LAPORAN PRAKTEK KERJA PROFESI APOTEKER
PT. KALBE FARMA, Tbk.
PERIODE 1 FEBRUARI – 30 MARET 2012
LAPORAN PRAKTEK KERJA PROFESI APOTEKER
CHRISTY CECILIA SN, S.Farm.
1106046761
ANGKATAN LXXIV
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM PROGRAM PROFESI APOTEKER – DEPARTEMEN FARMASI
LAPORAN PRAKTEK KERJA PROFESI APOTEKER
PT. KALBE FARMA, Tbk.
PERIODE 1 FEBRUARI – 30 MARET 2012
LAPORAN PRAKTEK KERJA PROFESI APOTEKER
Diajukan sebagai salah satu syarat untuk memperoleh gelar Apoteker
CHRISTY CECILIA SN, S.Farm.
1106046761
ANGKATAN LXXIV
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM PROGRAM PROFESI APOTEKER – DEPARTEMEN FARMASI
Laporan Praktek Kerja Profesi Apoteker diajukan oleh: Nama : Christy Cecilia SN, S.Farm.
NPM : 1106046761
Program Studi : Apoteker – Departemen Farmasi FMIPA UI Judul Laporan : Laporan Praktek Kerja Profesi Apoteker
PT. Kalbe Farma, Tbk.
Periode 1 Februari – 30 Maret 2012
Telah berhasil dipertahankan di hadapan Dewan Penguji dan diterima sebagai bagian persyaratan yang diperlukan untuk memperoleh gelar Apoteker pada program studi Apoteker – Departemen Farmasi, Fakultas Matematika dan Ilmu Pengetahuan Alam, Universitas Indonesia.
DEWAN PENGUJI
Pembimbing 1 : Mimi Yosiani, S.Si., Apt
Pembimbing 2 : Dr. Harmita, Apt.
Penguji I : (...)
Penguji II : (...)
Penguji III : (...)
Ditetapkan di : Depok
Segala puji dan syukur saya panjatkan kehadirat Tuhan Yesus Kristus atas kasih dan anugerah-Nya yang begitu besar sehingga penulis dapat menyelesaikan Praktek Kerja Profesi Apoteker (PKPA) Angkatan LXXIV Universitas Indonesia, yang diselenggarakan pada tanggal 1 Februari – 30 Maret 2012 di PT. Kalbe Farma, Tbk. Jalan Letnan Jenderal Suprapto, Kav. 4, Jakarta.
Kegiatan Praktek Kerja Profesi Apoteker dan penyusunan laporan Praktek Kerja Profesi Apoteker merupakan bagian dari kegiatan perkuliahan program pendidikan profesi Apoteker dengan tujuan untuk meningkatkan pemahaman, pengetahuan, dan keterampilan mahasiswa. Setelah mengikuti kegiatan Praktek Kerja Profesi Apoteker, diharapkan Apoteker yang lulus nantinya dapat mengaplikasikan pengetahuan dan keterampilan yang dimiliki kepada masyarakat pada saat memasuki dunia kerja. Dalam pelaksanaan kegiatan Praktek Kerja Profesi Apoteker ini, penulis mendapat banyak bantuan, bimbingan, dan saran-saran dari berbagai pihak. Oleh karena itu, pada kesempatan ini dengan penuh ketulusan dan kerendahan hati penulis ingin menyampaikan terima kasih kepada : 1. Bapak Drs. Sie Johan selaku Director of Corporate Business Development
and Management System yang telah memberikan kesempatan untuk
melaksanakan Praktek Kerja Profesi Apoteker di PT. Kalbe Farma, Tbk. 2. Ibu Dra. Nurul Hidayah Yusuf, MM., Apt. selaku General Manager of
Corporate Regulatory Affairs yang telah memberikan kesempatan untuk
melaksanakan Praktek Kerja Profesi Apoteker di PT. Kalbe Farma, Tbk. 3. Ibu Metty Susanti, S.Si., Apt. selaku Senior Regulatory Manager yang telah
memberikan kesempatan untuk melaksanakan Praktek Kerja Profesi Apoteker di PT. Kalbe Farma, Tbk dan banyak membimbing dalam pelaksanaan tugas selama Praktek kerja.
4. Ibu Mimi Yosiani, S.Si., Apt. selaku Regulatory Manager sekaligus pembimbing penyusunan laporan di PT. Kalbe Farma, Tbk. yang telah memberikan bimbingan, pengarahan, saran, dan informasi yang sangat bermanfaat selama pelaksanaan Praktek Kerja Profesi Apoteker dan
Farmasi FMIPA Universitas Indonesia.
6. Bapak Dr. Harmita, Apt. selaku Ketua Program Profesi Apoteker Departemen Farmasi FMIPA Universitas Indonesia sekaligus pembimbing yang telah memberikan bimbingan, pengarahan, dan saran selama pelaksanaan Praktek Kerja Profesi Apoteker dan penyusunan laporan ini.
7. Seluruh staf dan karyawan di PT. Kalbe Farma, Tbk. khususnya Corporate
Regulatory Affairs yang telah banyak membantu dan memberikan informasi
yang bermanfaat selama pelaksanaan Praktek Kerja Profesi Apoteker.
8. Seluruh dosen pengajar, staf, dan karyawan Departemen Farmasi FMIPA Universitas Indonesia.
9. Semua pihak yang telah memberikan bimbingan, pengarahan, informasi yang sangat bermanfaat, dan dukungan kepada penulis selama pelaksanaan Praktek Kerja Profesi Apoteker dan penyusunan laporan ini.
Penulis menyadari bahwa masih terdapat kekurangan dan kesalahan dalam penyusunan laporan ini. Oleh karena itu, penulis mengharapkan kritik dan saran yang membangun. Penulis berharap semoga pengetahuan dan pengalaman yang penulis dapatkan selama pelaksanaan Praktek Kerja Profesi Apoteker ini dapat memberikan manfaat bagi rekan-rekan sejawat dan semua pihak yang membutuhkan.
Penulis 2012
HALAMAN SAMPUL ... i
HALAMAN JUDUL ... ii
LEMBAR PENGESAHAN ... iii
KATA PENGANTAR ... iv
DAFTAR ISI ... vi
DAFTAR GAMBAR ... viii
DAFTAR TABEL ... ix
DAFTAR LAMPIRAN ... x
BAB 1 PENDAHULUAN ... 1
1.1 Latar Belakang ... 1
1.2 Tujuan ... 2
BAB 2 TINJAUAN UMUM ... 3
2.1 PT. Kalbe Farma, Tbk ... 3
2.2 Corporate Business Development ... 6
BAB 3 TINJAUAN KHUSUS ... 13
3.1 Registrasi ... 13
3.2 Pendaftar ... 14
3.3 Registrasi Obat ... 15
3.4 Registrasi Obat Tradisional, Obat Herbal Terstandard, dan Fitofarmaka ... 22
3.5 Registrasi Suplemen Makanan ... 25
3.6 Registrasi Pangan ... 29
3.7 Notifikasi Kosmetika ... 33
3.8 Registrasi Alat Kesehatan dan Perbekalan Kesehatan Rumah Tangga ... 36
3.9 ASEAN Common Technical Dossier / ASEAN Common Technical Requirements ... 41
4.2 Registrasi Obat ... 45
4.3 Registrasi Obat Tradisional, Obat Herbal Terstandard, dan Fitofarmaka ... 48
4.4 Registrasi Suplemen Makanan ... 49
4.5 Registrasi Pangan ... 50
4.6 Notifikasi Kosmetika ... 51
4.7 Registrasi Alat Kesehatan dan Perbekalan Kesehatan Rumah Tangga ... 52
4.8 Dokumen Pra Registrasi dan ASEAN Common Technical Dossier (ACTD) ... 53
BAB 5 KESIMPULAN DAN SARAN ... 54
5.1 Kesimpulan ... 54
5.2 Saran ... 54
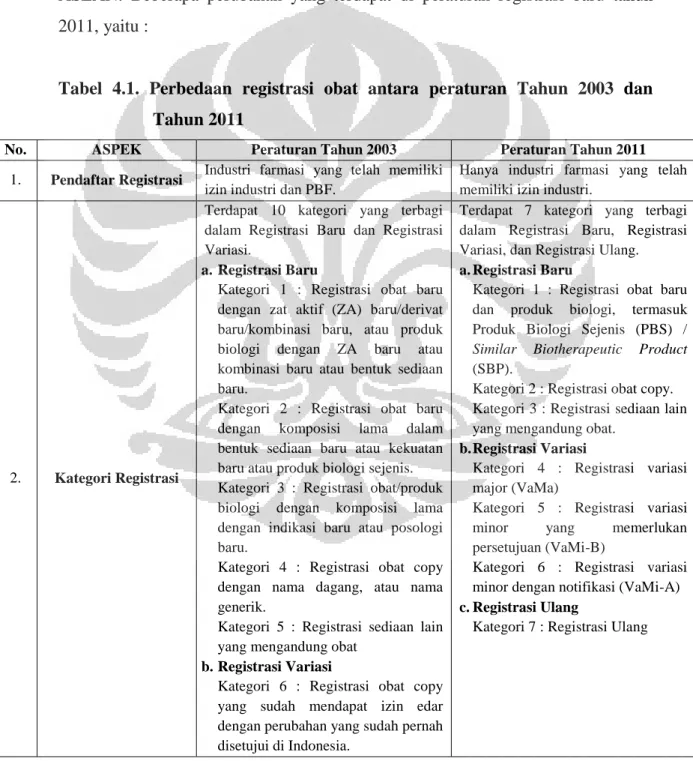
Tabel 4.1. Perbedaan Registrasi Obat Antara Peraturan Tahun 2003 dan Tahun 2011 ... 46 Tabel 4.2. Perbedaan Antara Sistem Registrasi dan Notifikasi Kosmetika . 51
Lampiran 1. Struktur Organisasi PT. Kalbe Farma, Tbk... 57
Lampiran 2. Struktur Kepemilikan Perseroan dan Anak Perusahaan PT. Kalbe Farma, Tbk ... 58
Lampiran 3. Isi Dokumen Pra Registrasi/Registrasi ... 59
Lampiran 4. Alur Registrasi ... 63
Lampiran 5. Alur Registrasi dan Evaluasi Obat ... 65
Lampiran 6. Kelengkapan Dokumen Registrasi Baru ... 66
Lampiran 7. Jenis Perubahan, Persyaratan, dan Kelengkapan Dokumen Registrasi Variasi ... 70
Lampiran 8. Persyaratan Registrasi Alat Kesehatan ... 99
1.1. Latar Belakang
Kehidupan manusia tidak terlepas dari kebutuhan baik jasmani maupun rohani. Kesehatan merupakan salah satu kebutuhan jasmani yang penting bagi manusia. Karena kesehatan sangat menunjang kehidupan, maka kebutuhan ini dilindungi oleh negara dengan dikeluarkannya undang-undang untuk menjamin kesehatan masyarakatnya. Kebutuhan akan kesehatan tersebut dapat diperoleh lebih lanjut melalui bahan, alat, maupun sarana penunjang seperti pangan dan bahan pangan; obat-obatan, baik obat-obatan sintesis, produk biologi, obat tradisional, obat herbal, dan fitofarmaka; suplemen makanan; kosmetika untuk menunjang, memelihara, dan mempertahankan kualitas hidup; dan alat kesehatan dan perbekalan kesehatan rumah tangga.
Salah satu yang mempengaruhi semakin meningkatnya kebutuhan manusia adalah modernisasi kehidupan dan kesadaran masyarakat. Selain itu, faktor lain seperti perkembangan ilmu pengetahuan dan teknologi juga memegang peranan penting dalam peningkatan kebutuhan, terutama dalam bidang kesehatan. Perkembangan dan modernisasi yang terwujud dalam produk yang dihasilkan oleh industri, haruslah diawasi keamanan dan kebenaran efikasinya sebelum digunakan masyarakat. Oleh karena itu, pemerintah sebagai pihak yang berwenang harus melakukan pengawasan terhadap peredaran bahan, alat, maupun sarana penunjang tersebut agar masyarakat dapat memperoleh produk yang bermutu, bermanfaat, mempunyai efikasi, dan aman jika digunakan. Hal tersebut sesuai dengan isi Undang-Undang Dasar 1945 Republik Indonesia yang menyebutkan bahwa segala aspek kehidupan yang berhubungan dengan hajat hidup orang banyak harus diatur oleh pemerintah. Pemerintah melalui Badan Pengawas Obat dan Makanan (BPOM) memiliki tanggung jawab dan wewenang penuh untuk melaksanakan pengawasan peredaran produk, baik sebelum produk tersebut diedarkan maupun sesudah produk tersebut diedarkan di masyarakat.
Setiap industri, baik industri dalam negeri maupun industri luar negeri, yang akan memasarkan produknya di Indonesia harus melalui proses registrasi terlebih dulu di BPOM untuk mendaftarkan produknya. Dengan adanya prosedur registrasi ini diharapkan masyarakat terlindung dari produk-produk yang tidak jelas mutu, efikasi, manfaat, dan keamanannya, sehingga produk-produk yang berkualitas saja yang beredar. Dalam pelaksanaan prosedur pendaftaran atau registrasi tersebut, industri ini memerlukan bagian yang dapat mengetahui, mengerti, dan memahami secara jelas pelaksanaan prosedur registrasi, persyaratan yang diperlukan, dan peraturan-peraturan pemerintah yang mengaturnya. Oleh karena itu, masing-masing industri harus memiliki bagian yang disebut dengan
Regulatory Affairs yang merupakan penghubung antara pihak industri dengan
pihak pemeritah, dalam hal ini BPOM sebagai badan pengawas.
Regulatory Affairs akan melaksanakan dan bertanggung jawab untuk
meregistrasikan setiap produk yang akan diedarkan oleh industri yang bersangkutan di masyarakat. Oleh karena itu, bagian Regulatory Affairs harus dapat menjamin bahwa produk yang akan diregistrasikannya tersebut benar-benar bermutu, aman, dan memiliki efikasi, serta manfaat. Hal tersebut dibuktikan melalui dokumen mutu yang dibawa saat akan melakukan proses registrasi produk. Apoteker sebagai salah satu tenaga kesehatan profesional, tentunya sangat berperan dalam bagian Regulatory Affairs ini karena tanggung jawabnya yang salah satunya adalah menjamin kesehatan masyarakat. Selain itu, Apoteker juga memiliki pengetahuan dan kompetensi di bagian Regulatory Affairs.
Oleh karena itu, sebelum para calon Apoteker mengaplikasikan kompetensi dan kemampuannya, maka para calon apoteker perlu diberikan pembekalan ilmu dan informasi yang lebih lagi untuk dapat lebih memahami mengenai tugas dan fungsinya. Oleh karena itu, Praktek Kerja Profesi Apoteker (PKPA), khususnya di Regulatory Affairs, perlu dilakukan oleh para calon Apoteker sehingga dapat lebih mengetahui dan sebagai gambaran di kemudian hari mengenai peranannya terhadap masyarakat di bidang industri, khususnya di bagian Regulatory Affairs sehingga masyarakat memperoleh produk-produk yang benar-benar bermutu, berefikasi, bermanfaat, dan aman.
1.2. Tujuan
Praktek Kerja Profesi Apoteker (PKPA) di PT. Kalbe Farma, Tbk., khususnya di Regulatory Affairs, bertujuan agar para calon Apoteker :
1. Memahami peranan apoteker di industri yang bergerak di bidang farmasi, khususnya pada bagian Regulatory Affairs.
2. Mengetahui mekanisme registrasi produk, baik obat dan produk biologi; obat tradisional, obat herbal terstandard, dan fitofarmaka; suplemen makanan, pangan, kosmetika, maupun alat kesehatan dan perbekalan kesehatan rumah tangga.
3. Mengetahui dan memahami lingkup kerja, tugas, dan fungsi seorang
2.1. PT. Kalbe Farma, Tbk. (PT. Kalbe Farma, Tbk., 2010)
2.1.1. Sejarah dan Perkembangan
Kalbe Farma didirikan oleh dr. Boenyamin Setiawan pada tanggal 10 September 1966 di Jalan Simpang I No. 1 Tanjung Priok, Jakarta Utara. Kalbe Farma memulai usaha di bidang produksi dan distribusi produk perawatan kesehatan dengan memproduksi Bioplacenton sebagai produk pertamanya yang dipasarkan tahun 1967. Pada tahun 1971, Kalbe Farma menempati pabrik yang lebih luas di Jalan Ahmad Yani, Pulomas, Jakarta Timur dan memperoleh status PMDN pada tahun 1974.
Kemajuan demi kemajuan dalam bidang bisnis diraih Kalbe Farma melalui semangat, tekad kuat, dan kerja keras. Dimulai pada tahun 1977, Kalbe Farma mengakuisisi PT. Dankos Laboratories. Pada tahun 1981, bisnis distribusi dialihkan kepada PT. Enseval sesuai dengan ketentuan pemerintah. Selanjutnya pada tahun 1985, Kalbe Farma mengakuisisi pula PT. Bintang Toedjoe dan PT. Hexpharm Jaya. Kemajuan bisnis Kalbe Farma semakin luas dan pesat dengan diakuisisinya PT. Sanghiang Perkasa pada tahun 1993 dan konsolidasi bisnis nutrisi dilakukan dalam anak perusahaan ini. Kalbe Farma group kembali mempertajam fokus bisnisnya pada produk kesehatan lainnya yang memiliki tingkat pertumbuhan menjanjikan seperti produk suplemen makanan dan obat tradisional melalui akuisisi 80% saham PT. Saka Farma pada tahun 1997.
Pada tahun 1998, pabrik Kalbe Farma berpindah lokasi ke Cikarang Bekasi dengan luas area dan bangunan yang lebih luas sehingga dapat menampung kegiatan industri yang makin berkembang secara menyeluruh. Pada tanggal 16 Desember 2005, dilakukan penggabungan usaha antara PT. Kalbe Farma, Dankos Laboratories (yang telah berubah nama menjadi Dankos Farma), dan PT. Enseval menjadi satu perusahaan terintegrasi dalam rangka menciptakan suatu perusahaan farmasi terbesar di kawasan Asia Tenggara. Fokus PT. Kalbe Farma di tahun berikutnya yaitu untuk memperluas cakupan regional, membangun
merek dan infrastruktur global, meningkatkan pengembangan penemuan obat, dan membangun jaringan dan kemitraan global. Hal ini terlihat di awal tahun 2007, Kalbe Farma memperkenalkan logo baru perusahaan menggunakan simbol DNA Helix yang menunjukkan bahwa perusahaan memfokuskan diri untuk masyarakat, peduli, dan berbagi juga warna hijau yang diasosiasikan sebagai lambang kehidupan, pertumbuhan dan inovasi. Di tahun yang sama, PT. Kalbe Farma, Tbk. juga mendirikan Stem Cell and Cancer Institute (SCI) yang bergerak di bidang riset sel punca dan kanker, yang memiliki potensi besar menjadi terapi masa depan menggantikan obat-obatan konvensional saat ini. Di tahun 2010, Manajemen Perseroan juga telah mengambil satu langkah besar yakni divestasi divisi kemasan Kalbe yaitu PT. Kageo Igar Jaya, Tbk. beserta anak perusahaannya. Hal ini dilakukan sehingga Kalbe dapat kembali fokus pada bisnis inti serta dapat mengalokasikan sumber daya yang ada ke bisnis inti Kalbe yakni menyediakan solusi kesehatan yang lengkap.
Hingga saat ini terdapat 9 SBU (Strategic Business Unit) yang dijalankan oleh PT. Kalbe Farma, Tbk., yaitu Pharmaceutical untuk obat-obatan yang diresepkan, Kesehatan Konsumen untuk obat-obat OTC, Nutrisi yang dijalankan oleh Sanghiang Perkasa, Distribusi dan Logistik dijalankan oleh PT. Enseval Putra Mitragading Tbk., Biofarma yang berhubungan dengan produk biologi seperti Laboratorium BE dan SCI, Eye care yang dijalankan oleh Kalbe Vision, Alat Kesehatan yang dijalankan oleh Enseval Medica Prima, Pelayanan Kesehatan melalui Klinik dan Apotek Mitra Sana, dan SBU Kalbe International. Dengan adanya 9 SBU ini, Kalbe Group telah berhasil memposisikan merek-mereknya sebagai pemimpin di dalam berbagai kategori terapi di pasar Indonesia dan juga internasional.
Kemajuan PT. Kalbe Farma, Tbk., yang begitu pesat dihasilkan dari penetapan tata kelola perusahaan yang baik oleh seluruh karyawan dan manajemen, termasuk pemegang saham. Tata kelola perusahaan yang baik berdasarkan Good Corporate Governance (GCG) telah diterapkan sejak tahun 2001, dan hingga saat ini kebijakan dan prakteknya terus diperkuat. Selain itu, strategi pertumbuhan Kalbe Group juga bertumpu pada tiga pilar penyempurnaan
2009. Strategi ini terus dikembangkan ke berbagai aspek yang lebih mendalam dan lebih meluas di dalam organisasi Kalbe Group. Penyempurnaan PIC terus ditingkatkan melalui mobilisasi dan rangkaian konvensi CONIM (Continous
Improvement) di seluruh jenjang karyawan. Visi, Misi dan Nilai-nilai yang
dipegang teguh oleh PT. Kalbe Farma, Tbk. juga menjadi pedoman untuk menjalankan roda perekonomiannya hingga menjadi perusahaan besar seperti saat ini.
2.1.2. Visi dan Misi
PT. Kalbe Farma, Tbk. mempunyai visi yaitu "To be the Best Indonesian
Health Care Company Driven by Innovation, Strong Brands, and Excellent Management". Visi tersebut apabila diartikan dalam Bahasa Indonesia berarti
“untuk menjadi perusahaan terbaik di Indonesia yang bergerak dalam pelayanan kesehatan melalui inovasi, merek dagang yang kuat, dan manajemen yang baik”. Adapun visi tersebut dicapai melalui misi perusahaan, yaitu "To improve health
for a better life" yang diartikan menjadi “meningkatkan kesehatan untuk hidup
yang lebih baik”.
2.1.3. Kalbe Panca Sradha
PT. Kalbe Farma, Tbk. memiliki lima kepercayaan atau nilai dalam menjalankan perusahaannya yang dikenal dengan Kalbe Panca Sradha. Lima kepercayaan tersebut adalah :
1. Trust is the glue of life
Saling percaya adalah perekat di antara kami 2. Mindfulness is the foundation of our action
Kesadaran penuh adalah dasar setiap tindakan kami 3. Innovation is the key to our success
Inovasi adalah kunci keberhasilan kami 4. Strive to be the best
Bertekad untuk menjadi yang terbaik
5. Interconnectedness is a universal way of life Saling keterkaitan adalah panduan hidup kami
2.1.4. Motto Perusahaan
Motto perusahaan adalah "The Scientific Pursuit of Health for a Better Life", yang menunjukkan bahwa perusahaan melakukan usaha pencarian di bidang kesehatan melalui ilmu pengetahuan sains dan teknologi untuk meraih kehidupan yang lebih baik.
2.2. Corporate Business Development
2.2.1. Product Planning
Product Planning (PPO) berperan dalam menangani persiapan peluncuran
produk baru dan melakukan pengawasan status perkembangan produk baru, serta mengevaluasi kelayakannya sehingga dapat diluncurkan pada jadwal yang ditetapkan dan sesuai dengan strategi perusahaan. Persyaratan yang dibutuhkan untuk menjadi PPO adalah kompetensi dalam hal negosiasi dan komunikasi. Hal tersebut dikarenakan peranannya yang berhubungan dengan produk baru. Peran dan fungsi PPO dalam perusahaan antara lain :
1. Melakukan koordinasi persiapan peluncuran produk baru dengan merencanakan rapat penjadwalan peluncuran produk dan rapat koordinasi produk baru.
2. Melakukan pengawasan dan menindaklanjuti status perkembangan produk baru mulai dari penjadwalan sampai produk tersebut dapat diluncurkan ke pasar sesuai jadwal yang telah direncanakan bersama dengan departemen terkait.
3. Melakukan koordinasi review sales, delisting product (pelaksanaan produk yang akan dimatikan peredarannya), transfer (pemindahan), dan rejuvinasi. 4. Berkomitmen terhadap implementasi kebijakan mutu, kesehatan, dan
keselamatan kerja dan lingkungan.
2.2.2. Business Development
Business Development (BD) berperan dalam memberikan layanan
pengembangan produk, bisnis dan servis baru untuk meningkatkan pertumbuhan perusahaan. Business Development dapat melakukan pendampingan dan membantu produktivitas perusahaan terhadap produk baru. Business Development
merupakan unit bisnis yang bertujuan untuk mengembangkan berbagai produk obat, alat kesehatan, nutrisi, dan suplemen kesehatan yang tepat dan untuk memastikan bahwa produk baru yang dikembangkan sejalan dengan kebijakan dan strategi bisnis.
Business Development (BD) dipimpin oleh seorang Manager BD yang harus
memiliki jiwa pengembangan bisnis yang baik. Hal ini sesuai dengan tujuan jabatannya, yaitu mampu mengembangkan bisnis dengan melihat trend perkembangan pengobatan sehingga dapat memberikan kontribusi penjualan untuk produk baru dan memaksimalkan nilai jual suatu produk. Peranan dan tugas BD antara lain :
1. Menganalisis peluang usaha terhadap produk-produk yang akan dikembangkan oleh PT. Kalbe Farma, Tbk. sesuai dengan kondisi marketing perusahaan dan kebijakan Badan Pengawas Obat dan Makanan (BPOM) selaku pihak evaluator.
2. Menganalisis paten, profil produk, penerimaan konsep produk oleh dokter dan konsumen, kompetitor, potensi pasar, dan pengembangannya ke depan.
3. Melakukan studi pre-marketing termasuk survei dari business case.
4. Melakukan negosiasi dan persetujuan dengan principal baru yang potensial, baik dalam bentuk lisensi maupun impor bahan baku material dan produk jadi. 5. Mengikuti trend penyakit, pengobatan, tindakan pencegahan, gaya hidup, dan
lain-lain yang berhubungan dengan kesehatan.
6. Menentukan dan mengusulkan kriteria delisting product.
7. Berkomitmen terhadap implementasi kebijakan mutu, kesehatan, dan keselamatan kerja dan lingkungan.
2.2.3. Regulatory Affairs 2.2.3.1. Struktur Organisasi
Regulatory Affairs (RA) adalah unit yang melakukan registrasi produk ke
Badan Pengawas Obat dan Makanan (BPOM) ataupun Kementerian Kesehatan Republik Indonesia (KemenKes RI). Produk-produk yang diregistrasikan oleh RA adalah produk dari PT. Kalbe Farma, Tbk., PT. Dankos Farma, PT. Hexpharm Jaya, dan PT. Finusolprima Farma Internasional. RA dipimpin oleh seorang
General Manager yang membawahi seorang Senior Regulatory Manager dan
seorang Regulatory Manager. Struktur organisasi dari RA dapat dilihat pada Gambar 2.1.
Gambar 2.1. Struktur Organisasi Regulatory Affairs
Dari gambar tersebut di atas terlihat bahwa terdapat dua kategori produk yang ditangani oleh RA Officer, yaitu :
1. Drug Kalbe
Beberapa RA Officer yang dipimpin langsung oleh Regulatory Manager menangani registrasi produk-produk obat dari PT. Kalbe Farma, Tbk. (produk dengan zat aktif baru, produk biologis, dan Branded Generic/BG) dan alat kesehatan.
2. Non Drug Kalbe
Para RA Officer yang bertanggung jawab atas Non Drug Kalbe menangani registrasi produk-produk obat dari PT. Hexpharm Jaya, PT. Dankos Farma, dan PT. Finusolprima Farma Internasional. Selain itu, juga melakukan registrasi obat tradisional dan suplemen kesehatan (TMHS), kosmetik, dan pangan. RA Officer yang menangani Non Drug Kalbe dipimpin oleh Senior Regulatory Manager.
2.2.3.2. Uraian Jabatan
Berdasarkan standard yang ditetapkan oleh PT. Kalbe Farma, Tbk., maka para pemangku jabatan di Regulatory Affairs (Regulatory Officer) harus memiliki pendidikan minimal Sarjana Farmasi. Selain itu, harus memiliki kompetensi seperti interpersonal skill yang baik, mampu berbahasa inggris, menguasai komputer, kemampuan komunikasi yang baik, memiliki pengetahuan mengenai registrasi, mampu memecahkan masalah, dan memiliki self leadership. Peran dan fungsi Regulatory Officer (RO) yaitu :
1. Mengkoordinir dan memonitor kegiatan pendaftaran dan memperoleh persetujuan produk dan perizinan lain yang dilakukan agar sesuai dengan jadwal yang ditetapkan.
2. Mengevaluasi dan menindaklanjuti proses pendaftaran produk dan perizinan melalui koordinasi dengan bagian terkait di dalam perusahaan maupun dengan pihak luar.
3. Memonitor pendaftaran SMF (Site Master File) dan memperoleh perizinan SMF tepat waktu.
4. Memonitor perolehan CPP (Certificate of Pharmaceutical Product), GMP (Good Manufacturing Practice), PPUB (Persetujuan Pelaksanaan Uji Bioekivalensi), PPUK (Persetujuan Pelaksanaan Uji Klinik), pemasukan obat jalur khusus atau SAS (Special Access Scheme), dan izin impor.
5. Memonitor perolehan persetujuan rancangan iklan dan promosi lainnya. 6. Membina hubungan baik dengan semua instansi terkait, yaitu BPOM,
KemenKes RI, dan Direktorat Jenderal Hak Kekayaan Intelektual (Ditjen HAKI).
7. Memastikan perusahaan update mengenai peraturan terbaru di bidang kesehatan yang berhubungan dengan bisnis perusahaan.
2.2.3.3. Hubungan Kerja
Regulatory Officer (RO) tidak dapat dipisahkan dari bagian lain dalam
melaksanakan tugasnya. Bagian-bagian yang bekerja sama secara internal dengan RO untuk memperoleh data dalam penyusunan dokumen registrasi, antara lain : 1. Bagian Medical, untuk memperoleh data package insert/brosur.
2. Bagian Marketing Ethical, Over The Counter, dan ekspor yang berhubungan dengan pemasaran produk obat.
3. Bagian Research and Development (R&D) atau Process Development (Proc. Dev.), untuk memperoleh informasi mengenai pengembangan produk dan kemasan, metode analisa, dan sebagainya.
4. Bagian Business Development (BD), berhubungan dengan perencanaan produk atau bisnis baru.
5. Bagian Pepustakaan, untuk memperoleh literatur yang diperlukan.
6. Bagian Purchasing, untuk memperoleh data sumber bahan baku obat, DMF (Drug Master File), dan spesifikasi masing-masing bahan baku yang dinyatakan dalam sertifikat analisis.
7. Bagian Pabrik seperti bagian Produksi, Production Planning Inventory
Control (PPIC), Quality Assurance (QA), dan Quality Control (QC), untuk
mendapatkan informasi mengenai perencanaan, pelaksanaan, dan pengawasan yang dilakukan selama proses produksi; data stabilitas; hasil validasi proses produksi; dan lain-lain dalam tujuannya menjamin kualitas obat.
8. Bagian Legal, untuk memperoleh trademark dari produk yang diregistrasikan.
Selain melaksanakan hubungan kerja sama internal, RO juga menjalin hubungan kerja sama eksternal dengan beberapa instansi terkait, seperti BPOM, Ditjen HAKI, KemenKes RI, Institusi Penelitian dan Pengembangan, Universitas, dan berbagai pihak terkait yang lainnya.
2.2.3.4. Alur Internal Proses Registrasi Obat
Proses registrasi obat dimulai dengan penyusunan FUPB/FUPP. FUPB adalah Formulir Usulan Produk Baru, sedangkan FUPP adalah Formulir Usulan Perubahan Produk. Kedua formulir ini diserahkan kepada RA oleh Bagian
Marketing yang bekerja sama dengan bagian BD berdasarkan ide yang mereka
dapatkan mengenai suatu produk. FUPB/FUPP berisi spesifikasi produk yang akan diproduksi, dan berbagai pertimbangan dari masing-masing bagian dalam perusahaan seperti bagian Marketing, bagian Medical, R&D, dan RA. Selanjutnya RA akan mengisi tabel registrasi dalam formulir tersebut. Setelah itu, RA akan
mengisi PDP (Permintaan Dokumen Pendaftaran) untuk tujuan pra registrasi dan mengirimkannya ke bagian BD, R&D atau Proc. Dev. Dokumen yang terkumpul akan direview oleh RA Manager, dan kemudian RO akan membawa dokumen tersebut ke BPOM untuk dievaluasi oleh evaluator.
Pada tahap pra registrasi setelah dokumen diperiksa, pendaftar akan diberikan formulir konsultasi yang harus dilengkapi jika masih terdapat kekurangan atau diberikan SPB (Surat Perintah Bayar) jika dokumen dinyatakan lengkap. Pendaftar harus membayar sesuai ketentuan dan menyerahkan bukti bayar beserta dokumen lengkap ke loket. Setelah hasil pra registrasi keluar, RA akan mengisi PDP untuk keperluan registrasi dan dokumen yang terkumpul diproses seperti tahap pra registrasi. Pada tahap ini kelengkapan yang harus diserahkan ke loket, yaitu bukti bayar, dokumen registrasi, dan disket registrasi yang berisi dokumen administratif, informasi umum mengenai produk, dan dokumen mutu mengenai bahan baku dan produk. Nomor izin edar (NIE) produk akan keluar dalam 120 - 360 hari kerja tergantung dari kategori registrasi produk yang didaftarkan. Selama menunggu NIE produk keluar dapat dilakukan konsultasi untuk mengetahui perkembangan hasil registrasi produk.
BAB 3
TINJAUAN KHUSUS
3.1. Registrasi (Kepala Badan Pengawas Obat dan Makanan Republik Indonesia,
2011a)
Registrasi adalah prosedur pendaftaran dan evaluasi suatu produk untuk mendapatkan izin edar. Izin edar adalah suatu bentuk persetujuan registrasi untuk dapat diedarkan di wilayah Indonesia. Registrasi bertujuan untuk melindungi masyarakat dari peredaran produk yang tidak memenuhi persyaratan khasiat, keamanan, dan mutu. Registrasi terdiri atas registrasi baru, registrasi variasi, dan registrasi ulang. Registrasi baru adalah registrasi produk yang belum mendapatkan izin edar di Indonesia. Registrasi variasi adalah registrasi perubahan aspek apapun pada produk yang telah memiliki izin edar di Indonesia. Registrasi ulang adalah registrasi perpanjangan masa berlaku izin edar.
3.2. Pendaftar
Pendaftar adalah industri farmasi yang telah mendapat izin industri farmasi sesuai ketentuan perundang-undangan (Kepala Badan Pengawas Obat dan Makanan Republik Indonesia, 2011a). Setiap pendaftar bertanggung jawab atas kelengkapan dokumen yang diserahkan, kebenaran semua informasi yang tercantum dalam dokumen registrasi, kebenaran dan keabsahan dokumen yang dilampirkan untuk kelengkapan registrasi, dan perubahan data dan informasi dari produk yang sedang dalam proses registrasi atau sudah memiliki izin edar (Kepala Badan Pengawas Obat dan Makanan Republik Indonesia, 2003). Jenis pendaftar dibagi menjadi beberapa kategori sesuai produk yang didaftarkan, yaitu (Kepala Badan Pengawas Obat dan Makanan Republik Indonesia, 2011a) :
3.2.1. Pendaftar Produk yang Diproduksi di Dalam Negeri
Produk dalam negeri meliputi produk tanpa lisensi, produk lisensi, dan produk kontrak. Pendaftar produk tanpa lisensi adalah pendaftar yang memiliki izin industri farmasi dan memiliki sertifikat Cara Pembuatan Obat yang Baik
(CPOB) yang masih berlaku sesuai dengan jenis dan bentuk sediaan yang diregistrasikan. Pendaftar produk lisensi adalah penerima lisensi yang memiliki ketentuan seperti pendaftar tanpa lisensi dan dokumen perjanjian lisensi. Pendaftar obat kontrak adalah pemberi kontrak yang memiliki izin industri farmasi, paling sedikit satu fasilitas produksi sediaan lain yang telah memenuhi persyaratan CPOB, dan dokumen perjanjian kontrak. Produk lisensi dan produk kontrak yang didaftarkan hanya berupa obat dan suplemen makanan.
3.2.2. Pendaftar Produk Impor
Pendaftar produk impor adalah industri farmasi dalam negeri yang mendapat persetujuan tertulis dari industri farmasi di luar negeri. Industri pemilik produk di luar negeri wajib memiliki izin industri farmasi dan memenuhi persyaratan CPOB yang dibuktikan dengan sertifikat CPOB yang masih berlaku atau dokumen lain yang setara, dan data inspeksi terakhir atau perubahan terkait paling lama dua tahun yang dikeluarkan oleh otoritas pengawas obat setempat dan/atau otoritas pengawas obat negara lain. Pendaftar juga harus menyerahkan dokumen SMF (Site Master File) terbaru jika induistri farmasi di luar negeri belum mempunyai produk dengan jenis dan bentuk sediaan yang sama dengan yang disetujui beredar di Indonesia atau industri tersebut mempunyai produk yang beredar di Indonesia dengan jenis dan bentuk sediaan yang sama namun terjadi perubahan pada fasilitas produksi.
3.2.3. Pendaftar Produk Khusus Ekspor
Pendaftar produk khusus ekspor adalah industri farmasi terdiri dari pendaftar produk dalam negeri yang ditujukan khusus ekspor dan produk impor khusus ekspor. Produk khusus ekspor dilarang diedarkan di wilayah Indonesia. Produk ekspor yang didaftarkan hanya berupa obat.
3.2.4. Pendaftar Produk yang Dilindungi Paten
Pendaftar produk yang dilindungi paten adalah pemilik hak paten atau yang ditunjuk oleh pemilik hak paten. Pendaftaran produk yang masih dilindungi paten dapat dilakukan oleh pendaftar yang bukan pemilik hak paten sesuai dengan
ketentuan perundang-undangan. Pendaftaran dapat diajukan mulai dua tahun sebelum berakhirnya perlindungan paten dengan melampirkan informasi tanggal berakhirnya perlindungan paten dan data ekivalensi untuk menjamin kesetaraan khasiat, keamanan, dan mutu.
3.3. Registrasi Obat (Kepala Badan Pengawas Obat dan Makanan Republik
Indonesia, 2011a)
3.3.1. Kategori Registrasi Obat
Registrasi obat terdiri atas registrasi baru, registrasi variasi, dan registrasi ulang.
3.3.1.1. Registrasi Baru
Permohonan registrasi baru diawali dengan proses pra registrasi. Kelengkapan dokumen dan persyaratan untuk registrasi baru dapat dilihat pada Lampiran 6. Registrasi baru terdiri atas tiga kategori, yaitu :
a. Kategori 1 : registrasi obat baru dan produk biologi, temasuk produk biologi sejenis (PBS) / Similar Biotherapic Product (SBP), meliputi :
1. Registrasi obat baru dengan zat aktif baru atau produk biologi 2. Registrasi obat baru atau produk biologi dengan kombinasi baru
3. Registrasi obat baru atau produk biologi dengan bentuk sediaan baru atau kekuatan baru
4. Registrasi obat baru atau produk biologi dengan rute pemberian baru 5. Registrasi produk biologi sejenis (PBS)/Similar Biotherapic Product
(SBP)
b. Kategori 2 : registrasi obat copy, meliputi :
1. Registrasi obat copy yang memerlukan uji klinik 2. Registrasi obat copy yang tidak memerlukan uji klinik c. Kategori 3 : registrasi sediaan lain yang mengandung obat 3.3.1.2. Registrasi Variasi
Registrasi variasi dilakukan apabila terjadi perubahan terhadap obat yang telah mendapat NIE. Kelengkapan dokumen, persyaratan, jenis perubahan dari registrasi variasi dapat dilihat pada Lampiran 7. Registrasi variasi terdiri atas tiga kategori, yaitu :
a. Kategori 4 : registrasi variasi major (VaMa)
b. Kategori 5 : registrasi variasi minor yang memerlukan persetujuan (VaMi-B) c. Kategori 6 : registrasi variasi minor dengan notifikasi (VaMi-A)
3.3.1.3. Registrasi Ulang
Permohonan pengajuan registrasi ulang dilakukan paling cepat 120 hari sebelum berakhir masa berlaku izin edarnya. Permohonan ini diajukan dengan mengisi formulir registrasi dan melampirkan dokumen registrasi ulang. Persetujuan atas permohonan registrasi ulang secara otomatis berlaku sejak berakhir masa izin edarnya, kecuali untuk registrasi ulang dengan informasi terbaru yang terkait aspek keamanan obat, khasiat obat, dan/atau kerasionalan formula obat. Registrasi ulang termasuk dalam kategori 7.
3.3.2. Tata Laksana Registrasi Obat
Permohonan pra registrasi dan registrasi diajukan oleh pendaftar secara tertulis kepada Kepala BPOM dan dilampiri dengan dokumen pra registrasi atau dokumen registrasi. Proses registrasi dibagi ke dalam dua tahap, yaitu tahap pra registrasi dan tahap registrasi. Kelengkapan persyaratan untuk proses pra registrasi atau registrasi dapat dilihat pada Lampiran 3 dan alur proses registrasi dapat dilihat pada Lampiran 4.
3.3.2.1 Tahap Pra Registrasi
Permohonan pra registrasi dilakukan untuk penapisan registrasi obat, penentuan kategori registasi, penentuan jalur evaluasi, penentuan biaya evaluasi, dan penentuan dokumen registrasi obat. Permohonan ini diajukan dengan mengisi formulir pra registrasi, menyerahkan bukti pembayaran biaya pra registrasi, dan melampirkan dokumen lengkap pra registrasi.
Pada tahap ini, paling lama dalam jangka waktu 40 hari sejak diterimanya permohonan pra registrasi Kepala BPOM memberikan surat hasil pra registrasi (HPR) kepada pendaftar yang berlaku satu tahun sejak tanggal dikeluarkan. Apabila sebelum jangka waktu yang dimaksud diperlukan penambahan data atas dokumen administratif dan/atau teknis, maka pendaftar akan diberikan surat permintaan tambahan data. Perhitungan jangka waktu pengeluaran HPR diberhentikan (clock off) sampai pendaftar menyampaikan tambahan data yang
diterima dan penyerahan tambahan data tersebut harus disampaikan paling lama 20 hari setelah surat dikeluarkan.
Jalur evaluasi untuk tahap pra registrasi terdiri atas :
1. Jalur 40 hari, meliputi registrasi variasi minor yang memerlukan persetujuan dan registrasi obat khusus ekspor
2. Jalur 100 hari meliputi :
a. Registrasi baru obat baru dan produk biologi yang diindikasikan untuk terapi penyakit serius yang mengancam nyawa manusia (life saving), dan/atau mudah menular pada orang lain, dan/atau belum ada atau kurangnya pilihan terapi lain yang aman dan efektif
b. Registrasi baru obat baru dan produk biologi yang berdasarkan justifikasi diindikasikan untuk penyakit serius dan langka (orphan drug)
c. Registrasi baru obat baru dan produk biologi ditujukan untuk program kesehatan masyarakat
d. Registrasi baru obat baru dan produk biologi yang telah melalui proses obat pengembangan baru yang dikembangkan oleh industri farmasi atau institusi riset di Indonesia dan seluruh tahapan uji kliniknya dilakukan di Indonesia
e. Registrasi baru obat copy esensial generik yang dilengkapi dengan dokumen penunjang kebutuhan program atau data pendukung sebagai obat esensial
f. Registrasi baru obat copy dengan standar informasi elektronik (Stinel) g. Registrasi variasi major indikasi baru/posologi baru untuk obat yang
ditujukan sebagaimana dimaksud pada huruf a-d.
h. Registrasi variasi major yang tidak termasuk pada huruf g. 3. Jalur 150 hari meliputi :
a. Registrasi baru obat baru, produk biologi, dan registrasi variasi major indikasi baru/posologi baru yang telah disetujui di negara yang telah menerapkan sistem evaluasi terharmonisasi dan di negara dengan sistem evaluasi yang telah dikenal baik
b. Registrasi baru obat baru, produk biologi, dan registrasi variasi major indikasi baru/posologi baru yang telah disetujui paling sedikit di tiga
negara dengan sistem evaluasi yang telah dikenal baik c. Registrasi baru obat copy tanpa Stinel
4. Jalur 300 hari meliputi registrasi baru obat baru, produk biologi, produk biologi sejenis, atau registrasi variasi major indikasi baru/posologi baru yang tidak termasuk dalam jalur evaluasi sebagaimana dimaksud pada jalur 100 dan 150 hari.
3.3.2.2 Tahap Registrasi
Pengajuan registrasi dilakukan dengan menyerahkan berkas registrasi dengan mengisi formulir registrasi dan disket disertai bukti pembayaran biaya evaluasi dan pendaftaran, dan HPR. Berkas registrasi terdiri atas formulir registrasi dengan dokumen administratif dan dokumen penunjang. Dokumen tersebut disusun sesuai format ASEAN Common Technical Dossier (ACTD) dan merupakan dokumen rahasia yang dipergunakan hanya untuk keperluan evaluasi oleh yang berwenang. Dokumen registrasi yang diserahkan harus dilengkapi dengan rancangan kemasan dan brosur. Rancangan kemasan, meliputi etiket, dus/bungkus luar, strip/blister, catch over, ampul atau vial, dan kemasan lain sesuai ketentuan tentang pembungkusan luar dan penandaan yang berlaku, yang merupakan rancangan kemasan obat yang akan diedarkan, dan dilengkapi dengan rancangan warna.
3.3.3. Evaluasi dan Pemberian Keputusan 3.3.3.1. Evaluasi
Evaluasi dilakukan terhadap dokumen registrasi yang telah dinyatakan lengkap. Alur registrasi dan evaluasi obat dapat dilihat pada Lampiran 5. Evaluasi dilaksanakan sesuai jalur evaluasi 40 hari kerja, 100 hari kerja, 150 hari kerja, atau 300 hari kerja yang dihitung sejak penyerahan dokumen registrasi obat. Untuk melakukan evaluasi dibentuk Komite Nasional (KOMNAS) Penilai Obat, Panitia Penilai Khasiat Keamanan, Panitia Penilai Mutu, dan Panitia Penilai Informasi Produk dan Penandaan.
Evaluasi data khasiat dan keamanan dilakukan berdasarkan pembuktian ilmiah dan pedoman penilaian khasiat dan keamanan oleh Penilai Khasiat Keamanan. Hasil evaluasi khasiat dan keamanan disampaikan kepada pendaftar
paling lambat 30 hari. Berdasarkan hasil evaluasi terebut, KOMNAS Penilai Obat dapat memberikan rekomendasi kepada Kepala BPOM. Apabila diperlukan klarifikasi dan/atau penjelasan teknis secara rinci dari dokumen yang diserahkan, KOMNAS Penilai Obat dapat merekomendasikan untuk dilakukan dengar pendapat oleh pendaftar. Untuk dengar pendapat, BPOM akan menyampaikan surat pemberitahuan kepada pendaftar. Evaluasi informasi produk dan penandaan dilakukan oleh Penilai Informasi Produk dan Penandaan sesuai kriteria yang lengkap, objektif, tidak menyesatkan yang menjamin penggunaan obat secara tepat, rasional, dan aman.
Jika diperlukan tambahan data, maka permintaan tambahan data akan disampaikan kepada pendaftar secara tertulis. Tambahan data ini harus disampaikan paling lama 100 hari setelah tanggal permintaan, sementara itu waktu perhitungan waktu evaluasi dihentikan. Perhitungan waktu evaluasi dilanjutkan setelah pendaftar menyerahkan tambahan data dan jika pendaftar tidak dapat memenuhi maka Kepala BPOM mengeluarkan surat penolakan.
3.3.3.2 Pemberian Keputusan
Keputusan terhadap registrsai obat dapat berupa pemberian persetujuan atau penolakan yang dipertimbangkan berdasarkan hasil evaluasi dokumen registrasi dan hasil pemeriksaan pada pabrik pembuatan obat.
1. Persetujuan
Persetujuan diberikan secara tertulis kepada pendaftar berupa peretujuan izin edar, persetujuan impor dalam bentuk ruahan, persetujuan impor khusus ekspor, dan persetujuan khusus ekspor.
2. Penolakan
Penolakan registrasi disampaikan secara tertulis oleh Kepala BPOM berupa surat penolakan dan biaya registrasi yang telah dibayarkan tidak dapat ditarik kembali. Registrasi yang ditolak dapat diajukan kembali dengan mengikuti tata cara sesuai ketentuan.
3. Dengar pendapat
Jika terdapat keberatan terhadap hasil evaluasi khasiat dan keamanan dari KOMNAS Penilai Obat maka pendaftar dapat mengajukan permohonan dengar
pendapat secara tertulis dalam jangka waktu 20 hari sejak tanggal surat pemberitahuan kepada Kepala BPOM.
4. Peninjauan kembali
Jika keputusan hasil registrasi berupa penolakan maka pendaftar dapat mengajukan permohonan peninjauan kembali kepada Kepala BPOM. Peninjauan kembali ini dapat diajukan paling lama enam bulan setelah tanggal surat penolakan dan hanya dapat dilakukan satu kali. Permohonan ini harus dilengkapi dengan data baru dan/atau data yang sudah pernah diajukan dengan dilengkapi justifikasi. Pembahasan terhadap surat permohonan ini dilakukan paling lama 100 hari sejak dokumen diterima.
5. Pengajuan kembali registrasi
Apabila registrasi ditolak, pendaftar dapat mengajukan permohonan registrasi kembali sesuai ketentuan. Akan tetapi jika registrasi ditolak karena alasan tidak memenuhi kriteria khasiat dan keamanan, selain harus mengikuti tata cara sesuai ketentuan, registrasi kembali hanya dapat diajukan dengan data baru dan paling cepat satu tahun setelah tanggal surat penolakan.
3.3.4. Masa Berlaku dan Pelaksanaan Izin Edar 3.3.4.1. Masa Berlaku Izin Edar
Izin edar obat berlaku paling lama lima tahun selama masih memenuhi ketentuan yang berlaku termasuk persetujuan impor dalam bentuk ruahan, persetujuan impor khusus ekspor, dan persetujuan khusus ekspor. Jika obat yang diregistrasikan berdasarkan perjanjian/penunjukkan dengan masa kerja sama kurang dari lima tahun, maka masa berlaku izin edar disesuaikan dengan masa berlaku kerja sama dalam dokumen perjanjian. Dalam hal perjanjian/penunjukkan kerja sama dihentikan sebelum masa izin edar berakhir, izin edar obat yang bersangkutan dibatalkan. Obat yang telah habis masa berlaku izin edarnya dapat diperpanjang selama memenuhi kriteria melalui mekanisme registrasi ulang. Apabila obat yang telah habis masa berlaku izin edarnya dan tidak diperpanjang maka dianggap sebagai obat yang tidak memiliki izin edar.
3.3.4.2. Pelaksanaan Izin Edar
Pendaftar wajib memproduksi atau mengimpor, dan mengedarkan obat yang telah mendapatkan izin edar selambatnya satu tahun setelah tanggal persetujuan dikeluarkan dan harus melapor kepada Kepala BPOM dengan menyerahkan kemasan siap edar. Kemasan siap edar yang diserahkan berupa kemasan primer, kemasan sekunder, dan informasi produk. Penyerahan kemasan dilakukan paling lambat satu bulan sebelum pelaksanaan peredaran obat. Pemilik izin edar obat wajib melakukan pemantauan khasiat, keamanan, dan mutu selama obat diedarkan dan melaporkan hasilnya kepada Kepala BPOM.
3.3.5. Evaluasi Kembali dan Sanksi
Evaluasi kembali dapat dilakukan terhadap obat yang telah mendapat izin edar. Evaluasi ini dilakukan jika berdasarkan hasil pemantauan terdapat perkembangan baru mengenai khasiat, keamanan, dan mutu obat yang berbeda dari data penunjang saat registrasi. Keputusan hasil evaluasi kembali dapat berupa perubahan penandaan, perbaikan komposisi/formula, pemberian batasan penggunaan, penarikan obat dari peredaran, dan/atau pembekuan izin edar dan/atau pembatalan izin edar.
Pendaftar yang tidak memenuhi ketentuan dapat dikenakan sanksi administratif berupa peringatan tertulis, pembatalan proses registrasi obat, pembekuan izin edar obat yang bersangkutan, pembatalan izin edar obat yang bersangkutan, atau sanksi administratif lain sesuai ketentuan perundang-undangan. Pemberian sanksi berupa pembatalan atau pembekuan izin edar terjadi jika tidak melaksanakan kewajiban memproduksi/mengimpor/mengedarkan obat yang telah mendapat izin edar, selama 12 bulan berturut-turut tidak memproduksi/mengimpor/mengedarkan obat, izin industri farmasi pemilik izin edar dicabut, dan/atau pemilik izin edar melakukan pelanggaran di bidang produksi dan/atau distribusi obat. Pembekuan dan pembatalan izin edar dilakukan secara tertulis kepada pemilik izin edar.
3.4. Registrasi Obat Tradisional, Obat Herbal Terstandard, dan Fitofarmaka (Kepala Badan Pengawas Obat dan Makanan Republik
Indonesia, 2005c)
3.4.1. Persyaratan dan Kriteria
Obat tradisional, obat herbal terstandard (OHT), dan fitofarmaka yang dibuat dan/atau diedarkan di wilayah Indonesia wajib memiliki izin edar dari Kepala BPOM oleh karena itu diperlukan proses pendaftaran. Pendaftaran tersebut tidak termasuk untuk obat yang digunakan untuk penelitian, obat tradisional impor yang digunakan sendiri dalam jumlah terbatas, obat tradisional impor yang telah terdaftar dan beredar di negara asal untuk tujuan pameran dalam jumlah terbatas, obat tradisional tanpa penandaan yang dibuat oleh usaha jamu racikan dan jamu gendong, dan bahan baku berupa simplisia dan sediaan galenik.
Kriteria yang harus dipenuhi obat tradisional, OHT, dan fitofarmaka adalah harus menggunakan bahan berkhasiat dan bahan tambahan yang memenuhi persyaratan mutu, keamanan dan kemanfaatan/khasiat; obat dibuat sesuai dengan ketentuan tentang Pedoman Cara Pembuatan Obat Tradisional yang Baik (CPOTB) yang berlaku, dan penandaan berisi informasi lengkap dan obyektif yang dapat menjamin penggunaan obat tradisional, OHT, dan fitofarmaka yang tepat, rasional dan aman sesuai dengan hasil evaluasi dalam rangka pendaftaran.
Pendaftar obat tradisional, obat herbal terstandard, dan fitofarmaka memiliki tanggung jawab sebagai berikut :
1. Memastikan dokumen yang diserahkan lengkap.
2. Menjamin kebenaran semua informasi yang tercantum dalam dokumen pendaftaran.
3. Menjamin kebenaran dan keabsahan dokumen yang dilampirkan untuk kelengkapan pendaftaran.
4. Bertanggung jawab atas perubahan data dan informasi dari produk yang sedang dalam proses.
3.4.2. Kategori Pendaftaran
Pendaftaran OT, OHT, dan fitofarmaka dikategorikan menjadi pendaftaran baru dan pendaftaran variasi. Pendaftaran baru terdiri dari :
a. Kategori 1 : Pendaftaran OT yang hanya mengandung simplisia berasal dari Indonesia (indigenous) dalam bentuk sediaan sederhana (rajangan, serbuk, parem, pilis, dodol, tapel, cairan obat luar). b. Kategori 2 : Pendaftaran obat tradisional yang hanya mengandung simplisia
berasal dari Indonesia (indigenous) dalam bentuk sediaan modern (pil, tablet, kapsul, krim, gel, salep, supositoria anal, cairan obat dalam).
c. Kategori 3 : Pendaftaran obat tradisional dari kategori 1 dan 2 dengan klaim indikasi baru, bentuk sediaan baru, posologi dan dosis baru. d. Kategori 4 : Pendaftaran OHT.
e. Kategori 5 : Pendaftaran fitofarmaka.
f. Kategori 6 : Pendaftaran kategori 4 dan 5 dengan klaim indikasi baru, bentuk sediaan baru, dan dosis baru.
g. Kategori 7 : Pendaftaran obat tradisional yang mengandung simplisia berasal bukan dari Indonesia (non-indigenous) dan/atau simplisia yang profil keamanannya belum diketahui dengan pasti.
h. Kategori 8 : Pendaftaran obat tradisional dari kategori 7 dengan klaim indikasi baru, bentuk sediaan baru, posologi baru, bentuk sediaan baru, posologi dan dosis baru.
Sedangkan untuk pendaftaran variasi dilakukan terhadap OT, OHT, dan fitofarmaka yang telah mendapat NIE dengan perubahan, seperti :
a. Kategori 9 : Pendaftaran OT, OHT, dan fitofarmaka yang telah mendapat izin edar dengan :
9.1. Perubahan nama produk tanpa perubahan komposisi 9.2. Perubahan atau penambahan ukuran kemasan
9.3. Perubahan klaim pada penandaan yang tidak merubah manfaat 9.4. Perubahan desain kemasan
9.5. Perubahan nama pabrik atau nama pemberi lisensi, tanpa perubahan status kepemilikan
9.6. Perubahan nama importir, tanpa perubahan status kepemilikan
b. Kategori 10 : Pendaftaran OT, OHT, dan fitofarmaka yang telah mendapat izin edar dengan :
10.1. Perubahan spesifikasi dan/atau metoda analisis bahan baku. 10.2. Perubahan spesifikasi dan/atau metoda analisis produk jadi 10.3. Perubahan stabilitas
10.4. Perubahan teknologi produksi 10.5. Perubahan tempat produksi
10.6. Perubahan atau penambahan jenis kemasan
c. Kategori 11 : Pendaftaran OT, OHT, dan fitofarmaka yang telah mendapat izin edar dengan perubahan formula atau komposisi termasuk bahan tambahan yang tidak mengubah khasiat.
3.4.3. Tata Laksana Memperoleh Izin Edar
Pendaftaran OT, OHT, dan fitofarmaka dilakukan dalam 2 (dua) tahap yaitu pra penilaian dan penilaian. Pra penilaian merupakan tahap pemeriksaan kelengkapan, keabsahan dokumen, dan dilakukan penentuan kategori pendaftaran (baru atau variasi). Penilaian merupakan proses evaluasi terhadap dokumen dan data pendukung. Hasil pra penilaian diberitahukan selambat-lambatnya 10 hari kerja (HK) untuk pendaftaran variasi dan 20 HK untuk pendaftaran baru.
Pengajuan pendaftaran dilakukan dengan menyerahkan berkas pendaftaran yang terdiri dari formulir atau disket pendaftaran yang telah diisi, dilengkapi dengan dokumen pendukung. Dokumen pendukung yang dimaksud adalah dokumen mutu dan teknologi, serta dokumen yang mendukung klaim indikasi sesuai jenis dan tingkat pembuktian. Berkas pendaftaran harus dilengkapi dengan rancangan kemasan dan brosur yang mencantumkan informasi mengenai OT, OHT, dan fitofarmaka.
Berkas yang diserahkan pada pendaftaran baru terdiri dari formulir TA, TB, TC, dan TD. Formulir TA berisi keterangan mengenai dokumen administrasi, formulir TB berisi dokumen yang mencakup formula dan cara pembuatan, formulir TC berisi dokumen yang mencakup cara pemeriksaan mutu bahan baku dan produk jadi, dan formulir TD berisi dokumen yang mencakup klaim indikasi, dosis, cara pemakaian, dan bets. Untuk pendaftaran variasi berkas yang diserahkan terdiri dari formulir pendaftaran variasi dan kelengkapan pendaftaran variasi untuk masing-masing kategori.
Jika dokumen pendaftaran OT, OHT, dan fitofarmaka telah memenuhi ketentuan, akan dilaksanakan penilaian oleh Panitia Penilai Obat Tradisional (PPOT) dan Komite Penilai Obat Tradisional (KOMNAS POT) yang dilakukan melalui :
a. Jalur 1
1.1. Untuk produk kategori 1 dan 2 yang menggunakan nama umum dengan komposisi tunggal atau komposisi sederhana (maksimum 5 jenis bahan). 1.2. Untuk produk kategori 9 yang variasinya tidak mempengaruhi mutu dan
keamanan. b. Jalur 2
2.1.Untuk produk kategori 1 dan 2 yang menggunakan nama dagang dengan komposisi tunggal atau kompleks.
2.2.Untuk produk kategori 10 yang variasinya mempengaruhi mutu. c. Jalur 3
3.1. Untuk produk kategori 3.
3.2. Untuk produk kategori 11 yang variasinya mempengaruhi mutu. d. Jalur 4 : Untuk produk kategori 6 dan 8.
e. Jalur 5 : Untuk produk kategori 4, 5, dan 7.
Hasil penilaian mutu, keamanan, dan khasiat dapat berupa memenuhi syarat, belum memenuhi syarat, atau tidak memenuhi syarat. Jika hasil penilaian memenuhi syarat, Kepala BPOM akan memberikan surat keputusan persetujuan pendaftaran. Jika belum memenuhi persyaratan dan memerlukan tambahan data, pendaftar akan diberitahukan keterangan permintaan tambahan data. Tambahan data ini selambat-lambatnya harus diserahkan 3 (tiga) bulan terhitung tanggal pemberitahuan dan bila dilampaui maka berkas pendaftaran akan dikembalikan. Berkas yang dikembalikan dapat diajukan kembali sebagai pendaftaran baru dan dilengkapi dengan tambahan data. Keputusan hasil penilaian diberikan mulai dari 7 - 90 HK tergantung dari jalur pendaftarannya.
3.5. Registrasi Suplemen Makanan (Kepala Badan Pengawas Obat dan
Makanan, 2005a dan 2005b)
kebutuhan zat gizi makanan, mengandung satu atau lebih bahan berupa vitamin, mineral, asam amino atau bahan lain (berasal dari tumbuhan atau bukan tumbuhan) yang mempunyai nilai gizi dan/atau efek fisiologis dalam jumlah yang terkonsentrasi. Suplemen makanan yang dapat didaftarkan berupa suplemen makanan dalam negeri (suplemen makanan tanpa lisensi, suplemen makanan dengan lisensi, suplemen makanan kontrak), suplemen makanan impor, dan suplemen makanan yang dilindungi oleh paten.
Suplemen makanan yang akan diedarkan harus memiliki beberapa kriteria, seperti menggunakan bahan yang memenuhi standard mutu dan persyaratan keamanan, serta standard dan persyaratan lain yang ditetapkan; kemanfaatan yang dinilai dari komposisi dan/atau didukung oleh data pembuktian; hanya dapat diproduksi oleh industri farmasi/industri obat tradisional/industri pangan dengan menerapakan Cara Pembuatan yang Baik (CPOB/CPOTB/CPPB); kemanfaatan suplemen makanan harus disesuaikan dengan jumlah dan komposisi bahan yang dikandungnya; bahan yang berasal dari tumbuhan/hewan/mikroorganisme non patogen yang digunakan dalam bentuk kombinasi harus memiliki kesesuaian khasiat yang didukung dengan data pembuktian.
Suplemen makanan harus dikemas dalam wadah yang dapat melindungi isi terhadap pengaruh dari luar selama masa peredaran dan menjamin mutu, keutuhan, dan keaslian isinya, serta wadah harus dibuat dengan mempertimbangkan keamanan pemakai dan dibuat dari bahan yang tidak mengeluarkan atau menghasilkan bahan berbahaya atau bahan yang dapat mengganggu kesehatan dan tidak mempengaruhi mutu. Dalam memberikan penandaan pada wadah dan pembungkus harus mencantumkan informasi yang lengkap, obyektif, benar, tidak menyesatkan, dan sesuai dengan penandaan yang telah disetujui pada saat pendaftaran.
3.5.1 Kategori Pendaftaran
Pendaftaran suplemen makanan dikategorikan menjadi dua, yaitu pendaftaran baru dan pendaftaran variasi.
Pengajuan pendaftaran baru dilakukan dengan menyerahkan berkas yang terdiri dari formulir SA, SB, dan SC. Formulir SA berisi keterangan mengenai dokumen administrasi; formulir SB berisi dokumen yang mencakup formula dan cara pembuatan; dan formulir SC berisi dokumen yang mencakup cara pemeriksaan mutu bahan baku dan produk jadi.
Pendaftaran baru dibagi menjadi tiga kategori, yaitu :
a. Kategori 1 : Pendaftaran suplemen makanan yang mengandung satu atau lebih bahan berupa vitamin, mineral, asam amino, karbohidrat, protein, lemak atau bahan lain berupa isolat.
b. Kategori 2 : Pendaftaran suplemen makanan yang mengandung satu atau lebih bahan berupa vitamin, mineral, asam amino, karbohidrat, protein, lemak, isolat lain, dan bahan lain berupa bahan alam. c. Kategori 3 : Pendaftaran suplemen makanan dari kategori 1 dan 2 dengan
klaim penggunaan baru, bentuk sediaan baru, posologi, dan dosis baru.
3.5.1.2 Pendaftaran Variasi
Pengajuan pendaftaran variasi dilakukan dengan menyerahkan berkas yang terdiri dari formulir pendaftaran variasi dan kelengkapan pendaftaran variasi untuk masing-masing kategori. Pendaftaran variasi dibagi menjadi tiga kategori, yaitu :
1. Kategori 4 : Pendaftaran suplemen makanan yang telah mendapat izin edar dengan :
4.1.Perubahan nama produk tanpa perubahan komposisi. 4.2.Perubahan atau penambahan ukuran kemasan.
4.3.Perubahan klaim pada penandaan yang tidak mengubah manfaat. 4.4.Perubahan desain kemasan.
4.5.Perubahan nama pabrik atau nama pemberi lisensi tanpa perubahan status kepemilikan.
4.6.Perubahan nama importir, tanpa perubahan status kepemilikan.
b. Kategori 5 : pendaftaran suplemen makanan yang telah mendapat izin edar dengan :
5.2.Perubahan spesifikasi dan/atau metode analisis produk jadi. 5.3.Perubahan stabilitas.
5.4.Perubahan teknologi produksi. 5.5.Perubahan tempat produksi.
5.6.Perubahan atau penambahan jenis kemasan.
c. Kategori 6 : pendaftaran suplemen makanan yang telah mendapat izin edar dengan :
6.1.Perubahan formula atau komposisi yang bahan utamanya tergolong dalam satu kelompok.
6.2.Perubahan bahan tambahan yang tidak mengubah manfaat.
3.5.2 Tata Laksana Memperoleh Izin Edar 3.5.2.1 Pendaftaran
Pendaftaran untuk suplemen makanan diajukan kepada Kepala BPOM dan dilakukan dalam dua tahap, yaitu tahap pra penilaian dan tahap penilaian. Pra penilaian adalah tahap dimana dilakukan pemeriksaan kelengkapan dan keabsahan dokumen, serta untuk menentukan kategori pendaftaran. Sedangkan, tahap penilaian merupakan proses evaluasi terhadap dokumen dan data pendukung. Hasil pra penilaian diberitahukan kepada pendaftar secara tertulis paling lambat sepuluh hari kerja untuk pendaftaran variasi dan dua puluh hari kerja untuk pendaftaran baru terhitung sejak tanggal diterimanya berkas pendaftaran. Hasil pra penilaian bersifat mengikat.
Pengajuan pendaftaran untuk suplemen makanan dilakukan dengan menyerahkan berkas pendaftaran yang terdiri dari formulir atau disket pendaftaran yang telah diisi. Berkas pendaftaran tersebut harus dlengkapi dengan rancangan kemasan suplemen makanan yang akan diedarkan yang meliputi etiket, dus, pembungkus, strip, blister, catch over, dan kemasan lain sesuai ketentuan tentang pembungkus dan penandaan yang berlaku, dan dilengkapi dengan rancangan warna, serta brosur yang mencantumkan informasi mengenai suplemen makanan. Selain itu, pendaftar juga harus melengkapinya dengan dokumen administrasi dan dokumen pendukung yang terdiri dari dokumen mutu dan teknologi, serta dokumen yang mendukung klaim kegunaan sesuai jenis dan tingkat pembuktian.
3.5.2.2 Penilaian
Dokumen pendaftaran suplemen makanan yang telah memenuhi ketentuan dan persyaratan, selanjutnya akan dilakukan penilaian terhadap suplemen makanan yang akan didaftarkan sesuai kriteria yang harus dimiliki pada masing-masing suplemen makanan. Hasil penilaian mutu, keamanan, dan kemanfaatan dapat berupa memenuhi syarat, belum memenuhi syarat, atau tidak memenuhi syarat. Untuk melakukan penilaian dibentuk Panitia Penilai Suplemen Makanan (PPSM) dan Komite Nasional Penilai Suplemen Makanan (KOMNAS PSM). Pelaksanaan penilaian yang dilakukan melalui:
1. Jalur 1 (7 HK)
a. Untuk suplemen makanan kategori 1 yang menggunakan nama generik. b.Untuk suplemen makanan kategori 4.
2. Jalur 2 (15 HK)
a. Untuk suplemen makanan kategori 1 yang menggunakan nama dagang. b.Untuk suplemen makanan kategori 5.
3. Jalur 3 (30 HK)
a. Untuk suplemen makanan kategori 2 yang profil keamanannya telah diketahui dengan pasti.
b.Untuk suplemen makanan kategori 6. 4. Jalur 4 (60 HK)
a. Untuk suplemen makanan kategori 2 dengan profil keamanan belum diketahui dengan pasti dan kategori 3.
3.6. Registrasi Pangan (Kepala Badan Pengawas Obat dan Makanan Republik
Indonesia, 2011b dan 2011c)
Pangan adalah segala sesuatu yang berasal dari sumber hayati dan air, baik yang diolah maupun yang tidak diolah, yang digunakan sebagai makanan atau minuman untuk dikonsumsi, termasuk bahan tambahan pangan (BTP), bahan baku pangan, dan bahan lain yang digunakan dalam proses penyiapan, pengolahan, dan/atau pembuatan makanan atau minuman. Sedangkan, pangan olahan adalah makanan atau minuman hasil proses dengan cara atau metode tertentu dengan atau tanpa bahan tambahan, termasuk pangan olahan tertentu,
bahan tambahan pangan, pangan produk rekayasa genetika, dan pangan iradiasi. Setiap pangan olahan, baik yang diproduksi di dalam negeri ataupun yang dimasukkan ke wilayah Indonesia, dengan maksud untuk diperdagangkan wajib memiliki surat persetujuan pendaftaran yang dikeluarkan oleh Kepala BPOM. Pangan olahan yang didaftarkan dibedakan menjadi pangan olahan produksi sendiri, pangan olahan berlisensi, pangan olahan yang dikemas kembali, dan pangan olahan yang diproduksi berdasarkan kontrak. Akan tetapi, terdapat pangan olahan yang tidak wajib memiliki surat persetujuan pendaftaran, seperti :
1. Pangan olahan yang diproduksi oleh industri rumah tangga yang memiliki sertifikat produksi pangan industri rumah tangga.
2. Pangan olahan yang mempunyai masa simpan kurang dari tujuh hari pada suhu kamar.
3. Pangan olahan yang dimasukkan ke wilayah Indonesia dalam jumlah kecil untuk sampel pangan olahan yang digunakan untuk keperluan pendaftaran, penelitian, dan konsumsi sendiri.
4. Pangan olahan yang digunakan lebih lanjut sebagai bahan baku dan tidak dijual secara langsung kepada konsumen akhir.
Pangan olahan yang akan didaftarkan di wilayah Indonesia harus memenuhi kriteria, seperti keamanan yang meliputi batas maksimum cemaran mikroba, cemaran fisik, dan cemaran kimia; pemenuhan persyaratan mutu sesuai standar dan persyaratan yang berlaku, serta cara produksi pangan yang baik untuk pangan olahan yang diproduksi di dalam negeri atau cara distribusi pangan yang baik untuk pangan olahan yang dimasukkan ke wilayah Indonesia; gizi yang sesuai dengan persyaratan yang ditetapkan; dan harus memenuhi persyaratan label.
3.6.1 Pendaftaran Umum
Permohonan pendaftaran diajukan secara tertulis dengan mengisi formulir pendaftaran yang disertai dengan kelengkapan dokumen pendaftaran sebanyak dua rangkap (asli dan fotokopi) kepada Kepala BPOM dan Direktur untuk dilakukan pemeriksaan dokumen yang sesuai kriteria dan persyaratannya. Hasil dari pemeriksaan dokumen tersebut dapat berupa diterima untuk dinilai lebih
lanjut, dikembalikan untuk dilengkapi, ataupun ditolak. Apabila dokumen yang diajukan diterima, maka kemudian akan dilakukan penilaian dan hasilnya berupa surat persetujuan pendaftaran atau surat penolakan pendaftaran. Jangka waktu dikeluarkannya surat tersebut bergantung masing-masing jenis pangan yang didaftarkan, yaitu :
1. Untuk pangan olahan tertentu dikeluarkan paling lama 150 hari.
2. Untuk pangan fungsional/pangan berklaim, pangan dengan herbal dikeluarkan paling lama 120 hari.
3. Untuk pangan iradiasi, pangan hasil rekayasa genetika, BTP perisa, pangan organik, susu dan hasil olahnya, daging dan hasil olahnya, ikan dan hasil olahnya, serta minuman beralkohol dikeluarkan paling lama 100 hari.
4. BTP selain perisa dan pangan lainnya dikeluarkan paling lama 60 hari.
Apabila pada hasil penilaian diperlukan tambahan data dan/atau kajian lebih lanjut, maka akan dikeluarkan surat permintaan tambahan data. Pendaftar harus menyerahkan tambahan data yang diminta tersebut paling lambat 50 hari setelah tanggal surat permintaan tambahan data. Jika kelengkapan data yang diserahkan belum memenuhi persyaratan sesuai permintaan maka pendaftar wajib menyerahkan tambahan data paling lambat 15 hari setelah tanggal surat permintaan tambahan data. Jika pendaftar menganggap waktu tersebut tidak mencukupi, maka pendaftar dapat mengajukan permintaan perpanjangan waktu untuk melengkapi tambahan data sebanyak satu kali untuk waktu 25 hari. Pendaftar yang tidak menyerahkan tambahan data selama waktu yang telah ditetapkan akan diberikan surat penolakan pendaftaran dan berkas permohonannya akan dimusnahkan.
Apabila terdapat keberatan terhadap hasil penilaian atas kriteria keamanan pangan olahan, maka pendaftar dapat mengajukan permohonan dengar pendapat kepada Kepala BPOM paling lama 25 hari sejak tanggal surat tambahan data. Akan tetapi, apabila terdapat keberatan terhadap penolakan pendaftaran, maka pendaftar dapat mengajukan permohonan peninjauan kembali sebanyak satu kali dalam waktu paling lama 50 hari setelah tanggal surat penolakan. Permohonan peninjauan kembali harus dilengkapi dengan data baru dan/atau data yang sudah pernah diajukan yang dilengkapi dengan justifikasi. Keputusan atas permohonan
peninjauan kembali akan diberikan dalam waktu paling lama 150 hari sejak tanggal permohonan peninjauan kembali.
Surat persetujuan pendaftaran berkaku selama lima tahun dan dapat diperpanjang melalui pendaftaran kembali. Surat persetujuan pendaftaran yang telah habis masa berlakunya dinyatakan tidak berlaku dan pangan olahan tersebut dilarang untuk diedarkan. Untuk melakukan pendaftaran kembali pangan olahan yang telah habis masa berlakunya dapat dilakukan paling cepat enam bulan sebelum masa berlaku surat peretujuan pendaftaran berakhir. Dalam mengajukan pendaftaran kembali, pendaftar dapat melakukan perubahan data pangan olahan. Surat persetujuan pendaftaran atau surat penolakan pendaftaran pada pendafaran kembali yang tidak mengalami perubahan dapat dikeluarkan paling lama :
a. 75 hari untuk pangan olahan tertentu.
b. 50 hari untuk pangan fungsional/pangan berklaim, pangan dengan herbal. c. 45 hari untuk pangan iradiasi, pangan hasil rekayasa genetika, BTP perisa, dan
pangan organik.
d. 30 hari untuk BTP selain perisa dan pangan lainnya.
Pangan olahan yang telah mendapat surat persetujuan pendaftaran dapat dilakukan penilaian kembali oleh Kepala BPOM jika terdapat data dan/atau informasi baru terkait keamanan, mutu, gizi, dan label pangan olahan yang bersangkutan. Hasil penilaian kembali disampaikan kepada perusahaan pemegang surat persetujuan pendaftaran dan perusahaan tersebut wajib melakukan tindakan sesuai hasil penilaian kembali.
3.6.2 Perubahan Data
Permohonan perubahan data dapat diajukan kepada Kepala BPOM dan Direktur untuk dilakukan pemeriksaan dokumen. Hasil pemeriksaan tersebut dapat berupa diterima untuk dinilai lebih lanjut, dikembalikan untuk dilengkapi, dan ditolak. Apabila dokumen yang diajukan diterima, maka kemudian akan dilakukan penilaian dan hasilnya berupa surat persetujuan perubahan data atau surat penolakan perubahan data. Apabila keputusan hasil penilaian berupa penolakan perubahan data, maka akan dikeluarkan surat penolakan yang disertai dengan alasan penolakan.