xi
OPTIMASI PEMISAHAN DAN PENETAPAN KADAR CAMPURAN PARASETAMOL DAN NATRIUM FENOBARBITAL DENGAN METODE
KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK
INTISARI
Saat ini, karena terbatasnya bentuk sediaan dan kombinasi zat aktif yang tersedia di pasaran, maka tenaga medis di Rumah Sakit X mengombinasikan parasetamol dan natrium fenobarbital dalam bentuk pulveres untuk pasien anak-anak.
Pulveres dibuat dengan mengubah sediaan tablet menjadi pulveres dan dilakukan dalam jumlah besar. Maka untuk menjamin patient safety, diperlukan kontrol kualitas terhadap pulveres tersebut. Salah satu metode yang pernah digunakan untuk menetapkan kadar campuran parasetamol dan natrium fenobarbital adalah Kromatografi Cair Kinerja Tinggi (KCKT) fase terbalik.
Penelitian ini bertujuan untuk mengetahui akurasi, presisi, linearitas, LOD dan LOQ dari metode KCKT fase terbalik untuk menetapkan kadar campuran parasetamol dan natrium fenobarbital. Jenis penelitan ini adalah noneksperimental deskriptif. Sistem KCKT menggunakan kolom C18 (30 cm); perbandingan fase gerak metanol:buffer fosfat pH 3,2 yang dioptimasi 30:70 dan 10:90; kecepatan alir yang dioptimasi 1 dan 1,5 ml/menit; detektor UV pada ? pengamatan 236 nm.
Hasil yang diperoleh menunjukkan bahwa metode KCKT fase terbalik dengan kondisi optimum komposisi fase gerak 90:10 dan kecepatan alir 1,5 ml/menit memiliki akurasi, presisi dan linearitas yang baik untuk menetapkan kadar campuran parasetamol dan natrium fenobarbital. Dengan LOD dan LOQ untuk parasetamol berturut-turut 0,006 mg/ml dan 0,02 mg/ml sedangkan untuk natrium fenobarbital berturut-turut 0,124 mg/ml dan 0,414 mg/ml.
xii
MIXTURE OF PARACETAMOL AND SODIUM PHENOBARBITAL WITH HIGH PERFORMANCE LIQUID CHROMATOGRAPHY METHOD
REVERSED PHASE
ABSTRACT
Nowadays, because of dossage form and combination of active ingredient in market are limited, then the medical staff in hospital X combines paracetamol and sodium phenobarbital in pulveres form for pediatri. Pulveres made by changing tablet form into pulveres, in a large quantity. So in order to guarantee patient safety, quality control is needed towards those pulveres. One of the methods that ever used to determine the amount of paracetamol and sodium phenobarbital is reversed phase High Performance Liquid Chromatoraphy (HPLC).
This research’s aims are to know the accuracy, precision, linearity, LOD and LOQ from reversed phase HPLC to determine the amount of paracetamol and sodium phenobarbital in combination. Kind of this research is descriptive nonexperimental. HPLC system uses C18 column (30 cm); combination of mobile phase methanol:phosphat buffer pH 3.2 that have been optimated are 30:70 and 10:90; flow rate that have been optimated are 1 and 1.5 ml/minutes; UV detector in observative ? 236 nm.
The result shows that HPLC method reversed phase with optimum condition of mobile phase composition 90:10 and flow rate 1.5 ml/minutes has good accuracy, precision and linearity to determine the amount of paracetamol and sodium phenobarbital. With LOD and LOQ for paracetamol are 0.006 mg/ml and 0.02 mg/ml, while LOD and LOQ for sodium phenobarbital are 0.124 mg/ml and 0.414 mg/ml.
OPTIMASI PEMISAHAN DAN PENETAPAN KADAR CAMPURAN PARASETAMOL DAN NATRIUM FENOBARBITAL DENGAN METODE
KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm)
Program Studi Ilmu Farmasi
Oleh :
Marischa Novita Lissanta NIM : 048114044
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA
YOGYAKARTA
ii
PARASETAMOL DAN NATRIUM FENOBARBITAL DENGAN METODE KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm)
Program Studi Ilmu Farmasi
Oleh :
Marischa Novita Lissanta NIM : 048114044
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA
YOGYAKARTA
iii
Skripsi berjudul
PEMISAHAN DAN PENETAPAN KADAR CAMPURAN PARASETAMOL
DAN NATRIUM FENOBARBITAL DENGAN METODE KROMATOGRAFI
CAIR KINERJA TINGGI FASE TERBALIK
Oleh :
Marischa Novita Lissanta NIM : 048114044
Telah disetujui oleh :
Pembimbing
v
U ntuk yang terCI NTA…
Keluargaku karena cinta dan dukungan kalian yang begitu besar
Sahabat-sahabatku, yang
membuat hidupku lebih berarti
vi
PUBLIKASI KARYA ILMIAH UNTUK KEPENTINGAN AKADEMIS
Yang bertanda tangan di bawah ini, saya mahasiswa Universitas Sanata Dharma: Nama : Marischa Novita Lissanta
Nomor Mahasiswa : 048114044
Demi pengembangan ilmu pengetahuan, saya memberikan kepada Perpustakaan Universitas Sanata Dharma karya ilmiah saya yang berjudul:
“OPTIMASI PEMISAHAN DAN PENETAPAN KADAR CAMPURAN PARASETAMOL DAN NATRIUM FENOBARBITAL DENGAN METODE
KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK”
beserta perangkat yang diperlukan (bila ada). Dengan demikian saya memberikan kepada Perpustakaan Universitas Sanata Dharma hak untuk menyimpan, mengalihkan dalam bentuk media lain, mengelolanya dalam bentuk pangkalan data, mendistribusikan secara terbatas, dan mempublikasikannya di Internet atau media lain untuk kepentingan akademis tanpa perlu meminta ijin dari saya maupun memberikan royalti kepada saya selama tetap mencantumkan nama saya sebagai penulis.
Demikian pernyataan ini yang saya buat dengan sebenarnya: Dibuat di Yogyakarta
Pada tanggal 24 April 2008 Yang menyatakan
vii
PRAKATA
Puji syukur penulis panjatkan kepada Bapa YAHWEH di Sorga, karena hanya karena hikmat, kekuatan, mukjizat serta penyertaanNYA maka skripsi yang berjudul ”Optimasi Pemisahan dan Penetapan Kadar Campuran Parasetamol dan Natrium Fenobarbital dengan Metode Kromatografi Cair Kinerja Tinggi Fase Terbalik” ini dapat diselesaikan oleh penulis. Skripsi ini disusun untuk memenuhi salah satu syarat memperoleh gelar Sarjana Farmasi (S.Farm) di Fakultas Farmasi Universitas Sanata Dharma Yogyakarta.
Selama penyusunan skripsi ini, banyak pihak yang telah begitu luar biasanya membant u penulis, maka pada kesempatan ini penulis mengucapkan terima kasih yang sebesar-besarnya kepada :
1. Ibu Rita Suhadi M,Si., Apt. selaku Dekan Fakultas Farmasi Universitas Sanata Dharma Yogyakarta dan ketua penelitian payung yang penulis ikuti. Terima kasih banyak atas bantuan dan semangatnya.
2. Ibu Christine Patramurti, M.Si., Apt. selaku pembimbing yang telah begitu sabar membimbing penulis, memberikan masukan, arahan, kritikan dan dukungan selama penyusunan skripsi ini.
viii
kasih sayang, doa, dukungan dan semua yang penulis butuhkan. Tanpa kalian, Novi tidak akan jadi seperti ini.
5. Bapak Yohanes Dwiatmaka, S.Si., M.Si. yang telah membantu penulis membelikan alat yang pecah.
6. Seluruh staf laboratorium di Fakultas Farmasi Universitas Sanata Dharma Yogyakarta : Pak Mukmin, Pak Prapto, Pak Parlan, Mas Sarwanto, Mas Kunto, Mas Otok, Mas Heru yang telah banyak membantu penulis selama penelitian di laboratorium dan mendukung kelancarannya.
7. Ai, ishakku, yang telah begitu banyak memberikan kasih sayang, perhatian, dukungan yang luar biasa, semangat, doa, berkat, nasehat, kritik sebelum, selama dan sesudah penyusunan skripsi ini (IDLU4E!)
8. Tika, Rissa dan Nur sebagai teman dan sahabat sejak penulis memasuki Fakultas Farmasi Universitas Sanata Dharma Yogyakarta, terima kasih atas semua dukungan, semangat dan doanya.
9. Rian dan Tika terima kasih untuk semua dukungan, semangat, bantuan dan solusi masalah yang dihadapi selama penyusunan skripsi ini.
10.A-Cu dan Reni, terima kasih atas team work yang solid, terima kasih untuk membantu kerja di lab sampai malam, dukungan doa dan semangatnya.
ix
12.Semua saudara seiman di GAIN Alfa Omega Yogyakarta, Tante dan Om, Yosafat, Viktor, Pak dan Bu Joko, Komang, Diana, Doni atas semua dukungan doa dan berkatnya. YAHWEH berkati.
13.Teman-teman FST yang luar biasa kekompakannya, kalian memberi warna di hidupku.
14.Setiap orang yang mungkin tidak dapat disebutkan satu per satu, terima kasih karena baik atau buruk kalian telah membentukku menjadi pribadi ya ng seperti ini. Lanjutkan hidup kalian.
Penulis menyadari bahwa skripsi ini masih memiliki banyak kekurangan. Maka penulis sangat terbuka terhadap kritik dan saran yang akan membantu penulis dalam perkembangan selanjutnya. Akhir kata, semoga skripsi ini berguna bagi kemajuan ilmu pengetahuan.
x
Saya menyatakan dengan sesungguhnya bahwa skripsi yang saya tulis ini tidak memuat karya, atau bagian karya orang lain, kecuali yang telah disebutkan dalam kutipan dan daftar pustaka, sebagaimana layaknya karya ilmiah.
Yogyakarta, Mei 2008 Penulis
xi
OPTIMASI PEMISAHAN DAN PENETAPAN KADAR CAMPURAN PARASETAMOL DAN NATRIUM FENOBARBITAL DENGAN METODE
KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK
INTISARI
Saat ini, karena terbatasnya bentuk sediaan dan kombinasi zat aktif yang tersedia di pasaran, maka tenaga medis di Rumah Sakit X mengombinasikan parasetamol dan natrium fenobarbital dalam bentuk pulveres untuk pasien anak-anak.
Pulveres dibuat dengan mengubah sediaan tablet menjadi pulveres dan dilakukan dalam jumlah besar. Maka untuk menjamin patient safety, diperlukan kontrol kualitas terhadap pulveres tersebut. Salah satu metode yang pernah digunakan untuk menetapkan kadar campuran parasetamol dan natrium fenobarbital adalah Kromatografi Cair Kinerja Tinggi (KCKT) fase terbalik.
Penelitian ini bertujuan untuk mengetahui akurasi, presisi, linearitas, LOD dan LOQ dari metode KCKT fase terbalik untuk menetapkan kadar campuran parasetamol dan natrium fenobarbital. Jenis penelitan ini adalah noneksperimental deskriptif. Sistem KCKT menggunakan kolom C18 (30 cm); perbandingan fase gerak metanol:buffer fosfat pH 3,2 yang dioptimasi 30:70 dan 10:90; kecepatan alir yang dioptimasi 1 dan 1,5 ml/menit; detektor UV pada ? pengamatan 236 nm.
Hasil yang diperoleh menunjukkan bahwa metode KCKT fase terbalik dengan kondisi optimum komposisi fase gerak 90:10 dan kecepatan alir 1,5 ml/menit memiliki akurasi, presisi dan linearitas yang baik untuk menetapkan kadar campuran parasetamol dan natrium fenobarbital. Dengan LOD dan LOQ untuk parasetamol berturut-turut 0,006 mg/ml dan 0,02 mg/ml sedangkan untuk natrium fenobarbital berturut-turut 0,124 mg/ml dan 0,414 mg/ml.
xii
MIXTURE OF PARACETAMOL AND SODIUM PHENOBARBITAL WITH HIGH PERFORMANCE LIQUID CHROMATOGRAPHY METHOD
REVERSED PHASE
ABSTRACT
Nowadays, because of dossage form and combination of active ingredient in market are limited, then the medical staff in hospital X combines paracetamol and sodium phenobarbital in pulveres form for pediatri. Pulveres made by changing tablet form into pulveres, in a large quantity. So in order to guarantee patient safety, quality control is needed towards those pulveres. One of the methods that ever used to determine the amount of paracetamol and sodium phenobarbital is reversed phase High Performance Liquid Chromatoraphy (HPLC).
This research’s aims are to know the accuracy, precision, linearity, LOD and LOQ from reversed phase HPLC to determine the amount of paracetamol and sodium phenobarbital in combination. Kind of this research is descriptive nonexperimental. HPLC system uses C18 column (30 cm); combination of mobile phase methanol:phosphat buffer pH 3.2 that have been optimated are 30:70 and 10:90; flow rate that have been optimated are 1 and 1.5 ml/minutes; UV detector in observative ? 236 nm.
The result shows that HPLC method reversed phase with optimum condition of mobile phase composition 90:10 and flow rate 1.5 ml/minutes has good accuracy, precision and linearity to determine the amount of paracetamol and sodium phenobarbital. With LOD and LOQ for paracetamol are 0.006 mg/ml and 0.02 mg/ml, while LOD and LOQ for sodium phenobarbital are 0.124 mg/ml and 0.414 mg/ml.
xiii
DAFTAR ISI
HALAMAN JUDUL... ii
HALAMAN PERSETUJUAN PEMBIMBING... iii
HALAMAN PENGESAHAN...iv
HALAMAN PERSEMBAHAN...v
PERNYATAAN PERSETUJUAN PUBLIKASI...vi
PRAKATA...vii
PERNYATAAN KEASLIAN KARYA... x
INTISARI... xi
ABSTRACT...xii
DAFTAR ISI...xiii
DAFTAR TABEL...xvii
DAFTAR GAMBAR... xviii
DAFTAR LAMPIRAN...xx
BAB I. PENGANTAR... 1
A. Latar Belakang... 1
1. Permasalahan... 3
2. Keaslian Penelitian... 3
3. Manfaat Penelitian... 4
xiv
A. Parasetamol... 5
B. Natrium fenobarbital... 6
C. Buffer... 7
D. Analisis Kualitatif dan Kuantitatif... 8
E. Kromatografi Cair Kinerja Tinggi... 9
1. Definisi dan instrumentasi... 9
2. Pembagian jenis kromatografi... 15
3. Kromatografi partisi... 16
4. Pemisahan puncak dalam kromatografi... 18
F. Spektrofotometri Ultraviolet... 21
G. Kesahihan Metode Analisis Instrumental... 24
H. Keterangan Empiris... 27
BAB III. METODE PENELITIAN... 28
A. Jenis Penelitian... 28
B. Definisi Operasional... 28
C. Bahan Penelitian... 28
D. Alat penelitian... 29
E. Tata Cara Penelitian... 30
1. Pembuatan fase gerak... 30
2. Pembuatan larutan baku parasetamol dan natrium fenobarbital…………. 31
xv
4. Optimasi pemisahan parasetamol dan natrium fenobarbital dalam campuran parasetamol dan natrium fenobarbital dengan perbandingan
11:1 dengan KCKT fase terbalik……… 33
5. Optimasi penetapan kadar parasetamol dan natrium fenobarbital dalam campuran parasetamol dan natrium fenobarbital dengan perbandingan 11 : 1 dengan KCKT fase terbalik………... 35
6. Validasi metode penetapan kadar parasetamol dan natrium fenobarbital dalam campuran parasetamol dan natrium fenobarbital dengan perbandingan 11 : 1 dengan KCKT fase terbalik………36
F. Analisis Hasil... 38
BAB IV. HASIL DAN PEMBAHASAN... 39
A. Penyiapan Fase Gerak... 39
B. Pembuatan Larutan Baku... 40
C. Penentuan Panjang Gelombang Pengamatan Parasetamol dan Natrium Fenobarbital dengan Spektrofotometer UV……….. 41
D. Optimasi Pemisahan Parasetamol dan Natrium Fenobarbital dengan KCKT Fase Terbalik……….46
E. Optimasi Penetapan Kadar Parasetamol dan Natrium Fenobarbital dengan KCKT Fase Terbalik………. 56
xvi
A. Kesimpulan... 65
B. Saran... 66
DAFTAR PUSTAKA... 67
LAMPIRAN...70
xvii
DAFTAR TABEL
Tabel I. Nilai indeks polaritas pelarut... 13 Tabel II. Rentang rata-rata persen perolehan kembali yang dapat diterima
(Anonimc, 2004)...25 Tabel III. Presisi yang dapat diterima (Anonimc, 2004)... 26 Tabel IV. Resolusi pemisahan parasetamol 0,21 mg/ml dan asam
xviii
Gambar 1. Struktur Parasetamol... 5
Gambar 2. Struktur Natrium Fenobarbital... 6
Gambar 3. Peralatan KCKT... 11
Gambar 4. Detektor ultraviolet untuk KCKT... 15
Gambar 5. Reaksi silanisasi... 17
Gambar 6. Gugus kromofor dan auksokrom parasetamol... 43
Gambar 7. Gugus kromofor dan auksokrom natrium fenobarbital...43
Gambar 8. Spektrum serapan parasetamol (?maks = 245 nm)... 44
Gambar 9. Spektrum serapan natrium fenobarbital (?maks = 236 nm)... 45
Gambar 10. Gabungan spektrum serapan parasetamol konsentrasi 0,009 mg/ml dan natrium fenobarbital konsentrasi 0,09 mg/ml... 46
Gambar 11. Reaksi antara natrium fenobarbital dengan asam fosfat... ... 47
Gambar 12. Gugus nonpolar dari parasetamo l dan asam fenobarbiturat yang berinteraksi dengan fase diam...48
Gambar 13. Puncak parasetamol dan asam fenobarbiturat dengan fase gerak buffer fosfat pH 3,2 : metanol (70 : 30), fase diam oktadesilsilan, kecepatan alir 1 ml/menit, deteksi UV 236 nm...50
xix
Gambar 15. Puncak parasetamol dan asam fenobarbiturat dengan fase gerak buffer fosfat pH 3,2 : metanol (90 : 10), fase diam oktadesilsilan, kecepatan alir 1 ml/menit, deteksi UV 236 nm...52 Gambar 16. Puncak parasetamol dan asam fe nobarbiturat dengan fase gerak
xx
Lampiran 1. Sertifikat analisis parasetamol...70 Lampiran 2. Sertifikat analisis natrium fenobarbital... 71 Lampiran 3. Data penimbangan bahan... 72 Lampiran 4. Spektra panjang gelombang serapan maksimum parasetamol...73 Lampiran 5. Spektra panjang gelombang serapan maksimum natrium
fenobarbital... 75 Lampiran 6. Kromatogram baku parasetamol... 77 Lampiran 7. Kromatogram baku natrium fenobarbital...82 Lampiran 8. Kromatogram sampel parasetamol dan natrium fenobarbital ... 87 Lampiran 9. Contoh perhitungan kadar larutan baku parasetamol...99 Lampiran 10. Contoh perhitungan kadar larutan baku natrium fenobarbital... 101 Lampiran 11. Perhitungan CV parasetamol dan natrium fenobarbital dalam
1
BAB I
PENGANTAR
A. Latar Belakang
Saat ini terbatasnya bentuk sediaan dan kombinasi zat aktif yang tersedia di pasaran seringkali menyulitkan proses terapi untuk pasien anak-anak. Maka cara yang dapat dilakukan adalah mengombinasikan beberapa zat aktif untuk mencapai tujuan terapi tertentu seperti yang dilakukan tenaga medis di Rumah Sakit X. Salah satunya adalah kombinasi parasetamol dan natrium fenobarbital. Parasetamol digunakan sebagai penghilang nyeri dan penurun panas sedangkan natrium fenobarbital yang memiliki khasiat antiepilepsi dimanfaatkan efek sampingnya untuk menena ngkan pasien anak-anak yang terkena demam.
Kombinasi parasetamol dan natrium fenobarbital dibuat dalam bentuk sediaan pulveres yang dapat diterima oleh anak-anak karena pasien anak pada umumnya sulit menerima obat dalam bentuk sediaan tablet. Selain itu, keterbatasan sediaan tablet adalah dosisnya ditujukan untuk pasien dewasa sehingga perlu dilakukan pengubahan bentuk sediaan tablet menjadi bentuk sediaan pulveres dengan tujuan membagi dosis supaya sesuai untuk anak-anak. Pulveres adalah serbuk yang dibagi dalam bobot yang lebih kurang sama, dibungkus dengan kertas perkamen atau bahan pengemas lain yang cocok (Anief, 2000).
kuantitatif sehingga tidak ada jaminan keseragaman zat aktif, keamanan, dan khasiat penggunaannya. Sedangkan obat jadi adalah produk final artinya tidak layak direfo rmulasikan apalagi dicampurkan dengan sediaan jadi lainnya.
Bagi apoteker yang lebih mengutamakan pasien, patient safety merupakan isu kritis dan harus ditangani dengan tepat karena menyangkut keselamatan pasien. Maka salah satu upaya yang dapat dilakukan adalah dengan melakukan kontrol kualitas baik secara kualitatif maupun kuantitatif terhadap pulveres yang telah dibuat. Hal ini penting sebab proses pengubahan bentuk sediaan dapat mengakibatkan perubahan sifat obat, yang dapat membahayakan pasien seperti beberapa kasus yang terjadi di Indonesia.
Untuk dapat melakukan analisis kualitatif dan kuantitatif diperlukan suatu metode yang tepat. Akan tetapi karena kombinasi zat aktif dalam sediaan pulveres
yang dibuat merupakan obat-obat yang diresepkan oleh dokter, maka metode penetapan kadar yang ada saat ini belum ada yang telah tervalidasi. Untuk itu, peneliti hendak melakukan penelitian mengenai optimasi dan validasi metode untuk menetapkan kadar campuran parasetamol dan natrium fenobarbital.
Metode yang dipilih adalah kromatografi cair kinerja tinggi (KCKT) fase terbalik karena metode ini pernah digunakan untuk menetapkan kadar parasetamol dan natrium fenobarbital dalam sampel biologis (Lunn dan Schmuff, 1997). Oleh karena itu pada penelitian ini dilakukan optimasi dan validasi metode KCKT untuk penetapan kadar campuran parasetamol dan natrium fenobarbital dalam sediaan
Parasetamol dan natrium fenobarbital yang telah diubah menjadi bentuk asamnya, memiliki gugus polar dan nonpolar yang berbeda sehingga dapat berinteraksi dengan fase diam dan fase gerak pada KCKT. Selain itu metode KCKT merupakan metode yang cocok untuk analisis kualitatif dan kuantitatif campuran dua senyawa atau lebih (multikomponen) tanpa perlu melakukan pemisahan senyawa-senyawa tersebut terlebih dahulu (Johnson dan Stevenson, 1978).
1. Permasalahan
Berdasarkan latar belakang tersebut, permasalahan yang muncul.
a. Bagaimana kondisi optimum untuk melakukan pemisahan parasetamol dan natrium fenobarbital menggunakan metode KCKT fase terbalik?
b. Bagaimana validitas metode KCKT fase terbalik untuk menetapkan kadar campuran parasetamol dan natrium fenobarbital dalam sediaan pulveres?
2. Keaslian penelitian
3. Manfaat penelitian
Penelitian ini dapat memberikan manfaat.
a. Manfaat teoritis. Diharapkan penelitian ini dapat memberikan sumbangan terhadap ilmu pengetahuan tentang metode KCKT yang dapat digunakan untuk menetapkan kadar campuran parasetamol dan natrium fenobarbital dalam sediaan obat.
b. Manfaat metodologis. Diharapkan hasil penelitian ini dapat digunakan sebagai dasar untuk melakukan analisis kualitatif maupun kuantitatif terhadap campuran parasetamol dan natrium fenobarbital dalam resep racikan pulveres di Rumah Sakit X.
c. Manfaat praktis. Diharapkan penelitian ini dapat meningkatkan kualitas pelayanan kefarmasian di Rumah Sakit X dan menjamin patient safety.
B. Tujuan Penelitian
Maka berdasarkan latar belakang dan permasalahan tersebut, tujuan dilakukannya penelitian ini.
1. Untuk mengetahui bagaimana kondisi yang optimum untuk memisahkan parasetamol dan natrium fenobarbital dengan metode KCKT fase terbalik.
2. Untuk mengetahui bagaimana validitas metode KCKT fase terbalik untuk menetapkan kadar campuran parasetamol dan natrium fenobarbital dalam sediaan
5
BAB II
PENELAAHAN PUSTAKA
A. Parasetamol
Parasetamol atau 4’-hidroksiasetanilida dengan bobot molekul 151,16 mengandung tidak kurang dari 98,0 % dan tidak lebih dari 101,0 % C8H9NO2, dihitung terhadap zat anhidrat. Parasetamol merupakan serbuk hablur putih, tidak berbau, dengan rasa sedikit pahit (Anonima, 1995). Rumus bangun parasetamol dapat dilihat pada gambar 1.
OH HN
C O
CH3
Gambar 1. Struktur Parasetamol (Anonima, 1995)
Satu bagian parasetamol larut dalam 70 bagian air, 7-10 bagian etanol dan 13 bagian aseton, agak sukar larut dalam kloroform, praktis tidak larut dalam eter (Clarke, 1986). Larut dalam natrium hidroksida 1 N (Anonima, 1995).
serapan jenis adalah serapan dari larutan 1% zat terlarut dalam sel dengan ketebalan 1 cm (Anonima, 1995).
Parasetamol diindikasikan untuk sakit kepala, nyeri muskoloskeletal sementara, dismenore dan demam. Parasetamol tidak memiliki aktivitas antiinflamasi yang berarti dan kurang mengiritasi lambung dibanding dengan asetosal (Anonimb, 2000).
B. Natrium Fenobarbital
Natrium fenobarbital dengan BM 254,22 mengandung tidak kurang dari 98 % dan tidak lebih dari 101,0 % C12H11N2NaO3, dihitung terhadap zat yang telah dikeringkan (Anonima, 1995). Rumus bangun natrium fenobarbital dapat dilihat pada gambar 2.
Gambar 2. Struktur Natrium Fenobarbital (Anonima, 1995)
Sangat mudah larut dalam air, larut dalam etanol, praktis tidak larut dalam eter dan dalam kloroform (Anonima, 1995).
Asam fenobarbiturat memiliki nilai pKa 7,4. Serapan maksimumnya pada daerah ultraviolet dalam NaOH pH 13 adalah 254 nm (A 1%, 1 cm = 342) (Clarke, 1986).
Fenobarbital diindikasikan untuk mengobati semua jenis epilepsi kecuali petit mal. Efek sampingnya mengantuk, depresi mental, resah dan bingung (Anonimb, 2000).
C. Buffer
Buffer merupakan larutan yang dapat mempertahankan pH saat sejumlah kecil asam atau basa ditambahkan, atau jika larutan diencerkan. Larutan buffer merupakan campuran antara asam lemah dan basa konjugasinya atau basa lemah dengan asam konjugasinya dalam perbandingan atau konsentrasi tertentu (Christian, 2004).
Mekanisme pendaparan campuran asam lemah dan garamnya dapat dijelaskan sebagai berikut. Nilai pH merupakan logaritma dari rasio antara garam dan asam :
mencapai kesetimbangan HA H+ + A - sesuai dengan asas Le Châtelier, di mana reaksi akan bergeser ke kiri. Karena perubahan rasio
[ ]
A−[ ]
HA kecil, maka perubahan pH yang terjadi juga kecil (Christian, 2004).Kapasitas, atau kemampuan buffer untuk mempertahankan pH saat larutan dimasuki asam/basa dengan pH berlainan, mencapai 100% saat pH buffer sama dengan pKa asamnya. Maka supaya fase gerak memiliki kontrol pH yang baik, range pH yang dapat digunakan adalah + 1 unit pH dari nilai pKa asam lemah. Untuk buffer fosfat kapasitas buffer yang paling baik adalah pada pH 2,1 + 1; pH 7,2 + 1; dan pH 12,3 + 1 (Heyrman dan Henry, 2006).
D. Analisis kualitatif dan kuantitatif
Dua langkah utama yang dilakukan dalam analisis adalah identifikasi dan estimasi komponen-komponen suatu senyawa. Langkah identifikasi dikenal sebagai analisis kualitatif sedangkan langkah estimasinya adalah analisis kuantitatif. Analisis kuantitatif dapat diklasifikasikan dengan dasar perbedaan metode analisis atau diklasifikasikan dengan dasar skala analisisnya (Khopkar, 1990).
lebih besar dari 0,100 gram, semimikro antara 0,100-0,001 gram, sedangkan yang kurang dari 0,001 gram adalah sampel mikro (Khopkar, 1990).
Konsentrasi analit merupakan hal yang penting karena kesulitan dalam metode analisis seringkali berkaitan dengan rentang konsentrasinya. Pada penentuan kadar rendah, derau dan alunan yang disebabkan oleh pencemaran-pelarut, naik-turunnya suhu maupun keragaman aliran dapat mengurangi ketepatan dibanding dengan penetapan kadar tinggi (Johnson dan Stevenson, 1978).
Metode yang paling umum digunakan untuk menetapkan konsentrasi suatu senyawa dalam suatu sampel adalah dengan menggunakan kurva kalibrasi menggunakan baku eksternal. Disebut sebagai baku eksternal karena disiapkan dan dianalisis secara terpisah denga n senyawa yang ada dalam sampel. Selanjutnya, sampel yang akan ditetapkan konsentrasinya dianalisis dengan cara yang sama. Konsentrasi senyawa kemudian ditentukan dengan metode grafik dari kurva kalibrasi secara numerik (Rohman dan Gandjar, 2007).
E. Kromatografi Cair Kinerja Tinggi (KCKT)
1. Definisi dan instrumentasi KCKT
dan tetap dibiarkan dalam fase diam kemudian ditentukan untuk analisis (Mulja dan Suharman, 1995).
Kemajuan dalam teknologi kolom, sistem pompa tekanan tinggi dan detektor yang sensitif telah menyebabkan perubahan kromatografi kolom cair menjadi suatu sistem pemisahan dengan kecepatan dan efisiensi yang tinggi. Metode ini dikenal sebagai kromatografi cair kinerja tinggi (Anonima, 1995). Kromatografi Cair Kinerja Tinggi (KCKT) merupakan salah satu metode kromatografi cair yang fase geraknya dialirkan secara cepat dengan bantuan tekanan dan hasilnya dideteksi dengan instrument. Tidak seperti kromatografi gas, KCKT tidak dibatasi oleh volatilitas analit atau ketahanan analit terhadap panas. KCKT memiliki fase diam yang lebih banyak jenisnya sehingga memungkinkan lebih banyak interaksi spesifik untuk terjadinya pemisahan senyawa (Willard, Merrit, Dean, dan Settle, 1988).
Gambar 3. Peralatan KCKT (Kazakevich dan Nair, 1996)
Tiga variabel utama yang harus diperhatikan untuk proses pemisahan dan analisis menggunakan KCKT, yaitu :
a. Fase gerak
Pemisahan dengan fase gerak tunggal disebut elusi isokratik, sedangkan elusi gradien menggunakan dua fase gerak dengan berbagai perubahan komposisi. Suatu KCKT yang baik seharusnya mempunyai lebih dari dua penampung fase gerak. Fase gerak dialirkan ke botol penyampur pada berbagai laju aliran. Sebagian besar pompa KCKT mempunyai keluaran tekanan 70-400 atm, dan mampu menghasilkan aliran sampai 20 ml/menit. Sampel dimasukkan dalam sistem injeksi dengan penyuntik hiperdermik. Sampel sejumlah 2-100 µl dapat ditampung dalam sistem injeksinya (Khopkar, 1990).
memungkinkan untuk memperoleh kembali analit dengan mudah (jika diperlukan), tidak mudah terbakar dan toksisitasnya rendah, memiliki harga yang wajar (Skoog, Holler, dan Nieman, 1985). Fase gerak KCKT juga harus bebas dari gas yang terlarut karena dapat mempengaruhi respon detektor sehingga memuculkan sinyal palsu dan akan mempengaruhi kolom (Gritter, Bobbit, Schwarting, 1985). Maka peralatan
degassing diperlukan untuk menghilangkan gas yang terlarut di dalam fase gerak (Dean, 1995).
Fase gerak yang paling sering digunakan untuk pemisahan dengan fase terbalik adalah campuran larutan buffer dengan metanol atau campuran air dengan asetonitril (Rohman dan Gandjar, 2007).
Variasi retensi analit untuk pemisahan yang optimum dicapai dengan mengubah komposisi fase gerak. Snyder mendefinisikan parameter solvent strength, eo, sebagai energi adsorbsi per unit area dari adsorbent. Kenaikan nilai eo berbanding lurus dengan kenaikan nilai log k’. Semakin besar solvent strength maka kekuatan fase gerak untuk mengelusi semakin besar dan menyebabkan semakin kecilnya nilai k’ (faktor pemisahan) untuk kurva analit. Dengan demikian, fase gerak dapat dipilih dengan mencocokkan polaritas relatif dari fase gerak dengan komponen sampel (Willard, Merrit, Dean, dan Settle, 1988).
n
dengan F merupakan fraksi pelarut dalam campuran dan n adalah jenis pelarut yang digunakan (Skoog et al., 1985).
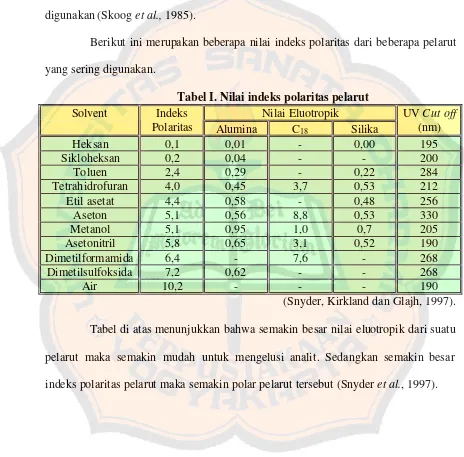
Berikut ini merupakan beberapa nilai indeks polaritas dari beberapa pelarut yang sering digunakan.
Tabel I. Nilai indeks polaritas pelarut
Nilai Eluotropik Solvent Indeks
Polaritas Alumina C18 Silika
UV Cut off
b. Fase diam
Kolom pada KCKT dapat berupa gelas atau baja tidak berkarat. Kolom gelas dapat menahan tekanan sampai 50 atm. Panjang kolom bervariasi antara 15-150 cm. pengisi kolom biasanya adalah silika gel, alumina dan elit (Khopkar, 1990).
Diameter kolom dibuat 3-5 mm dengan tujuan supaya kepekaannya lebih teliti, menghemat fase gerak, memperluas kemampuan detektor, dan mengurangi jumlah sampel yang dianalisis. Untuk mendapatkan fase yang nonpolar silika gel direaksikan dengan klorosilan Cl-Si-(R)n (Mulja dan Suharman, 1995). Oktadesil silika (ODS) merupakan fase diam yang paling banyak dipakai karena mampu memisahkan senyawa-senyawa dengan kepolaran yang rendah, sedang, maupun tinggi (Rohman dan Gandjar, 2007).
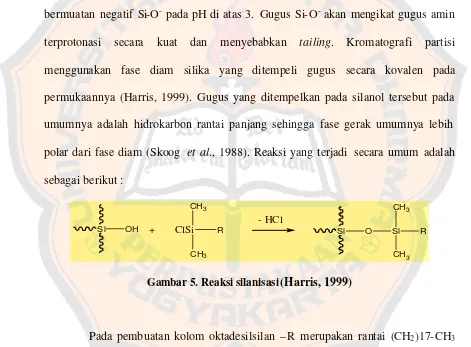
Analit yang polar, terutama yang bersifat basa atau memiliki gugus amin akan memberikan puncak yang mengekor (tailing peak) pada penggunaan fase diam silika fase terikat. Hal ini disebabkan oleh adanya interaksi adsorbsi antara gugus amin pada analit dengan residu silanol dan pengotor logam yang terdapat pada silika. Hal ini dapat diatasi dengan end-capping yakni suatu proses menutup residu silano l dengan gugus trimetilsilil dan menggunakan silika dengan kemurnian tinggi (kandungan logam < 1 ppm) (Rohman dan Gandjar, 2007).
c. Detektor
yang linier terhadap konsentrasi analit; dapat bekerja pada temperatur kamar sampai 400oC; tidak terpengaruh oleh perubahan temperatur dan kecepatan fase gerak; mudah didapat dan mudah dioperasikan; selektif terhadap berbagai macam analit di dalam fase gerak; tidak merusak analit; dapat menghilangkan ”zone broadening” dengan adanya pengaruh minimal internal volume (Mulja dan Suharman, 1995).
Detektor UV umumnya digunakan untuk analisis bahan organik bergugus fungsi (Khopkar, 1990). Detektor ini didasarkan pada adanya penyerapan radiasi ultraviolet oleh spesies analit yang mempunyai struktur atau gugus kromoforik. Detektor dengan panjang gelombang yang bervariasi lebih berguna karena seorang analis dapat memilih panjang gelombang dengan sensitifitas yang paling tinggi (Rohman dan Gandjar, 2007). Diagram skematik detektor UV tampak pada gambar di bawah ini.
Gambar 4. Detektor ultraviolet untuk KCKT (Skoog et al., 1985).
2. Pembagian jenis kromatografi
gerak dengan memanfaatkan perbedaan kecil sifat-sifat fisik komponen-komponen yang yang hendak dipisahkan (Mulja dan Suharman, 1995).
Kromatografi dapat dibagi menjadi lima jenis berdasarkan mekanisme pemisahannya yaitu kromatografi partisi, kromatografi adsorbsi, kromatografi pertukaran ion, kromatografi pasangan ion, kromatografi eksklusi dan kromatografi afinitas (Harris, 1999).
3. Kromatografi Partisi
Prinsip kromatografi partisi didasarkan pada partisi analit di antara dua fase yang tidak saling campur, karena adanya perbedaan koefisien distribusi dari masing-masing senyawa (Johnson dan Stevenson, 1978). Sebagai fase gerak adalah campuran metanol atau asetonitril dengan air atau dengan larutan buffer. Untuk analit yang bersifat asam atau basa lemah, peranan pH sangat penting karena jika pH fase gerak tidak diatur maka analit akan mengalami ionisasi. Terbentuknya spesies yang terionisasi ini menyebabkan ikatannya dengan fase diam menjadi lemah dibanding jika analit dalam bentuk spesies yang tidak terionisasi, karena spesies yang terionisasi akan terelusi lebih cepat (Rohman dan Gandjar, 2007).
m s
C C
D= (3)
Penggunaan temperatur kolom hanya beberapa derajat di bawah temperatur kamar akan meningkatkan reprodusibilitas waktu retensi dan meningkatkan presisi analisis kuantitatif. Permukaan silika pada kolom memiliki gugus silanol (Si-OH) sampai 8 µmol per meter persegi. Gugus silanol akan mengalami disosiasi menjadi bermuatan negatif Si-O- pada pH di atas 3. Gugus Si-O- akan mengikat gugus amin terprotonasi secara kuat dan menyebabkan tailing. Kromatografi partisi menggunakan fase diam silika yang ditempeli gugus secara kovalen pada permukaannya (Harris, 1999). Gugus yang ditempelkan pada silanol tersebut pada umumnya adalah hidrokarbon rantai panjang sehingga fase gerak umumnya lebih polar dari fase diam (Skoog et al., 1988). Reaksi yang terjadi secara umum adalah sebagai berikut :
Si OH + ClSi
Gambar 5. Reaksi silanisasi (Harris, 1999)
4. Pemisahan puncak dalam kromatografi
a. Efisiensi kolom
Berdasarkan teori lempeng, jumlah lempeng (N) yang didasarkan pada konsep lempeng teoritis pada distilasi kolom digunakan sebagai ukuran efisiensi kolom. N didefinisikan sebagai berikut.
2
W merupakan lebar setengah puncak kromatogram (Rohman dan Gandjar, 2007).
Suatu ukuran alternatif yang tergantung pada panjang kolom kromatografi adalah tinggi lempeng (H) atau yang biasa disebut dengan tinggi setara pelat teori (HETP = Height Equivalent Theoritical Plate). Hubungan antara HETP dan jumlah lempeng (N) serta panjang kolom (L) dapat dirumuskan dengan :
N L
H = (5)
Kolom yang memberikan jumlah lempeng (N) yang besar dan nilai HETP yang kecil akan mampu memisahkan kompone n-komponen dalam suatu campuran, yang berarti efisiens i kolom adalah besar (Rohman dan Gandjar, 2007).
transfer massa tidak seimbang. Sedangkan parameter-parameter yang menentukan berlangsungnya proses-proses tersebut adalah : laju aliran, ukuran partikel, laju difusi dan ketebalan stasioner. Van Deemter menghubungkan ketiga proses di atas dengan efisiensi kolom dalam suatu persamaan. Menurut Van Deemter hubungan antara laju aliran (µ) dengan tinggi piringan dapat dinyatakan dengan : H = A +
µ B
+ C. µ
(Khopkar, 1990).
Persamaan Van Deemter dapat juga dituliskan sebagai berikut.
(
)
µDi mana ? = tetapan ukuran ketidakteraturan kemasan dp = diameter rata-rata partikel penyangga DM = kedifusian analit dalam fase gerak k’ = faktor kapasitas
µ = kecepatan alir
? = faktor koreksi kelikuan saluran dalam kolom
setimbang. Hubungan antara diameter partikel rata-rata dengan difusi Eddy (A) adalah A = 2?dp. Dimana ? adalah faktor penjejalan kolom. Sedangkan difusi longitudinal (B) ditimbulkan sebagai akibat dari kecenderungan molekul untuk berpindah dari bagian tengah penampang piringan kolom yang konsentrasinya lebih tinggi, ke bagian tepi piringan yang konsentrasinya lebih rendah. Besarnya difusi longitudinal adalah B = 2?DM, dimana ? adalah faktor yang merupakan ukuran rentangan suatu molekul bebas untuk berdifusi, sedangkan DM adalah koefisien difusi zat terlarut dalam fase bergerak. Transfer massa tidak setimbang (C) akan melebarkan puncak akibat gerakan fase bergerak yang tinggi (Khopkar, 1990).
b. Waktu tambat (tR) dan resolusi
Waktu tambat atau waktu retensi (retention time) adalah selang waktu yang diperlukan oleh analit mulai saat injeksi sampai keluar dari kolom dan sinyalnya ditangkap detektor, dinyatakan sebagai tR (Mulja dan Suharman, 1995).
Di samping waktu tambat untuk analit, dikenal pula waktu tambat untuk pelarut pengembang atau pengembang campur yang dinyatakan sebagai tM (Mulja dan Suharman, 1995).
yang berarti analit akan lebih lama tinggal di dalam fase gerak dan memiliki waktu retensi lebih cepat (Mulja dan Suharman, 1995).
Faktor resolusi (R) adalah ukuran pemisahan dari dua puncak berdekatan yang dapat diukur dengan persamaan :
2
Harga tR1 dan tR2 merupakan waktu retensi senyawa yang diukur pada titik maksimum puncak, harga w1 dan w2 merupakan lebar alas puncak (Johnson dan Stevenson, 1978). Untuk pemisahan yang baik R harus > 1,5 karena berarti pemisahan kedua senyawa > 99,7% (Sastrohamidjojoa, 2002).
F. Spektofotometri Ultraviolet
Teknik spektroskopik merupakan salah satu teknik analisis fisiko-kimia yang mengamati interaksi atom atau molekul dengan suatu radiasi elektromagnetik (REM). Spektrofotometri ultraviolet adalah anggota teknik analisis spektroskopik yang menggunakan sumber radiasi elektromanetik ultraviolet dekat (190-380 nm) dengan menggunakan instrumen spektrofotometer. Radiasi ultraviolet jauh (100-190 nm) tidak dipakai, sebab pada daerah tersebut REM diabsorbsi oleh udara (Mulja dan Suharman, 1995).
yang dikenal sebagai orbital elektron antiikatan. Ada empat tipe transisi elektronik yang mungkin terjadi yaitu σ→σ*, π→π*, n →π*, dan n →σ*. Eksitasi elektron (σ→ σ*) memberikan energi yang terbesar dan terjadi pada daerah ultraviolet jauh yang diberikan oleh ikatan tungal, misalnya alkana. Eksitasi elektron (π → π*) diberikan oleh ikatan rangkap dua dan rangkap tiga, juga terjadi pada daerah ultraviolet jauh. Sedangkan eksitasi elektron (n → σ*) terjadi pada gugus karbonil yang terjadi pada ultraviolet jauh (Mulja dan Suharman, 1995).
Dalam praktek spektrofotometri ultraviolet digunakan terbatas pada sistem terkonjugasi. Meskipun demikian terdapat keuntungan yang selektif dari serapan ultraviolet. Yaitu gugus-gugus karakteristik dapat dikenal dalam molekul yang sangat kompleks (Sastrohamidjojob, 2002).
Suatu molekul dapat menyerap radiasi elektromagnetik jika memiliki kromofor, yaitu gugus tak jenuh kovalen sebaga i penyerap dalam molekul. Pada senyawa organik dikenal pula gugus auksokrom, yaitu gugus yang tidak menyerap radiasi namun bila terikat bersama kromofor dapat meningkatkan penyerapan oleh kromofor atau mengubah panjang gelombang serapan maksimum (Christian, 2004). Auksokrom merupakan heteroatom yang langsung terikat pada kromofor, misalnya gugus -OCH3, -Cl, -OH dan -NH2 (Sastrohamidjojob, 2002).
akan menyebabkan penurunan tingkat energi p* dan memberikan pengurangan karakter antiikatan. Sebagai konsekuensinya, panjang gelombang molekul ya ng mempunyai ikatan rangkap terkonjugasi akan mengalami pergeseran batokromik (Rohman dan Gandjar, 2007).
b
Io = intensitas radiasi yang datang It = intensitas radiasi yang diteruskan e = daya serap molar (Liter.mol-1.cm-1) c = konsentrasi (mol.Liter-1)
b = tebal larutan (cm) A= serapan
(Mulja dan Suharman, 1995).
G. Kesahihan Metode Analisis Instrumental
Kesahihan metode analisis merupakan suatu prosedur untuk membuktikan bahwa metode analisis yang digunakan dapat memberikan hasil seperti yang diharapkan, dengan kecermatan dan ketelitian yang memadai sesuai dengan standar yang berlaku (Mulja dan Suharman, 1995).
1. Akurasi
Akurasi suatu metode merupakan keterdekatan nilai pengukuran dengan nilai sebenarnya dari analit dalam sampel (Mulja dan Hanwar, 2003). Penentuan akurasi metode analisis biasanya dinyatakan dengan persen perolehan kembali terhadap analit yang kadarnya telah diketahui dengan pasti (Mulja dan Suharman, 1995).
Persen perolehan kembali yang dapat diterima bergantung pada matriks analit, prosedur pengolahan analit dan konsentrasi analit (Anonimc, 2004). Rentang rata-rata perolehan kembali ya ng dapat diterima, harus memenuhi ketentuan dalam tabel II berikut.
Tabel II. Rentang rata-rata persen perolehan kembali yang dapat diterima (Anonimc, 2004)
Kandungan zat aktif (%b/v)
Rentang rata-rata persen perolehan kembali yang
diterima
Hanwar, 2003). Suatu metode dinyatakan memenuhi syarat presisi jika memenuhi kriteria pada tabel III berikut.
Tabel III. Presisi yang dapat diterima (Anonimc, 2004)
Kandungan zat aktif (%b/v)
4. Limit kuantifikasi (LOQ) dan limit deteksi (LOD)
Sensitivitas suatu metode analisis harus diketahui batas kadar terkecil yang masih dapat ditentukan untuk analisis kuantitatif ya ng dikenal sebagai LOD (Limit of Detection). LOD merupakan suatu parameter untuk penetuan suatu analit dengan kadar yang terkecil tetapi masih memberikan tanggap detektor yang berbeda dengan pembanding (tanpa analit) (Mulja dan Suharman, 1995). Batas deteksi yang umum digunakan dalam kimia analisis adalah bahwa batas deteksi merupakan kadar analit yang memberian respon sebesar respon blangko (Yb) ditambah dengan 3 simpangan baku blangko (3 Sb) (Rohman dan Gandjar, 2007).
Sedangkan LOQ (Limit of Quantification) adalah kadar terkecil dari suatu analit yang masih dapat dianalisis dengan hasil yang tetap memenuhi syarat akurasi dan presisi (Mulja dan Suharman, 1995). LOQ ditentukan dengan rasio signal to noise 10 : 1 (Rohman dan Gandjar, 2007).
H. Keterangan Empiris
28
METODE PENELITIAN
A. Jenis Penelitian
Jenis penelitian dalam “O ptimasi Pemisahan dan Penetapan Kadar Campuran Parasetamo l dan Natrium Fenobarbital dengan Metode Kromatografi Cair Kinerja Tinggi Fase Terbalik” adalah penelitian noneksperimental deskriptif.
B. Definisi Operasional
1. Campuran parasetamol dan natrium fenobarbital adalah campuran antara parasetamol dan natrium fenobarbital dengan perbandingan 11 : 1
2. Sistem Kromatografi Cair Kinerja Tinggi (KCKT) fase terbalik yang digunakan adalah seperangkat alat KCKT dengan fase diam kolom reversed phase C18 dengan fase gerak campuran metanol : buffer fosfat pH 3,2 dengan perbandingan optimum 3. Kadar parasetamol dan natrium fenobarbital dalam sampel ditetapkan dalam satuan mg/ml
4. Parameter kesahihan metode analisis yang digunakan yaitu akurasi, presisi, LOD, LOQ dan linearitas
C. Bahan Penelitian
dari destilasi aquadest (laboratorium Kimia Organik, Fakultas Farmasi Universitas Sanata Dharma), dinatrium hidroksi fosfat, dan asam asetat glassial p.a (E.Merck).
D. Alat Penelitian
1. Spektrofotometer UV/Vis merk Genessis UV 10. 2. Sistem KCKT, yang terdiri dari :
a. Pompa merk Shimadzu model LC-10 AD No.C20293309457 J2.
b. Detektor UV-Vis merk Shimadzu model SPD-10 AV No. C20343502697 KG.
c. Injektor jenis katup suntik, model 7725i.
d. Kolom oktadesilsilan, merk Waters BondapacT M C18 (panjang 30 cm; P61271B02 P/N 27324; diameter 5-10 mm).
e. CBM-101 merk Shimadzu, Cat No.223-03750-94, serial No. C50363502311 SA.
f. Seperangkat komputer merk COMPAQ.
3. Syringe No. 046-00038-01, jarum syringe No. 228-18216-91. 4. Alat degassing merk Retsch, Tipe T 460 No. V935922013 EY. 5. Vakum merk Gast, Model DOA-P104-BN.
6. Penyaring Whatman
b. Anorganic Solvent Membrane Filter dengan ukuran pori 0,45 µm, diameter 47 mm, Cat. No. 7184 004.
7. Membrane Filter Holder merk Whatman dengan kapasitas 300 ml, Cat. No. 1960 004.
8. Potensiometer 9. Penyaring Milipore
10.Mikropipet Socorex ukuran 200-1000 µl dan 100-500 µl
11.Neraca analitik merk Scaltec SBC 22 max 60/210 g; d = 0,01/0,1 mg; e = 1 mg 12.Seperangkat alat-alat gelas merk Pyrex.
E. Tata Cara Penelitian
1. Pembuatan fase gerak
Fase gerak yang digunakan dalam penelitian menggunakan menggunakan campuran metanol dan buffer fosfat pH 3,2 dengan perbandingan 30 : 70 dan 10 : 90. Buffer dibuat dengan melarutkan lebih kurang 7,5 gram Na2HPO4 dalam 500,0 mL aquabidest kemudian pH dibuat sampai 3,20 dengan asam asetat glassial p.a menggunakan alat potensiometer kemudian ditambah aquabidest sampai 1000,0 ml.
2. Pembuatan larutan baku parasetamol dan natrium fenobarbital
a. Pembuatan larutan stok parasetamol. Menimbang seksama lebih kurang 50 mg serbuk parasetamol dan dilarutkan dengan 3,0 ml metanol p.a kemudian ditambah buffer fosfat pH 3,2 dalam labu ukur 10,00 ml sampai tanda.
b. Pembuatan larutan intermediet parasetamol. Mengambil 5,0 ml larutan stok dimasukkan dalam labu ukur 10,00 ml kemudian diencerkan dengan buffer fosfat pH 3,2 sampai tanda.
c. Pembuatan larutan stok natrium fenobarbital. Menimbang seksama lebih kurang 55 mg serbuk natrium fenobarbital dan dilarutkan dengan 5,0 ml metanol p.a kemudian ditambah dengan buffer fosfat pH 3,2 dalam labu ukur 25,0 ml sampai tanda.
d. Pembuatan seri kurva baku parasetamol. Memipet sebanyak 0,280 ml dan 0,560 ml larutan intermediet parasetamol, dan memipet sebanyak 0,420; 0,560; dan 0,700 ml larutan stok parasetamol. Masing- masing larutan tersebut kemudian diencerkan dengan buffer fosfat pH 3,2 dalam labu takar 10,00 ml sampai tanda. Hingga diperoleh lima seri larutan baku parasetamol (0,07; 0,14; 0,21; 0,28 dan 0,35 mg/ml). Disaring dengan milipore dan didegassing selama 15 menit. Replikasi dilakukan sebanyak 3 kali.
diperoleh lima seri larutan baku natrium fenobarbital (1; 1,25; 1,5; 1,75 dan 2,2 mg/ml). Disaring dengan milipore dan didegassing selama 15 menit. Replikasi dilakukan sebanyak 3 kali.
f. Pembuatan campuran parasetamol dan natrium fenobarbital untuk penetapan kadar. Menimbang seksama lebih kurang 15 mg natrium fenobarbital dan dicampur homogen dengan 166,7 mg parasetamol (ditimbang seksama lebih kurang), dilarutkan dengan 3 ml metanol kemudian ditambah buffer fosfat pH 3,2 dalam labu takar 10,00 ml sampai tanda.
3. Pengamatan panjang gelombang pengamatan parasetamol dan natrium
fenobarbital dengan spektrofotometer UV
Menimbang seksama lebih kurang 10 mg parasetamol, dilarutkan dengan 3,0 ml metanol kemudian ditambah dengan buffer fosfat pH 3,2 dalam labu takar 10,00 ml sampai tanda. Memipet sebanyak 50,0; 70,0 dan 90,0 µl larutan stok kemudian diencerkan dengan buffer fosfat pH 3,2 dalam labu takar 10,00 ml sampai tanda. Larutan ini dibaca absorbansinya pada panjang gelombang 200-300 nm dengan spektrofotometer UV. Kemudian diperoleh kurva hubungan panjang gelombang dan absorbansi parasetamol.
tanda. Larutan ini dibaca absorbansinya pada panjang gelombang 200-300 nm dengan spektrofotometer UV. Kemudian diperoleh kurva hubungan panjang gelombang dan absorbansi natrium fenobarbital.
Selanjutnya dari kurva parasetamol dan natrium fenobarbital tersebut, spektra ditumpangtindihkan untuk mengetahui panjang gelombang pengamatan pada deteksi dengan KCKT fase terbalik.
4. Optimasi pemisahan parasetamol dan natrium fenobarbital dalam
campuran parasetamol dan natrium fenobarbital dengan perbandingan 11 : 1
dengan KCKT fase terbalik
a. Pengamatan waktu retensi parasetamol. Mengambil 0,420 ml larutan stok parasetamol kemudian diencerkan dengan buffer fosfat pH 3,2 dalam labu takar 10,00 ml sampai tanda, hingga mencapai konsentrasi 0,21 mg/ml. Disaring dengan
milipore dan didegassing selama 15 menit. Kemudian sebanyak 50,0 µl larutan disuntikkan ke dalam sistem KCKT dengan kolom ODS (5 mm x 30 cm). Optimasi dilakukan pada panjang gelombang pengamatan. Perbandingan fase gerak dan kecepatan alir pada sistem KCKT diubah-ubah hingga waktu retensi parasetamol dan natrium fenobarbital berbeda jauh. Perbandingan fase gerak buffer fosfat pH 3,2 : metanol yang digunakan adalah 70 : 30 dan 90 : 10 (point 1). Kecepatan alir yang digunakan adalah 1 dan 1,5 ml/menit.
3,2 dalam labu takar 10,00 ml sampai tanda, hingga mencapai konsentrasi 1,5 mg/ml. Disaring dengan milipore dan didegassing selama 15 menit. Kemudian sebanyak 50,0 µl larutan disuntikkan ke dalam sistem KCKT dengan kolom ODS (5 mm x 30 cm). Optimasi dilakukan pada panjang gelombang pengamatan. Perbandingan fase gerak dan kecepatan alir pada sistem KCKT diubah- ubah hingga waktu retensi natrium fenobarbital berbeda jauh dengan parasetamol. Perbandingan fase gerak buffer fosfat pH 3,2 : metanol yang digunakan adalah 70 : 30 dan 90 : 10 (point 1). Kecepatan alir yang digunakan adalah 1 dan 1,5 ml/menit. Kemudian berdasarkan kromatogram 2 senyawa, harga tR dari masing- masing kromatogram dibandingkan untuk melihat pemisahan puncak parasetamol dan natrium fenobarbital. Replikasi dilakukan sebanyak 3 kali.
10,00 ml. Disaring dengan milipore dan didegassing selama 15 menit. Kemudian sebanyak 50,0 µl larutan disuntikkan ke dalam sistem KCKT dengan kolom ODS (5 mm x 30 cm). Menggunakan fase gerak dan kecepatan alir hasil optimasi, kemudian mengamati kromatogram parasetamol yang terjadi pada panjang gelombang pengamatan. Melakukan penghitungan nilai resolusi dari pemisahan campuran parasetamol dan natrium fenobarbital.
5. Optimasi penetapan kadar parasetamol dan natrium fenobarbital dalam
campuran parasetamol dan natrium fenobarbital dengan perbandingan 11 : 1
dengan KCKT fase terbalik
a. Pembuatan persamaan kurva baku parasetamol. Sebanyak 50,0 µl masing-masing seri larutan baku disuntikkan ke dalam sistem KCKT dengan kolom ODS (5 mm x 30 cm), menggunakan perbandingan fase gerak dan kecepatan alir yang sudah dioptimasi. Replikasi dilakukan sebanyak 3 kali dan dipilih persamaan kurva baku untuk parasetamol yang paling baik.
b. Pembuatan persamaan kurva baku natrium fenobarbital. Sebanyak 50,0 µl masing- masing seri larutan baku disuntikkan ke dalam sistem KCKT dengan kolom ODS (5 mm x 30 cm), menggunakan perbandingan fase gerak dan kecepatan alir yang sudah dioptimasi. Replikasi dilakukan sebanyak 3 kali dan dipilih persamaan kurva baku untuk natrium fenobarbital ya ng paling baik.
dan natrium fenobarbital dengan perbandingan 11 : 1 untuk penetapan kadar (point 2.e). Disaring dengan milipore dan didegassing selama 15 menit. Kemudian sebanyak 50,0 µl larutan disuntikkan ke dalam sistem KCKT dengan kolom ODS (5 mm x 30 cm) menggunakan fase gerak dan kecepatan alir hasil optimasi. Menghitung kadar natrium fenobarbital yang ada dalam campuran dengan menggunakan persamaan kurva baku. Mengambil sebanyak 0,125 ml larutan campuran tersebut kemudian dilarutkan dengan buffer fosfat pH 3,2 dalam labu takar 10 ml. Disaring dengan milipore dan didegassing selama 15 menit. Kemudian sebanyak 50,0 µl larutan disuntikkan ke dalam sistem KCKT dengan kolom ODS (5 mm x 30 cm) menggunakan fase gerak dan kecepatan alir hasil optimasi. Menghitung kadar parasetamol yang ada dalam campuran dengan menggunakan persamaan kurva baku. Replikasi dilakukan sebanyak 6 kali.
6. Validasi metode penetapan kadar parasetamol dan natrium fenobarbital
dalam campuran parasetamol dan natrium fenobarbital dengan perbandingan
11 : 1 dengan KCKT fase terbalik
a. Akurasi
Akurasi metode analisis dinyatakan dengan recovery.
100%
Jika nilai % recovery dari 6 kali replikasi parasetamol berada pada rentang 90-110 %, dan nilai % recovery dari 6 kali replikasi natrium fenobarbital berada pada rentang 80-120 % maka metode ini dinilai memiliki akurasi yang baik (Anonimc, 2004).
b. Presisi
Presisi metode analisis dinyatakan dengan % koefisien variasi (KV).
100%
Jika nilai % koefisien variasi parasetamol kurang dari sama dengan 5 %, dan % koefisien variasi natrium fenobarbital kurang dari sama dengan 10 %, maka metode ini dinilai memiliki presisi yang baik (Anonimc, 2004).
c. Limit of Detection (LOD)
ditentukan dengan mengukur Ys yaitu kadar analit yang memberikan respon sebesar respon blangko (Yb) ditambah dengan 3 simpangan baku blangko (3Sb). Kemudian LOD merupakan nilai x dengan memasukkan Ys sebagai AUC.
d. Limit of Quantification (LOQ)
Jumlah terkecil analit dalam sampel yang dapat terkuantifikasi dan memenuhi standar presisi di bawah kondisi penelitian yang ditentukan, dapat dinyatakan dalam LOQ. Nilainya ditentukan dengan mengukur Ys yaitu kadar analit yang memberikan respon sebesar respon blangko (Yb) ditambah dengan 10 simpangan baku blangko (10Sb). Kemudian LOQ merupakan nilai x dengan memasukkan Ys sebagai AUC.
F. Analisis Hasil
1. Luas area kromatogram (AUC = Area Under The Curve) dari berbagai seri baku digunakan untuk membuat kurva baku menggunakan persamaan regresi linear y = bx + a yang merupakan hubungan antara kadar dengan luas area yang dihasilkan.
39
BAB IV
HASIL DAN PEMBAHASAN
A. Penyiapan Fase Gerak
Fase gerak yang digunakan pada penelitian ini mengacu pada metode Lunn dan Schmuff (1997) yang menggunakan metode KCKT untuk analisis farmasetik parasetamol dan natrium fenobarbital dalam sampel biologis. Fase gerak yang digunakan pada metode tersebut adalah campuran asetonitril dan buffer fosfat pH 3,2 dengan menggunakan sistem elusi gradien perbandingan 5 : 95 sampai 22 : 78 selama 24 menit. Pada penelitian ini, fase gerak asetonitril diganti dengan metanol dengan perbandingan metanol dan buffer fosfat pH 3,2 yang akan dioptimasi adalah 10 : 90 dan 30 : 70. Komposisi pelarut organik pada penelitian ini lebih banyak dibanding metode Lunn dan Schmuff (1997) supaya kepolaran campuran fase gerak mirip dengan kepolaran campuran fase gerak yang digunakan oleh Lunn dan Schmuff (1997), karena kepolaran metanol lebih rendah dibanding asetonitril. Sedangkan optimasi dilakukan untuk mengetahui komposisi mana yang optimum digunakan pada KCKT dengan sistem elusi isokratik.
memiliki tingkat keasaman yang cukup untuk mengubah natrium fenobarbital menjadi asam fenobarbiturat, sesuai dengan metode yang telah dilakukan Lunn dan Schmuff (1997).
Metanol digunakan sebagai salah satu campuran fase gerak pada penelitian ini karena kelarutan parasetamol dan natrium fenobarbital yang baik dalam metanol, selain itu metanol memiliki viskositas yang rendah 0,54 cP, sehingga penggunaan metanol dapat mengurangi tekanan pada kolom dan meningkatkan efisiensi kolom untuk memisahkan parasetamol dan natrium fenobarbital.
Campuran fase gerak metanol dan buffer fosfat pH 3,2 bersifat polar, sedangkan fase diam yang digunakan adalah kolom oktadesilsilan (C18) yang bersifat nonpolar sehingga sistem kromatografi yang digunakan adalah kromatografi partisi fase terbalik.
B. Pembuatan Larutan Baku
Larutan baku dibuat dalam konsentrasi tertentu dengan menggunakan pelarut campuran metanol p.a dan buffer fosfat pH 3,2. Pelarut tersebut memenuhi syarat pelarut yang dapat digunakan dalam sistem KCKT yaitu memiliki kemurnian yang tinggi, dapat bercampur dengan fase gerak serta mudah terelusi.
Larutan baku parasetamol dan natrium fenobarbital dibuat dalam 5 seri konsentrasi. Untuk parasetamol konsentrasinya 0,07 mg/ml, 0,14 mg/ml, 0,21 mg/ml, 0,28 mg/ml dan 0,35 mg/ml. Sedangkan untuk natrium fenobarbital konsentrasinya 1 mg/ml, 1,25 mg/ml, 1,5 mg/ml, 1,75 mg/ml dan 2,2 mg/ml.
Pemilihan seri larutan baku ini didasarkan pada perbandingan konsentrasi parasetamol dan natrium fenobarbital dalam sampel yaitu 11 : 1 (16,67 mg/ml : 1,5 mg/ml). Secara teoritis probabilitas transisi elektron parasetamol lebih besar dibanding asam fenobarbiturat. Dengan demikian parasetamol akan memiliki daya serap molar yang lebih besar dibanding natrium fenobarbital. Berdasarkan alasan tersebut maka dilakukan pengenceran sebanyak + 80 kali terhadap konsentrasi parasetamol 16,67 mg/ml dalam sampel hingga mencapai konsentrasi 0,21 mg/ml supaya respon absorbansinya tidak jauh berbeda dengan natrium fenobarbital.
Dengan demikian masing- masing konsentrasi sampel merupakan konsentrasi tengah dari kurva baku, hal ini bertujuan supaya persamaan kurva baku yang diperoleh nantinya dapat digunakan untuk menetapkan kadar sampel.
C. Optimasi Penentuan Panjang Gelombang Pengamatan Parasetamol dan
Natrium Fenobarbital dengan Spektrofotometer UV
natrium fenobarbital menggunakan spektrofotometer UV, yang bertujuan untuk mengetahui panjang gelombang di mana parasetamol dan natrium fenobarbital dalam pelarut metanol dan buffer fosfat pH 3,2 memberikan serapan yang maksimum saat dikenai sinar UV.
Penentuan ?maks dilakukan menggunakan 3 seri kadar dengan tujuan untuk meyakinkan hasil yang didapat benar-benar panjang gelombang serapan maksimum dari senyawa tersebut. Sehingga nantinya ?maks dapat digunakan untuk analisis kualitatif senyawa tersebut dalam pelarut metanol dan buffer fosfat pH 3,2, karena ?maks bersifat khas untuk suatu senyawa. Penggunaan 3 seri kadar juga diperlukan untuk memastikan kebenaran senyawa yang digunakan, dengan membandingkan panjang gelombang serapan maksimum yang diperoleh dengan panjang gelombang serapan maksimum dari literatur. Hal ini diperlukan karena parasetamol dan natrium fenobarbital yang digunakan memiliki kualitas working standard.
Pembacaan serapan dilakukan pada rentang panjang gelombang 200-300 nm karena berdasarkan literatur parasetamol dan natrium fenobarbital memiliki panjang gelombang serapan maksimum pada rentang tersebut.
auksokrom yang terikat langsung pada gugus kromofor. Gugus auksokrom merupakan suatu gugus yang memiliki pasangan elektron bebas pada orbital n yang kemudian dapat berinteraksi dengan elektron p pada kromofor. Denga n demikian, gugus auksokrom ini berperan dalam pergeseran panjang gelombang dan intensitas serapan maksimum dari parasetamol dan natrium fenobarbital. Gambar gugus kromofor dan auksokrom dari masing- masing senyawa dapat dilihat pada gambar berikut.
Gambar 6. Gugus kromofor dan auksokrom parasetamol Keterangan : --- = kromofor
Gambar 7. Gugus kromofor dan auksokrom natrium fenobarbital Keterangan : --- = kromofor
Dalam Farmakope Indonesia Edisi IV (1995) disebutkan bahwa pengujian panjang gelombang serapan maksimum mempunyai makna jika serapan maksimum tersebut tepat atau dalam batas 2 nm dari panjang gelombang yang ditentukan. Spektrum serapan yang dihasilkan oleh parasetamol dan natrium fenobarbital dapat dilihat pada gambar berikut.
Gambar 8. Spektrum serapan parasetamol (?maks = 245 nm)
Keterangan : A = konsentrasi 0,005 mg/ml; B = konsentrasi 0,007 mg/ml; C = konsentrasi 0,009 mg/ml
parasetamol karena memenuhi ketentuan yang disebutkan dalam Farmakope Indonesia Edisi IV.
Gambar 9. Spektrum serapan natrium fenobarbital (?maks = 236 nm)
Keterangan : A = konsentrasi 0,05 mg/ml; B = konsentrasi 0,07 mg/ml; C = konsentrasi 0,09 mg/ml
Pada gambar tersebut terlihat bahwa ketiga seri kadar natrium fenobarbital dalam pelarut metanol dan buffer fosfat pH 3,2 yang diuji memiliki panjang gelombang serapan maksimum 236 nm. Hal ini menunjukkan bahwa panjang gelombang 236 nm benar-benar merupakan panjang gelombang di mana natrium fenobarbital memberikan serapan yang maksimum.
Gambar 10. Gabungan spektrum serapan parasetamol (A) konsentrasi 0,009 mg/ml dan natrium fenobarbital (B) konsentrasi 0,09 mg/ml
Pada gambar 10 dapat terlihat bahwa pada perbandingan 1:10 natrium fenobarbital baru dapat memberikan absorbansi yang mirip dengan parasetamol. Sehingga berdasarkan spektrum tumpang tindih tersebut, panjang gelombang pengamatan yang dipilih adalah 236 nm yang merupakan panjang gelombang serapan maksimum dari natrium fenobarbital. Panjang gelombang ini dipilih supaya natrium fenobarbital dapat terdeteksi karena absorbansi natrium fenobarbital sangat kecil dibandingkan parasetamol. Selain itu pada gambar 10 dapat terlihat juga bahwa parasetamol dapat memberikan absorbansi pada panjang gelombang 236 nm.
D. Optimasi Pemisahan Parasetamol dan Natrium Fenobarbital dengan
KCKT Fase Terbalik
adalah campuran metanol dan buffer fosfat pH 3,2 yang bersifat polar. Dengan demikian, sampel yang bersifat lebih polar akan terelusi lebih dulu sedangkan sampel yang lebih nonpolar akan terelusi belakangan karena tertambat pada fase diam (Willard dkk, 1988).
Pada kromatografi fase terbalik, ionisasi analit harus diminimalkan supaya analit tidak terlalu polar dan tetap dapat berinteraksi dengan fase diam oktadesilsilan (C18) yang bersifat nonpolar. Maka pada penelitian ini digunakan buffer asam sebagai pelarut dan fase gerak, untuk mengubah garam natrium fenobarbital menjadi bentuk asamnya, supaya tidak mengalami ionisasi dan dapat berinteraksi dengan fase diam. Apabila digunakan aquabidest sebagai fase gerak, garam natrium fenobarbital akan larut dan berubah menjadi spesies ion yang tidak dapat berinteraksi dengan fase diam akibatnya kromatogram natrium fenobarbital tidak dapat diamati.
Dengan demikian untuk selanjutnya natrium fenobarbital akan disebut sebagai asam fenobarbiturat dalam penelitian ini, kecuali dalam perhitungan kurva baku dan sampel. Reaksi antara buffer fosfat pH 3,2 dengan natrium fenobarbital, membentuk asam fenobarbiturat (fenobarbital) adalah sebagai berikut.
+ NH
natrium fenobarbital asam fenobarbiturat
P O
asam fosfat natrium dihidroksi fosfat
Pemisahan pada KCKT dipengaruhi oleh interaksi suatu analit dengan fase diam dan fase gerak. Parasetamol dan asam fenobarbiturat yang terbentuk memiliki gugus polar dan nonpolar. Gugus polar akan berinteraksi dengan fase gerak melalui ikatan hidrogen, sedangkan gugus nonpolar akan berinteraksi dengan fase diam oktadesilsilan melalui ikatan van der Waals. Gambar gugus nonpolar dari parasetamol dan asam fenobarbiturat yang akan membentuk ikatan van der Waals dengan fase diam adalah sebagai berikut.
HO HN C
Gambar 12. Gugus nonpolar dari parasetamol (A) dan asam fenobarbiturat (B) yang berinteraksi dengan fase diam
Keterangan : = gugus nonpolar
Pada gambar di atas dapat terlihat bahwa asam fenobarbiturat memiliki gugus nonpolar yang lebih banyak dibanding parasetamol. Dengan demikian asam fenobarbiturat akan lebih tertahan pada fase diam dibandingkan parasetamol, sehingga waktu retensinya lebih lama. Waktu retensi (tR) merupakan waktu yang dibutuhkan suatu senyawa untuk keluar dari kolom.
disebutkan sebelumnya, pada penelitian ini perbandingan fase gerak buffer fosfat pH 3,2 : metanol yang dioptimasi adalah 90 : 10 dan 70 : 30, sedangkan kecepatan alir yang dioptimasi adalah 1 dan 1,5 ml/menit.
Gambar 13. Puncak parasetamol (a) dan asam fenobarbiturat (b) Keterangan : Fase gerak buffer fosfat pH 3,2 : metanol (70 : 30)
Gambar 14. Puncak parasetamol (a) dan asam fenobarbiturat (b) Keterangan : Fase gerak buffer fosfat pH 3,2 : metanol (70 : 30)
Gambar 15. Puncak parasetamol (a) dan asam fenobarbiturat (b) Keterangan : Fase gerak buffer fosfat pH 3,2 : metanol (90 : 10)
Gambar 16. Puncak parasetamol (a) dan asam fenobarbiturat (b) Keterangan : Fase gerak buffer fosfat pH 3,2 : metanol (90 : 10)
Berdasarkan kromatogram optimasi pemisahan tersebut, terlihat bahwa peak
parasetamol mengalami tailing yang disebabkan karena parasetamol memiliki gugus amin yang akan berinteraksi dengan gugus silanol di dalam kolom. Adanya interaksi ini dapat menyebabkan pengekoran pada kurva parasetamol, karena sebagian analit terlambat keluar dari kolom. Tailing juga terjadi pada kromatogram asam fenobarbiturat karena asam fenobarbiturat juga memiliki gugus amin. Tailing juga dapat terjadi karena parasetamol dan asam fenobarbituratmemiliki kelarutan rendah dalam fase gerak yang sebagian besar komposisinya adalah buffer fosfat pH 3,2. Hal ini menyebabkan analit yang sama-sama memiliki gugus nonpolar tertahan pada kolom lebih lama, baru kemudian larut pada fase gerak sedikit demi sedikit sehingga menyebabkan pengekoran kromatogram. Sedangkan munculnya double peak pada kromatogram parasetamol dan natrium fenobarbital disebabkan karena pelarut yang digunakan (buffer fosfat pH 3,2:metanol 70:30) memiliki kekuatan ionik yang lebih besar dibandingkan fase gerak (buffer fosfat pH 3,2:metanol 90:10).
Tabel IV. Resolusi pemisahan parasetamol 0,21 mg/ml dan asam fenobarbiturat 1,5 mg/ml pada KCKT
Buffer fosfat pH 3,2 : metanol
Kecepatan alir (ml/menit)
Rep Resolusi Keterangan
1 0,35
Berdasarkan data yang diperoleh, komposisi buffer fosfat pH 3,2 : metanol yang dipilih adalah komposisi 90 : 10 karena dapat memisahkan kromatogram parasetamol dan asam fenobarbiturat dengan resolusi yang baik sedangkan pada komposisi 70 : 30 parasetamol dan asam fenobarbiturat tidak dapat memisah.
fenobarbiturat memiliki waktu retensi yang lebih cepat, karena sebagian analit kemungkinan masih berada dalam bentuk garam/ ion yang lebih menyukai fase gerak, sehingga menyebabkan pemisahannya dengan parasetamol tidak sempurna.
Berdasarkan tabel IV, pada komposisi fase gerak 90 : 10, kedua kecepatan alir yang dioptimasi memberikan hasil yang memenuhi persyaratan resolusi > 1,5. Akan tetapi kecepatan alir yang dipilih adalah 1,5 ml/menit. Alasan pemilihan ini adalah karena kecepatan alir 1,5 ml/menit memiliki tingkat reprodusibilitas yang lebih baik daripada kecepatan alir 1 ml/menit, hal ini terlihat pada besarnya standar deviasi dari resolusi yang dihasilkan dari tiga kali replikasi. Selain itu, waktu retensi asam fenobarbiturat lebih pendek pada kecepatan alir 1,5 ml/menit dibanding kecepatan alir 1 ml/menit karena adanya tekanan yang lebih besar pada kolom. Hal ini sangat penting untuk efisiensi waktu kerja dalam analisis.
Dengan demikian, selanjutnya analisis campuran parasetamol dan natrium fenobarbital dengan metode KCKT menggunakan fase gerak metanol : buffer fosfat pH 3,2 dengan perbandingan 90 : 10 dan kecepatan alir 1,5 ml/menit.
E. Optimasi Penetapan Kadar Parasetamol dan Natrium Fenobarbital
dengan KCKT Fase Terbalik
koefisien korelasi (r), yang menunjukkan korelasi atau hubungan antara konsentrasi dengan AUC. Dari ketiga replikasi kurva baku yang diperoleh, kemudian dipilih satu kurva yang kemudian akan digunakan untuk analisis kuantitatif senyawa tersebut. Pemilihan kurva baku didasarkan pada nilai r- nya yang lebih besar dari r tabel untuk lima data dengan derajat bebas (db) = 3 yaitu sebesar 0,878 (taraf kepercayaan 95%).
Data persamaan kurva baku yang diperoleh dari parasetamol dan natrium fenobarbital dapat dilihat pada tabel-tabel berikut.
Tabel V. Data kurva baku parasetamol
Replikasi I Replikasi II Replikasi III
C
Tabel VI. Data kurva baku natrium fenobarbital
Replikasi I Replikasi II Replikasi III