PENETAPAN KADAR SULFADOKSIN DAN PIRIMETAMIN
DALAM SEDIAAN TABLET SECARA
KROMATOGRAFI CAIR KINERJA TINGGI
SKRIPSI
OLEH:
RIZKA RAMADHANI
NIM: 071524060
PROGRAM SARJANA EKSTENSI
FAKULTAS FARMASI
UNIVERSITAS SUMATERA UTARA
MEDAN
PENETAPAN KADAR SULFADOKSIN DAN PIRIMETAMIN
DALAM SEDIAAN TABLET SECARA
KROMATOGRAFI CAIR KINERJA TINGGI
SKRIPSI
Diajukan untuk melengkapi salah satu syarat untuk memperoleh gelar Sarjana Farmasi pada Fakultas Farmasi
Universitas Sumatera Utara
OLEH:
RIZKA RAMADHANI
NIM: 071524060
PROGRAM SARJANA EKSTENSI
FAKULTAS FARMASI
UNIVERSITAS SUMATERA UTARA
MEDAN
PENGESAHAN SKRIPSI
PENETAPAN KADAR SULFADOKSIN DAN PIRIMETAMIN
DALAM SEDIAAN TABLET SECARA
KROMATOGRAFI CAIR KINERJA TINGGI
OLEH:RIZKA RAMADHANI
NIM: 071524060
Dipertahankan di hadapan Panitia Penguji Fakultas Farmasi
USU
Pada tanggal : Maret 2010
Pembimbing I, Panitia penguji,
(Dra. Salbiah M.Si., Apt.) Prof. Dr. Rer. Nat. Effendy De Lux Putra, SU., Apt. NIP. 194810031987012001 NIP. 195306191983031001
Pembimbing II, Drs. Syafruddin, MS., Apt. NIP. 194811111976031003
(Drs. Fathur Rahman Harun, M.Si., Apt.) Dra. Muclisyam, Msi., Apt. NIP. 195201041980031002 NIP. 195006221980021001
Medan, Maret 2010
Fakultas Farmasi Universitas Sumatera Utara
Dekan,
PENETAPAN KADAR SULFADOKSIN DAN PIRIMETAMIN DALAM SEDIAAN TABLET SECARA
KROMATOGRAFI CAIR KINERJA TINGGI
Abstrak
Campuran Sulfadoksin dan Pirimetamin merupakan salah satu jenis
kombinasi dalam sediaan tablet yang berkhasiat untuk pengobatan penyakit malaria.
Pirimetamin efektif digunakan pada P. Malariae dan kombinasinya dengan
Sulfadoksin dapat meningkatkan efektifitas Pirimetamin.
United States Pharmacopoeia 30 (2007) merekomendasikan Kromatografi
Cair Kinerja Tinggi pada penetapan kadar campuran Sulfadoksin dan Pirimetamin
dalam sediaan tablet dengan metode baku dalam, menggunakan fase gerak asetonitril
dan asam asetat glasial dalam air 1% (1:4). Pada penelitian ini telah dicoba
menggunakan metode baku luar dengan memodifikasi berbagai perbandingan fase
gerak asetonitril dan asam asetat glasial dalam air 1%. Metode baku luar lebih praktis
bila dibandingkan dengan baku dalam.
Bahan baku Sulfadoksin dan Pirimetamin sebelum digunakan terlebih dahulu
diidentifikasi menggunakan spektrofotometer FTIR, sedangkan terhadap sampel
dilakukan identifikasi secara KCKT. Dari hasil uji identifikasi menunjukkan bahwa
bahan baku dan sampel yang ditentukan adalah Sulfadoksin dan Pirimetamin.
Hasil uji linieritas dari kurva kalibrasi diperoleh koefisien korelasi, untuk
Sulfadoksin 0,9996 dan untuk Pirimetamin 0,9997 dengan masing-masing persamaan
regresi Y=30931,2X + 317656 dan Y=19319,1X + 37454,2. Dari hasil uji validasi
metode yang digunakan memberikan hasil akurasi dan presisi yang dapat diterima
dengan persen perolehan kembali untuk Sulfadoksin = 103,13% (RSD=2,093%) dan
Pirimetamin = 93,17% (RSD=1,66%).
Hasil penetapan kadar dari tiga sampel dengan nama dagang dan generik,
terdapat satu sampel nama dagang yang tidak memenuhi persyaratan tablet menurut
USP 30 (2007), yaitu tidak kurang dari 90,0% dan tidak lebih dari 110,0% dari
jumlah yang tertera pada etiket.
DETERMINATION DEGREE OF SULFADOXIN AND PYRIMETHAMIN IN TABLETS USING HIGH PERFORMANCE LIQUID
CHROMATOGRAPHY
Abstract
Sulfadoxin and Pyrimethamin is a kind of combination tablet that have
function for malaria treatment. Pyrimethamin is efectife to use P. Malariae and that
combination with Sulfadoxin can improve the Pyrimethamin effectiviness.
United States Pharmacopoeia 30th edition 2007 to recommendate High Performance Liquid Chromatography in determination amount of combination
Sulfadoxin and Pyrimethamin in tablet by internal standard method and using
acetonitril-glasial acetic acid in water 1 % (1:4) mobile phase. In this research have
tried external standard method and modification of mobile phase.External standard
method is more practical when compared with the internal standard.
Before using the Sulfadoxin and Pyrimethamin raw meterial they have
identified with spectrophotometer FTIR, while for the sample is identified by HPLC.
From that identification test showed that raw material and samples are Sulfadoxin and
Pyrimethamin.
Linearity test obtained correlation coefficien 0,9996 and 0,9997 with
regression equation Y=30931,2X + 317656 and Y=19319,1X + 37454,2 for
Sulfadoxin and Pyrimethamin respectively. From the validation method it was giving
good accuracy and precision with percent recovery for Sulfadoxin 103,13% (RSD =
2,03%) and Pyrimethamin 93,17% (RSD = 1,66%).
The results of the three samples determination with trade and generic name,
there is one sample with trade name did meet the requirements of USP 30th edition 2007, which is not less than 90,0% and not more than 110% of the labeled amount.
DAFTAR ISI
Halaman
JUDUL ... i
LEMBAR PENGESAHAN ... ii
ABSTRAK ... iii
ABSTRACT ... iv
DAFTAR ISI ... v
DAFTAR TABEL ... vii
DAFTAR GAMBAR ... viii
DAFTAR LAMPIRAN ... ix
BAB I PENDAHULUAN ... 1
1.1 Latar Belakang ... 1
1.2 Perumusan Masalah ... 2
1.3 Hipotesis ... 3
1.4 Tujuan ... 3
BAB II METODOLOGI PENELITIAN ... 4
2.1 Waktu dan Tempat Penelitian ... 4
2.2 Alat - alat ... 4
2.3 Bahan - bahan ... 4
2.4 Pengambilan Sampel ... 4
2.5 Prosedur penelitian ... 5
2.5.1 Uji identifikasi Sulfadoksin dan Pirimetamin baku pabrik (PT. Ifars) secara spektrofotometer IR ... 5
2.5.2 Penentuan kualitatif dan kuantitatif Sulfadoksin dan Pirimetamin menggunakan KCKT ... 5
2.5.2.1 Pembuatan fase gerak asetonitril-asam asetat Glasial dalam air 1% ... .. 5
2.5.2.3 Pembuatan larutan induk baku Suladoksin dan
Pirimetamin BPFI ... 6
2.5.2.4 Penyiapan alat Kromatografi Cair Kinerja Tinggi . 6
2.5.2.5 Penentuan perbandingan fase gerak ... 6
2.5.2.6 Uji kualitatif Sulfadoksin dan Pirimetamin menggunakan KCKT ... 7
2.5.2.6.1 Menentukan waktu tambat Sulfadoksin dan Pirimetamin BPFI... 7
2.5.2.6.2 Identifikasi sampel ... 7
2.5.2.7 Penentuan kuantitatif Sulfadoksin dan Pirimetamin Menggunakan KCKT ... 8
2.5.2.7.1 Pembuatan kurva kalibrasi Sulfadoksin BPFI ... 8
2.5.2.7.2 Pembuatan kurva kalibrasi Pirimetamin . 8
2.5.2.7.3 Penetapan kadar sampel ... 8
2.5.3 Penetuan Uji validasi dengan parameter akurasi dan Presisi ... 9
2.5.3.1 Uji akurasi dengan persen perolehan kembali ... 10
2.5.3.2 Uji Presisi ... 10
2.5.3.3 Penentuan Batas Deteksi (LOD) dan Batas Kuantitasi (LOQ) ... 10
2.5.3.4 Analisis data secara statistik ... 11
BAB III HASIL DAN PEMBAHASAN ... 12
BAB IV KESIMPULAN DAN SARAN... 24
4.1 Kesimpulan ... 24
4.2 Saran ... 24
DAFTAR PUSTAKA ... 25
DAFTAR TABEL
Halaman
Tabel 1 Hasil optimasi fase gerak dengan parameter waktu tambat
Resolusi, tailing factor dan teoritical plate ... 16
Tabel 2 Data hasil penetapan kadar Sulfadoksin dan Pirimetamin
dalam sediaan tablet ... 21
Tabel 3 Data hasil perolehan kembali Sulfadoksin dan Pirimetamin dengan metode penambahan bahan baku
DAFTAR GAMBAR
Halaman
Gambar 1 Spektrum Inframerah dari baku pabrik (PT. Ifars) Sulfadoksin .. 12
Gambar 2 Spektrum Inframerah baku pabrik Pirimetamin (PT. Ifars) ... 13
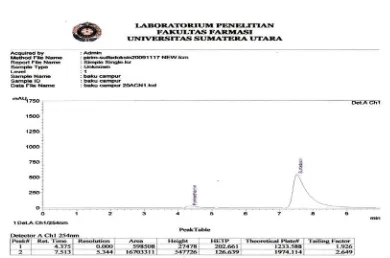
Gambar 3 Kromatogram hasil penyuntikkan larutan BPFI dengan Konsentrasi 500 mcg/ml Sulfadoksin dan 25 mcg/ml Pirimetamin, menggunakan perbandingan fase gerak Asetonitril-asam asetat glasial dalam air 1% (20:80) ... 15
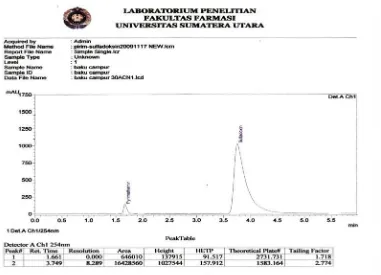
Gambar 4 Kromatogram hasil penyuntikkan larutan BPFI dengan Konsentrasi 500 mcg/ml Sulfadoksin dan 25 mcg/ml Pirimetamin, menggunakan perbandingan fase gerak Asetonitril-asam asetat glasial dalam air 1%(30:70) ... 15
Gambar 5 Kromatogram hasil penyuntikkan larutan BPFI dengan Konsentrasi 500 mcg/ml Sulfadoksin dan 25 mcg/ml Pirimetamin, menggunakan perbandingan fase gerak Asetonitril-asam asetat glasial dalam air 1%(40:60) ... 16
Gambar 6 Kromatogram hasil penyuntikkan larutan BPFI Pirimetamin 25 mcg/ml dengan perbandingan asetonotril-asam asetat glasial dalam air 1% (40:60) ... 17
Gambar 7 Kromatogram hasil penyuntikan larutan BPFI Sulfadoksin 25 mcg/ml dengan perbandingan asetonotril-asam asetat glasial dalam air 1% (40:60) ... 18
Gambar 8 Kurva kalibrasi larutan Sulfadoksin BPFI versus luas puncak ... 19
Gambar 9 Kurva kalibrasi larutan Pirimetamin BPFI versusu luas puncak . 20
Gambar 10 Alat KCKT (Shimadzu) ... 26
Gambar 11 Syringe 100 μl (SGE)... 26
Gambar 12 Sonifikator (Branson 1510) ... 27
Gambar 13 Pompa vakum dan Penyaring fase gerak ... 27
Gambar 14 Spektrum Inframerah Sulfadoksin ... 28
DAFTAR LAMPIRAN
Lampiran 1 Gambar alat KCKT dan Syringe ... 26
Lampiran 2 Gambar Alat ultrasonic cleaner dan penyaring ... 27
Lampiran 3 Spektrum Inframerah Sulfadoksin pada literatur
Pharmaceutical Sub stance (UV/IR) ... 28
Lampiran 4 Spektrum Inframerah Pirimetamin pada literatur
Pharmaceutical Sub stance (UV/IR) ... 29
Lampiran 5 Perhitungan persamaan regresi dari kurva kalibrasi
Sulfadoksin ... 30
Lampiran 6 Perhitungan Batas Deteksi (LOD) dan Batas Kuantitasi (LOQ) Sulfadoksin ... 31
Lampiran 7 Perhitungan persamaan regresi dari kurva kalibrasi
Pirimetamin ... 32
Lampiran 8 Perhitungan Batas Deteksi (LOD) dan Batas Kuantitasi (LOQ) Pirimetamin ... 33
Lampiran 9 Contoh perhitungan kadar kombinasi Sulfadoksin dan
Pirimetamin dari tablet Fansidar (PT. Roche) ... 34
Lampiran 10 Kromatogram hasil dari penyuntikkan dari larutan tablet
Fansifar (PT. Roche) ... 35
Lampiran 11 Analisis data statistik untuk mencari kadar sebenarnya
dari penyuntikan larutan tablet Fansidar (PT. Rhoce) ... 38
Lampiran 12 Kromatogram hasil penyuntikkan dari larutan tablet
Sulfadoksin-Pirimetamin Generik (PT. Ifars) ... 40
Lampiran 13 Analisis data statistik untuk mencari kadar sebenarnya Dari penyuntikan larutan tablet Sulfadoksin-Pirimetamin
Generik (PT. Ifars) ... 43
Lampiran 14 Kromatogram hasil penyuntikkan dari larutan tablet
Suldox (PT. Actavis)... 45
Lampiran 15 Analisis data statistik untuk mencari kadar sebenarnya
Dari penyuntikan larutan tablet Suldox (PT Actavis) ... 48
Lampiran 16 Hasil pengolahan data dari tablet Fansidar, Generik dan
Suldox ... 52
Lampiran 17 Kromatogram hasil penyuntikkan dari larutan tablet Fansidar (PT. Roche ) dan bahan baku, persen perolehan kembali
Lampiran 18 Contoh perhitungan persen perolehan kembali ... 58
Lampiran 19 Perhitungan berat sampel dari lampiran 18 setelah penimbangan ... 60
Lampiran 20 Analisis data statistik persen perolehan kembali pada tablet Fansidar (PT. Roche) ... 62
Lampiran 21 Data hasil perolehan kembali Sulfadoksin dan Pirimetamin Pada tablet Fansidar (PT. Roche) ... 63
Lampiran 22 Contoh perhitungan % Recovery dengan metode penambahan Bahan baku (Standard Adition Method) dari tablet Fansidar ... 64
Lampiran 23 Sertifikat pengujian Sulfadoksin BPFI ... 65
Lampiran 24 Sertifikat pengujian Pirimetamin BPFI ... 66
Lampiran 25 Sertifikat bahan baku Sulfadoksin dari PT. Ifars... 67
Lampiran 26 Sertifikat bahan baku Pirimetamin dari PT. Ifars ... 68
PENETAPAN KADAR SULFADOKSIN DAN PIRIMETAMIN DALAM SEDIAAN TABLET SECARA
KROMATOGRAFI CAIR KINERJA TINGGI
Abstrak
Campuran Sulfadoksin dan Pirimetamin merupakan salah satu jenis
kombinasi dalam sediaan tablet yang berkhasiat untuk pengobatan penyakit malaria.
Pirimetamin efektif digunakan pada P. Malariae dan kombinasinya dengan
Sulfadoksin dapat meningkatkan efektifitas Pirimetamin.
United States Pharmacopoeia 30 (2007) merekomendasikan Kromatografi
Cair Kinerja Tinggi pada penetapan kadar campuran Sulfadoksin dan Pirimetamin
dalam sediaan tablet dengan metode baku dalam, menggunakan fase gerak asetonitril
dan asam asetat glasial dalam air 1% (1:4). Pada penelitian ini telah dicoba
menggunakan metode baku luar dengan memodifikasi berbagai perbandingan fase
gerak asetonitril dan asam asetat glasial dalam air 1%. Metode baku luar lebih praktis
bila dibandingkan dengan baku dalam.
Bahan baku Sulfadoksin dan Pirimetamin sebelum digunakan terlebih dahulu
diidentifikasi menggunakan spektrofotometer FTIR, sedangkan terhadap sampel
dilakukan identifikasi secara KCKT. Dari hasil uji identifikasi menunjukkan bahwa
bahan baku dan sampel yang ditentukan adalah Sulfadoksin dan Pirimetamin.
Hasil uji linieritas dari kurva kalibrasi diperoleh koefisien korelasi, untuk
Sulfadoksin 0,9996 dan untuk Pirimetamin 0,9997 dengan masing-masing persamaan
regresi Y=30931,2X + 317656 dan Y=19319,1X + 37454,2. Dari hasil uji validasi
metode yang digunakan memberikan hasil akurasi dan presisi yang dapat diterima
dengan persen perolehan kembali untuk Sulfadoksin = 103,13% (RSD=2,093%) dan
Pirimetamin = 93,17% (RSD=1,66%).
Hasil penetapan kadar dari tiga sampel dengan nama dagang dan generik,
terdapat satu sampel nama dagang yang tidak memenuhi persyaratan tablet menurut
USP 30 (2007), yaitu tidak kurang dari 90,0% dan tidak lebih dari 110,0% dari
jumlah yang tertera pada etiket.
DETERMINATION DEGREE OF SULFADOXIN AND PYRIMETHAMIN IN TABLETS USING HIGH PERFORMANCE LIQUID
CHROMATOGRAPHY
Abstract
Sulfadoxin and Pyrimethamin is a kind of combination tablet that have
function for malaria treatment. Pyrimethamin is efectife to use P. Malariae and that
combination with Sulfadoxin can improve the Pyrimethamin effectiviness.
United States Pharmacopoeia 30th edition 2007 to recommendate High Performance Liquid Chromatography in determination amount of combination
Sulfadoxin and Pyrimethamin in tablet by internal standard method and using
acetonitril-glasial acetic acid in water 1 % (1:4) mobile phase. In this research have
tried external standard method and modification of mobile phase.External standard
method is more practical when compared with the internal standard.
Before using the Sulfadoxin and Pyrimethamin raw meterial they have
identified with spectrophotometer FTIR, while for the sample is identified by HPLC.
From that identification test showed that raw material and samples are Sulfadoxin and
Pyrimethamin.
Linearity test obtained correlation coefficien 0,9996 and 0,9997 with
regression equation Y=30931,2X + 317656 and Y=19319,1X + 37454,2 for
Sulfadoxin and Pyrimethamin respectively. From the validation method it was giving
good accuracy and precision with percent recovery for Sulfadoxin 103,13% (RSD =
2,03%) and Pyrimethamin 93,17% (RSD = 1,66%).
The results of the three samples determination with trade and generic name,
there is one sample with trade name did meet the requirements of USP 30th edition 2007, which is not less than 90,0% and not more than 110% of the labeled amount.
BAB I
PENDAHULUAN
1.1Latar Belakang
Malaria adalah penyakit menular yang disebabkan oleh parasit (protozoa) dari
genus plasmodium yang dapat ditularkan melalui gigitan nyamuk anopeles. Biasanya
malaria menyerang penduduk yang tinggal didaerah endemis atau orang-orang yang
berpergian ke daerah yang angka penularannya tinggi. Kegiatan pemberantasan
penyakit ini sudah dilakukan sejak lama, adanya parasit malaria yang resisten
terhadap obat-obatan, menambah sulit pemberantasan penyakit ini (Prabowo, 2008).
Tablet Fansidar merupakan salah satu obat kombinasi Sulfadoksin dan
Pirimetamin dengan pemberian peroral, Pirimetamin bekerja dengan menghambat
enzim dihidrofolat reduktase plasmodium, penghambatan ini menyebabkan protozoa
kekurangan tetrahidrofolat, suatu kofaktor yang diperlukan dalam biosintesis purin
dan pirimidin. Pirimethamin efektif digunakan pada P. Malariae yang
dikombinasikan dengan Sulfadoksin sehingga dapat meningkatkan efektivitas dari
Pirimetamin (Harvey, 2001).
Menurut Undang-undang No. 36 tahun 2009 pasal 105 ayat 1 tentang
kesehatan bahwa obat dan bahan obat harus memenuhi standar Farmakope dan buku
standar lain. Salah satu parameter obat tersebut dikatakan memenuhi standar apabila
kadar zat berkhasiat yang terkandung didalamnya memenuhi persyaratan Farmakope
Indonesia dan buku standar lainnya.
Menurut USP edisi 30 (2007) penetapan kadar tablet kombinasi Sulfadoksin
standar) menggunakan kolom VP-ODS (3,9 mm x 30 cm) dengan perbandingan fase
gerak Asetonitril-Asam asetat glasial dalam air 1% (1:4), dengan laju alir 2 ml/menit
dan dideteksi pada panjang gelombang 254 nm,.
Dalam penelitian ini dilakukan penetapan kadar dengan metode yang berbeda
yaitu metode baku luar (Eksternal standar), menggunakan kolom VP-ODS (4,6 mm x
25 cm), fase gerak Asetonitril dan Asam asetat glasial dalam air 1% dengan berbagai
perbandingan.
Metode baku luar (Eksternal standar) lebih praktis dibanding dengan metode
baku dalam (Internal standar). Pemilihan metoda KCKT ini karena memiliki banyak
keuntungan antara lain cepat, resolusi baik, mudah melaksanakannya, detektor yang
sensitif dan beragam sehingga mampu menganalisa berbagai cuplikan secara
kualitatif dan kuantitatif (Rohman, 2007).
Untuk menguji keabsahan dari metode yang digunakan dilakukan uji akurasi
dengan parameter persen recovery dan uji presisi dengan relatif standar deviasi
(RSD).
1.2Perumusan Masalah
- Apakah metode Kromatografi Cair Kinerja Tinggi dengan baku luar
(Eksternal standard) dapat diterapkan pada penetapan kadar Sulfadoksin dan
Pirimetamin dalam sediaan tablet dengan uji validasi metode yang memenuhi
syarat.
- Apakah kadar Sulfadoksin dan Pirimetamin dalam sediaan tablet di pasaran
memenuhi persyaratan kadar menurut The United States Pharmacopeia 30
1.3Hipotesa
- Metode Kromatografi Cair Kinerja Tinggi dengan baku luar (Eksternal
standard) dapat diterapkan pada penetapan kadar Sulfadoksin dan
Pirimetamin dalam sediaan tablet dengan uji validasi metode yang memenuhi
syarat.
- Kombinasi Sulfadoksin dan Pirimetamin dalam sediaan tablet yang beredar di
pasaran memenuhi persyaratan kadar menurut The United States
Pharmecopeia 30 (2007) yaitu tidak kurang dari 90,0% dan tidak lebih dari
110,0%.
1.4Tujuan Penelitian
- Menentukan kadar Sulfadoksin dan Pirimetamin dalam sediaan tablet secara
Kromatografi Cair Kinerja Tinggi menggunakan baku luar (Eksternal
standard) dengan uji validasi metode yang memenuhi syarat.
- Mengetahui kadar kombinasi Sulfadoksin dan Pirimetamin dalam sediaan
tablet yang beredar di pasaran memenuhi persyaratan kadar menurut The
United States Pharmacopoeia 30 (2007) yaitu tidak kurang dari 90,0% dan
BAB II
TINJAUAN PUSTAKA
Tablet adalah sediaan padat kompak, dibuat secara kempa cetak, dalam
bentuk tabung pipih atau siskuler, kedua permukaannya rata atau cembung,
mengandung satu jenis obat atau lebih dengan atau tanpa bahan tambahan (Depkes
RI,1979). Sedangkan menurut Farmakope Indonesia (1995) tablet adalah sediaan
padat mengandung bahan obat dengan atau tanpa bahan pengisi.
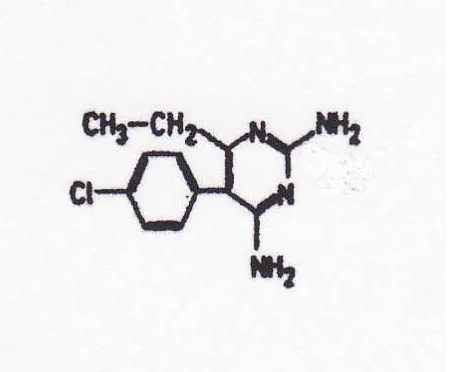
1. Sulfadoksin
Rumus struktur :
Gambar 1. struktur Sulfadoxin
Nama Kimia : N1-(5,6-Dimetoksi-4-pirimidinil)sulfanilamida
Rumus Molekul : C12H14N4O4S
Berat Molekul : 310,33 (Depkes RI, 1995)
Pemerian : Serbuk kristal putih
Kelarutan : Sangat sukar larut dalam air, sukar larut dalam etanol dan
metanol, praktis tidak larut dalam eter.
2. Pirimetamin
Rumus Struktur :
Gambar 2. Struktur Pirimetamin
Nama Kimia : 2,4-Diamino-5-(p-klorofenil)-6-etilpirimidina
Rumus Molekul : C12H13ClN4
Berat Molekul : 248,71 (Depkes RI, 1995)
Pemerian : Serbuk kristal putih.
Log P : 2,7 (octanol/water)
pKa : 7,3
Kelarutan : Praktis tidak larut dalam air, larut dalam 200 bagian etanol,
3 Kromatografi
Kromatografi adalah suatu terminologi umum yang digunakan untuk bermacam-
macam teknik pemisahan yang didasarkan atas partisi cuplikan diantara suatu fase
gerak yang bisa berupa gas ataupun cairan dan fase diam yang juga bisa berupa cairan
atau padatan.
Kromatografi dapat dibedakan atas berbagai macam tergantung pada
pengelompokannya. Berdasarkan pada mekanisme pemisahannya, kromatografi
dibedakan menjadi :
a) Kromatografi adsorbsi
b) Kromatografi partisi
c) Kromatografi pasangan ion
d) Kromatografi penukar ion
e) Kromatografi eksklusi ukuran, dan
f) Kromatografi afinitas
3.1 Penggunaan Kromatografi
1. Pemakaian untuk tujuan kualitatif mengungkapkan ada atau tidak adanya
senyawa tertentu dalam cuplikan
2. Pemakaian untuk tujuan kuantitatif menunjukkan banyaknya masing-masing
komponen campuran
3. Pemakaian untuk tujuan preparatif untuk memperoleh komponen campuran
dalam jumlah memadai dalam keadaan murni.
Selama pemisahan kromatografi, solut individual akan membentuk profil
konsentrasi yang simetris atau dikenal juga dengan profil Gaussian dalam arah aliran
fase gerak. Profil dikenal juga dengan punak atau pita, secara perlahan - lahan akan
melebar dan sering juga membentuk profil yang asimetrik karena solut – solut
melanjutkan migrasinya ke fase diam.
3.3 Puncak asimetris
Profil konsentrasi solut yang bermigrasi akan simetris jika rasio distribusi
solut (D) konstan selama dikisaran konsentrasi keseluruhan puncak, sebagaimana
ditunjukkan oleh isoterm sorpsi yang linear yang merupakan plot konsentrasi solut
dalam fase diam (Cs) terhadap konsentrasi solut dalam fase gerak(Cm). Meskipun
demikian, kurva isot erm akan berubah menjadi 2 jenis puncak asimetris yakni
membentuk puncak yang berekor (tailing) dan adanya puncak pendahulu (fronting)
jika ada perubahan rasio distribusi solut yang lebih besar.
Untuk kromatografi yang melibatkan kolom, kuantifikasi dapat dilakukan
dengan luas puncak atau tinggi puncak. Tinggi puncak atau luas puncak berbanding
langsung dengan banyaknya solut yang dikromatografi, jika dilakukan pada kisaran
detektor yang linier.
1. Metode tinggi puncak
Metode yang paling sederhana untuk pengukuran kuantitatif adalah dengan
tinggi puncak. Tinggi puncak diukur sebagai jarak dari garis dasar ke puncak
maksimum seperti puncak 1, 2, dan 3 pada gambar 3. Penyimpangan garis dasar
Gambar 1. Pengukuran tinggi puncak
Metode tinggi puncak hanya digunakan jika perubahan tinggi puncak linier
dengan konsentrasi analit. Kesalahan akan terjadi jika metode ini digunakan pada
puncak yang mengalami penyimpangan (asimetris) atau jika kolom mengalami
kelebihan muatan.
2. Metode luas puncak
Prosedur penentuan luas puncak serupa dengan tinggi puncak. Suatu teknik
untuk mengukur luas puncak adalah dengan mengukur luas puncak sebagai hasil kali
tinggi puncak dan lebar pada setengah tinggi (W1/2). Teknik ini hanya dapat
digunakan untuk kromatografi yang simetris atau yang mempunyai bentuk serupa
(Johnson, 1991).
Saat ini integrator elektronik telah banyak digunakan untuk mengukur luas
puncak pada kromatografi cair kinerja tinggi dan pada kromatografi gas. Integrator
digital mengukur luas puncak dan mengubahnya dalam bentuk angka (Rohman,
2007).
Baik tinggi puncak maupun luasnya dapat dihubungkan dengan konsentrasi.
yang disebabkan oleh variasi suhu dan komposisi pelarut. Oleh karena itu, luas
puncak dianggap merupakan parameter yang lebih akurat untuk pengukuran
kuantitatif (Ditjen POM, 1995).
3.4 Kromatografi Cair Kinerja Tinggi (KCKT)
Kromatografi cair kinerja tinggi (KCKT) merupakan sistem pemisahan
dengan kecepatan dan efisiensi yang tinggi. Hal ini karena didukung oleh kemajuan
dalam teknologi kolom, sistem pompa tekanan tinggi, dan detektor yang sangat
sensitif dan beragam. KCKT mampu menganalisa berbagai cuplikan secara kualitatif
maupun kuantitatif, baik dalam komponen tunggal maupun campuran (Ditjen POM,
1995).
KCKT merupakan teknik pemisahan yang diterima secara luas untuk analisis
dan pemurnian senyawa tertentu dalam suatu sampel pada sejumlah bidang antara
lain; farmasi, lingkungan dan industri-industri makanan.
Kegunaan umum KCKT adalah untuk pemisahan sejumlah senyawa organik,
anorganik, maupun senyawa biologis, analisis ketidakmurnian (impurities) dan
analisis senyawa-senyawa yang tidak mudah menguap (nonvolatil). KCKT paling
sering digunakan untuk: menetapkan kadar senyawa-senyawa tertentu seperti
asam-asam amino, asam-asam-asam-asam nukleat dan protein-protein dalam cairan fisiologis,
menentukan kadar senyawa-senyawa aktif obat dan lain-lain.
Kelebihan KCKT antara lain:
−Mampu memisahkan molekul-molekul dari suatu campuran
−Mudah melaksanakannya
−Kecepatan analisis dan kepekaannya tinggi
−Dapat dihindari terjadinya dekomposisi/kerusakan bahan yang dianalisis −Dapat digunakan bermacam-macam detektor
−Kolom dapat digunakan kembali
−Mudah melakukan rekoveri cuplikan
−Tekniknya tidak begitu tergantung pada keahlian operator dan reprodusibilitasnya
lebih baik
−Instrumennya memungkinan untuk bekerja secara automatis dan kuantitatif
−Waktu analisis umumnya singkat
−Kromatografi cair preparatif memungkinkan dalam skala besar
- Ideal untuk molekul besar dan ion.
Keterbatasan metode KCKT adalah untuk identifikasi senyawa, kecuali jika
KCKT dihubungkan dengan spektrometer massa (MS). Keterbatasan lainnya adalah
jika sampelnya sangat kompleks, maka resolusi yang baik sulit diperoleh (Munson,
1991).
3.4.1 Cara Kerja KCKT
Kromatografi merupakan teknik yang mana solut atau zat-zat terlarut terpisah
oleh perbedaan kecepatan elusi, dikarenakan solut-solut ini melewati suatu kolom
kromatografi. Pemisahan solut-solut ini diatur oleh distribusi dalam fase gerak dan
fase diam. Penggunaan kromatografi cair membutuhkan penggabungan secara tepat
diameter kolom, kecepatan alir fase gerak, suhu kolom, dan ukuran sampel (Rohman,
2007).
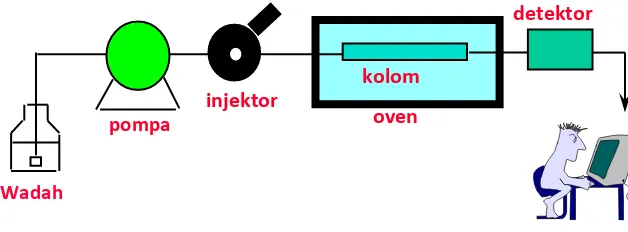
3.4.2 Komponen KCKT
Gambar 2. Bagan alat KCKT
3.4.2.1 Wadah Fase Gerak
Wadah fase gerak harus bersih dan lembam (inert). Wadah pelarut kosong
ataupun labu laboratorium dapat digunakan sebagai wadah fase gerak. Wadah ini
biasanya dapat meampung fase gerak antara 1 sampai 2 liter pelarut.Fase gerak
sebelum digunakan harus dilakukan degassing(penghilangan gas) yang ada pada fase
gerak, sebab adanya gas akan berkumpul dengan komponen lain terutama dipompa
dan detektor sehingga akan mengacaukan analisis. (Rohman, 2007)
3.4.2.2 Pompa
Pompa yang cocok digunakan untuk KCKT adalah pompa yang mempunyai
syarat sebagaimana syarat wadah pelarut yakni pompa harus inert terhadap fase
gerak. Bahan yang umum dipakai untuk pompa adalah gelas, baja tahan karat, Teflon, pompa
injektor
kolom oven
detektor
Wadah
dan batu nilam. Pompa yang dgunakan sebaiknya mampu memberikan tekanan
sampai 5000 psi dan mampu mengalirkan fase gerak dengan kecepatan alir 3 ml/
menit.
3.4.2.3 Injektor
Cuplikan harus dimasukkan kedalam pangkal kolom (kepala kolom),
diusahakan agas sesedikit mungkin terjadi gangguan pada kemasan kolom.
Ada tiga jenis dasar injektor, yaitu:
a. Hentikan aliran/stop flow: aliran dihentikan, injeksi dilakukan pada kinerja
atmosfir, sistem tertutup, dan aliran dilanjutkan lagi. Teknik ini bisa digunakan
karena difusi di dalam aliran kecil dan resolusi tidak dipengaruhi.
b. Septum: injektor-injektor langsung ke aliran fase gerak umumnya sama dengan
yang digunakan pada kromatografi gas. Injektor ini dapat digunakan pada kinerja
sampai 60-70 atmosfir. Tetapi septum ini tidak tahan dengan semua
pelarut-pelarut kromatografi cair. Disamping itu, partikel kecil dari septum yang
terkoyak (akibat jarum injektor) dapat menyebabkan penyumbatan.
c. Katup putaran (loop valve): ditunjukkan secara skematik dalam Gambar 5, tipe
injektor ini umumnya digunakan untuk menginjeksi volume lebih besar daripada
10 µl dan sekarang digunakan dengan cara otomatis (dengan adaptor khusus,
volume-volume lebih kecil dapat diinjeksikan secara manual). Pada posisi LOAD,
sampel loop (cuplikan dalam putaran) diisi pada tekanan atmosfir. Bila katup
Gambar 3. Tipe injektor katup putaran
3.4.2.4 Kolom
Kolom adalah jantung kromatografi. Berhasil atau gagalnya suatu analisis
tergantung pada pemilihan kolom dan kondisi percobaan yang sesuai. Kolom dapat
dibagi menjadi dua kelompok:
1. Kolom analitik: diameter khas adalah 2-6 mm. Panjang kolom tergantung pada
jenis kemasan. Untuk kemasan pelikel biasanya panjang kolom 50-100 cm. Untuk
kemasan mikropartikel berpori, umumnya 10-30 cm. Dewasa ini ada yang 5 cm.
2. Kolom preparatif: umumnya memiliki diameter 6 mm atau lebih besar dan
panjang kolom 25-100 cm.
Kolom umumnya dibuat dari stainless steel dan biasanya dioperasikan pada
temperatur kamar, tetapi bisa juga digunakan temperatur lebih tinggi, terutama untuk
kromatografi penukar ion dan kromatografi eksklusi. Kemasan kolom tergantung
pada mode KCKT yang digunakan.
3.4.2.5 Detektor
Suatu detektor dibutuhkan untuk mendeteksi adanya komponen cuplikan
sensitifitas yang tinggi, gangguan (noise) yang rendah, kisar respons linier yang luas,
dan memberi tanggapan/respon untuk semua tipe senyawa. Suatu kepekaan yang
rendah terhadap aliran dan fluktuasi temperatur sangat diinginkan, tetapi tidak selalu
dapat diperoleh.
Detektor yang paling banyak digunakan dalam kromatografi cair modern
kecepatan tinggi adalah detektor spektrofotometer UV 254 nm. Bermacam-macam
detektor dengan variasi panjang gelombang UV-Vis sekarang menjadi populer karena
mereka dapat digunakan untuk mendeteksi senyawa-senyawa dalam rentang yang
luas. Detektor indeks refraksi juga secara luas digunakan, terutama dalam
kromatografi eksklusi, tetapi umumnya kurang sensitif dari pada detektor
spektrofotometer UV. Detektor lainnya, antara lain: detektor fluometer, detektor
ionisasi nyala, detektor elektrokimia dan lain-lain juga telah digunakan.
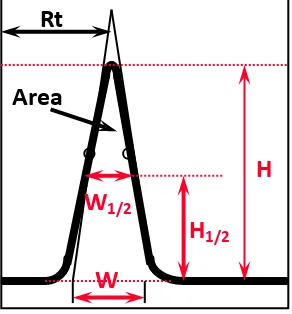
3.4.2.6 Pengolahan Data
Komponen yang terelusi mengalir ke detektor dan dicatat sebagai
puncak-puncak yang secara keseluruhan disebut sebagai kromatogram.
Gambar 4. Kromatogram W
W1/2
H1/2 H
Rt
Guna kromatogram:
1. Kualitatif
Waktu retensi selalu konstan dalam setiap kondisi kromatografi yang sama
dapat digunakan untuk identifikasi.
2. Kuantitatif
Luas puncak proporsional dengan jumlah sampel yang diinjeksikan dan dapat
digunakan untuk menghitung konsentrasi.
3. Kromatogram dapat digunakan untuk mengevaluasi efisiensi pemisahan dan
kinerja kolom (kapasitas ‘k’, selektifitas ‘α’, jumlah pelat teoritis ‘N’, jarak
setara dengan pelat teoritis ‘HETP’ dan resolusi ‘R’).
3.4.2.7 Fase Gerak
Fase gerak atau eluen biasanya terdiri atas campuran pelarut yang dapat
bercampur yang secara keseluruhan berperan dalam daya elusi dan resolusi. Daya
elusi dan resolusi ini ditentukan oleh polaritas keseluruhan pelarut, polaritas fase
diam, dan sifat komponen-komponen sampel (Johnson & Stevenson, 1991).
Dalam kromatografi cair komposisi pelarut atau fase gerak adalah satu
variabel yang mempengaruhi pemisahan. Terdapat keragaman yang luas dari fase
gerak yang digunakan dalam semua mode KCKT, tetapi ada beberapa sifat-sifat yang
diinginkan yang mana umumnya harus dipenuhi oleh semua fase gerak.
Fase gerak harus:
• Murni; tidak ada pencemar/kontaminan
• Sesuai dengan detektor
• Melarutkan cuplikan
• Mempunyai viskositas rendah
• Mudah rekoveri cuplikan, bila diinginkan
• Tersedia diperdagangan dengan harga yang pantas
Umumnya, pelarut-pelarut dibuang setelah digunakan karena prosedur
pemurnian kembali membosankan dan mahal. Dari semua persyaratan di atas, 4
persyaratan pertama adalah yang paling penting.
Gelembung udara (degassing) yang ada harus dihilangkan dari pelarut, karena
udara yang terlarut keluar melewati detektor dapat menghasilkan banyak noise
sehingga data tidak dapat digunakan (Putra, 2007).
Elusi Gradien dan Isokratik
Elusi pada KCKT dapat dibagi menjadi dua sistem yaitu:
1. Sistem elusi isokratik. Pada sistem ini, elusi dilakukan dengan satu macam atau
lebih fase gerak dengan perbandingan tetap (komposisi fase gerak tetap selama
elusi).
2. Sistem elusi gradien. Pada sistem ini, elusi dilakukan dengan campuran fase gerak
yang perbandingannya berubah-ubah dalam waktu tertentu (komposisi fase gerak
berubah-ubah selama elusi).
Elusi gradien didefinisikan sebagai penambahan kekuatan fase gerak selama
suatu analisis kromatografi berlangsung. Digunakan untuk meningkatkan resolusi
luas. Pengaruh yang menguntungkan dari elusi gradien adalah memperpendek waktu
analisis senyawa-senyawa yang secara kuat ditahan di dalam kolom (Putra, 2007).
Tipe kromatografi
a. Kromatografi fase normal
Kromatografi fase normal (fase diam lebih polar daripada fase gerak),
kemampuan elusi meningkat dengan meningkatnya polaritas pelarut. Fase gerak
ini biasanya tidak polar. Dietil eter, benzen, hidrokarbon lurus seperti pentana,
heksana, heptana maupun iso-oktana sering digunakan. Halida alifatis seperti
dikloro metana, dikloroetana, butilklorida dan kloroform juga digunakan.
Umumnya gas terlarut tidak menimbulkan masalah pada fase normal. Tekanan
rendah diperlukan untuk menjaga kecepatan aliran yang memadai, karena pelarut
ini kebanyakan kurang kental (Munson, 1991).
Fase diam yang digunakan dapat dilihat pada gambar dibawah ini:
Gambar 5. Jenis-jenis fase diam untuk tipe kromatografi fase normal
Kromatografi fase terbalik (fase diam kurang polar daripada fase gerak),
kemampuan elusi menurun dengan meningkatnya polaritas pelarut. Kandungan
utama fase gerak fase terbalik adalah air. Pelarut yang dapat campur dengan air
seperti metanol, etanol, asetonitril, dioksan, tetrahirofuran dan dimetilformamida
ditambahkan untuk mengatur kepolaran fase gerak. Dapat ditambahkan pula
asam, basa, dapar dan/atau surfaktan. Mutu air harus tinggi baik air destilasi
maupun awamineral.
Fase diam yang digunakan dapat dilihat pada gambar dibawah ini :
Gambar 6. Jenis-jenis fase diam untuk tipe kromatografi fase terbalik
Adsorpsi solut oleh fase diam atau adsorben sangat tergantung pada:
1. Struktur kimia solut atau adanya gugus aktif tertentu yang berinteraksi dengan
adsorben.
2. Ukuran partikel adsorben. Semakin kecil ukuran partikel adsorben, maka luas
permukaannya semakin luas sehingga interaksinya dengan solut juga semakin
luas.
3. Kelarutan solut dalam fase gerak. Solut yang makin mudah larut dalam fase gerak
Validasi
Validasi adalah suatu tindakan terhadap parameter tertentu pada prosedur
penetapan yang dipakai untuk membuktikan bahwa parameter tersebut memenuhi
persyaratan untuk penggunaannya (WHO, 1992).
Validasi metode menurut United States Pharmacopeia (USP) dilakukan untuk
menjamin bahwa metode analisis akurat, spesifik, reprodusibel dan tahan pada
kisaran analit yang akan dianalisis. Suatu metode analis harus divalidasi untuk
verifikasi bahwa parameter-parameter kinerjanya cukup mampu untuk mengatasi
masalah dalam analisis. Parameter analisis yang ditentukan pada validasi adalah
akurasi, presisi, batas deteksi, batas kuantitasi, spesifikasi, linieritas dan rentang,
kekasaran (Ruggedness) dan ketahanan (Robutness).
Presisi merupakan ukuran keterulangan metode analisis dan biasanya
diekspresikan sebagai relatif standar deviasi (RSD) dari sejumlah sampel yang
berbeda secara signifikan secara statistik.
Batas deteksi (limit of detection, LOD) didefinisikan sebagai konsentrasi
analit terendah dalam sampel yang masih dapat terdeteksi.
Batas kuantitasi (limit of quantitation, LOQ) didefinisikan sebagai konsentrasi
analit terendah dalam sampel yang dapat ditentukan dengan presisi dan akurasi yang
dapat diterima pada kondisi operasional metode yang digunakan.
Linieritas merupakan kemampuan suatu metode untuk memperoleh hasil-hasil
uji yang secara langsung proporsional dengan konsentrasi analit pada kisaran yang
BAB III
METODOLOGI PENELITIAN
3.1 Waktu dan tempat penelitian
Penelitian dilakukan di Laboratorium Penelitian Fakultas Farmasi Universitas
Sumatera Utara pada bulan Oktober sampai November 2009.
3.2 Alat-alat
Alat-alat yang digunakan dalam penelitian ini adalah satu unit alat KCKT
(Shimadzu) yang terdiri dari Vacum degasser, pompa, detektor UV/Vis, printer,
kolom shimpac VP-ODS (4,6 mm x 25 cm), wadah fase gerak, penyuntik mikroliter
(100 µl), neraca analitik (Mettler Toledo), membran filter PTFE 0,5 µ m dan 0,2 µ m,
cellulose nitrat membran filter 0,45 µ m, Spektrofotometer IR (Shimadzu IR
Prestige-21).
3.3 Bahan-bahan
Bahan-bahan yang digunakan dalam penelitian yaitu Asetonitril (BDH),
metanol p.a (Merck), akuabidestilata (PT. Ikapharmindo putramas), Sulfadoksin dan
Pirimetamin BPFI (Badan POM RI), Sulfadoksin baku pabrik (Nanhu Farms
Chongshu Jiangshu China), Pirimetamin baku pabrik (Hesni Town, Changshu,
Jiangshu, China) dan serbuk KBr (Kyoto Japan).
3.4 Pengambilan sampel
Pengambilan sampel secara purposif yaitu tanpa membandingkan antara satu
homogen. Sampel yang digunakan adalah tablet Fansidar (PT. Rhoce), tablet generik
(Ifars), tablet Suldox (PT. Actavis).
3.5 Prosedur penelitian
3.5.1 Uji Identifikasi Sulfadoksin dan Pirimetamin baku pabrik (PT. Ifars)
secara spektrofotometri Inframerah
Dicampur 1 mg serbuk Sulfadoksin dengan 100 mg serbuk KBr dalam
lumpang digerus hinggga halus dan homogen, campuran tersebut diletakkan pada
sampel pan, kemudian dipasangkan pada DRS 8000 dan dianalisa pada bilangan
gelombang 4000 – 500 cm-1. Spektrum Inframerah yang diperoleh dibandingkan
dengan literatur.
Baku Pirimetamin juga di analisa dengan perlakuan yang sama seperti
Sulfadoksin.
3.5.2 Penentuan Kualitatif dan Kuantitatif Sulfadoksin dan Pirimetamin
menggunakan KCKT
3.5.2.1 Pembuatan fase gerak Asetonitril–Asam asetat glasial dalam air (1%)
Asetonitril 500 ml disaring dengan menggunakan membran filter PTFE 0,5
µ m.
Sebanyak 10 ml asam asetat glasial diencerkan dengan akuabidestilata dalam
dengan menggunakan celllulosa nitrat membran filter 0,45 µ m. Masing-masing
diawaudarakan selama 20 menit.
3.5.2.2 Pembuatan pelarut
Dicampur 100 ml asetonitril dan 400 ml larutan campuran asam asetat glasial
dalam air (1%). Kemudian pelarut disaring menggunakan membran filter PTFE 0,5
µ m dan diawaudarakan selama 20 menit.
3.5.2.3 Pembuatan Larutan Induk Baku Sulfadoksin dan Pirimetamin BPFI
Sejumlah 50 mg Baku Pembanding Sulfadoksin ditimbang seksama,
dimasukkan kedalam labu tentukur 50 ml, ditambah asetonitril 35 ml aduk hingga
larut, lalu dicukupkan sampai garis tanda dengan pelarut dan dikocok hingga
homogen, maka diperoleh larutan dengan konsentrasi 1000 mcg/ml (LIB).
Sejumlah 25 mg Baku Pembanding Pirimetamin ditimbang seksama,
dimasukkan kedalam labu tentukur 100 ml, ditambah asetonitril 35 ml aduk hingga
larut, lalu dicukupkan sampai garis tanda dengan pelarut dan dikocok hingga
homogen, maka diperoleh larutan dengan konsentrasi 250 mcg/ml (LIB).
3.5.2.4 Penyiapan alat Kromatografi Cair Kinerja Tinggi
Masing-masing unit diatur, kolom yang digunakan Shimpac VP-ODS (4,6
mm x 25 cm), detektor UV/Vis, dengan laju alir 2 ml/menit, sensitifitas 1.000 AUFS
dan dideteksi pada panjang gelombang 254 nm.
Setelah alat KCKT dihidupkan, maka pompa dijalankan dan fase gerak
dibiarkan mengalir selama 30 menit sampai diperoleh garis alas yang datar,
menandakan sistem tersebut telah stabil.
Dipipet 5 ml Larutan Induk Baku Sulfadoksin dan 1 ml Larutan Induk Baku
Pirimetamin masukkan dalam labu tentukur 10 ml, dicukupkan dengan pelarut hingga
garis tanda, kocok sehingga diperoleh larutan Sulfadoksin dengan konsentrasi 500
mcg/ml dan Pirimetamin dengan konsentrasi 25 mcg/ml, disaring dengan membran
filter PTFE 0,2 µl dan diawaudarakan selama 5 menit, kemudian diinjeksikan ke
dalam sistem KCKT dengan volum penyuntikan 20 µ l, menggunakan fase gerak
asetonitril - asam asetat glasial dalam air (1%) dengan perbandingan (20:80), (30:70),
(40:60), laju alir 2 ml/menit dan dideteksi pada panjang gelombang 254 nm.
Kemudian dipilih perbandingan fase gerak yang memberikan data yang terbaik.
3.5.2.6 Uji Kualitatif Sulfadoksin dan Pirimetamin menggunakan KCKT
3.5.2.6.1 Menentukan waktu tambat Sulfadoksin dan Pirimetamin BPFI
Larutan Induk Baku Sulfadoksin dipipet 5 ml masukkan dalam labu 10 ml,
dicukupkan dengan pelarut sampai garis tanda, dikocok. Maka diperoleh larutan
Sulfadoksin dengan konsentrasi 500 mcg/ml.
Larutan Induk baku Pirimetamin dipipet 1 ml masukkan dalam labu 10 ml,
dicukupkan dengan pelarut samapi garis tanda, dikocok. Maka diperoleh larutan
Pirimetamin dengan konsentrasi 25 mcg/ml.
Masing-masing larutan disaring dengan membran filter PTFE 0,2 µ l dan
diawaudarakan selama 5 menit, diinjeksikan ke dalam sistem KCKT sebanyak 20 µ l,
dengan laju alir 2 ml/menit, menggunakan perbandingan fase gerak yang telah dipilih,
kemudian dicatat masing-masing waktu tambatnya.
Masing-masing larutan sampel Sulfadoksin dan Pirimetamin dalam sediaan
tablet dengan konsentrasi Sulfadoksin 500 mcg/ml dan Pirimetamin 25 mcg/ml,
disuntikkan kesistem KCKT dengan volume penyuntikan 20 µ l pada kondisi
kromatogafi yang sama dengan BPFI. Kemudian waktu tambat masing-masing tablet
dibandingkan dengan waktu tambat Sulfadoksin dan Pirimetamin BPFI. Apabila
waktu tambat sampel hampir sama dengan waktu tambat BPFI, maka sampel tablet
mengandung Sulfadoksin dan Pirimetamin.
3.5.2.7 Penentuan Kuantitatif Sulfadoksin dan Pirimetamin menggunakan
KCKT
3.5.2.7.1 Pembuatan Kurva Kalibrasi Sulfadoksin BPFI
Larutan Induk Baku Sulfadoksin dipipet sebanyak 3 ml, 4 ml, 5 ml, 6 ml dan
7 ml, dimasukkan dalam labu tentukur 10 ml, diencerkan dengan pelarut hingga garis
tanda, kocok. Maka diperoleh konsentrasi 300 mcg/ml, 400 mcg/ml, 500 mcg/ml, 600
mcg/ml, 700 mcg/ml. Kemudian masing-masing larutan disaring dengan membran
filter PTFE 0,2 µ m dan diawaudarakan selama 5 menit, diinjeksikan kesistem KCKT
sebanyak 20 µ l, dengan laju alir 2 ml/menit dan dideteksi pada panjang gelombang
254 nm. Selanjutnya dari luas area yang diperoleh dibuat kurva kalibrasi, dihitung
persamaan regresi dan faktor korelasinya.
3.5.2.7.2 Pembuatan Kurva Kalibrasi Pirimetamin BPFI
Larutan Induk Baku Pirimetamin dipipet sebanyak 1 ml, 2 ml, 3 ml, 4 ml dan
garis tanda, kocok. Maka diperoleh konsentrasi 10 mcg/ml, 20 mcg/ml, 30 mcg/ml,
40 mcg/ml, 50 mcg/ml. Kemudian masing-masing larutan disaring dengan membran
filter PTFE 0,2 µ m dan diawaudarakan selama 5 menit, diinjeksikan kesistem KCKT
sebanyak 20 µ l, dengan laju alir 2 ml/menit dan dideteksi pada panjang gelombang
254 nm. Selanjutnya dari luas area yang diperoleh dibuat kurva kalibrasi, dihitung
persamaan regresi dan faktor korelasinya.
3.5.2.7.3 Penetapan kadar sampel
Ditimbang 20 tablet yang mengandung Sulfadoksin dan Pirimetamin
kemudian digerus, ditimbang sejumlah serbuk tablet setara dengan 500 mg
Sulfadoksin dan 25 mg Pirimetamin (sebanyak 6 kali perlakuan). Masing-masing
dimasukkan kedalam labu tentukur 50 ml, dilarutkan dengan 35 ml acetonitril,
diencerkan dengan pelarut sampai garis tanda, kocok. Maka diperoleh larutan dengan
konsentrasi 10000 mcg/ml Sulfadoksin dan 500 mcg/ml Pirimetamin, kemudian
saring dengan kertas saring, 10 % filtrat pertama dibuang. Dari keenam larutan
masing-masing dipipet 0,5 ml dimasukkan kedalam labu tentukur 10 ml, diencerkan
dengan pelarut sampai garis tanda, kocok. Maka diperoleh larutan dengan konsentrasi
500 mcg/ml Sulfadoksin dan 25 mcg/ml Pirimetamin. Masing-masing larutan tersebut
disaring dengan membran filter PTFE 0,2 µ l dan diawaudarakan selama 5 menit,
kemudian diinjeksikan ke sistem KCKT sebanyak 20 µl, dengan laju alir 2 ml/menit
dan dideteksi pada panjang gelombang 254 nm.
Kadar dapat dihitung dengan mensubtitusikan luas area sampel pada Y dari
persamaan regresi :
3.5.3 Penentuan Uji Validasi dengan Parameter Akurasi dan Presisi
3.5.3.1 Uji akurasi dengan persen perolehan kembali (% Recovery)
Uji akurasi dengan parameter persen perolehan kembali (% Recovery)
dilakukan secara Standard Addition Method dengan membuat 3 konsentrasi analit
Sulfadoksin-Pirimetamin dan baku pembanding dengan rentang spesifik 80%, 100%,
120%, setiap rentang mengandung 70% analit sampel dan 30% bahan baku, pada
perlakuan yang sama dengan perlakuan sampel.
Menurut WHO (1992) persen perolehan kembali dapat dihitung dengan
rumus:
% Perolehan kembali x100%
C B A−
=
Keterangan :
A = Konsentrasi sampel yang diperoleh setelah penambahan bahan baku
B = Konsentrasi sampel sebelum penambahan bahan baku
C = Konsentrasi baku yang ditambahkan
3.5.3.2 Uji Presisi
Uji presisi ditentukan dengan parameter Relatif Standar Deviasi (RSD)
dengan rumus:
% 100
x X SD RSD=
Keterangan:
RSD = Standar Deviasi Relatif (%)
X = Kadar rata-rata sampel (Rohman, 2007)
3.5.3.3 Penentuan Batas Deteksi (LOD) dan Batas Kuantitasi (LOQ)
Untuk menentukan batas deteksi (LOD) dan batas kuantitasi (LOQ)
digunakan rumus: 2 ) ( 2 − − = n Yi Y SB Slope SB x LOD=3
Slope SB x LOQ=10
Keterangan:
SB = Simpangan baku
LOD = Batas Deteksi
LOQ = Batas Kuantitasi (WHO, 1992)
3.5.3.4 Analisis Data Secara Statistik
Untuk menghitung Standar Deviasi (SD) digunakan rumus:
1 ) ( − − =
∑
n X X SD Keterangan :SD = Standar deviasi
X = Kadar sampel
X = Kadar rata-rata sampel
Kadar dapat dihitung dengan persamaan garis regresi dan untuk menentukan
data diterima atau ditolak digunakan rumus:
t hitung
n SD
X X
/ − =
Dengan dasar penolakan apabila t hitung ≥ t tabel
Untuk mencari kadar sebenarnya dengan α = 0,01, dk = n - 1, dapat digunakan rumus:
n SD x t
X (1 1/2α)dk
µ= ± −
Keterangan:
μ = Kadar sebenarnya
X = Kadar sampel
n = Jumlah perlakuan
t = Suatu harga tergantung pada derajad kebebasan dan tinggkat kepercayaan
dk= Derajad kebebasan. (Wibisono, 2005)
BAB IV
HASIL DAN PEMBAHASAN
4.1 Uji Identifikasi menggunakan Inframerah
Baku Sulfadoksin dan Pirimetamin yang diperoleh dari PT. Ifars sebelum
digunakan sebagai pembanding terlebih dahulu diidentifikasi menggunakan
Spektrofotometer IR pada rentang bilangan gelombang 4000 – 500 cm-1.
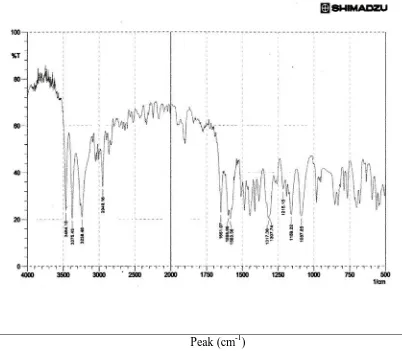
[image:42.612.116.518.309.660.2]Spektrum Inframerah baku Sulfadoksin dan Pirimetamin dapat dilihat pada
Gambar 1 dan 2 di bawah ini:
Peak (cm-1)
Gambar 1. Spektrum Inframerah dari baku pabrik Sulfadoksin (PT. Ifars)
Peak (cm-1)
810.1
833.2
5
1085.9
2
1573.9
1
1625.9
9
1645.2
8
3130.4
7
3305.9
9
3468.0
1
Gambar 2. Spektrum Inframerah baku pabrik Pirimetamin (PT. Ifars)
Dari hasil spektrum Sulfadoksin dan Pirimetamin diperoleh bentuk spektrum
yang hampir sama dengan spektrum pembanding yang terdapat pada library (dapat
dilihat pada Lampiran 3 dan 4). Bilangan gelombang pada daerah sidik jari juga
Sulfadoksin pada bilangan gelombang 1596, 1583, 1315, 1161, 1091, 1305 cm-1.
Sedangkan untuk Pirimetamin pada bilangan gelombang 1640, 1628, 1575, 1075,
835, 805 cm-1 (Clarke’s, 2005).
Pada daerah gugus fungsi dari spektrum Sulfadoksin terlihat beberapa peak,
yaitu pada bilangan gelombang 3464,15 - 3375,43 cm-1 menunjukkan gugus amin
primer dan pada bilangan gelombang 3238,48 cm-1 menunjukan gugus amin
sekunder. Sedangkan pada daerah gugus fungsi spektrum Pirimetamin terlihat
beberapa peak, yaitu pada bilangan gelombang 3468,01 - 3305,99 cm-1 menunjukkan
gugus amin primerdan pada bilangan gelombang 3100 cm-1 menunjukkan stretching
═C─H aromatis. Dari data spektrum yang diperoleh, dapat diambil kesimpulan
bahwa baku yang diidentifikasi adalah Sulfadoksin dan Pirimetamin.
4.2 Uji Kualitatif dan Kuantitatif Sulfadoksin-Pirimetamin menggunakan
KCKT
4.2.1 Penentuan perbandingan fase gerak
Pada kromatografi fase terbalik, fase diamnya kurang polar dari pada fase
gerak, kemampuan elusi menurun dengan meningkatnya polaritas pelarut. Pemisahan
dua zat tergantung dari perbedaan polaritas solut yang dipisahkan. Besarnya polaritas
dari suatu zat meningkat dengan bertambahnya gugus fungsional dan harga pKa serta
berkurang dengan bertambahnya berat molekul (Adnan, 1997). Dari rumus bangun
Pirimetamin mempunyai BM rendah, harga log P dan pKa lebih tinggi bila
dibandingkan dengan Sulfadoksin, dengan demikian dapat disimpulkan bahwa
Menurut Munson tahun 1991, zat terlarut yang kuat berinteraksi dengan fase
gerak akan terelusi lebih cepat. Hal ini berarti interaksi Pirimetamin dengan fase diam
lebih singkat dibanding interaksi Sulfadoksin dengan fase diam. Sehingga waktu
tambat Pirimetamin lebih cepat dibanding Sulfadoksin.
Untuk mendapatkan kondisi fase gerak yang optimum dilakukan dengan
menyuntikkan campuran Sulfadoksin dan Pirimetamin masing-masing dengan
konsentrasi 500 mcg/ml dan 25 mcg/ml, diinjeksikan ke dalam sistem KCKT dan
divariasikan perbandingan fase gerak asetonitril dan asam asetat asetat glasial dalam
air 1%, yaitu (20:80), (30:70), (40:60). Hasil kromatogram dapat dilihat pada Gambar
[image:45.612.118.508.346.604.2]3, 4 dan 5.
Gambar 3 Kromatogram hasil penyuntikan larutan BPFI dengan konsentrasi 500
perbandingan fase gerak asetonitril-asam asetat glasial dalam air 1%
[image:46.612.123.504.163.438.2](20:80)
Gambar 4. Kromatogram hasil penyuntikan larutan BPFI dengan konsentrasi 500
mcg/ml Sulfadoksin dan 25 mcg/ml Pirimetamin dengan menggunakan
perbandingan fase gerak asetonitril-asam asetat glasial dalam air 1%
Gambar 5. Kromatogram hasil penyuntikan larutan BPFI dengan konsentrasi 500
mcg/ml Sulfadoksin dan 25 mcg/ml Pirimetamin, menggunakan
perbandingan fase gerak asetonitril - asam asetat glasial dalam air 1%
(40:60)
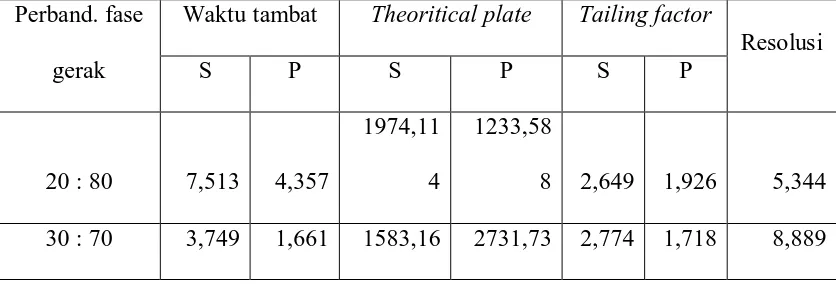
Tabel 1. Hasil optimasi fase gerak dengan parameter data waktu tambat, resolusi,
tailing dan theoretical plate
Perband. fase
gerak
Waktu tambat Theoritical plate Tailing factor
Resolusi
S P S P S P
20 : 80 7,513 4,357
1974,11
4
1233,58
8 2,649 1,926 5,344
[image:47.612.114.532.549.692.2]4 1
40 : 60 2,631 1,329
1549,06
0 884,796 2,466 0,841 5,841
Dari tabel diatas perbandingan fase gerak asetonitril-asam asetat glasial dalam
air (1%) yang terbaik yaitu dengan perbandingan (40:60), karena waktu tambatnya
dari kedua komponen relatif lebih singkat dan memberikan faktor tailing yang lebih
kecil.
Setelah didapatkan perbandingan fase gerak yang terbaik selanjutnya
dilakukan uji identifikasi.
4.2.2 Uji Kualitatif Sulfadoksin-Pirimetamin menggunakan KCKT
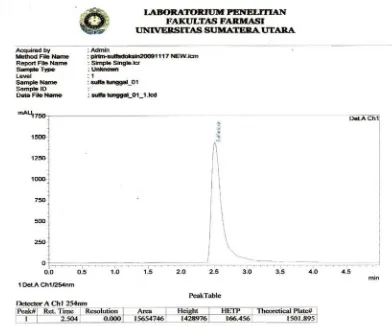
Hasil uji kualitatif Sulfadoksin dan Pirimetamin BPFI pada penyuntikan
terpisah masing-masing dengan konsentrasi 500 mcg/ml dan 25 mcg/ml diperoleh
kromatogram dengan waktu tambat Sulfadoksin 2,504 menit dan Pirimetamin 1,271
menit dapat dilihat pada Gambar 6 dan 7. Waktu tambat yang diperoleh dari
pengujian BPFI dibandingkan dengan waktu tambat yang diperoleh sampel yang akan
Gambar 6. Kromatogram hasil penyuntikan larutan 25 mcg/ml Pirimetamin BPFI,
dengan perbandingan asetonitril-asam asetat glasial dalam air 1%
Gambar 7. Kromatogram hasil penyuntikan larutan 500 mcg/ml Sulfadoksin BPFI,
dengan perbandingan asetonitril-asam asetat glasial dalam air 1%
(40:60)
Hasil pengujian untuk sampel diperoleh waktu tambat yang hampir sama
dengan Sulfadoksin-Pirimetamin BPFI. Waktu tambat rata-rata tablet Fansidar (PT.
Roche) Sulfadoksin 2,542 menit dan Pirimetamin 1,285 menit. Tablet Generik (PT.
Ifars) Sulfadoksin 2,548 menit dan Pirimetamin 1,311. Tablet Suldox (PT. Actavis)
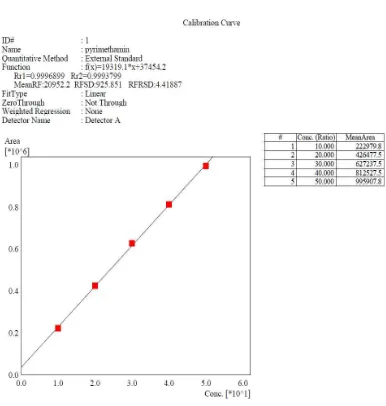
4.2.3 Penentuan linieritas kurva kalibrasi
Penentuan linieritas kurva kalibrasi Sulfadoksin BPFI ditentukan berdasarkan
luas puncak, pada konsentrasi 300, 400, 500, 600, 700 mcg/ml, diperoleh hubungan
yang linier dengan koefisien korelasi (r) = 0,9996 dan persamaan regresi Y = 30931,2
X + 317656.
Gambar 8. Kurva kalibrasi larutan Sulfadoksin BPFI konsentrasi versus luas puncak
Penentuan linieritas kurva kalibrasi Pirimetamin BPFI ditentukan berdasarkan
luas puncak, pada konsentrasi 10, 20, 30, 40, 50 mcg/ml, diperoleh hubungan yang
linier dengan koefisien korelasi (r) = 0,9997 dan persamaan regresi Y = 19319,1 X +
37454,2.
Gambar 9. Kurva kalibrasi larutan Pirimetamin BPFI konsentrasi versus luas
puncak.
[image:53.612.122.508.140.546.2]Sesuai dengan teori yang dikemukakan oleh Snyder dan Kirland (1979) yaitu
pengukuran kadar lebih baik dengan cara pengukuran luas puncak dibandingkan
dengan pengukuran tinggi puncak. Maka dalam hal ini yang digunakan adalah nilai
dari luas puncak.
Kadar dapat dihitung dengan mensubtitusikan luas puncak pada Y dari
persamaan regresi Sulfadoksin Y = 30931,2 X + 317656 dan Pirimetamin Y =
19319,1 X + 37454,2.
Hasil perhitungan kadar setelah dilakukan uji statistik dapat dilihat pada Tabel
2.
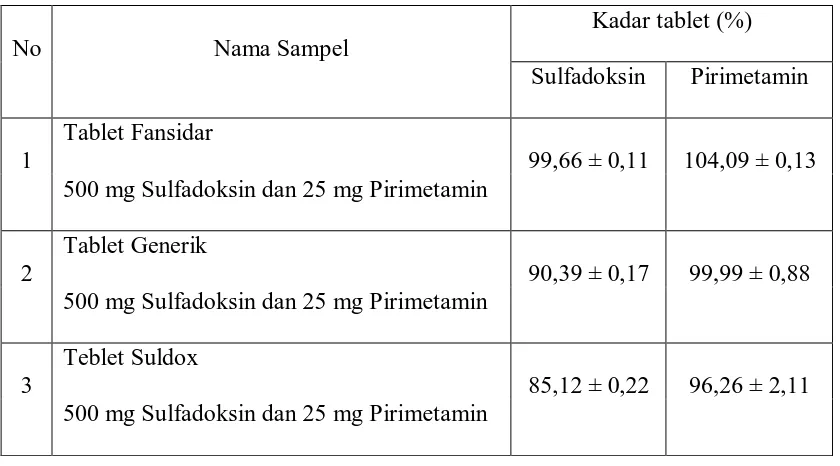
Tabel 2. Kadar hasil penetapan kadar Sulfadoksin dan Pirimetamin dalam tablet
No Nama Sampel
Kadar tablet (%)
Sulfadoksin Pirimetamin
[image:54.612.112.532.386.619.2]1
Tablet Fansidar
99,66 ± 0,11 104,09 ± 0,13 500 mg Sulfadoksin dan 25 mg Pirimetamin
2
Tablet Generik
90,39 ± 0,17 99,99 ± 0,88 500 mg Sulfadoksin dan 25 mg Pirimetamin
3
Teblet Suldox
85,12 ± 0,22 96,26 ± 2,11 500 mg Sulfadoksin dan 25 mg Pirimetamin
Dari tabel diatas terlihat bahwa dari ketiga sampel yang diteliti terdapat satu
diperoleh Sulfadoksin kurang dari 90%, sedangkan persyaratan kadar yang tertera
dalam USP edisi 30 tahun 2007 adalah tablet Sulfadoksin-Pirimetamin tidak kurang
dari 90,0% dan tidak lebih dari 110,0% dari jumlah yang tertera pada etiket.
4.4 Hasil uji validasi
Pada penelitian ini dilakukan uji validasi metode, dengan metode
penambahan bahan baku (Standard Addition Method) terhadap sampel tablet Fansidar
(PT. Roche). Uji ini meliputi uji akurasi dengan parameter persen perolehan kembali
(% recovery) dan uji presisi dengan parameter RSD (Relatif Standar Deviasi), batas
deteksi (LOD) dan batas kuantitasi (LOQ).
Uji akurasi dengan parameter persen perolehan kembali dilakukan dengan
membuat 3 konsentrasi analit dengan rentang spesifik 80%, 100%, 120%,
masing-masing dengan 3 replikasi dan setiap rentang spesifik mengandung 70% analit dan
30% bahan baku. Data hasil pengujian perolehan kembali tablet Fansidar dengan
metode penambahan bahan baku (Standar Addition Method) dapat dilihat pada Tabel
[image:55.612.114.507.552.699.2]3 di bawah ini.
Tabel 3. Data hasil perolehan kembali Sulfadoksin-Pirimetamin dengan metode
penambahan bahan baku (Standard Addition Method)
No Rentang Spesifik (%)
Luas area % Recovery
S P S P
1
80
12899960 428949 105.79 91.49
2 12897714 427830 105.73 90.55
4
100
15908128 527126 103.39 92.56
5 15873431 529186 102.64 93.94
6 15899481 528702 103.20 93.62
7
120
18841766 628138 100.47 94.54
8 18872960 629466 101.03 95.28
9 18859348 628000 100.78 94.46
Kadar rata-rata (%) 103.13 93.17
Standar Deviasi (SD) 2.09 1.65
Relatif Standar Deviasi 2.03 1.66
Keterangan :
- S = Sulfadoksin - P = Pirimetamin
Dari tabel diatas diperoleh persen perolehan kembali Sulfadoksin 103,13%
dengan standar deviasi (SD) 2,09%, sedangkan Pirimetamin 93,17% dengan standar
deviasi 1,65%. Syarat persen perolehan kembali yang diizinkan terletak pada rentang
90 – 107 % (Harmita, 2004).
Dari hasil uji presisi dengan parameter Relatif Standar Deviasi (RSD) untuk
Sulfadoksin 2,03% dan Pirimetamin 1,66%. Nilai RSD yang diizinkan adalah ≤ 2,5%.
Batas deteksi (LOD) dan batas kuantitasi (LOQ) yang diperoleh dari
penelitian ini untuk Sulfadoksin sebasar 35,99 mcg/ml dan 119,95 mcg/ml,
BAB V
KESIMPULAN DAN SARAN
5.1 Kesimpulan
Penetapan kadar Sulfadoksin dan Pirimetamin dalam sediaan tablet dapat
dilakukan secara Kromatografi Cair Kinerja Tinggi (KCKT) dengan metoda baku
luar (Eksternal standar), menggunakan fase gerak asetonitril-asam asetat glasial
dalam air 1% dengan perbandingan (40:60), laju alir 2 ml/menit, pada panjang
gelombang 254 nm. Dari hasil uji validitas menunjukkan bahwa metode yang
dilakukan memenuhi persyaratan validasi, meliputi uji akurasi dan presisi.
Hasil penetapan kadar tablet Sulfadoksin dan Pirimetamin dari tiga sampel
dengan nama dagang dan generik, terdapat satu sampel dengan nama dagang yaitu
Suldox yang tidak memenuhi persyaratan kadar yang tertera dalam USP edisi 30
tahun 2007, yaitu tidak kurang dari 90,0% dan tidak lebih dari 110,0% dari jumlah
yang tertera pada etiket.
5.2 Saran
Disarankan agar dilakukan penelitian lebih lanjut menggunakan kolom fase
DAFTAR PUSTAKA
Anonim. (2008). Pedoman Penulisan Skripsi. Fakultas Farmasi. Universitas
Sumatera Utara.
Anonim. (2007). The United States Pharmacopeia 30th Edition. National Formulary.
United States Pharmacopeia Convention. Hal. 3243.
Harmita. (2004). Petunjuk Pelaksanaan Validasi Metode dan Cara Perhitungannya.
Reviw Artikel. Majalah Ilmu Kefarmasian, Volume I (3). Hal.117-135.
Harvey, R.A, Mycek, M. J. (2001) Farmakologi Ulasan Bergambar. Edisi II. Editior
Pamela C. Champe. Penerbit Widya Medika. Jakarta. Hal. 357.
Moffat, A.C. (2005). Clarke’S Analysis Of Drug And Poisons. Thirth edition.
London: Pharmaceutical Press. Electronic version.
Munson, J. W. (1991). Analisis Farmasi Metode Modern. Penerbit Air langga
Univercity Press. Surabaya. Hal. 46.
Prabowo, A. (2004). Malaria mencegah dan mengatasinya. Penerbit: Puspa Swara,
Jakarta. Hal 2-5.
Rohman, A. (2007). Kimia Faramasi Analisis. Yogyakarta. Penerbit: Pustaka pelajar.
Hal.18.
Snyder, L. And Kirkland, J. (1979). Introduction to Modern Liquid Chromatography.
2nd edition, By Jhon Wiley and Son. London. Page. 554.
WHO. (1992). The International Pharmacopoeia. Fourth Edition. Electronic Version
Geneva: World Health Organization.
Wibisono, Y. (2005). Metode Statistik. Yogyakarta. Gajah mada. Univercity Press.
Lampiran 1. Gambar alat KCKT dan syringe 100 µ l
Gambar 11. Syringe 100 µ l (SGE)
Lampiran 2. Gambar Sonifikator (Branson 1510) dan Penyaring
[image:60.612.177.484.373.643.2]Gambar Penyaring
Gambar 13. Pompa Vakum (Gast DO A-PG04-BN) dan alat
Lampiran 3. Spektrum Inframerah Sulfadoksin pada literatur Pharmaceutical Sub
Gambar 14. Spektrum Inframerah Sulfadoksin
Lampiran 4. Spektrum Inframerah Pirimetamin pada literatur Pharmaceutical Sub
Lampiran 5. Perhitungan persamaan regresi dari kurva kalibrasi Sulfadoksin
N
o X Y XY X² Y²
1 300 9,692,420.8 2,907,726,240 90,000 93,943,020,964,272.7
2 400 12,707,450.4 5,082,980,160 160,000 161,479,295,668,460.0
3 500 15,556,742.6 7,778,371,300 250,000 242,012,240,322,655.0
4 600 18,895,768.3
11,337,460,98
0 360,000 357,050,059,647,285.0
5 700 22,063,852.9
15,444,697,03
0 490,000 486,813,604,792,838.0
∑ 2,500 78,916,235.0
42,551,235,71 0 1,350,00 0 6,227,772,146,575,220. 0 b aX Y= +
( ) ( )( )
( )
2( )
2 X X n Y X XY n a Σ − Σ Σ Σ − Σ =(
)
(
)
(
) (
)
22500 000 . 350 . 1 5 235 . 916 . 78 500 . 2 710 . 235 . 551 . 42 5 − − =
=30.931,18 aX Y b= −
=15.783.247−
(
30.931,18)( )
500 =317.656( ) ( )( )
( ) ( )
[
2 2]
[
( )
2( )
2]
Y Y n X X n Y X XY n r Σ − Σ Σ − Σ Σ Σ − Σ =
(
) (
)(
)
(
) (
)
[
2]
[
(
) (
)
2]
235 . 916 . 78 510 . 395 . 221 . 298 . 341 . 1 5 500 . 2 000 . 350 . 1 5 235 . 916 . 78 2500 710 . 235 . 551 . 42 5 − − − = r 9996344 , 0 = r
317656 2 , 30931 + = X Y
Lampiran 6. Perhitungan Batas Deteksi (LOD) dan Batas Kuantitasi (LOQ)
Sulfadoksin
No Consentrasi
Luas
Puncak Yi Y – Yi ( Y - Yi )²
X (mcg/ml) Y
1 300 9692420.8 9597016 95404.8 9102075863.04
2 400 12707450.4 12372480 334970.4 112205168876.16
3 500 15556742.6 15465600 91142.6 8306973534.76
4 600 18895768.3 18558720 337048.3 113601556532.89
5 700 22063852.9 21651840 412012.9 169754629766.41
∑ 412970404573.26
2 ) ( 2 − − =
∑
n Yi Y SB 2 5 ) 26 , 73 4129704045 ( − = SB 29 , 371021 = SB Slope SB x LOD=32 , 30931 29 , 371021 10 x LOQ= 95 , 119 = LOQ mcg/ml
Lampiran 7. Perhitungan Persamaan Regresi dari kurva kalibrasi Pirimetamin
No X Y XY X² Y²
1 10 222.979,80 2.229.798 100 49.719.991.208,04
2 20 426.477,50 8.529.550 400 181.883.058.006,25
3 30 627.237,50 18.817.125 900 393.426.881.406,25
4 40 812.527,50 32.501.100 1.600 660.200.938.256,25
5 50 995.907,80 49.795.390 2.500 991.832.346.100,84
∑ 150 3.085.130,10 111.872.963 5.500 2.277.063.214.977,63
b aX Y= +
( ) ( )( )
( )
2( )
2 X X n Y X XY n a Σ − Σ Σ Σ − Σ =(
)
(
)
(
) ( )
2150 500 . 5 5 1 , 130 . 085 . 3 150 963 . 872 . 111 5 − − =
=19.319,1 aX Y b= −
=617.026,02−
(
19.319)( )
30 =37454,2( ) ( )( )
( ) ( )
[
2 2]
[
( )
2( )
2]
Y Y n X X n Y X XY n r Σ − Σ Σ − Σ Σ Σ − Σ =
(
) ( )(
)
(
) ( )
[
2]
[
(
) (
)
2]
Jadi Persamaannya didapat : 2 , 37454 1 , 19319 + = X Y
Lampiran 8. Perhitungan Batas Deteksi (LOD) dan Batas Kuantitasi (LOQ)
Pirimetamin
No
Consentrasi Luas Puncak
Yi Y - Yi ( Y - Yi )² X (mcg/ml) Y
1 10 222979.8 230645.2 -7665.4 58758357.16
2 20 426477.5 423836.2 2641.3 6976465.69
3 30 627237.5 617027.2 10210.3 104250226.09
4 40 812527.5 810218.2