalam HPLC (Rod McIlwrick, 2007).
4.Injeksi sampel
Injeksi sampel seluruhnya otomatis, tidak akan dapat mengetahui apa
yang terjadi pada tingkat dasar. Karena proses ini meliputi tekanan, tidak sama
halnya dengan kromatografi gas (Rod McIlwrick, 2007).
5. Waktu retensi
akan berpengaruh pada laju alir dari pelarut), kondisi dari fase diam (tidak hanya terbuat dari material apa, tetapi juga pada ukuran partikel), komposisi yang tepat
dari pelarut, dan temperatur pada kolom. Itu berarti bahwa kondisi harus dikontrol secara hati-hati jika menggunakan waktu retensi sebagai sarana untuk mengidentifikasi senyawa-senyawa (Rod McIlwrick, 2007).
6. Profil puncak dan pelebaran puncak
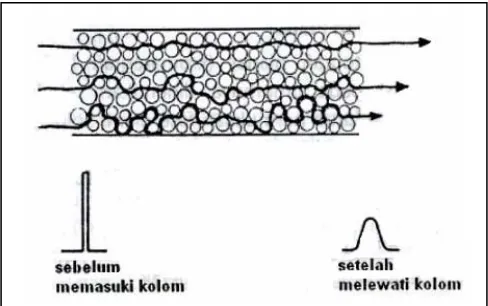
Selama pemisahan kromatografi, solut secara individual akan membentuk profil konsentrasi yang simetri atau dikenal juga dengan profil Gaussian dalam arah aliran fase gerak. Profil, dikenal juga dengan puncak atau pita, secara perlahan-lahan akan melebar dan sering juga membentuk profil yang asimetrik karena solut-solut melanjutkan migrasinya ke fase diam. Penyebab terjadinya pelebaran puncak kromatografi, yaitu :
Gambar 9. Difusi Eddy dalam kromatografi kolom
Gambar 10. Difusi distribusi aliran dalam kromatografi
c. Penyebab ketiga : Difusi molekul sampel dalam fase gerak. Molekul-molekul sampel menyebar di dalam pelarut tanpa adanya pengaruh luar apapun (perhatikan bagaimana suatu gula melarut dalam air secara perlahan-lahan bahkan tanpa diaduk). Hal ini merupakan difusi longitudinal (gambar 11). Difusi ini mempunyai pengaruh yang tidak menguntungkan pada tinggi puncak jika partikel-partikel fase diamnya kecil, kecepatan alir fase gerak terlalu rendah (dihubungkan dengan diameter partikel), koefisien difusi sampel relatif besar.
Kecepatan alir fase gerak harus dipilih sedemikian rupa sehingga difusi longitudinal tidak mempunyai efek yang merugikan. Pada gambar 11, pelebaran pita oleh difusi longitudinal. Kiri : daerah sampel sesaat setelah diinjeksikan. Sampel akan menyebar dalam ruangan ke 3 arah (arah anak panah). Kanan : daerah sampel setelah beberapa saat. Daerah sampel saat ini lebih luas disebabkan oleh difusi. Sampel ini juga akan pindah oleh aliran fase gerak (Rohman, 2009).
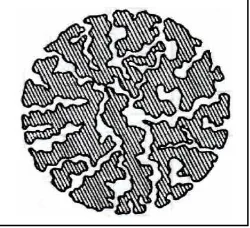
d.Penyebab keempat : perpindahan massa antara fase gerak, fase gerak yang stagnan, dan fase diam. Gambar 12 menunjukkan struktur partikel fase diam. Salurannya ada yang sempit dan ada yang luas. Pori-pori itu terisi oleh fase gerak yang tidak bergerak (stagnan). Suatu molekul sampel yang masuk ke dalam pori akan berhenti untuk dipindahkan dengan aliran fase gerak dan posisinya berubah hanya dengan difusi. Meskipun demikian, ada dua kemungkinan yang terjadi yang pertama molekul sampel berdifusi balik ke aliran fase gerak. Keadaan ini, yang mana molekul sampel keluar bersama aliran fase gerak, membutuhkan waktu. Yang kedua molekul berinteraksi dengan fase diam dan akan teradsorbsi. Untuk sementara waktu, molekul sampel tetap menempel pada fase diam. Sekali lagi, perpindahan massa ini membutuhkan waktu yang cukup lama (gambar 13). Fase diam mempunyai pusat adsorbsi C (dalam kerapatan yang luas) yang akan menarik molekul-molekul di sekitarnya.
Dalam kedua kasus di atas, pelebaran puncak meningkat seiring dengan meningkatnya kecepatan alir fase gerak (Rohman, 2009).
Gambar 13. Perpindahan massa antara fase diam dan fase gerak
7. Persamaan Van Deemter
Tinggi lempeng teoritis (H), yang merupakan ukuran efisiensi kolom, dapat diekspresikan sebagai fungsi kecepatan alir fase gerak u (Gambar.14). Kurva H/u juga disebut dengan kurva Van Deemter. Kecepatan alir optimum (Uopt) tergantung pada sifat-sifat analit. Dalam kurva Van Deemter no.1 adalah difusi Eddy, no.2 adalah difusi longitudinal, no.3 komponen perpindahan massa, no.4 resultan atau hasil kurva Van Deemter (Rohman, 2009).
Gambar 14. Kurva Van Deemter
Di mana :
H adalah ukuran efisiensi kolom; semakin kecil nilai H, maka kolom akan semakin efisien.
u merupakan kecepatan alir fase gerak A adalah difusi Eddy
B adalah difusi longitudinal. Dalam kromatografi cair, difusi longitudinal ini kontribusinya sangat kecil. Sumbangan difusi longitudinal ini dalam pelebaran pita akan menurun. Jika kecepatan alir meningkat, dan hanya akan bermakna jika kecepatan alir fase gerak sangat rendah.
Cs merupakan resistensi terhadap perpindahan atau transfer massa molekul dalam fase diam, dan nilainya tergantung pada koefisien difusinya (Ds) dalam fase diam dan tergantung pula pada ketebalan fase diam (d)
Cs =
Cm merupakan resistensi terhadap transfer massa yang disebabkan oleh diameter dan bentuk partikel fase diam (d) dan kecepatan difusi molekul dalam fase gerak.
Semakin teratur partikel-partikel fase diam, maka kontribusinya terhadap pelebaran pita semakin kecil (Rohman, 2009).
7. Faktor-faktor yang digunakan untuk evaluasi kinerja kolom
Kualitas pemisahan dengan kromatografi kolom dapat dikontrol dengan melakukan serangkaian uji kesesuaian sistem di antaranya adalah efisiensi kolom dan simetrisitas puncak.
a. Efisiensi kolom. Salah satu karakteristik sistem kromatografi yang paling penting adalah efisiensi atau jumlah lempeng teoritis (N). Ukuran efisiensi kolom adalah jumlah lempeng (plate number, N) yang didasarkan pada konsep lempeng teoritis pada distilasi. Jumlah lempeng (N) dihitung dengan :
Nilai N juga dapat dihitung dengan :
Yang mana :
tR = waktu retensi solut
σt = simpangan baku lebar puncak
Wh/2 = lebar setengah tinggi puncak Wb = lebar dasar puncak
Gambar 15. Cara mengukur tR, σt, Wh/2, Wb suatu puncak kromatogram.
Persamaan berikut digunakan untuk menggambarkan hubungan antara panjang kolom (L) dengan efisiensi kolom (H):
suhu yang lebih tinggi, molekul-molekul sampel yang lebih kecil, pengaruh di luar kolom yang minimal (Rohman, 2009).
b. Faktor asimetri (Faktor pengekoran). Suatu situasi yang menunjukkan kinerja kromatografi yang kurang baik adalah ketika ditemukan suatu puncak yang mengalami pengekoran (tailing) sehingga menyebabkan puncak tidak simetri. Jika puncak yang akan dikuantifikasi adalah asimetri, maka suatu perhitungan asimetrisitas merupakan cara yang paling berguna untuk mengontrol atau mengkarakterisasi sistem kromatografi. Puncak asimetri muncul karena bebagai faktor. Peningkatan puncak yang asimetri akan menyebabkan penurunan resolusi, batas deteksi, dan presisi. Gambar 16 menunjukkan bagaimana menghitung nilai faktor pengekoran (tailing factor, TF). Kromatogram yang memberikan harga TF=1 menunjukkan bahwa kromatogram tersebut bersifat setangkup atau simetris. Harga TF >1 menunjukkan bahwa kromatogram mengalami pengekoran (tailing). Semakin besar harga TF maka kolom yang dipakai semakin kurang efisien. Dengan demikian harga TF dapat digunakan untuk melihat efisiensi kolom kromatogram (Rohman, 2009).
N. Validitas Metode Analisis Instrumental
Validasi metode analisis adalah suatu tindakan penilaian terhadap parameter tertentu, berdasarkan percobaan laboratorium, untuk membuktikan bahwa parameter tersebut memenuhi persyaratan untuk penggunaannya (Harmita, 2004). Validasi metode analisis diartikan sebagai suatu prosedur yang digunakan untuk membuktikan bahwa metode analisis tersebut dapat memberikan hasil seperti yang diharapkan dengan kecermatan dan ketelitian yang memadai. Metode analisis instrumen merupakan metode yang terpilih dan memadai untuk mengantisipasi persoalan analisis yaitu sangat kecilnya kadar senyawa yang dianalisis dan kompleksnya matriks sampel yang dianalisis (Mulja dan Suharman, 1995). Untuk itu diperlukan suatu pedoman mengenai kesahihan metode analisis yang didukung oleh parameter-parameter dibawah ini:
1. Akurasi
Tabel IV. Kriteria rentang recovery yang dapat diterima (Harmita, 2004)
Presisi atau keseksamaan adalah ukuran yang menunjukkan derajat kesesuaian antara hasil uji individual, diukur melalui penyebaran hasil individual dari rata-rata jika prosedur diterapkan secara berulang pada sampel-sampel yang diambil dari campuran yang homogen (Harmita, 2004). Presisi biasanya dinyatakan dalam koefisien variasi (KV). Suatu metode dapat dinyatakan memiliki presisi yang baik apabila memiliki KV < 2 % tetapi kriteria ini fleksibel tergantung dari kondisi analit yang diperiksa, jumlah sampel dan kondisi laboratorium. Berikut ketentuan nilai KV yang dapat diterima (Harmita, 2004) :
Tabel V. Kriteria KV yang dapat diterima
3. Linieritas dan rentang
Linieritas merupakan kemampuan suatu metode (pada rentang tertentu) untuk mendapatkan hasil uji yang secara langsung proporsional dengan konsentrasi (jumlah) analit di dalam sampel. Rentang adalah jarak antara level terbawah dan teratas dari metode analisis yang telah dipakai untuk mendapatkan presisi, linieritas dan akurasi yang bisa diterima (Anonim, 2007). Persyaratan data linearitas yang bisa diterima jika memenuhi nilai koefisien korelasi (r) > 0,99 atau r2≥ 0,997 (Anonim, 2004; Chan et al, 2004).
4. Spesifisitas
Spesifisitas suatu metode adalah kemampuannya yang hanya mengukur zat tertentu saja secara cermat dan seksama dengan adanya komponen lain yang mungkin ada dalam matriks sampel. Selektivitas metode ditentukan dengan membandingkan hasil analisis sampel yang mengandung cemaran, hasil urai, senyawa sejenis, senyawa asing lainnya atau pembawa plasebo dengan hasil analisis sampel tanpa penambahan bahan-bahan tadi. Penyimpangan hasil merupakan selisih dari hasil uji keduanya (Harmita, 2004).
USP 28 mencantumkan beberapa kategori uji umum yang harus memenuhi validitas data, yaitu :
a. Kategori I. Metode analitik yang digunakan untuk mengukur secara kuantitatif sejumlah besar komponen dari serbuk obat atau senyawa aktif (termasuk preservarif).
c. Kategori III. Metode analitik yang digunakan untuk penentuan sifat-sifat khusus seperti kecepatan disolusi dan pelepasan obat.
d. Kategori IV. Metode analitik yang digunakan untuk mengidentifikasi sediaan farmasi.
O. Landasan Teori
Gel merupakan sediaan setengah padat yang terdiri dari suspensi yang dibuat dari partikel anorganik yang kecil atau molekul organik yang besar terpenetrasi oleh suatu cairan. Dalam sediaan gel terdapat gelling agent yang merupakan senyawa pembentuk gel yaitu karaginan.
Karaginan dapat membentuk gel secara reversibel artinya dapat membentuk gel pada saat pendinginan dan kembali cair pada saat dipanaskan serta akan larut di air panas pada suhu > 60ºC.
Pemanasan menyebabkan rusak atau terbongkarnya ikatan silang (cross link) antar rantai polimer gelling agent karagenin sehingga akan mengakibatkan polimer karaginan dalam larutan menjadi random coil (acak) dengan lepasnya ikatan glikosidik α-1,3 dan β-1,4 menyebabkan susunan molekul polimer mengalami perubahan dan bahkan rusak menjadi unit-unit monomer D-galaktosa 4-sulfat dan 3,6-anhidro-D-galaktosa. Rusaknya sistem gel menyebabkan bahan tambahan dalam jelly mudah untuk diisolasi dari sediaan termasuk senyawa parasetamol di dalamnya.
Suhu dan pH lingkungan mempengaruhi kadar parasetamol dalam sediaan. Karena adanya pengaruh suhu yang tinggi yaitu 60ºC, dikhawatirkan dapat mendegradasi zat aktif sehingga kadar parasetamol yang ada didalam sediaan dapat berkurang.
bantuan pompa bertekanan dan hasilnya dideteksi dengan detektor. Analisis dengan HPLC mempunyai sensitivitas yang tinggi dan diharapkan didapatkan pemisahan yang baik dalam waktu yang relatif singkat.
Sebelumnya, metode penetapan kadar parasetamol dalam jelly dengan menggunakan teknik preparasi pemanasan perlu di validasi terlebih dahulu untuk membuktikan bahwa metode yang digunakan memberikan hasil seperti yang diharapkan berdasarkan parameter akurasi, presisi, spesifisitas, dan linearitas yang memadai.
P. Hipotesis
1. Metode HPLC fase terbalik mempunyai validitas yang baik untuk menetapkan kadar parasetamol dalam jelly parasetamol yang didasarkan pada parameter validitas akurasi, presisi, sensitivitas , dan linearitas.
BAB III
METODE PENELITIAN
A. Jenis dan Rancangan Penelitian
Penelitian ini merupakan jenis penelitian non eksperimental dengan rancangan penelitian deskriptif, karena dalam penelitian ini tidak dilakukan manipulasi terhadap subjek uji, yaitu jelly parasetamol. Penelitian ini hanya mendeskripsikan keadaan yang ada.
B. Variabel dan Definisi Operasional 1. Klasifikasi Variabel
a. Variabel Bebas
Suhu dan waktu pemanasan untuk memecah sistem jelly. b. Variabel Tergantung
Kadar parasetamol dalam jelly. c. Variabel Pengacau Terkendali
Kualitas dari bahan yang digunakan, ataupun pelarut. 2. Definisi Operasional
a. Jelly merupakan sistem semipadat terdiri dari suspensi yang dibuat dari partikel anorganik yang kecil atau molekul organik yang besar terpenetrasi oleh suatu cairan.
b. High Performance Liquid Chromatography (HPLC) fase terbalik yang digunakan adalah seperangkat alat HPLC dengan fase diam kolom
reserved phase C18 dan fase gerak campuran metanol dan aquabides (90:10).
c. Kadar parasetamol dalam jelly di tetapkan dengan satuan mg/kemasan. d. Parameter validasi metode yang digunakan adalah akurasi, presisi,
sensitivitas, dan linearitas.
C. Bahan Penelitian
Bahan-bahan yang digunakan dalam penelitian ini meliputi “Jelly” (Nutrijel), baku parasetamol kualitas working standard (ANQUI LU’AN
PHARMACEUTICAL CO., LTD), Metanol p.a. (E. Merck), Aquabidestilata
(Ikapharmindo Pharmaceutical).
D. Alat Penelitian
Alat-alat yang digunakan dalam penelitian ini meliputi: Spektrofotometer UV/Vis merk Perkin-Elmer Lambda 20, Kuvet, Sistem HPLC yang terdiri dari : pompa merk Shimadzu model LC-10 AD No. C20293309457 J2, detector UV-VIS merk Shimadzu, model SPD-10 AV No. C20343502697 KG, seperangkat computer merk Compaq, printer Hewlett Packard Deskjet 670 C, injector jenis katup suntik model 77251, kolom C18 Merek KNAUER dengan packing Kromasil 100-5, panjang kolom 25 cm, internal diameter 4,6mm, syringe merk Hamilton
Pat No. 2933087, degassing ultrasonicator merk Retsch tipe T460 no V935922013 EY, vaccum merk Gast model DOA-P104-BN, organic solvent
membrane filter merk Whatman ukuran pori (0,5 µm ; diameter 47 mm),
volume, beker glass, labu takar, gelas ukur, buret 10,0 ml, kompor listrik beserta
magnetic stirrer.
E. Tata Cara Penelitian 1. Pembuatan fase gerak
Fase gerak dibuat dalam campuran metanol : aquabides (90:10) sebanyak 500,0 ml, campuran tersebut digojog dan disaring dengan kertas Whatman organik dengan bantuan pompa vakum dan di degassing selama 15 menit.
2. Pembuatan larutan baku parasetamol
Lebih kurang 10 mg parasetamol ditimbang seksama, dilarutkan dengan fase gerak kemudian dimasukkan ke dalam labu takar 10 ml dan diencerkan dengan fase gerak sampai volumenya tepat 10,0 ml (larutan stok). Larutan ini kemudian dipipet 0,5 ml lalu dimasukkan dalam labu takar 50 ml dan diencerkan dengan fase gerak hingga volumenya tepat 50,0 ml (larutan intermediet). Dari larutan intermediet ini kemudian dibuat larutan baru dengan konsentrasi 3,0 ; 4,0 5,0 ; 6,0 ; 7,0 ; 8,0 ppm. Masing-masing seri larutan baku disaring dengan
miliphore dengan ukuran diameter 0,45 μm dan di degassing selama 15 menit.
3. Penetapan λ maksimum parasetamol
UV. Kemudian diperoleh kurva hubungan panjang gelombang dan absorbansi parasetamol.
4. Pembuatan kurva baku dan penentuan waktu retensi parasetamol
Masing-masing seri konsentrasi larutan baku parasetamol yang telah dibuat pada point 2 disuntikkan dalam injector port pada sistem HPLC menggunakan instrumen Shimadzu LC-10 AD, kolom C18 Merek KNAUER dengan packing Kromasil 100-5 panjang kolom 25 cm, internal diameter 4,6mm, fase gerak campuran metanol:aquabides (90:10), flow rate 1 ml/menit, AUFs/Attenuation 0,01/7, detektor UV pada 247,4 nm, dengan volume injeksi 20
µl (Rheodyne Loop Injection). Kemudian diamati puncak kromatogram yang
muncul dan nilai AUC dari masing-masing puncak. Dengan metode regresi linier, memplotkan konsentrasi (ppm) terhadap nilai AUC dari masing-masing seri larutan baku sehingga didapat persamaan y = bx + a (y = nilai respon, x = konsentrasi senyawa baku, a = intersept, b = slope). Pembuatan kurva baku di replikasi 3x dan di ambil kurva baku terbaik dengan nilai r > 0,999. Selain itu, dilihat pula waktu retensi dari masing-masing seri larutan baku parasetamol.
5. Validasi metode analisis
konsentrasi 3,0 ; 5,0 ; 8,0 ppm. Masing-masing seri larutan baku direplikasi 3 kali lalu disaring dengan miliphore dengan ukuran diameter 0,45 μm dan di degassing
selama 15 menit. Larutan disuntikkan kedalam sistem HPLC. 6. Penetapan kadar sampel
a. Pembuatan larutan jelly tanpa parasetamol. Lebih kurang 6,4286 g serbuk jelly ditimbang, dilarutkan dengan 300 ml aquabides dalam beaker glass
500 ml dan dipanaskan sampai mendidih pada suhu 100ºC selama kurang lebih 5 menit sambil diaduk sampai semua serbuk larut merata dan homogen. Dinginkan hingga memadat. Selanjutnya larutan dimasukkan dalam 20 cetakan masing-masing cetakan berisi 15 ml jelly, diamkan selama beberapa saat hingga semua jelly memadat.
Salah 1 jelly kemudian dipanaskan pada suhu 50ºC selama 30 menit bersama 100 ml aquabidest hingga jelly mencair. Pipet larutan jelly sebanyak 0,5 ml dan encerkan dengan fase gerak ad 50,0 ml. Ambil 6,6 ml larutan intermediet dengan menggunakan buret 10,0 ml dan encerkan dengan fase gerak ad 10,0 ml (larutan blanko). Kemudian larutan blanko disaring dengan miliphore ukuran pori diameter 0,45 μm dan di degassing selama 15 menit. Larutan disuntikkan kedalam sistem HPLC
parasetamol hingga volumenya tepat 15,0 ml. Suhu saat pencampuran antara jelly dan parasetamol di atur dan di kontrol pada suhu < 50ºC diamkan selama beberapa saat hingga semua jelly memadat. Kemudian mengambil secara acak sebanyak 10 buah jelly (prinsip pengambilan sampel secara random/cuplikan random/cuplikan acak). 10 sampel jelly ini yang nantinya digunakan untuk penetapan kadar.
c. Destruksi jelly. Masing-masing jelly dipanaskan pada suhu 50ºC selama 30 menit dalam 100 ml aquabidest hingga jelly mencair dan di bantu dengan magnetic stirrer pada kecepatan 500 rpm. Pipet larutan sebanyak 0,5 ml dan di encerkan dengan fase gerak ad 50,0 ml. Ambil 6,6 ml larutan ini dengan menggunakan buret 10,0 ml dan encerkan dengan fase gerak ad 10,0 ml. Larutan kemudian disaring dengan miliphore ukuran pori diameter 0,45 μm dan di
degassing selama 15 menit.
F. Analisis Hasil 1. Validasi metode
a. Akurasi
Dinyatakan dalam persen perolehan kembali ( % recovery )
% recovery =
Metode ini dikatakan memiliki akurasi yang baik jika nilai % recovery berada pada rentang 98-102 % untuk analit pada matriks sampel 100% (Harmita, 2004).
d. Presisi
Presisi biasanya dinyatakan dengan coefficient of variation (CV)
x 100%
Suatu metode dapat dinyatakan memiliki presisi yang baik apabila memiliki CV < 2 % (Harmita, 2004).
e. Linearitas
Dinyatakan dalam koefisien korelasi (r) y = bx + a
Linieritas yang baik ialah nilai r yang lebih besar dari 0,999 untuk minimal 6 seri konsentrasi (Anonim, 2008).
d. Spesifisitas
2. Analisis Kuantitatif
Analisis kuantitatif yang dilakukan adalah penetapan kadar parasetamol dalam jelly berdasarkan analisi data AUC sampel dan kurva baku parasetamol. Satuan untuk menetapka kadar parasetamol dalam jelly adalah mg/kemasan. Data kemudian ditampilkan dalam bentuk % recovery. Rumus % recovery sebagai berikut :
BAB IV
HASIL DAN PEMBAHASAN
A. Penyiapan Fase Gerak
Fase gerak yang digunakan dalam penelitian adalah campuran antara metanol : aquabides dengan perbandingan (90:10). Campuran fase gerak ini bersifat polar. Pemilihan fase gerak sangat penting karena hal ini dapat mempengaruhi waktu retensi dan pemisahan dari komponen-komponen dalam sampel yang akan dianalisis. Fase diam yang digunakan adalah kolom C18 yang bersifat non polar sehingga sistem kromatografi yang digunakan adalah sistem kromatografi partisi fase terbalik. Pemilihan sistem kromatografi yang tepat dan sesuai dengan sampel yang dipisahkan, akan menghasilkan pemisahan yang baik. Berdasarkan bagan pendekatan umum dalam memilih jenis HPLC Johnson Stevenson (1978), dimana parasetamol memiliki BM < 2000 (yaitu 151,16), tidak larut dalam air, homolog, maka dipilih kromatografi partisi dengan fase diam non polar dan fase gerak bersifat polar. Selain itu pemilihan kondisi kromatografi juga didasarkan pada penuntun pemilihan kolom dan sistem HPLC menurut Gritter (1991) dimana kelarutan cuplikan (parasetamol) dalam pelarut organik dengan kepolaran yang tinggi maka dipilih kolom Oktadekil (C18) dengan sistem fase terbalik.
Harapan dari pemilihan kondisi kromatografi fase terbalik ini adalah dapat memisahkan analit dari sampel dengan waktu analisis yang cepat (waktu
retensi analit yang singkat) karena analit bersifat polar akan terelusi lebih cepat dengan fase gerak yang juga bersifat polar.
B. Optimasi Metode HPLC
1. Penentuan panjang gelombang serapan maksimum menggunakan spektrofotometer ultraviolet
Penentuan panjang gelombang serapan maksimum ini bertujuan untuk mendapatkan panjang gelombang serapan maksimum dari parasetamol. Analisis senyawa menggunakan HPLC memerlukan panjang gelombang dimana suatu senyawa memberikan absorbansi maksimum untuk dibaca pada detektor UV pada alat HPLC dimana dengan panjang gelombang maksimum parasetamol diharapkan semua kadar/konsentrasi parasetamol dalam sampel dapat terdeteksi oleh detektor UV
Penentuan panjang gelombang serapan maksimum ini dilakukan dengan menggunakan konsentrasi larutan baku dengan konsentrasi 5,0 ppm dan dilakukan dua kali pengamatan. Perbandingan dua spektrum serapan maksimum parasetamol pada konsentrasi yang sama ini perlu dilakukan karena senyawa baku parasetamol yang dipakai adalah parasetamol kualitas working standard dimana perlu di uji dan di pastikan bahwa senyawa baku yang digunakan adalah benar-benar parasetamol dan dapat memberikan spektrum serapan maksimum yang sama pada konsentrasi yang ditetapkan.
radiasi ultraviolet pada sampel yaitu parasetamol. Parasetamol dalam strukturnya memiliki gugus kromofor yang merupakan ikatan rangkap yang memiliki elektron π dimana elektron π ini jika dikenai sinar radiasi elektromagnetik akan mudah
tereksitasi ke tingkat yang lebih tinggi yaitu menuju ke orbital π*. Gugus kromofor dari senyawa parasetamol dapat dilihat pada gambar dibawah ini
Gambar 17. Gugus Kromofor Parasetamol
Keterangan : = kromofor
Selain gugus kromofor, parasetamol memiliki gugus auksokrom yang terikat langsung pada gugus kromofor. Gugus auksokrom memiliki pasangan elektron bebas pada elektron n yang dapat berinteraksi dengan elekron π pada kromofor. Dengan demikian, gugus auksokrom berperan dalam pengubahan/pergeseran panjang gelombang maksimum dan intensitas serapan maksimum dari parasetamol. Gugus auksokrom dari senyawa parasetamol dapat dilihat pada gambar dibawah ini :
Gambar 18. Gugus Auksokrom Parasetamol
Dalam Farmakope Indonesia Edisi IV (1995) disebutkan bahwa pengujian panjang gelombang serapan maksimum mempunyai makna jika serapan maksimum tersebut tepat atau dalam batas 2 nm dari panjang gelombang yang ditentukan. Spektrum serapan yang dihasilkan oleh senyawa parasetamol dapat dilihat pada gambar berikut :
Pelarut yang digunakan dalam seri larutan baku adalah campuran antara metanol : aquabides dengan perbandingan 90:10. Dalam proses scanning panjang gelombang parasetamol ini, pelarut baik metanol maupun aquabides tidak mempengaruhi hasil dari pengamatan karena panjang gelombang (λ) dari metanol 205 nm dan aquabides 180 nm tidak berdekatan atau bertumpukan dengan
Gambar 19. Spektrum serapan parasetamol (λmaks = 247,2 nm) pada konsentrasi 5,0 ppm replikasi 1
panjang gelombang maksimum teoritis dari parasetamol dengan campuran pelarut metanol dan aquabides yaitu 244 nm.
Berdasarkan hasil penentuan panjang gelombang serapan maksimum menggunakan spektrofotometer ultraviolet dapat dilihat bahwa pada konsentrasi 5,0 ppm replikasi 1 spektrum serapan maksimum parasetamol adalah 247,2 nm dan pada replikasi 2 spektrum serapan maksimum parasetamol adalah 247,4 nm. Pola absorbansi dari spektrum serapan yang dihasilkan dari dua kali pengamatan pada konsentrasi 5,0 ppm ini sama. Maka dapat di pastikan analit yang digunakan adalah parasetamol
Menurut Farmakope Indonesia Edisi IV (1995), parasetamol dalam campuran pelarut metanol dan air memiliki serapan maksimum pada 244 nm. Dan pada penentuan panjang gelombang serapan parasetamol ini terdapat pergeseran panjang gelombang. Pergeseran ini disebabkan oleh perbandingan jumlah air dan metanol yang digunakan dimana dalam Farmakope Indonesia Edisis IV, jumlah air yang digunakan lebih banyak daripada metanol. Namun dalam penelitian, jumlah metanol yang digunakan lebih banyak daripada air. Selain itu instrumentasi yang digunakan berbeda, kualitas baku parasetamol yang digunakanpun juga berbeda dengan yang digunakan berdasarkan farmankope. Pada pergeseran λmaks parasetamol, perbandingan jumlah pelarut metanol dengan λ 205 nm yang lebih banyak daripada air akan menggeser λmaks teoritis dari