xxii INTISARI
Telah dilakukan penelitian mengenai optimasi komposisi dan kecepatan alir fase gerak sistem Kromatografi Cair Kinerja Tinggi (KCKT) fase terbalik pada penetapan kadar nikotin dalam rokok “merek X” menggunakan standar internal asetanilida. Penelitian ini bertujuan untuk menentukan kondisi optimal sistem KCKT fase terbalik sehingga dapat digunakan untuk validasi metode dan penetapan kadar nikotin dalam ekstrak etanolik tembakau rokok.
Penelitian ini merupakan penelitian eksperimental deskriptif. Sistem KCKT fase terbalik yang dipakai dalam penelitian ini menggunakan fase diam oktil silika (C8) dan fase gerak metanol : ammonium asetat 10 mM + TEA 0,1% dengan detektor
UV pada panjang gelombang 260 nm. Optimasi dilakukan dengan mengubah komposisi fase gerak 50:50, 60:40, dan 70:30 serta kecepatan alir 0,5; 0,8 dan 1,0 mL/min untuk menentukan bentuk peak, retention time yang efisien, nilai resolusi, nilai tailing factor, dan reprodusibilitas retention time.
Kondisi optimum sistem KCKT fase terbalik hasil optimasi yakni fase gerak metanol : ammonium asetat 10 mM + TEA 0,1% (70:30) pada kecepatan alir 1,0 mL/min. Kondisi ini memenuhi parameter pemisahan yang baik yaitu Rs 3,915; tR
nikotin 4,638 menit; TF 1,983 dan tR asetanilida 3,645 menit; TF 1,699.
xxiii ABSTRACT
A research about optimization of reversed phase High Performance Liquid Chromatography (HPLC) to determine nicotine in cigarette “brand X” using acetanilide as internal standard have been done. The purpose of this research are finding an optimum mobile phase composition and flow rate of reversed phase HPLC system used for method validation and determination levels of nicotine in ethanolic extract of tobacco cigarettes.
This research is conducted with a descriptive experimental design. The reversed phase HPLC system uses in this research using octyl silica (C8) as stationary
phase, methanol:ammonium acetate 10 mM + TEA 0.1% as mobile phase, and UV detector at wavelength 260 nm. Optimization is done by changing the composition of the mobile phase 50:50, 60:40 and 70:30 with flow rate at 0.5; 0.8 and 1.0 mL/minute to determine a peak shape, efficient retention time, resolution value, tailing factor value, and retention time reproducibility.
This research resulted in an optimum condition of reversed phase HPLC system in mobile phase composition of methanol:ammonium acetate 10 mM + TEA 0.1% (70:30) and a flow rate of 1.0 mL/min. This conditions fulfills the parameters of good separation which contains resolution value 3.915; nicotine’s tR 4.638 minutes
i
OPTIMASI KOMPOSISI DAN KECEPATAN ALIR FASE GERAK SISTEM KROMATOGRAFI CAIR KINERJA TINGGI (KCKT) FASE TERBALIK
PADA PENETAPAN KADAR NIKOTIN DALAM ROKOK “MEREK X” MENGGUNAKAN STANDAR INTERNAL ASETANILIDA
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Farmasi
Oleh: Eric Antonius NIM : 098114108
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
ii
Persetujuan Pembimbing
OPTIMASI KOMPOSISI DAN KECEPATAN ALIR FASE GERAK SISTEM
KROMATOGRAFI CAIR KINERJA TINGGI (KCKT) FASE TERBALIK PADA PENETAPAN KADAR NIKOTIN DALAM ROKOK “MEREK X”
MENGGUNAKAN STANDAR INTERNAL ASETANILIDA
Skripsi yang diajukan oleh: Eric Antonius NIM : 098114108
telah disetujui oleh
Pembimbing Utama
iii
Pengesahan Skripsi berjudul
OPTIMASI KOMPOSISI DAN KECEPATAN ALIR FASE GERAK SISTEM KROMATOGRAFI CAIR KINERJA TINGGI (KCKT) FASE TERBALIK
PADA PENETAPAN KADAR NIKOTIN DALAM ROKOK “MEREK X” MENGGUNAKAN STANDAR INTERNAL ASETANILIDA
Oleh : Eric Antonius NIM : 098114108
Dipertahankan di hadapan Panitia Penguji Skripsi Fakultas Farmasi
Universitas Sanata Dharma Pada tanggal : 22 Februari 2013
iv
HALAMAN PERSEMBAHAN
Kehidupan terkadang menyakitkan saat kita tahu kebenarannya. Cinta terkadang menyakitkan saat kita tahu kejujurannya.
Pengetahuan terkadang menyakitkan saat kita tidak mampu meraihnya.
Tekad terkadang menjadi arang saat kegagalan melanda. TETAPI
Kehidupan terkadang menyenangkan saat kita mampu menghadapinya.
Cinta terkadang membahagiakan saat kita tahu pengorbanannya.
Pengetahuan terkadang menghibur saat kita mampu mendapat solusinya.
Tekad menjadi batu karang saat kita berani menggenapinya. BERMIMPILAH dan BERUSAHALAH
Kesuksesan merupakan buah kemauan yang dilakukan berulang
kali dengan sepenuh hati.
Karya ini kudedikasikan untuk orang tuaku, kekasih hatiku, teman-temanku dan almamaterku.
“You have to dream before your dreams can come true.”– Abdul Kalam
“Anyone who has never made a mistake has never tried anything new” – Albert Einstein
v
PERNYATAAN KEASLIAN KARYA
Saya menyatakan dengan sesungguhnya bahwa skripsi yang saya susun ini tidak memuat karya atau bagian dari pekerjaan orang lain, kecuali yang telah disebutkan dalam kutipan dan daftar pustaka, sebagaimana layaknya sebuah karya ilmiah.
Apabila dikemudian hari ditemukan adanya indikasi plagiarisme dalam naskah yang saya susun ini, maka saya bersedia menanggung segala resiko dan sanksi sesuai dengan peraturan perundang-undangan yang berlaku.
Yogyakarta, 18 Februari 2013 Penulis,
vi
LEMBAR PERNYATAAN PERSETUJUAN
PUBLIKASI KARYA ILMIAH UNTUK KEPENTINGAN AKADEMIS
Yang bertanda tangan di bawah ini, saya mahasiswa Universitas Sanata Dharma: Nama : Eric Antonius
Nomor Mahasiswa : 09811410
Demi pengembangan ilmu pengetahuan, saya memberikan kepada Perpustakaan Sanata Dharma karya ilmiah yang berjudul:
“OPTIMASI KOMPOSISI DAN KECEPATA ALIR FASE GERAK SISTEM KROMATOGRAFI CAIR KINERJA TINGGI (KCKT) FASE TERBALIK
PADA PENETAPAN KADAR NIKOTIN DALAM ROKOK “MEREK X” MENGGUNAKAN STANDAR INTERNAL ASETANILIDA”
Beserta perangkat yang diperlukan (bila ada). Dengan demikian saya memberikan kepada Perpustakaan Universitas Sanata Dharma hak untuk menyimpan, mengalihkan dalam bentuk media lain, mengelolanya dalam bentuk pangkalan data, mendistribusikannya secara terbatas, dan mempubliksaikannya di internet atau media lain untuk kepentingan akademis tanpa perlu meminta ijin dari saya maupun memberikan royalti kepada saya selama tetap mencantumkan nama saya sebagai penulis.
Dengan demikian pernyataan ini saya buat dengan sebenarnya.
Dibuat di Yogyakarta
Pada tanggal : 18 Februari 2013 Yang menyatakan
vii PRAKATA
Puji dan syukur kepada Tuhan Yang Maha Esa atas cinta kasih, berkat, ijin dan peryertaan-Nya yang begitu besar, sehingga penulis dapat menyelesaikan skripsi
yang berjudul “Optimasi Metode Kromatografi Cair Kinerja Tinggi (Kckt) Fase
Terbalik Pada Penetapan Kadar Nikotin Dalam Rokok “Merek X” Menggunakan
Standar Internal Asetanilida” sebagai salah satu syarat yang harus dipenuhi demi memperoleh gelar Sarjana Farmasi (S.Farm.) di Fakultas Farmasi Universitas Sanata Dharma Yogyakarta.
Penulis menyadari bahwa penelitian dan penyusunan skripsi ini dapat terselesaikan karena adanya masukan, kritikan, diskusi, arahan, saran, dan bimbingan dari berbagai pihak. Penulis mengucapkan terima kasih yang sebesar-besarnya kepada :
1. Ipang Djurnarko, M.Sc., Apt. Selaku Dekan Fakultas Farmasi Uninversitas Sanata Dharma Yogyakarta atas teladan seorang pemimpin yang diberikan
viii
3. Christine Patramurti, M.Si., Apt. selaku dosen pembimbing di laboratorium dan teman selama penelitian skripsi yang telah memberikan masukan, diskusi, saran, dan dukungan moral kepada penulis selama penelitian skripsi ini.
4. Jeffry Julianus, M.Si. selaku dosen penguji yang memberikan banyak kritik dan saran yang membangun untuk skripsi ini.
5. Prof. Dr. Sri Noegrohati, Apt. selaku dosen penguji yang memberikan banyak kritik dan saran yang membangun untuk skripsi ini.
6. C.M.Ratna Rini Nastiti, M.Pharm., Apt. sebagai Kaprodi Fakultas Farmasi Universitas Sanata Dharma Yogyakarta atas teladan kepemimpinan, masukan, dan saran yang diberikan selama penulis berkuliah dan menyusun naskah.
7. Rini Dwi Astuti, M.Sc., Apt. sebagai Kepala Laboratorium Fakultas Farmasi Universitas Sanata Dharma Yogyakarta
8. Prof. Dr. Sudibyo Martono, M.S., Apt. atas waktu yang diluangkan untuk memberikan sedikit masukan diawal penelitian
9. Bimo Adithya, Suparlan, dan Kunto dan segenap staf laboran yang senantiasa siap membantu dan meluangkan waktunya dalam penyediaan bahan dan alat selama penelitian.
10.Semua dosen dan karyawan Fakultas Farmasi Universitas Sanata Dharma atas pengalaman, masukan, keceriaan, semangat, dan persahabatan yang diberikan. 11.Demas dan Is Sumitro sebagai rekan kerja dalam penelitian skripsi ini. Terima
ix
12.Lucia Shinta R, Sisilia Mirsya A, Metri S.K., Agnes Mutiara, Victor Purnama Agung, dan Novia Sarwoningtyas sebagai teman seperjuangan dalam satu lantai Laboratorium Analisis Instrumental. Terima kasih atas diskusi, canda tawa, semangat selama kita bekerja bersama-sama.
13.Yenni Soenardi Tjokro terima kasih atas perhatian, kasih sayang, semangat, dan dukungannya yang diberikan selama penelitian yang dilakukan.
14.Sihendra Ze selaku kakak dan teman berbagai cerita selama penyusunan naskah skripsi, terima kasih atas masukan dan kritik yang diberikan.
15.Fendy, Joe dan Kenny selaku sahabat, teman berbagi cerita dan teman seperjuangan atas semangat, diskusi, canda tawa yang diberikan selama penulis berkuliah di Fakultas Farmasi Universitas Sanata Dharma Yogyakarta.
16.Teman-teman “Konco Dolan”, terima kasih atas dukungannya dan canda tawa yang diberikan selama penelitian.
17.Teman angkatan 2009 yang bersama-sama berjuang dan mengisi sebagian cerita hidupku, terima kasih atas kebersamaan, diskusi, dan bantuan selama perkuliahan. 18.Semua pihak yang tidak dapat disebutkan satu persatu, atas segala bantuan,
semangat dan doa yang menyertai penulis dari awalnya penelitian hingga diselesaikannya penulisan skripsi ini.
x
yang berarti bagi para pembaca. Akhir kata, penulis mempersembahkan skripsi ini demi majunya ilmu pengetahuan farmasi.
Yogyakarta, 18 Februari 2013
xii
BAB II PENELAAHAN PUSTAKA... 6
A. Rokok... 6
B. Nikotin... 7
C.Ekstrak Tumbuhan... 9
D.Spektrofotometri UV... 10
E. Kromatografi Cair Kinerja Tinggi (KCKT)... 14
1. Definisi dan Instrumentasi KCKT... 14
2. Optimasi Metode... 23
3. Pemisahan Puncak Dalam Kromatografi... 24
F. Standar Internal... 34
G.Landasan Teori... 36
H.Hipotesis... 36
BAB III METODE PENELITIAN... 38
A. Jenis dan Rancangan Penelitian... 38
B. Variabel Penelitian... 38
1. Variabel Bebas... 38
2. Variabel Tergantung... 38
3. Varianel Pengacau Terkendali... 38
C. Definisi Operasional... 39
D. Bahan Penelitian... 39
xiii
F. Tata Cara Penelitian... 40
1. Pembuatan Fase Gerak... 40
2. Pembuatan Larutan Internal Standar Asetanilida... 41
3. Pembuatan Larutan Baku Nikotin... 41
4. Preparasi Sampel... 42
5. Penentuan Panjang Gelombang Pengamatan Nikotin dan Asetanilida... 44
6. Optimasi Metode KCKT Fase Terbalik... 44
G. Analisis Hasil... 45
1. Bentuk Peak... 45
2. Retention time (tR)... 46
3. Resolusi (Rs)... 46
4. HETP... 46
5. Reprodusibilitas retention time... 46
BAB IV HASIL DAN PEMBAHASAN... 47
A.Penentuan Fase Gerak... 47
B. Pembutan Seri Larutan Baku Nikotin dan Asetanilida... 49
C.Preparasi Sampel... 50
D.Penentuan Panjang Gelombang Pengamatan Nikotin dan Asetanilida... 53
xiv
BAB V KESIMPULAN DAN SARAN... 75
A.Kesimpulan... 75
B.Saran... 75
DAFTAR PUSTAKA... 76
LAMPIRAN... 80
xv
DAFTAR TABEL
Tabel I. Ringkasan Sifat/Karakteristik yang dimiliki nikotin... 8 Tabel II. Deret elutropik dan indeks polaritas beberapa pelarut KCKT... 19 Tabel III. Komposisi fase gerak yang dibuat dalam penelitian... 40 Tabel IV. Perbandingan panjang gelombang maksimum nikotin hasil
pengukuran terhadap panjang gelombang maksimum teoritis... 55 Tabel V. Perbandingan panjang gelombang maksimum asetanilida hasil
pengukuran terhadap panjang gelombang maksimumteoritis... 57 Tabel VI. Perbandingan komposisi fase gerak dan indeks polaritas
masing-masing komposisi fase gerak... 63 Tabel VII. Hasil optimasi peak baku nikotin dan asetanilida pada berbagai
komposisi fase gerak dan kecepatan alir... 65 Tabel VIII. Perbandingan hasil parameter optimasi baku nikotin dan
asetanilida dengan fase gerak metanol : ammonium asetat 10 mM + TEA 0,1%... 70 Tabel IX. Tekanan pompa pada berbagai komposisi fase gerak dan
kecepatan alir... 71 Tabel X. Hasil perhitungan CV nilai retention time larutan baku nikotin
xvi
Tabel XI. Hasil perhitungan CV nilai retention time larutan baku asetanilida dengan fase gerak metanol : ammonium asetat 10mM + TEA 0,1% (70:30) pada kecepatan alir 1,0 mL/min... 72 Tabel XII. Hasil perhitungan CV nilai retention time larutan sampel dan
sampel yang ditambahkan adisi nikotin sebanyak 20 µg/mL dengan fase gerak metanol : ammonium asetat 10mM + TEA 0,1% (70:30) pada kecepatan alir 1,0 mL/min... 73 Tabel XIII. Hasil perhitungan CV resolusi sampel dengan fase gerak metanol
xvii
DAFTAR GAMBAR
Gambar 1. Struktur nikotin (3-(1-metil-pirolodinil)pirindin)... 7 Gambar 2. Struktur dari macam alkaloid yang terdapat dalam tembakau... 9 Gambar 3. Diagram tingkat energi elektronik... 11 Gambar 4. (A) Pengaruh pelarut polar terhadap – transisi π → π* dan (B) –
transisi n → π*... 13
Gambar 5. Dasar pemisahan kromatografi partisi... 15 Gambar 6. Skema instrumentasi KCKT... 16 Gambar 7. Skema sampler KCKT. (A) – posisi load, sampel diinjeksikan
dan terisolasi dari fase gerak. (B) – posisi inject, sampel terbawa fase gerak dan memasuki kolom... 21 Gambar 8. Gambaran peak kromatogram untuk mengukur resolusi... 27 Gambar 9. Ilustrasi gambar yang menyebabkan pelebaran puncak selama
pemisahan menggunakan KCKT. Simbol merepresentasifkan molekul analit sebelum migrasi, simbol menyatakan molekul analit setelah migrasi, dan ---> menyatakan pergerakan dari molekul analit... 29 Gambar 10. Ilustrasi difusi Eddy saat memasukin kolom dan menyebabkan
xviii
Gambar 12. Pengaruh perubahan nilai α, k’, dan N terhadap pemisahan peak kromatogram... 33 Gambar 13. Penentuan asymmetry factor (As) dan tailing factor (TF)... 34 Gambar 14. Gambaran interaksi yang teerjadi antara TEA dengan sisi silanol
bebas yang tidak tercapping oktil silika... 48 Gambar 15. Nikotin yang berada dalam bentuk terprotonasi diubah menjadi
bentuk molekulnya karena dikondisikan dalam suasana basa dengan penambahan KOH... 51 Gambar 16. Distribusi bentuk nikotin dalam posisi protonated atau
unprotonated berdasarkan variasi pH larutan... 53 Gambar 17. Gambaran struktur nikotin dan kromofor yang dimiliki... 54 Gambar 18. Spektra yang didapatkan dari hasil percobaan dan dibandingkan
dengan sumber acuan. (A) - Spektra teoritis asetanilida. (B) – Spektra pengujian dengan konsentrasi nikotin 0,02 µg/mL. (C) – Spektra pengujian dengan konsentrasi asetanilida 0,03 µg/mL. (D) – Spektra pengujian dengan konsentrasi asetanilida 0,04 µg/mL... 55 Gambar 19. Gambaran struktur asetanilida dan kromofor yang dimilikinya.... 56 Gambar 20. Spektra yang didapatkan dari hasil percobaan dan dibandingkan
xix
Spektra pengujian dengan konsentrasi asetanilida 5 µg/mL. (D) – Spektra pengujian dengan konsentrasi asetanilida 10 µg/mL... 58 Gambar 21. (A) – Perbedaan pengukuran absorbansi pada panjang gelombang
maksimum dan tidak pada panjang gelombang maksimum. (B) – Pengukuran pada panjang gelombang maksimum akan memberikan garis linear. (C) – Pengukuran tidak pada panjang gelombang maksimum memberikan garis yang tidak linear... 60 Gambar 22. A) – Kromatogram baku asetanilida konsentrasi 20 µg/mL. (B) –
Kromatogram baku nikotin konsentrasi 20 µg/mL. Komposisi fase gerak metanol : ammonium asetat 10 mM + TEA 0,1% (60:40) dengan kecepatan alir 0,5 mL/min... 61 Gambar 23. A) – Kromatogram baku nikotin konsentrasi 20 µg/mL. (B) –
Kromatogram baku asetanilida konsentrasi 20 µg/mL. Komposisi fase gerak metanol : ammonium asetat 10 mM + TEA 0,1% (50:50) dengan kecepatan alir 1,0 mL/min... 66 Gambar 24. (A) – Kromatogram baku nikotin konsentrasi 20µg/mL. (B) –
Kromatogram baku asetanilida konsentrasi 20 µg/mL. Komposisi fase gerak metanol : ammonium asetat 10 mM + TEA 0,1% (60:40) dengan kecepatan alir 0,8 mL/min... 67 Gambar 25. (A) – Kromatogram baku nikotin konsentrasi 20µg/mL. (B) –
xx
xxi
DAFTAR LAMPIRAN
Lampiran 1. Certificate of Analysis baku nikotin E.Merck... 80 Lampiran 2. Certificate of Analysis baku asetanilida E.Merck... 81 Lampiran 3. Perhitungan polaritas fase gerak yang dioptimasi... 82 Lampiran 4. Kromatogram hasil optimasi fase gerak metanol : ammonium
asetat + TEA 0,1% (50 : 50)... 83 Lampiran 5. Kromatogram hasil optimasi fase gerak metanol : ammonium
asetat + TEA 0,1% (60 : 40)... 89 Lampiran 6. Kromatogram hasil optimasi fase gerak metanol : ammonium
asetat + TEA 0,1% (70 : 30)... 95 Lampiran 7. Contoh kromatogram sampel dengan fase gerak yang telah
teroptimasi (metanol : ammonium asetat + TEA 0,1% (70:30)).. 101 Lampiran 8. Contoh kromatogram seri baku dengan fase gerak yang telah
xxii INTISARI
Telah dilakukan penelitian mengenai optimasi komposisi dan kecepatan alir fase gerak sistem Kromatografi Cair Kinerja Tinggi (KCKT) fase terbalik pada
penetapan kadar nikotin dalam rokok “merek X” menggunakan standar internal
asetanilida. Penelitian ini bertujuan untuk menentukan kondisi optimal sistem KCKT fase terbalik sehingga dapat digunakan untuk validasi metode dan penetapan kadar nikotin dalam ekstrak etanolik tembakau rokok.
Penelitian ini merupakan penelitian eksperimental deskriptif. Sistem KCKT fase terbalik yang dipakai dalam penelitian ini menggunakan fase diam oktil silika (C8) dan fase gerak metanol : ammonium asetat 10 mM + TEA 0,1% dengan detektor UV pada panjang gelombang 260 nm. Optimasi dilakukan dengan mengubah komposisi fase gerak 50:50, 60:40, dan 70:30 serta kecepatan alir 0,5; 0,8 dan 1,0 mL/min untuk menentukan bentuk peak, retention time yang efisien, nilai resolusi, nilai tailing factor, dan reprodusibilitas retention time.
Kondisi optimum sistem KCKT fase terbalik hasil optimasi yakni fase gerak metanol : ammonium asetat 10 mM + TEA 0,1% (70:30) pada kecepatan alir 1,0 mL/min. Kondisi ini memenuhi parameter pemisahan yang baik yaitu Rs 3,915; tR nikotin 4,638 menit; TF 1,983 dan tR asetanilida 3,645 menit; TF 1,699.
xxiii ABSTRACT
A research about optimization of reversed phase High Performance Liquid
Chromatography (HPLC) to determine nicotine in cigarette “brand X” using
acetanilide as internal standard have been done. The purpose of this research are finding an optimum mobile phase composition and flow rate of reversed phase HPLC system used for method validation and determination levels of nicotine in ethanolic extract of tobacco cigarettes.
This research is conducted with a descriptive experimental design. The reversed phase HPLC system uses in this research using octyl silica (C8) as stationary phase, methanol:ammonium acetate 10 mM + TEA 0.1% as mobile phase, and UV detector at wavelength 260 nm. Optimization is done by changing the composition of the mobile phase 50:50, 60:40 and 70:30 with flow rate at 0.5; 0.8 and 1.0 mL/minute to determine a peak shape, efficient retention time, resolution value, tailing factor value, and retention time reproducibility.
1
BAB I
PENGANTAR
A. Latar Belakang
Rokok adalah hasil olahan dari tanaman Nicotiana Tabacum, Nicotiana
Rustica dan spesies lainnnya atau sintetisnya yang mengandung nikotin dan tar
dengan atau tanpa bahan tambahan (Presiden Republik Indonesia, 2003). Perokok
aktif di Indonesia mencapai 60 juta penduduk, sehingga Indonesia menduduki
peringkat ketiga dunia jumlah perokok terbanyak dan nomor satu dalam lingkup
Asia Tenggara (Yahya, 2010). Pada tahun 2007, tembakau memberikan
sumbangan pendapatan negara sebanyak 52 triliun rupiah dan devisa ekspor
tembakau sebesar 1,9 triliun rupiah. (Anonima, 2012).
Bahaya rokok cukup beragam, terbanyak disebabkan oleh serangan
jantung koroner, diikuti penyakit stroke, kanker, radang saluran pernapasan dan
paru-paru. Rokok mengandung nikotin yang merupakan psikotropika stimulan,
karena menimbulkan perasaan tenang, dan bebas stress. Tetapi saat rokok dihisap,
selain nikotin terdapat 4000 macam senyawa kimia lain dan 20 macam racun maut
yang terdapat dalam tar yang ikut terhisap (Partodiharjo, 2010). Nikotin sendiri
dapat meningkatan sekresi air liur, mual, muntah. Keracunan nikotin dapat pula
terjadi pada sistem saraf pusat yang menyebabkan depresi, selain itu berefek
terhadap kelemahan otot, tremor, konvulsi yang pada akhirnya dapat
Dipandang dari tingkat keterbahayaan rokok, pemerintah sejauh ini
melakukan usaha pengamanan rokok, dengan pencantuman label informasi
kandungan kadar nikotin dan tar untuk setiap batang rokok yang diproduksi
(Presiden Republik Indonesia, 2003). Adanya label pencantuman kadar nikotin
dan tar terkait upaya penjaminan mutu rokok menjadi perhatian penting terkait
peraturan pemerintah dalam upaya pengamanan rokok. Menurut Peraturan
Pemerintah Republik Indonesia nomor 109 tahun 2012 tentang Pengamanan
Bahan Yang Mengandung Zat Adiktif Berupa Produk Tembakau Bagi Kesehatan,
Pasal 10 Ayat 1 menyatakan “Setiap orang yang memproduksi produk tembakau
berupa rokok harus melakukan pengujian kandungan kadar nikotin dan tar per
batang untuk setiap varian yang diproduksi” (Presiden Republik Indonesia, 2012).
Pada penelitian ini akan dilakukan penetapan kadar nikotin yang terkandung
dalam ekstrak etanolik tembakau rokok “merek X” melalui serangkaian proses
penelitian.
Penelitian ini akan melalui serangkaian proses yang mencakup optimasi
metode analisis, validasi metode analisis, dan penetapan kadar nikotin dalam
ekstrak etanolik tembakau rokok “merek X”. Pada penelitian ini penulis
mengupayakan optimasi metode penetapan kadar ekstrak etanolik tembakau rokok
“merek X” tersebut.
Metode yang dipilih dalam menetapkan kadar nikotin dalam ekstrak
etanolik tembakau rokok adalah metode kromatografi cair kinerja tinggi (KCKT)
fase terbalik. Metode ini secara selektif memisahkan senyawa multikomponen
penelusuran literatur, penelitian terhadap penetapan kadar nikotin dalam tembakau
menggunakan KCKT fase terbalik sudah pernah dilakukan oleh Putri (2011).
Penulis melakukan modifikasi terhadap sistem KCKT yang digunakan
Putri (2011), sistem KCKT fase terbalik yang digunakan Putri (2011) ialah buffer
asetat : metanol : asetonitril (40:54:6) sebagai fase geraknya, kolom oktadesil
silika (C18) sebagai fase diamnya dan standar eksternal sebagai baku
pembandingnya, sedangkan penulis menggunakan fase gerak metanol :
ammonium asetat 10 mM + TEA 0,1%, kolom oktil silika (C8) sebagai fase diam
dan digunakan standar internal asetanilida sebagai baku pembanding. Sejauh
penelusuran penulis terhadap penggunaan kolom oktil silika (C8) dalam analisis
nikotin dengan sampel rokok belum pernah dilakukan, dengan demikian penulis
ingin menunjukkan bahwa dengan kolom oktil silika nikotin dalam sampel rokok
dapat dipisahkan dengan baik dan menjadi salah satu alternatif pemilihan kolom
untuk memisahan nikotin dari matriksnya. Penggantian standar eksternal menjadi
internal terkait preparasi sampel yang dilakukan dimana standar internal
digunakan untuk sampel yang melewati proses preparasi panjang (Anonimc,
2012), dalam penelitian ini tembakau pada rokok diproses hingga menjadi ekstrak
etanolik dalam fraksi kloroform. Tujuan optimasi metode disini ialah
mendapatkan kondisi optimal dari instrumen KCKT untuk memisahkan nikotin
terhadap senyawa lain yang terdapat dalam ekstrak etanolik nikotin. Optimasi
dilakukan terhadap komposisi fase gerak dan variasi kecepatan alir. Perbandingan
komposisi fase gerak dan kecepatan alir yang optimal diharapkan dapat
time, bentuk peak, nilai resolusi, dan tailing factor, nilai resolusi yang dihasilkan,
reprodusibilitas retention time baku dan sampel serta reprodusibilitas resolusi
sampel. Retention time yang efisien untuk analisis rutin ialah < 10 menit (Smith,
2002), bentuk peak dengan TF ≤ 2 (Center for Drug Evaluation and Research,
1994), nilai resolusi ≥ 1,5 (base line resolution) akan mampu memisahkan
komponen lebih baik (Gandjar dan Rohman, 2010).
1. Permasalahan
Berdasarkan latar belakang, pemasalahan yang muncul adalah bagaimana
komposisi fase gerak dan kecepatan alir yang optimal untuk menghasilkan
pemisahan yang baik dengan metode KCKT fase terbalik pada penetapan kadar
nikotin ekstrak etanolik tembakau rokok “merek X” yang dilihat dari bentuk peak,
retention time yang efisien, resolusi, nilai tailing factor, nilai resolusi yang
dihasilkan, reprodusibilitas retention time baku dan sampel serta reprodusibilitas
resolusi sampel?
2. Keaslian Penelitian
Sejauh penelusuran literatur, penelitian optimasi metode kromatografi
cair kinerja tinggi (KCKT) fase terbalik pada penetapan kadar nikotin dalam
rokok “Merek X” belum pernah dilakukan sebelumnya. Penelitian mengenai
penetapan kadar nikotin dengan metode KCKT yang pernah dilakukan adalah
determinasi nikotin dan toksisitas umum dari rokok pada Market Jordan (Alali dan
Massadeh,2002); analisis nikotin dan beberapa metabolit lain dalam urine perokok
pasif menggunakan KCKT-Tandem Mass, penetapan kadar nikotin dalam sampel
kromatografi gas, spektrofotometri massa dan kromatografi cair-MS (LC-MS)
(Nakajima, Yamamoto, Kuroiwa dan Yokoi, 2000); analisis nikotin,
3-hidroksikotinin, kotinin dan kafein dalam urin pada perokok pasif dengan
KCKT-Tandem Mass (Tuomi, Johnson dan Reijula, 1999); studi kadar nikotin dan tar
sembilan merk rokok kretek filter yang beredar di wilayah Kabupaten Nganjuk
(Kusuma, Sudarminto dan Siti, 2012).
3. Manfaat Penelitian
a. Manfaat metodologis. Penelitian ini diharapkan menjadi
sumbangan ilmiah sebagai alternatif metode penelitian dalam upaya optimasi
metode kromatografi cair kinerja tinggi (KCKT) fase terbalik pada penetapan
kadar nikotin dalam ekstrak etanolik tembakau rokok.
b. Manfaat praktis. Penelitian ini diharapkan dapat memberikan
informasi terkait upaya pemilihan dan perbandingan komposisi fase gerak yang
paling baik pada penetapan kadar nikotin dalam rokok dengan metode
kromatografi cair kinerja tinggi (KCKT) fase terbalik.
B. Tujuan Penelitian
Menentukan kondisi optimal pada penetapan kadar nikotin ekstrak
etanolik tembakau rokok “merek X” dengan metode KCKT fase terbalik dilihat
dari bentuk peak, retention time yang efisien, nilai tailing factor, nilai resolusi
yang dihasilkan, reprodusibilitas retention time baku, reprodusibilitas resolusi
6
BAB II
PENELAAHAN PUSTAKA
A. Rokok
Rokok merupakan suatu produk yang dibungkus oleh kertas berbentuk
seperti silinder dengan panjang mendekati 90 mm, ketika dibakar dan dihisap
akan terhisap asap berupa rasa dari tembakau atau rokok tersebut dan mulailah
terjadinya absorpsi dari nikotin menuju tubuh (Stratton,2001). Terdapat sekitar
empat ribu macam zat kimia dalam rokok yang terdiri dari komponen gas (85%)
dan sisanya merupakan partikel. Diantara ribuan zat kimia tersebut setidaknya dua
ratus senyawa dinyatakan berbahaya bagi kesehatan. Beberapa zat kimia dari
sekitar empat ribu zat tersebut ialah nikotin, gas karbon monoksida, nitrogen
oksida, nitrogen sianida, amoniak, benzaldehid, benzen, dan metanol. Racun
utama pada rokok adalah tar, nikotin, dan karbon monoksida (Ma’arif, 2012).
Nikotin yang terdapat dalam asap rokok saat dibakar akan berada pada pH 8 dan
akan terabsorbsi lebih banyak melalui membran mukosa dimulut karena
peningkatan konsentrasi nikotin dalam bentuk molekulnya. Komponen adiktif
(biasanya disebut saus) yang ditambahkan ke dalam tembakau sebelum diproses
lebih lanjut menjadi rokok ditujukan untuk meningkatan aroma dan rasa.
Kandungan saus biasanya berisi gula, humektan dan komponen aromatis (Geiss
B. Nikotin
Gambar 1 Struktur Nikotin (3-(1-metil-2-pirolodinil)pirindin) (Anonimb,2012)
Nikotin yang terkandung dalam rokok berkisar 1,5% b/b dan rata-rata
antara 1-1,5 mg nikotin yang terabsorspi secara sistemik selama merokok
(Benowitz, Hukkanen dan Jacob, 2009). Nikotin dan senyawa alkaloida lainnya
ditemukan pada tanaman Nicotiana. Nikotin yang terkandung biasanya 2-8%
berat kering dari daun tembakaunya, terklasifikasi sebagai senyawa organik
dengan sifat basa. Nikotin dalam bentuk murni berupa cairan minyak yang hampir
tidak berwarna atau kuning pucat. Paparan cahaya atau udara serta penyimpanan
dalam waktu lama dapat menyebabkan nikotin teroksidasi dan berubah menjadi
warna kecoklatan (Domino, 1999).
Kunci dasar dalam penentuan absorpsi, eksresi, farmakologi, dan
toksikologi dari nikotin terlihat dari bentuk terion atau molekul utuh nikotin yang
berpengaruh dari pH. Nikotin bersifat dibasic karena adanya cincin pirolidin dan
nitrogen piridin, nilai pKa dari nitrogen piridin relatif lebih rendah karena adanya
efek hibridisasi sp2 pada cincin piridin sehingga elektron akan terikat lebih erat
dengan nukleus. Kondisi pH 7,4 dan suhu 37oC berkisar 69% cincin pirolidin
nikotin berada dalam posisi terionisasi (bermuatan positif) dan nitrogen piridin
mempengaruhi protonasi atom nitrogen dalam nikotin, bentuk molekul utuh/tak
terprotonasi mengindikasikan sifat lipofilik dan sebalikanya bentuk terprotonasi
akan mengidentifikasi sifat hidrofilik (Domino, 1999).
Tabel I Ringkasan sifat/karakterisitik yang dimiliki nikotin (Domino, 1999)
No. Sifat/Karakteristik Keterangan
Nikotin merupakan suatu basa lemah dan lebih sulit terabsorpsi melewati
membran saat berada dalam kondisi terprotonasi. Absorpsi cepat nikotin dapat
terjadi dari asap rokok karena terfasilitasi oleh luas permukaan yang luas dari
alveoli dan disolusi nikotin dalam cairan paru-paru (pH 7,4) (Benowitz, Hukkanen
dan Jacob,2009).
Keracunan nikotin menghasilkan efek yang lebih kurang sama seperti
keracunan tembakau, dimana para perokok akan merasakan peningkatan jumlah
saliva, mual muntah, dan peningkatan tekanan darah. Keracunan nikotin
menyerang sistem saraf pusat dan perifer yang mengakibatkan depresi, timbul
C. Ekstrak Tembakau
Ekstrak merupakan sediaan pekat yang telah diekstraksi dari simplisia
nabati atau hewani menggunakan pelarut yang sesuai, setelahnya dilakukan
penguapan terhadap pelarut hingga keseluruhan atau hampir keseluruhan pelarut
dan masa serbuk atau serbuk yang tersisa diperlakukan hingga memenuhi baku
yang telah ditetapkan (Departemen Kesehatan Republik Indonesia, 1995).
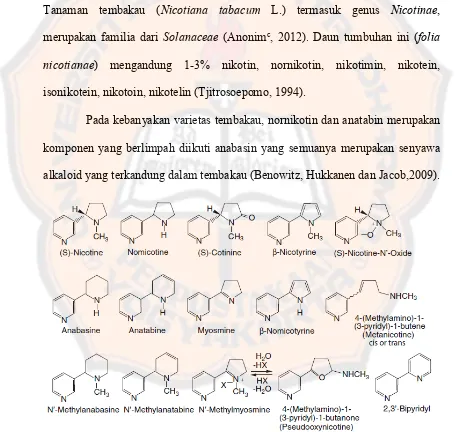
Tanaman tembakau (Nicotiana tabacum L.) termasuk genus Nicotinae,
merupakan familia dari Solanaceae (Anonimc, 2012). Daun tumbuhan ini (folia
nicotianae) mengandung 1-3% nikotin, nornikotin, nikotimin, nikotein,
isonikotein, nikotoin, nikotelin (Tjitrosoepomo, 1994).
Pada kebanyakan varietas tembakau, nornikotin dan anatabin merupakan
komponen yang berlimpah diikuti anabasin yang semuanya merupakan senyawa
alkaloid yang terkandung dalam tembakau (Benowitz, Hukkanen dan Jacob,2009).
D. Spektrofotometri UV
Spektrofotometeri UV merupakan salah satu teknik analisis
spektroskopik yang menggunakan radiasi elektromagnetik UV dekat dengan
menggunakan alat spektrofotometer (Skogg, West dan Holler, 1994). Radiasi
elektromagnetik pada daerah UV dan visibel biasanya ditulis dalam satuan
nanometer. Ketika sampel mengabsorbsi radiasi elektromagnetik (foton), terjadi
perubahan energi pada sampel tersebut. Energi yang diserap mempunyai
hubungan terhadap Persamaan Planck (Harvey,2000).
Molekul yang dikenakan gelombang radiasi elektromagnetik pada
frekuensi yang sesuai dapat terjadi penyerapan/absorpsi, adanya serapan tersebut
menghasilkan perbedaan energi serapan. Selisih energi tersebut setara dengan
energi foton yang diserap. Energi yang melompat dari keadaan dasar (ground
state) ke keadaan tereksitasi (excited state) disebut dengan transisi.
− = ℎ . � = ℎ.�
Dimana, E1= energi pada keadaan dasar/lebih rendah
E2= energi pada keadaan tereksitasi/lebih tinggi
h = konstanta Planck
υ = frekuensi foton yang diabsorpsi/diserap
= panjang gelombang
c = kecepatan
Transisi yang terjadi antar molekul tidaklah sama, hal ini menyebabkan perbedaan
spektra absorpsinya. Dengan demikian, spektra dapat digunakan sebagai bahan
gelombang tertentu setara dengan sinar yang diabsorpsi sehingga spektra juga
dapat digunakan sebagai bahan analisis kuantitatif (Gandjar dan Rohman, 2010).
Pada analisis dengan spektrofometer, dilakukan pembacaan absorbansi
yang disebut sebagai absorban (A) yang tidak memiliki satuan (Mulja dan
Suharman, 1995). Spektrum absorpsi merupakan plot absorbansi analit yang
merupakan fungsi dari panjang gelombang (Skogg, West dan Holler, 1994).
Gambar 3. Diagram tingkat energi elektronik (Gandjar dan Rohman, 2010)
Penyerapan foton yang dialami molekul mengakibatkan terjadinya
eksitasi elektron-elektron ikatan. Transisi elektronik yang terjadi antara tingkat
energi suatu molekul ada empat, yakni :
1. Transisi sigma–sigma star (σ → σ*)
Energi pada transisi ini terletak pada daerah < 180nm atau terjadi pada
daerah UV vakum dan kurang begitu bermanfaat untuk analisis spektrofotometri
UV-VIS (Gandjar dan Rohman, 2010).
2. Transisi non bonding elektron–sigma star (n → σ*)
Energi yang diperlukan untuk jenis transisi ini lebih kecil dibandingkan
transisi σ → σ*, sehingga sinar yang diserap memiliki panjang gelombang yang
lebih panjang (150–250nm). Kebanyakan transisi ini terjadi pada panjang
3. transisi n → π* dan transisi π → π*
Transisi ini terjadi pada molekul organik yang memiliki gugus fungsional
tidak jenuh, ikatan rangkap dalam gugus tersebut memberikan orbital phi yang
diperlukan. Transisi jenis ini paling cocok digunakan dalam analisis menggunakan
spektrofotometri UV–visibel karena berada diantara panjang gelombang 200–700
nm (Gandjar dan Rohman, 2010).
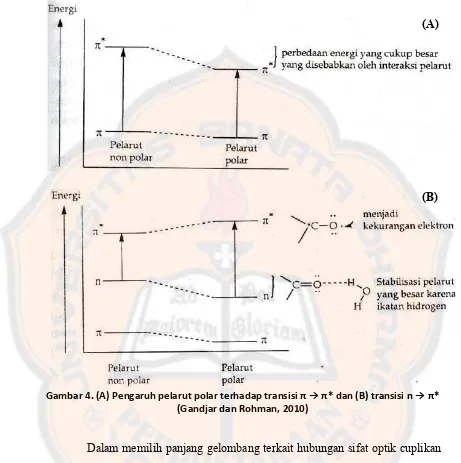
Pelarut dapat memberikan pengaruh transisi n → π* dan π → π*, hal ini
berkaitan dengan adanya perbedaan kemampuan dari pelarut untuk mensolvasi
antara keadaan dasar dengan keadaan tereksitasi. Pada transisi π → π*, molekul
yang berada dalam keadaan dasar akan relatif non polar dan keadaan
tereksitasinya lebih polar dibandingkan dari keadaan dasar. Penggunaan pelarut
polar akan menyebabkan interaksi lebih kuat saat keadaan tereksitasi
dibandingkan keadaan dasar sehingga perbedaan energi transisi π → π* lebih
kecil. Akibat yang ditimbulkan atas peristiwa ini ialah pergeseran ke panjang
gelombang yang lebih besar dari semula. Berbeda dengan transisi n → π*, pada
keadaan dasar molekul relatif lebih polar dibandingkan keadaan tereksitasi.
Pelarut yang berinteraksi hidrogen akan berinteraksi secara lebih kuat dengan
pasangan elektron yang tak berpasangan pada keadaan dasar dibandingkan
molekul pada keadaan tereksitasi. Hal ini mengakibatkan transisi n → π* akan
mempunyai energi yang lebih besar sehingga panjang gelombang akan bergeser
lebih pendek dibandingkan semula akibat kemampuan membentuk interaksi
Gambar 4. (A) Pengaruh pelarut polar terhadap tra sisi π → π* da B tra sisi → π* (Gandjar dan Rohman, 2010)
Dalam memilih panjang gelombang terkait hubungan sifat optik cuplikan
dan pelarut. Penyerapan radiasi UV atau visibel terkait dari elektron terluar atau
elektron valensi dari molekul dan tergantung pula pada jenis ikatan kimia dalam
molekul, adanya ikatan kimia penyebab terjadinya serapan sinar UV-Vis disebut
kromofor (Johnson dan Stevenson, 1978). Sinar UV mempunyai panjang
gelombang 200-400 nm, sedangkan sinar visibel mempunyai panjang gelombang
400-750 nm (Gandjar dan Rohman, 2010).
(A)
Kromofor merupakan ikatan rangkap tak jenuh selang-seling yang
menyerap radiasi pada daerah UV dan visibel, sedangkan aukosokrom merupakan
gugus jenuh yang terikat pada kromofor dapat menyebabkan adanya perubahan
panjang gelombang dan intensitas serapan maksimum. Ciri auksokrom adalah
gugusan heteroatom seperti –OCH3, -Cl, OH, dan NH2. Penambahan auksokrom
menyebabkan pergeseren batokromik. Pergeseran batokromik merupakan
pergeseran panjang gelombang ke arah yang lebih panjang akibat adanya subsitusi
gugus atau atom atau adanya pengaruh pelarut (Sastrohamidjojo, 2001).
E. Kromatografi Cair Kinerja Tinggi (KCKT)
1. Definisi Dan Instrumentasi KCKT
Kromatografi adalah proses pemisahan komponen dari campuran (solut)
yang terdistribusi diantara fase diam dan fase gerak berdasarkan kemampuan solut
terangkut melewati fase diamnya. Perpindahan solut melewati fase diam
tergantung pada afinitas relatif antar fase yang ditentukan dari parameter retensi
(Kealey dan Hainesm, 2005).
KCKT digunakan untuk senyawa yang tidak mudah menguap, contohnya
seperti terpenoid rantai panjang, golongan fenolik, alkaloid, lipid dan gula. KCKT
dapat menganalisis dengan sangat baik untuk senyawa yang dapat dibaca dengan
detektor UV-VIS (Harborne, 1998).
Teknik kromatografi membutuhkan adanya zat yang terlarut dan
terdistribusi diantara dua fase, yakni fase diam dan lainnya merupakan fase gerak.
lainnya yang terelusi lebih awal atau lebih akhir. Fase diam bertindak sebagai
penjerap (Departemen Kesehatan Republik Indonesia, 1995).
KCKT dapat menghasilkan pemisahan yang cepat, dengan keunggulan
zat yang tidak menguap atau zat yang tidak tahan panas dapat dipisahkan tanpa
terurai atau tanpa perlu diderivatisasi. Pada kromatografi partisi digunakan fase
gerak dan fase diam dengan polaritas yang berbeda. Jika fase gerak bersifat polar
dan fase diam bersifat nonpolar maka disebutlah sebagai kromatografi fase
terbalik, senyawa nonpolar yang larut dalam hidrokarbon dengan BM < 1000
dapat dipisahkan berdasarkan atas afinitasnya terhadap fase diam (Departemen
Kesehatan Republik Indonesia, 1995).
Gambar 5. Dasar Pemisahan Kromatografi Partisi (Lennan, 2010)
Kromatografi partisi merupakan metode pemisahan analit berdasarkan
kemampuan partisinya diantara fase diam dan fase gerak yang melewati fase
diam. Analit yang mempunyai afinitas lebih besar pada fase diam (gambar 5 -
bulatan merah) relatif lebih tertahan di fase diam daripada analit yang kurang
tertahan pada fase diam (gambar 5 - bulatan hijau) (Lennan, 2010). Urutan elusi
analit yang meningkat, analit yang mudah larut dalam air maka makin cepat analit
terelusi (bila digunakan fase gerak metanol-air) (Johnson dan Stevenson, 1978).
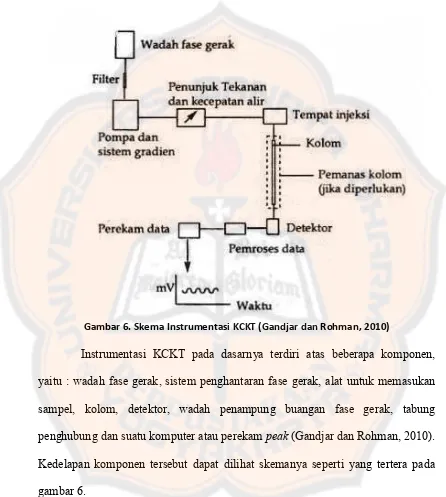
Gambar 6. Skema Instrumentasi KCKT (Gandjar dan Rohman, 2010)
Instrumentasi KCKT pada dasarnya terdiri atas beberapa komponen,
yaitu : wadah fase gerak, sistem penghantaran fase gerak, alat untuk memasukan
sampel, kolom, detektor, wadah penampung buangan fase gerak, tabung
penghubung dan suatu komputer atau perekam peak (Gandjar dan Rohman, 2010).
Kedelapan komponen tersebut dapat dilihat skemanya seperti yang tertera pada
gambar 6.
Wadah fase gerak harus bersih dan inert. Wadah fase gerak biasanya
merupakan wadah yang dapat menampung fase gerak antara 1 liter hingga 2 liter
terlebih dahulu untuk menghindari partikel kecil yang mungkin timbul sebagai
pengotor, adanya pengotor tersebut dapat menyebabkan gangguan pada sistem
kromatografi. Partikel pengotor yang terkumpul pada kolom atau pada selang
penghubung yang sempit dapat menimbulkan penyumbatan. Selain itu, fase gerak
yang digunakan haruslah diawaudarakan terlebih dahulu untuk menghilangkan
gas yang terdapat pada fase gerak, akibat gas yang berkumpul dengan komponen
lain pada pompa ataupun detektor dapat mengacaukan analisis. Saat membuat fase
gerak, sangat dianjurkan menggunakan pelarut dengan kemurnian yang sangat
tinggi agar tingkat pengotor yang ada rendah dan tidak mengacaukan sistem
kromatografi (Gandjar dan Rohman, 2010).
Fase gerak biasanya merupakan campuran pelarut yang dapat bercampur
secara keseluruhan dan berperan dalam daya elusi dan resolusi. Daya elusi dan
resolusi tersebut ditentukan berdasarkan polaritas keseluruhan pelarut fase gerak,
polaritas fase diam dan sifat komponen yang ada dalam sampel. Pada KCKT fase
normal (fase diam lebih polar daripada fase gerak), kemampuan elusi akan
meningkat seiring penurunan dari polaritas pelarut, sebaliknya untuk KCKT fase
terbalik (fase diam lebih nonpolar daripada fase gerak), kemampuan elusi akan
meningkat seiring peningkatan dari polaritas pelarut. Fase gerak yang paling
sering digunakan dalam pemisahan dengan sistem KCKT fase terbalik adalah
campuran metanol dengan buffer. Fase gerak yang sering digunakan biasanya
merupakan pelarut hidrokarbon atau menggunakan pelarut jenis alkohol (Gandjar
dipakai dalam pemisahan ialah air-metanol dan air-asetonitril (Johnson dan
Stevenson, 1978).
Pemilihan fase gerak dalam suatu metode pemisahan berdasarkan pada
indeks polaritas (P’) campuran fase gerak tersebut. Semakin besar nilai indeks
polaritasnya menyatakan semakin polar fase gerak yang digunakan. Fase gerak
yang sering digunakan merupakan kombinasi dari dua atau lebih campuran pelarut
yang saling bercampur secara keseluruhan. Campuran fase gerak tersebut akan
menghasilkan nilai polaritas tersendiri yang disebut indeks polaritas fase gerak
(Harvey, 2000).
�′ = Φ . �′ + Φ . �′
Dengan ΦA dan ΦB merupakan fraksi volume pelarut yang digunakan
pada pelarut A dan B, sedangkan P’Adan P’B merupakan indeks polaritas pelarut
yang digunakan pada pelarut A dan B (Harvey, 2000).
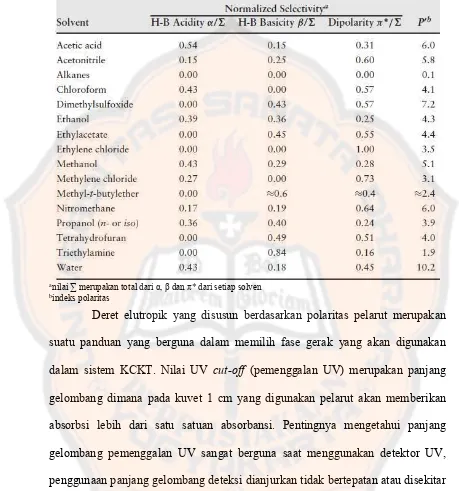
Tabel II. Indeks polaritas dan karakteristik solvent selectivity beberapa pelarut KCKT (Snyder, Kirkland dan Dolan, 2010)
anilai ∑ merupakan total dariα, β danπ* dari setiap solven bindeks polaritas
Deret elutropik yang disusun berdasarkan polaritas pelarut merupakan
suatu panduan yang berguna dalam memilih fase gerak yang akan digunakan
dalam sistem KCKT. Nilai UV cut-off (pemenggalan UV) merupakan panjang
gelombang dimana pada kuvet 1 cm yang digunakan pelarut akan memberikan
absorbsi lebih dari satu satuan absorbansi. Pentingnya mengetahui panjang
gelombang pemenggalan UV sangat berguna saat menggunakan detektor UV,
penggunaan panjang gelombang deteksi dianjurkan tidak bertepatan atau disekitar
panjang gelombang pemenggalan UV dari pelarut yang digunakan sebagai fase
gerak (Gandjar dan Rohman, 2010).
Pompa yang digunakan untuk memompa fase gerak pada sistem KCKT
yang digunakan sebaiknya memiliki kemampuan memberikan tekanan hingga 500
psi dan mampu mengalirkan fase gerak hingga 3 mL/min. Penggunaan pompa
atau sistem penghantaran fase gerak ialah agar dapat menjamin proses
penghantaran fase gerak yang berlangsung dengan tepat, reprodusibel, konstan
dan bebas ganggunan (Gandjar dan Rohman, 2010).
Metode pencampuran fase gerak dibedakan menjadi dua, yakni metode
isokratik dan metode gradien. Metode isokratik merupakan metode pencampuran
fase gerak secara manual dengan tangan dan saat memasuki sistem KCKT tidak
dibutuhkan adanya pencampuran fase gerak kembali dan dilakukan dengan satu
pompa. Metode gradien merupakan metode pencampuran fase gerak yang
dilakukan didalam sistem KCKT, beberapa pompa digunakan untuk memompa
pelarut ke dalam wadah pencampuran fase gerak dan hasil pencampuran fase
gerak tersebut yang dialirkan kedalam kolom (Snyder, Kirkland, dan Dolan,
2010).
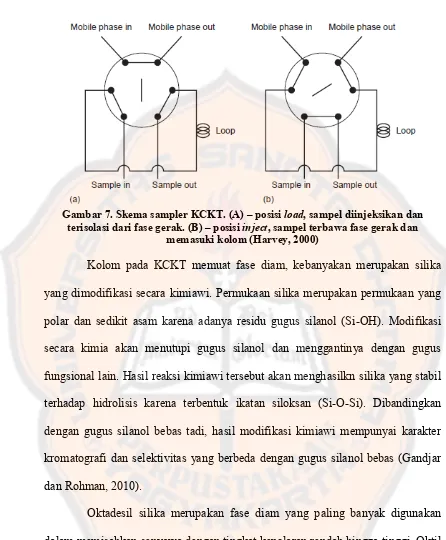
Penyuntikan sampel pada KCKT dilakukan secara langsung ke dalam
fase gerak yang mengalir menuju kolom. Penggunaan syringe ataupun
autosampler merupakan wadah yang digunakan dalam penyuntiksan sampel
(Gandjar dan Rohman, 2010). Pada sistem KCKT, penyuntikan sampel melalui
loop injector yang dapat menyimpan volume dari 0,5 µL – 2 mL. Pada posisi
load, loop sampler terisolasi dari fase gerak. Ketika katup dipindahkan ke posisi
loading, injektor berpindah ke posisi inject dan saat itu pula fase gerak mengaliri
Gambar 7. Skema sampler KCKT. (A) – posisi load, sampel diinjeksikan dan terisolasi dari fase gerak. (B) – posisi inject, sampel terbawa fase gerak dan
memasuki kolom (Harvey, 2000)
Kolom pada KCKT memuat fase diam, kebanyakan merupakan silika
yang dimodifikasi secara kimiawi. Permukaan silika merupakan permukaan yang
polar dan sedikit asam karena adanya residu gugus silanol (Si-OH). Modifikasi
secara kimia akan menutupi gugus silanol dan menggantinya dengan gugus
fungsional lain. Hasil reaksi kimiawi tersebut akan menghasilkn silika yang stabil
terhadap hidrolisis karena terbentuk ikatan siloksan (Si-O-Si). Dibandingkan
dengan gugus silanol bebas tadi, hasil modifikasi kimiawi mempunyai karakter
kromatografi dan selektivitas yang berbeda dengan gugus silanol bebas (Gandjar
dan Rohman, 2010).
Oktadesil silika merupakan fase diam yang paling banyak digunakan
dalam memisahkan senyawa dengan tingkat kepolaran rendah hingga tinggi. Oktil
atau rantai alkil yang lebih pendek lagi lebih sesuai untuk analit yang polar. Analit
polar terutama yang bersifat basa akan memberikan puncak yang mengekor
(tailing peak), hal ini terjadi karena adanya interaksi dengan residu silanol
mengalami mekanisme sorpsi partisi pada sistem KCKT fase terbalik dan analit
basa akan mengekor (Gandjar dan Rohman, 2010).
Deteksi pada KCKT dibagi menjadi empat secara umum, yakni bulk
property, sample specific, mobile-phase modification, dan hyphenated
techiniques.
a. Bulk property detector. Detektor ini dianggap sebagai detektor
universal yang dapat mengukur banyak komponen. Detektor ini memiliki
keuntungan karena dapat mendeteksi semua senyawa, sekaligus memiliki
kelemahan karena semua senyawa dari sampel yang terelusi akan terbaca sebagai
sinyal. Secara umum, detektor universal memiliki sensitivitas yang rendah
(Snyder dkk., 2010).
b. Sample specific detectors. Beberapa karakteristik sampel
mempunyai sifat unik yang mana tidak secara umum dimiliki oleh semua analit.
Detektor ini merespon terhadap keunikan karakteristik yang dimiliki analit
tersebut. Detektor UV merupakan detektor yang paling banyak digunakan dan
merespon analit yang mengabsorbsi sinar UV pada panjang gelombang tertentu.
Selain detektor UV, terdapat detektor lain seperti fluoresen dan detektor conduct
electricity (Snyder dkk., 2010). Detektor UV-VIS dapat mengukur analit yang
memiliki struktur kromoforik pada daerah panjang gelombang 190 – 800 nm.
Detektor UV-VIS ini dapat berupa detektor dengan panjang gelombang tetap
c. Mobile–phase modification detectors. Detektor ini mengubah fase
gerak setelah kolom KCKT menghasilkan pengubahan karakteristik analit, seperti
perubahan reaksi analit dan detektor spektometrik masa (Snyder dkk., 2010).
d. Hyphenated techniques. Teknik mengacu pada kopling dari analisis
KCKT yang dipadukan dengan teknik lain, seperti LC-MS dan LC-IR (Snyder
dkk., 2010).
Detektor pada KCKT idealnya memiliki beberapa karakteristik sebagai
berikut :
a. Respon terhadap analit cepat dan reprodusibel
b. Mampu mendeteksi analit hingga kadar yang sangat kecil
c. Stabil saat dioperasikan/digunakan
d. Memiliki sel volume yang kecil sehingga mampu meminimalkan pelebaran
pita.
e. Sinyal yang dihasilkan berbanding lurus dengan konsentrasi analit pada
kisaran luas/AUC
f. Tidak peka terhadap perubahan suhu dan kecepatan alir fase gerak.
Komputer atau integrator merupakan alat yang dihubungkan dengan
detektor unuk mengukur sinyal yang dihasilkan dan diplotkan sebagai suatu
kromatogram sehingga dapat dievaluasi oleh analis (Gandjar dan Rohman, 2010).
2. Optimasi Metode
Saat tahap optimasi, serangkaian kondisi awal yang mucul pada tahap
dimaksimalkan tersebut ialah resolusi, bentuk puncak, jumlah lempeng, asimetri,
kapasitas, dan retention time (Gandjar dan Rohman, 2010).
Kecepatan alir yang optimal biasanya 0,8 mL/min, 1,2 mL/min dan 2,5
mL/min untuk kolom dengan yang memiliki diameter internal 4,6 mm dan ukuran
partikel berkisar 3-10µm. Semakin kecil partikel menghasilkan pemisahan yang
lebih cepat dan optimal (Ahuja dan Rasmussen, 2007).
3. Pemisahan Puncak Dalam Kromatografi
Empat karakteristik umum yang biasanya digunakan untuk
mendeskripsikan kolom, sistem dan pemisahan kromatografi ialah faktor retensi
(k’), efisiensi (N), selektivitas(α), dan resolusi(R) (Ahuja dan Rasmussen, 2007).
a. Faktor retensi (k’). Faktor retensi (k’) merupakan pengukuran
retensi senyawa tertentu pada sistem kromatografi tertentu dan pada kondisi
tertentu. Faktor retensi didefinisikan dengan persamaan sebagai berikut :
�′= − = − =
Dimana VR merupakan retensi volume analit, V0 merupakan retensi volume fase
cair dalam sistem kromatografi dalam mL, tR merupakan retention time dalam
menit, t0 disebut sebagai retention time analit yang tidak tertahan dalam menit
(Ahuja dan Rasmussen, 2007), Vs merupakan volume sampel juga disebut sebagai
volume dari fase diam yang ada didalam kolom dalam µL dengan pendekatan
persamaan sebagai berikut :
= , √ �√ �
(3)
Dimana L merupakan panjang kolom dalam mm, dc merupakan diameter internal
kolom dan dp merupakan packing diameter parkikel penyusun kolom dalam µm.
Seluruh kondisi dari sistem kromatografi dapat mempengaruhi retensi analit.
(Snyder dkk, 2010).
Retention time analit yang tidak tertahan (t0) atau dihubungkan dengan
volume fase gerak dalam kolom sebagai hasil perkalian antara retention time
analit yang tidak tertahan terhadap kecepatan alir fase gerak yang melewati
kolom. Retention time analit yang tidak tertahan dapat dihitung berdasarkan
dimensi kolom dan kecepatan alirnya, persamaan matematika yang
menghubungkannya dituliskan sebagai berikut :
= � − �
L merupakan panjang kolom dalam mm, dc merupakan diameter internal kolom
dalam mm, F merupakan kecepatan alir fase gerak yang melewati kolom dalam
mL/min serta t0 sendiri dalam menit (Snyder dkk., 2010).
Faktor retensi mengukur waktu dari komponen sampel yang tinggal pada
fase diam dihubungkan terhadap waktu dari komponen sampel yang terbawa pada
fase gerak (Ettre,1993). Kecepatan migrasi analit melalui fase diam ditentukan
koefisen distribusinya (D), besarnya nilai D tersebut ditentukan oleh afinitas
relatif analit pada kedua fase (fase diam dan fase gerak). Dalam konteks
kromatografi, koefisien distribusi (D) didefinisikan sebagai perbandingan
konsentrasi analit dalam fase diam (Cs) dan fase gerak (Cm), yang dituliskan
sebagai berikut :
(6) =
Semakin besar nilai D, maka migrasi analit semakin lambat, sebaliknya semakin
kecil nilai D, maka migrasi analit semakin cepat. Jika perbedaan koefisien
distribusinya (D) dan faktor retensinya adalah 0 sehingga nilai tr=t0. Dalam sistem
kromatografi umumnya diatur agar nilai k’<20 untuk menghindari retention time
yang terlalu panjang (Gandjar dan Rohman, 2010). Retention time yang efisien
untuk analisis rutin ialah < 10 menit (Smith, 2002).
b. Resolusi (Rs). Resolusi dapat didefinisikan sebagai perbedaan
waktu antara retention time dua puncak peak yang saling berdekatan dibagi
dengan rata-rata lebar puncak, sehingga yang sangat berpengaruh terhadap
pemisahan komponen analit merupakan retention time (tR) masing-masing
analit dan lebar puncaknya (W). Nilai Rs harus mendekati atau lebih dari 1,5
untuk memberikan pemisahan yang baik (Gandjar dan Rohman, 2010).
= +−
(7)
Pemisahan komponen analit pada sistem KCKT diharapkan memiliki Rs
yang minimum dengan baseline separation. Baseline separation dapat dicapai
ketika detektor membaca peak kromatogram pertama secara utuh hingga baseline
sebelum detektor membaca peak kromatogram berikutnya. Baseline separation
dapat dicapai saat Rs > 1,5 atau mendekati 1,5 (Snyder, Kirkland dan Glajch,
1997).
Gambar 8. Gambaran peak kromatogram untuk mengukur resolusi (Snyder dkk., 1997)
c. Efisiensi kolom. Efisiensi merupakan karakteristik penting dalam
kolom. Efisiensi diekspresikan sebagai jumlah lempeng teoritis (N) yang dihitung
dengan persamaan sebagai berikut :
= ( )
Dimana tR merupakan retention time analit dan W merupakan lebar peak pada
posisi baseline (Ahuja dan Rasmussen, 2007).
Pengukuran terhadap efisiensi kolom membutuhkan faktor lebar peak
(W) karena waktu retensi berpengaruh terhadapnya, peningkatan nilai W akan
meningkatan retention time. Semakin tinggi nilai N mengindikasikan efisiensi
kolom yang lebih baik (Miller dan Crowther, 2010).
Terdapat parameter lain terkait efisiensi kolom, yakni plate height/tinggi
plat (H) yang dirumuskan dengan persamaan berikut :
� =
Dimana L merupakan panjang kolom. Plate height/tinggi plat (H) atau biasanya
disebut sebagai HETP (Height Equivalent to one Theoretical Plate) menggunakan
satuan unit panjang, pengukuran plate height lebih baik untuk penentuan efisiensi
kolom karena adanya faktor panjang kolom sebagai pembandingnya daripada
hanya dilihat dari jumlah lempeng teoritisnya (N). (Miller dan Crowther,2010).
HETP merupakan panjang kolom kromatografi (dalam mm) yang diperlukan
sampai terjadinya satu kali kesetimbangan molekul solut dalam fase gerak dan
fase diam) (Gandjar dan Rohman, 2010).
Pada saat pemisahan kromatografi, analit individual akan membentuk
profil konsentrasi yang simetri/profil Gaussian dalam arah aliran fase gerak. Peak
kromatogram secara perlahan akan melebar dan sering membentuk profil
asimetrik karena analit melanjutkan migrasinya ke fase diam (Gandjar dan
Rohman, 2010). Alasan timbulnya bentuk puncak dan pelebaran puncak
didasarkan pada difusi Eddy, difusi longitudinal, dan transfer massa (Snyder dkk.,
2010).
Difusi longitudinal ialah saat analit melewati kolom (gambar 9b). Proses
difusi ini menyebabkan pelebaran peak untuk meningkatan retention time (Snyder
dkk., 2010). Spesies analit menyebar kesegala arah dengan difusi ketika berada di
fase gerak sehingga berkontribusi terhadap pelebaran pita secara simetris (Gandjar
dan Rohman, 2010).
Gambar 9. Ilustrasi gambar yang menyebabkan pelebaran puncak selama pemisahan menggunakan KCKT. Simbol merepresentasifkan molekul analit sebelum migrasi,
simbol menyatakan molekul analit setelah migrasi, dan ---> menyatakan pergerakan dari molekul analit (Snyder dkk., 2010).
Difusi Eddy merepresentatifkan faktor lain yang menyebabkan pelebaran
pita. Molekul analit masuk ke dalam kolom melewati partikel fase diam dengan
arah yang berbeda–beda menuju ke luar kolom. Molekul yang bergerak lebih
lambat akan keluar lebih lambat dan molekul yang bergerak lebih cepat akan
keluar lebih dahulu. Pelebaran pita tidak bergantung dari kecepatan alir yang
digunakan dan hanya bergantung dari penyusunan dan ukuran partikel dalam
kolom. Pelebaran pita yang diakibatkan karena difusi Eddy akan semakin besar
Gambar 10. Ilustrasi difusi Eddy saat memasukin kolom dan menyebabkan pelebaran pita (Miller dan Crowther, 2010)
Transfer massa dapat menyebabkan pelebaran pita, terjadinya transfer
massa disebabkan oleh transfer massa fase gerak yang merupakan kecepatan alir
analit yang mempengaruhi pelebaran pita, diantara partikel fase diam terdapat
rongga yang bilamana analit melewatinya akan lebih cepat keluar terbaca detektor
dan bila analit cenderung lebih menyamping maka akan terjadi interaksi dahulu
terhadap partikel fase diam yang dapat dilihat dari gambar 9d. Transfer massa fase
diam merepresentatifkan analit yang terpenetrasi ke dalam partikel fase diam dan
tinggal lebih lama sebelum meninggalkan partikel fase diam. Perbedaan lama
waktu tinggal dan adanya analit yang terlebih dahulu terelusi keluar akan
menyebabkan pelebaran pita (Snyder dkk., 2010).
d. Selektivitas (α). Menurut Ahuja dan Rasmussen (2007),
selektivitas merupakan kemampuan sistem kromatografi untuk memisahkan dua
analit dan dapat digambarkan sebagai rasio faktor retensi, dengan persamaan
sebagai berikut :
� =�′�′
Kesempurnaan pemisahan pada kromatogram seperti yang pada
persamaan (6) belum memanfaatkan berbagai faktor, terdapat persamaan lain
yang menggunakan faktor – faktor lain seperti pada persamaan berikut :
= (√ ) [� −� ] [ − ��′ ′]
Persamaan (10) menjelaskan bahwa terdapat faktor lain yang turut menentukan
resolusi, yaitu jumlah lempeng (N), selektivitas (α), dan faktor retensi (k’)
(Gandjar dan Rohman, 2010).
Selektivitas pemisahan dapat dicapai dengan mengganti fase gerak,
parameter yang digunakan digambarkan oleh sebuah diagram solvent selectivity
triangle. Solvent selectivity triangle menggambarkan perbedaan antara solven
dengan melihat sifat keasaman (α), kebasaan (β), dan dipolar (π*). Perbedaan pola
elusi dari perbedaan solven yang besar dapat diharapkan dapat memisahkan peak
dengan baik. Solven A dalam kromatografi fase terbalik biasanya digunakan fase
gerak aquabidest dan solven B digunakan senyawa golongan alkohol (Meyer,
2004).
Gambar 11. Solvent Selectivity Triangle (Meyer, 2004)
Selektivitas dapat menghasilkan pergeseran satu puncak relatif terhadap
puncak lainnya dengan menaikan nilainya. Efisiensi pemisahan yang ditunjukan
oleh faktor N akan berubah dengan mengubah panjang kolom (L) atau mengubah
kecepatan alir fase gerak. Menaikan lempeng teoritis (N) suatu kolom akan
mengakibatkan penyempitan dua puncak sehingga lebar puncak (W) menjadi kecil
dan resolusi yang dihasilkan menjadi lebih besar. Pengubahan nilai k’ dengan
menurunkannya akan menghasilkan pemisahan yang jelas dan retention time yang
pendek, sebaliknya menaikan nilai k’ akan memberikan resolusi yang lebih baik
dengan konsekuensi tinggi puncak kromatogram akan turun dan waktu pemisahan
Gambar 12. Pe garuh perubaha ilai α, k’, da N terhadap pemisahan peak
kromatogram (Gandjar dan Rohman, 2010)
e. Faktor asimetri/pengekoran. Pita kromatogram biasanya tidak
menghasilkan bentuk yang Gaussian dan menimbulkan pengekoran. Pengekoran
pita/peak tailing dapat dihitung berdasarkan dua cara yakni menggunakan
asymmetry factor (As) atau menggunakan tailing factor (TF). Nilai As dapat
dikorelasikan dengan TF menggunakan persamaan sebagai berikut :
� ≈ + , −
Melihat dari persamaan (11), nilai As akan cenderung lebih besar dibandingkan
nilai TF (Snyder dkk., 2010).
Menurut Center for Drug Evalution and Research (1994), nilai tailing
factor yang baik ≤ 2.
Gambar 13. Penentuan asymmetry factor (As) dan tailing factor (TF) (Snyder dkk.,2010).
F. Standar Internal
Standar internal digunakan untuk menghasilkan presisi yang baik karena
masalah yang timbul akibat ketidakpastian jumlah analit saat preparasi sampel
dapat dihindari. Teknik kuantitatif dengan standar internal didasarkan pada jumlah
senyawa yang dimasukan pada baku dan sampel pada awal pengerjaan. Tinggi
peak atau area yang muncul pada analit uji dan senyawa yang berperan sebagai
internal standar akan dibandingkan untuk dianalisis (Chan, Lam, Lee, dan Zhang,
2004).
Senyawa yang dipilih sebagai standar internal bukan merupakan salah
salah satu komponen dari penyusun sampel dan tidak terjadi adanya overlapping
dengan peak yang ditimbulkan setiap analit. Standar internal digunakan untuk
diketahui secara tertentu dan harus memenuhi beberapa kriteria dalam analisis
kromatografi :
1. Senyawa yang digunakan sebagai standar internal harus terelusi didekat peak
yang diamati, tetapi terpisah dengan baik terhadap peak tersebut
2. Senyawa yang digunakan sebagai standar internal memiliki kemiripan sifat
kimia terhadap analit yang diteliti dan tidak menimbulkan reaksi dengan
komponen yang terdapat dalam sampel uji
3. Standar internal harus tersedia dalam kemurnian yang tinggi (Miller dan
Crowther, 2000).
Kehadiran standar internal dalam sistem kuantifikasi yang dilakukan
harus menjamin pemisahan antar peak dengan baik. Pemisahan yang baik
memiliki resolusi > 1,5 atau baseline separation (Chan, Lam, Lee, dan Zhang,
2004).
Standar internal yang digunakan pada sampel untuk memimic respon
analit. Standar internal harus ditambahkan sebelum dilakukannya preparasi
sampel/ekstraksi untuk melihat kehilangan atau kesalahan yang muncul selama
proses yang dilakukan, hal ini didasarkan pada proses preparasi yang panjang
maka kesalahan yang muncul akan semakin besar. Penggunaan internal standar
disini untuk meminimalisir kesalahan yang muncul tersebut (Chan, Lam, Lee, dan
Zhang, 2004).
Menurut penelitian yang dilakukan oleh Massadeh, Gharaibeh dan Omari
(2009) serta penelitian yang dilakukan oleh Nakajima, dkk (2000), asetanilida
G. Landasan Teori
Rokok merupakan produk tembakau yang terbungkus kertas dalam
bentuk seperti silinder dengan panjang mendekati 90 mm. Dalam tembakau rokok
terdiri dari sekitar 4000 macam zat kimia, salah satunya nikotin. Nikotin yang
terkandung dalam rokok antara 1-1,5 mg. Nikotin berupa cairan minyak yang
hampir tidak berwarna dan merupakan basa lemah yang bersifar dibasic (pKa
4,23 dan 9,15). Dengan adanya paparan cahaya atau udara nikotin dapat
teroksidasi dan berubah warna menjadi kecoklatan.
Ekstrak etanolik tembakau rokok merupakan senyawa multikomponen
yang mengandung nikotin dan beberapa metabolit lain. Metode KCKT fase
terbalik memiliki selektivitas dan sensitifitas yang tinggi sehingga mampu
memisahkan sampel multikomponen sekaligus mengkuantifikasinya.
Optimasi terhadap komposisi fase gerak dan kecepatan alir sistem KCKT
fase terbalik dengan penggunaan metode standar internal asetanilida dilakukan
untuk menghasilkan pemisahan analit uji yang baik dengan retention time yang
efisien yakni kurang dari 10 menit (Smith, 2002), bentuk peak dengan nilai TF ≤ 2
(Center for Drug Evaluation and Research, 1994), nilai resolusi > 1,5 (Gandjar
dan Rohman, 2010).
H. Hipotesis
Metode KCKT fase terbalik dengan menggunakan komposisi fase gerak